
Efficient photocatalytic synthesis of benzylhydrazine†
Yuhui Zhang‡
abc,
Fei Zeng‡abc,
Yangyang Liabc,
Meiqi Zhouabc,
Jiafan Wangabc and
Bingzhen Ma *abc
*abc
aSchool of Chemistry and Chemical Engineering, North Minzu University, Yinchuan 750021, P.R. China. E-mail: mbz0001@126.com
bNingxia Key Laboratory of Solar Chemical Conversion Technology, North Minzu University, Yinchuan 750021, P.R. China
cKey Laboratory for Chemical Engineering and Technology, State Ethnic Affairs Commission, North Minzu University, Yinchuan 750021, P.R. China
First published on 30th July 2025
Abstract
In this paper, an efficient synthesis of benzylhydrazine derivatives from unactivated phenylethanol analogues was successfully developed under mild redox-neutral reaction conditions. At the same time, it was also proved that a wide range of phenylethanol analogues and azodicarboxylates can be successfully applied in this fragment coupling transformation. Moreover, this method can be applied to the synthesis of active molecules of isocarboxazid.
Photoredox catalysis is a novel catalytic method based on the redox ability of photocatalysts, which converts the energy of visible light into chemical energy and achieves chemical transformation under mild and neutral conditions.1 In recent years, visible light catalysis has become a powerful tool for building molecular complexity, and the most prominent is the generation of free radicals centered on high-energy heteroatoms.2 Recently, many organometallic complexes have been developed as photocatalysts,3 such as Ir3+, Ru2+, etc. However, due to the high price of ruthenium and iridium polypyridine complexes, they cannot be used on a large scale. Therefore, it is urgent to develop economical and environmentally friendly photocatalysts. Zuo and co-workers reported that cerium complexes were used as photocatalysts to carry out various organic chemical reactions, such as dehydromethylation of alcohols,4 cycloaddition reactions of naphthols and olefins,5 etc. These cerium compounds in different oxidation states (Ce(III) and Ce(IV)) adopt different electronic transition mechanisms6 and have unique luminescence characteristics,7 providing an opportunity to obtain multiple activation modes in photoredox catalysis. The benzylhydrazine structure exists in many pharmacologically active compounds,8 such as procarbazine, benserazide, isocarboxazid, and atazanavir (Scheme 1). Procarbazine is a nonspecific antitumor drug that inhibits DNA and protein synthesis. Clinically, it is mainly used for the treatment of Hodgkin's disease, multiple myeloma, other malignant lymphomas and lung cancer.9 Isocarboxazid is a non-selective monoamine oxidase inhibitor and produces irreversible binding with monoamine oxidases A and B, affecting the oxidative metabolism of monoamine neurotransmitters.10 Because benzylhydrazine derivatives are widely used in clinical practice, more efficient synthesis methods are sorely needed. Moreover, benzylhydrazine can be further synthesized by simple conversion, which provides a new and mild method for the synthesis of benzylamine bioactive molecules.
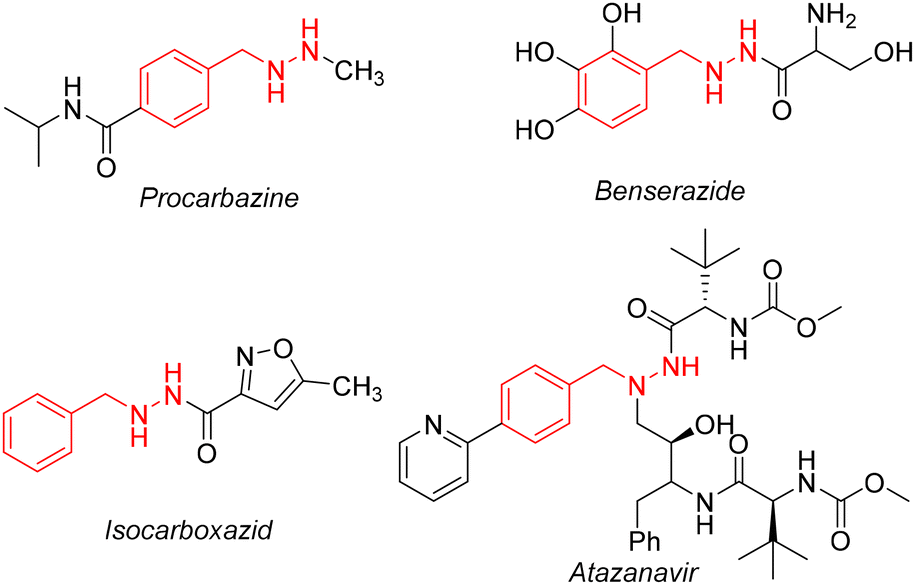 | ||
| Scheme 1 Pharmaceutically relevant benzylhydrazines. | ||
Since 2012, Nishibayashi et al.,11 Guan et al.,12 and Tunge et al.13 have successively reported the hydrazylation method of photocatalytic reactions. In 2016, Zuo's group14 discovered the unique redox properties of cerium complexes, causing the complexes formed with cycloalkanols to form highly reactive alkoxy radicals via ligand-to-metal charge transfer (LMCT); the alkoxy radical induces β-scission, and the alkyl radical formed reacts with DBAD to construct new carbon–nitrogen bonds and ketones (Scheme 2a). In 2019, Burkhard König's group15 reported the decarboxylative–hydrazination reaction of carboxylic acids with DBAD in the presence of cerium salt as a photocatalyst (Scheme 2b). With this chemistry in mind, we propose the synthesis of benzylhydrazine motifs by the photocatalytic dehydroxymethylation of cerium complexes from unactivated phenylethanol analogues (Scheme 2c).
 | ||
| Scheme 2 Synthesis of benzylhydrazines by dehydroxymethylation–hydrazination of unactivated phenylethanol analogues. | ||
With this mechanistic hypothesis in mind, we first selected phenylethanol as a substrate to test our hypothesis. In this reaction, phenylethanol (1.0 equiv.) and di-tert-butyl azodicarboxylate (DBAD, 3.0 equiv.) were used as substrates. Under the irradiation of 365 nm blue LED light, CeCl3 and DPA were used as dual photocatalysts to screen the solvents (entries 1–4, yields <1%–86%). It can be seen from Table 1 that when acetonitrile is used as the solvent, the target product can be obtained with an excellent yield of 86%. For solvents with large polarity such as 1,2-dichloroethane, the lone pair electrons on the solvent molecule will compete for coordination with metals, thus reducing the catalytic efficiency and making it difficult to fully react. Considering that bases may have an impact on the proton loss process of alcohols, we screened different bases in the presence of acetonitrile as solvent (entries 5 and 6, yields 56% and 12%). It can be seen from the table that the addition of base is more likely to cause the oxa-Michael reaction, and the yield is significantly reduced. Furthermore, we tried to use cerium bromide as the photocatalyst, but the yield was not significantly improved (entry 7, yield 33%). At the same time, a control experiment was also conducted. The experiments show that light, the cerium trichloride catalyst and DPA are necessary for this reaction. Meanwhile, the addition of (n-Bu)4NCl can significantly accelerate this reaction because the coordination between the chloride and the cerium center is crucial for the desired photoactivity.4
|
||
|---|---|---|
| Entry | Variations from standard conditions | Yield (%) |
| 1 | None | 86 |
| 2 | DCE instead of MeCN | 16 |
| 3 | 1,4-Dioxane instead of MeCN | 31 |
| 4 | THF instead of MeCN | <1 |
| 5 | Added NaHCO3 | 56 |
| 6 | Added Cs2CO3 | 12 |
| 7 | CeBr3 instead of CeCl3, (n-Bu)4NBr instead of (n-Bu)4NCl | 33 |
| 8 | No light | 0 |
| 9 | No CeCl3 and DPA | 0 |
| 10 | No CeCl3 and (n-Bu)4NCl | 0 |
After obtaining the optimal reaction conditions, we next turned our attention to examining the range of phenylethanol analogues. As shown in Scheme 3a, a series of phenylethanol analogues can be applied to this protocol under mild conditions. First, phenylethanol analogues with electron-withdrawing groups were tested, and phenylhydrazine was obtained in moderate to high yields (3b–3l, 39–73%). Notably, the efficacy of the reaction was hindered by halo substitution (3b, 3f, and 3i, 50–54%). In particular, bromine substitution has the most significant impact on the efficiency of this reaction (3d, 3g and 3j, 39%–57%). This might be caused by the increased possibility of the oxa-Michael reaction due to the substitution of halogens. It is worth noting that the trifluoromethyl substituted phenylethanol also gave good yield (3p, 65%). Then, methyl substituted phenylethanol was investigated. It was found that ortho-substituted, meta-substituted and para-substituted substrates could be obtained in moderate to good yields (3m–3o, 57–76%). Benzylhydrazine could also be obtained from methoxy-substituted substrates in moderate yield (3q, 53%). Benzylhydrazine was obtained in moderate yield by dehydroxymethylation–hydrazination of 1-naphthalene ethanol as a substrate (3r, 67%). 2-Thiopheneethanol was competent and afforded the desired product in 53% yield (3s). In order to further demonstrate the practicability of the synthetic protocol, as shown in Scheme 3b, dehydroxymethylation–hydrazination of 2-benzoxyethanol was shown to occur successfully under these mild redox-neutral conditions (3t, 21%). At the same time, N-Boc-phenylalaninol could also afford the dehydroxymethylation product under these reaction conditions (3u, 28%). However, due to the lack of the benzyl stabilizing effect, the reaction efficiency was poor. These successful examples provide a new method for the synthesis of other bioactive derivatives containing arylbenzylhydrazine and heteroarylbenzylhydrazine.
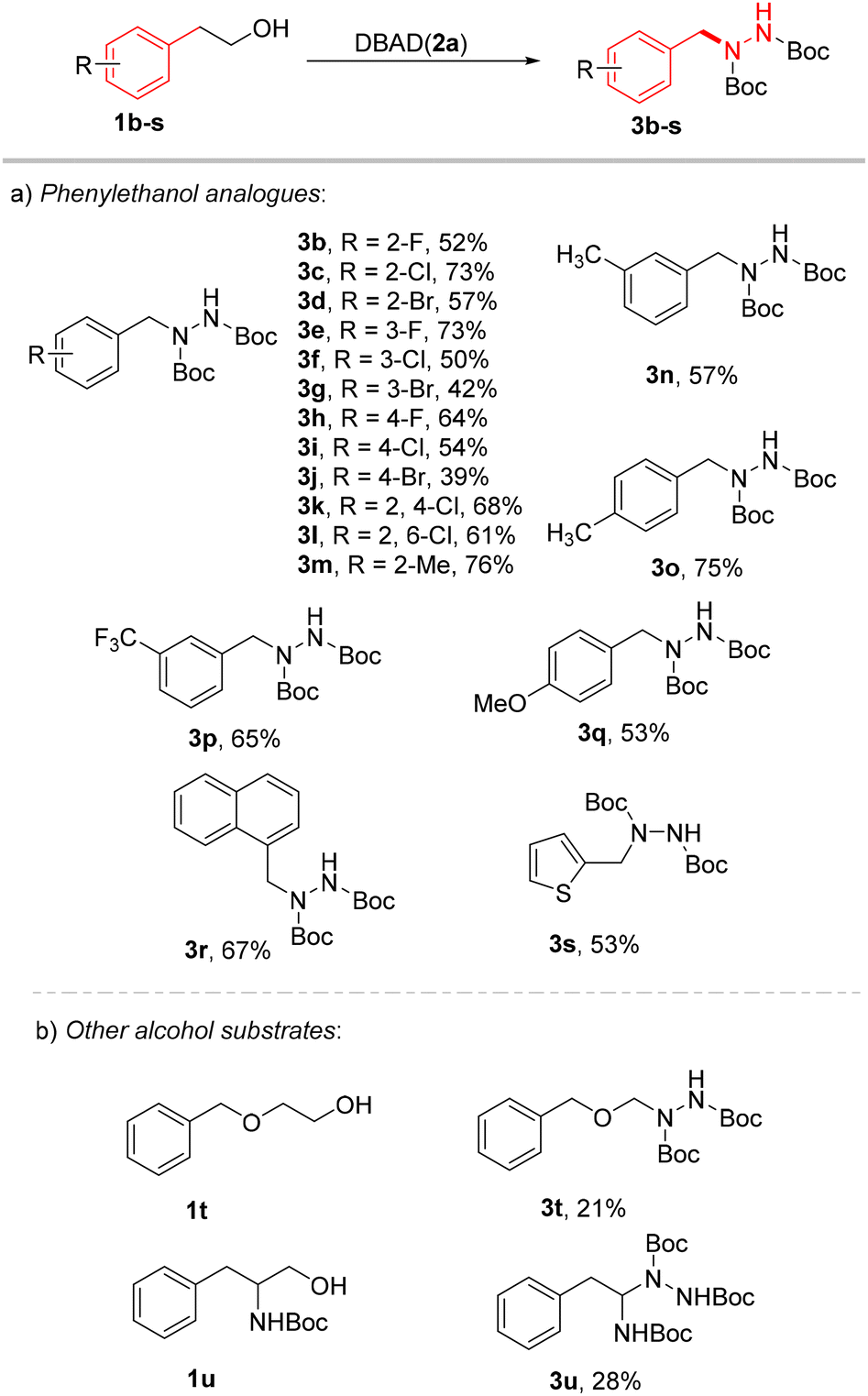 | ||
| Scheme 3 Alcohol scope. Reaction conditions: 1b–v (1.0 equiv.), 2a (3.0 equiv.), CeCl3 (5 mol%), DPA (5 mol%), (n-Bu)4NCI (15 mol%), MeCN (0.4 M), 24 h, and 365 nm LEDs. | ||
Next, we investigated the reactivity of different azodicarboxylate compounds with phenylethanol. As shown in Scheme 4a, both diethyl azodicarboxylate (3bb, 49%) and diisopropyl azodicarboxylate (3bc, 65%) could react with phenylethanol in good yield under these reaction conditions. Unfortunately, 4-(phenylazo)phenol and 4-(4-nitrophenylazo)resorcinol reacted with phenylethanol under these conditions without obtaining the expected benzylhydrazine products. Possibly due to the weak electron-withdrawing effect of the benzene ring, the double bond of azodicarboxylate lacks electrophilicity. To demonstrate the value of this process in the synthesis of biologically active molecules, the antidepressant isocarboxazid can be prepared in large quantities from compound 3a according to the literature (Scheme 4b). First, 1 g of phenylethanol (1a) reacted with DBAD (2a) under standard reaction conditions to afford benzylhydrazine (3a, 2.21 g) in 84% yield. According to the literature,15 3a can be deprotected under acidic conditions and coupled with 5-methylisoxazole-3-carboxylic acid to eventually synthesize isocarboxazid.
 | ||
| Scheme 4 Other azodicarboxylate substrates and synthesis of pharmacologically active scaffolds. Reaction conditions: 1a (1.0 equiv.), 2a–e (3.0 equiv.), CeCl3 (5 mol%), DPA (5 mol%), (n-Bu)4NCI (15 mol%), MeCN (0.4 M), 24 h, and 365 nm LEDs. | ||
From a design perspective, we envisioned that this dehydroxymethylation–hydrazination mechanism would proceed in the following manner (Scheme 5). First, phenylethanol (1a) is combined with cerium trichloride (A) to form a cerium complex (B), and then the excited 9,10-diphenylanthracene (DPA) strips an electron from the cerium complex. Given the relative oxidation potentials of the excited-state DPA catalyst (Ered1/2[DPA*/DPA] = 1.19 V vs. SCE in MeCN)16 and the in situ formed Ce(III) alkoxide complexes (B, Ered1/2[CeIII/CeIV] = 0.41 V vs. SCE in MeCN),17 we assumed that a rapid SET event would generate the Ce(IV) alkoxide (C) and a radical anion of DPA (H). Next, the resulting Ce(IV) alkoxide (C) is excited to a brief LMCT excited state, and then the homolytic cleavage of the coordination bond yields cerium(III) and primary alkoxy free radicals (D), while completing the cerium catalytic cycle. The alkoxy radical (D) is prone to β-scission, which generates a nucleophilic benzyl radical18 (E) and one molecule of formaldehyde. The conjugated addition of the benzyl radical (E) to di-tert-butyl azodicarboxylate (DBAD) eventually forms the desired C–N bond and a nitrogen-centered radical (F), an electrophilic radical that requires further single-electron reduction (SET) to produce the desired product. Single-electron reduction of this radical by the radical anion of DPA (H) would regenerate the DPA catalyst and deliver the desired product (3a). Meanwhile, the benzyl radical (E) could also be reduced to generate toluene.19
 | ||
| Scheme 5 Proposed mechanism for dehydroxymethylation–hydrazination. | ||
Conclusions
In summary, we use a cerium complex as a photocatalyst to catalyze dehydroxymethylation–hydrazination to synthesize benzylhydrazine derivatives from unactivated phenylethanol analogues. This atom- and step-economical transformation utilizes the ligand-to-metal charge transfer (LMCT) photocatalytic mode of cerium complexes. The method is simple and effective in the synthesis of benzylhydrazine derivatives and can be applied to the rapid synthesis of isocarboxazid. This method provides a new and simple way for the synthesis of phenylhydrazine derivatives, which is conducive to the discovery of new bioactive molecules.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
We thank the Natural Science Foundation of the Ningxia Hui Autonomous Region (2018AAC03116, 2022AAC03222 and 2023AAC03320), the Ningxia Key Research and Development Program (Special Talents) (No. 2020BEB04029), and the Fundamental Research Funds for the Central Universities of North Minzu University (2022XYZHG05) for financial support.References
-
(a) J. M. R. Narayanam and C. R. J. Stephenson, Visible light photoredox catalysis: applications in organic synthesis, Chem. Soc. Rev., 2011, 40(1), 102–113 RSC
; (b) Y. Han, L. Zhou, C. Wang, S. Feng, R. Ma and J.-P. Wan, Chin. Chem. Lett., 2024, 35, 108977 CrossRef CAS
-
(a) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC
- W. M. Cheng and R. Shang, Transition metal-catalyzed organic reactions under visible light: recent developments and future perspectives, ACS Catal., 2020, 10(16), 9170–9196 CrossRef CAS
- K. Zhang, L. Chang and Q. An, et al. Dehydroxymethylation of alcohols enabled by cerium photocatalysis, J. Am. Chem. Soc., 2019, 141(26), 10556–10564 CrossRef CAS PubMed
- A. Hu, Y. Chen and J. J. Guo, et al. Cerium-catalyzed formal cycloaddition of cycloalkanols with alkenes through dual photoexcitation, J. Am. Chem. Soc., 2018, 140(42), 13580–13585 CrossRef CAS PubMed
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis, Chem. Rev., 2013, 113(7), 5322–5363 CrossRef CAS PubMed
- Y. Qiao and E. J. Schelter, Lanthanide photocatalysis, Acc. Chem. Res., 2018, 51(11), 2926–2936 CrossRef CAS PubMed
-
(a) Hydrazine and its Derivatives, in Kirk-Othmer – Encylopedia Chemical Technology, ed. R. E. Kirk and D. F. Othmer, Wiley, New York, 4th edn, 1995, vol. 13 Search PubMed
- T. S. Gardner, J. Rutherford Lee, E. Fells and E. Wenis, US Patent, US2908688, 1959 Chem. Abstr., 1962, 56, 25114 Search PubMed
- M. B. H. Youdim, D. Edmondson and K. F. Topton, Nat. Rev. Neurosci., 2006, 7, 295 CrossRef CAS PubMed
- Y. Miyake, K. Nakajima and Y. Nishibayashi, Chem. – Eur. J., 2012, 18, 16473 CrossRef CAS PubMed
- M.-J. Zhang, G. M. Schroeder, Y.-H. He and Z. Guan, RSC Adv., 2016, 6, 96693 RSC
- S. B. Lang, K. C. Cartwright, R. S. Welter, T. M. Locascio and J. A. Tunge, Eur. J. Org. Chem., 2016, 3331 CrossRef CAS PubMed
- J.-J. Guo, A. Hu, Y. Chen, J. Sun, H. Tang and Z. Zuo, Angew. Chem., Int. Ed., 2016, 55, 15319 CrossRef CAS PubMed
- V. R. Yatham, P. Bellotti and B. König, Decarboxylative hydrazination of unactivated carboxylic acids by cerium photocatalysis, Chem. Commun., 2019, 55, 3489 RSC
- Y. Chen, X. Wang, X. He, Q. An and Z. Zuo, Photocatalytic Dehydroxymethylative Arylation by Synergistic Cerium and Nickel Catalysis, J. Am. Chem. Soc., 2021, 143, 4896–4902 CrossRef CAS PubMed
- A. Hu, J.-J. Guo, H. Pan, H. Tang, Z. Gao and Z. Zuo, δ-Selective Functionalization of Alkanols Enabled by Visible-Light-Induced Ligand-to-Metal Charge Transfer, J. Am. Chem. Soc., 2018, 140, 1612–1616 CrossRef CAS PubMed
-
(a) F. Li, D. Tian, Y. Fan, R. Lee, G. Lu, Y. Yin, B. Qiao, X. Zhao, Z. Xiao and Z. Jiang, Nat. Commun., 2019, 10, 1774 CrossRef PubMed
- K. Hironaka, S. Fukuzumi and T. Tanaka, J. Chem. Soc., Perkin Trans. 2, 1984, 1705 RSC
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ob00771b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |