
Click-like ortho-quinone methide trapping using rare 3-methylene isochromanones for a series of multifunctional pseudo-natural products†
Yi
Wang‡
a,
Dan
Li‡
a,
Hongping
Long‡
b,
Furong
Liu
a,
Guitao
Bai
a,
Kangping
Xu
a and
Xia
Yu
 *a
*a
aXiangya School of Pharmaceutical Sciences, Central South University, Changsha, Hunan 410013, People's Republic of China. E-mail: xyu226@csu.edu.cn
bCenter for Medical Research and Innovation, The First Hospital of Hunan University of Chinese Medicine, Changsha, Hunan 410007, P. R. China
First published on 14th July 2025
Abstract
3-Methylene isochromanones represent a unique subclass of isocoumarins with very limited synthetic accessibility. Effectively utilizing a highly reactive precursor as a reactive linchpin could provide a practical and flexible pathway to access this valuable scaffold and facilitate its diversification. Here, we demonstrate that the fermentation-accessible precursor hydroxyophioisocoumarin (HOI) serves as a uniquely reactive linchpin. We developed a novel click-like ortho-quinone methide platform that utilizes reactive HOI and various nucleophiles, allowing for a wide substrate scope in the synthesis of a series of multifunctional 3-methylene isocoumarins under mild and convenient conditions. The synthesized compounds exhibited notable biological activity, providing new chemical space for research in related fields.
Introduction
Natural products have long served as significant scaffolds in medicinal chemistry,1,2 yet their structural complexity often challenges synthetic accessibility. This limitation has driven the employment of pseudo-natural products (PNPs) – hybrid architectures combining natural product fragments with synthetic motifs – to expand chemical space while retaining bioactivities.3 Furthermore, PNPs can possess new properties and advantages, such as improved efficacy, reduced toxicity, and enhanced stability.4,53-Methylene isochromanones represent a distinctive and synthetically elusive subclass within the isocoumarin family. Characterized by a unique non-conjugated lactone ring featuring an exocyclic double bond at C-3 (Fig. 1),6–9 these structures defy conventional aromatic stabilization (Hückel's rule) due to the orientation of the double bond away from conjugation with the aromatic ring.
 | ||
| Fig. 1 3-Methylene isochromanones in this study and core structure of typical isocoumarin. | ||
Compared to their conjugated counterparts, the synthetic gap of 3-methylene isochromanones arises mainly from the significant thermodynamic difficulty in forming the non-conjugated, exocyclic double bond system, making their preparation a formidable challenge in synthetic chemistry.10–12 Notably, certain natural systems offer potential entry points. For instance, ophioisocoumarin (OI) and hydroxyophioisocoumarin (HOI),13 key biosynthetic intermediates leading to ascochin13 and possibly lycopodiellactone (1b),14 can be accessed through microbial fermentation and represent naturally occurring compounds that embody the challenging core structure of 3-methylene isochromanone. The existence of fermentation-accessible intermediates presents a compelling opportunity to develop chemical methods that efficiently transform these biosynthetic building blocks into diverse 3-methylene isochromanone derivatives and their pseudo-natural product hybrids. Effectively utilizing these intermediates could offer a practical and flexible pathway to access this valuable scaffold and facilitate its diversification, thus avoiding the need for direct de novo synthesis of the thermodynamically unfavorable core.
Herein, we draw mechanistic insights from the in vivo formation of lycopodiellactone and demonstrate that hydroxyophioisocoumarin (HOI) serves as a uniquely reactive linchpin. We establish that HOI spontaneously generates an unprecedented ortho-quinone methide (o-QM) species under mild conditions – a reactivity profile previously unrecognized and central to its role in methylene bridge formation. Capitalizing on this intrinsic reactivity, we developed a PNP-driven strategy to access structurally diverse 3-methylene isochromanones. Through site-selective trapping of the o-QM precursor with a variety of nucleophiles, we achieved a systematic expansion of 3-methylene isochromanone derivatives featuring functionalized motifs. Bioactivity assays indicate that these synthesized compounds exhibit a multifunctional bioactivity profile, including antibacterial, α-glucosidase inhibitory, and antitumor activities.
Results and discussion
The natural product lycopodiellactone (1b) features a methylene bridge connecting 4-hydroxy-6-methyl-2-pyranone (1a) to C-11 of the 3-methylene isochromanone ophioisocoumarin (OI), suggesting an in vivo C–C coupling mechanism. We suspect that an unidentified aldo/keto reductase AscB13 within the biosynthetic cluster of ophioisocoumarin is responsible for this reaction (Scheme 1A, detailed bioinformatic analysis of the asc cluster and the corresponding speculations were given in ESI†). However, heterologous expression of the asc cluster in Aspergillus nidulans revealed that exogenous supplementation of 1a to strains Aspergillus nidulans A1145 expressing ascABCD or ascACD yielded 1b (Fig. S53, structure was validated by NMR analysis as shown in Table S1 and Fig. S6–S9 along with HRMS analysis in Fig. S4†), indicating that the formation of 1b is independent of the uncharacterized reductase AscB. To decouple host-specific factors, Escherichia coli (E. coli) was fed with ascochin/1a or HOI/1a, where HOI and 1a produced 1b, thus excluding fungal enzymatic requirements. Based on the ortho-hydroxybenzyl alcohol motif of HOI, we hypothesized that the formation of lycopodiellactone occurs via an o-QM-driven spontaneous Michael addition.15,16 This hypothesis was confirmed by a water-mediated reaction of HOI/1a at room temperature for 4 days yielding 1b (Fig. S53†). Increasing the reaction temperature to 55 °C overnight resulted in a 32.6% yield (Fig. S53†) demonstrating a temperature-dependent relationship in product formation.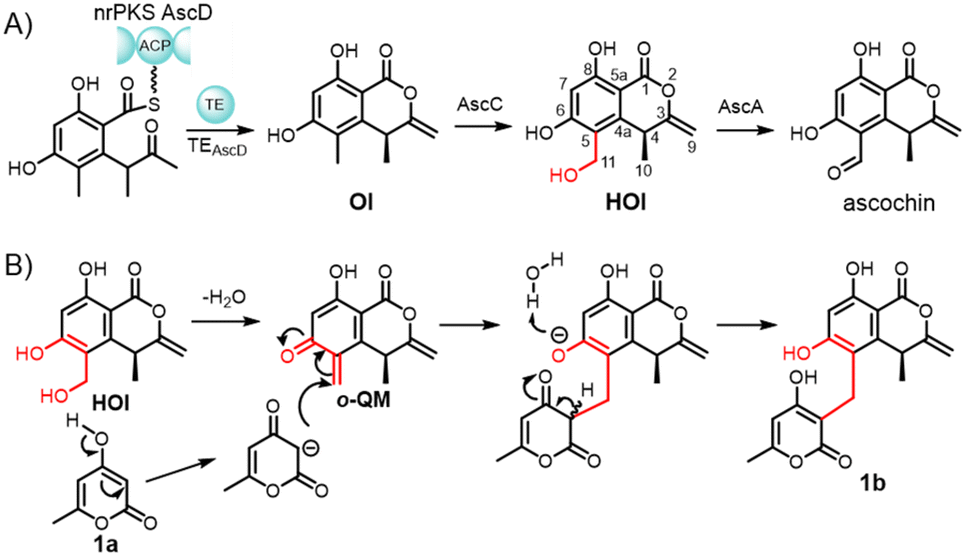 | ||
| Scheme 1 (A) Biosynthetic pathway of OI, HOI, and ascochin by the asc cluster. (B) Proposed formation mechanism of 1b through Michael addition reaction between HOI and 1a. | ||
To achieve systematic expansion of the PNP-driven strategy, a wide range of nucleophiles were investigated. Based on the widespread occurrence of polyphenol and indole scaffolds in pharmaceuticals and bioactive molecules, we opted for naphtholic compounds (2a–7a), phenolic compounds (8a–11a), indolic compounds (12a–17a) (Fig. S1 and S2†). Moreover, we screened organic substrates containing nitrogen heteroatoms, including quinoline derivatives (18a, 19a) and pyridine derivatives (20a) (Fig. S2†).
A series of experiments were conducted by subjecting HOI and selected substrates (2a–8a, each at 0.4 mM) to a 16-hour reaction in water at 55 °C, with a control containing only HOI. HPLC analysis revealed that 7 substrates (2a–8a) were detected to yield 9 products (2b–8b, 2c, 7c). The impact of the pH of the reaction system was also investigated using phosphate buffers at pH 2, 5, 7, 8, or 10 as reaction media, and the reaction time was extended to 20 hours. Then, the total yields of the products increased to 55.8%–95.6% (Table S27 and Fig. S52†). This result indicates that the pH of the reaction system significantly affects the yields of the coupling products. Therefore, the remaining substrates (9a–20a) were tested in water and in buffers at the aforementioned pH values, under identical reaction time. HPLC analysis revealed reactions occurring between 12 substrates (9a–20a) and HOI, resulting in the formation of 14 new products (9b–20b, 9c, 12c) with product yields varying from 34.3% to 88.0% (Table S28 and Fig. S52†). The results in Tables S27 and S28† showed that most products exhibited favourable yields in acidic phosphate buffer. Moreover, the self-dimerization product 21b of HOI was observed in the control group, with optimal results achieved at pH 7, yielding 82.1%. In contrast, no obvious formation of 21b occurred under acidic conditions, which explained the enhancement of generating non-self-dimerized coupling products under acidic conditions.
The facts we observed regarding the dependence of pH on the reaction rate (conversion) of the coupling between HOI and other phenolic compounds indicate that acid and base play important roles in this process, which may include the acid/base-assisted dehydration process in HOI to form the active o-QM intermediate (Scheme 1B), the acidic activation of the keto group of the o-QM and the deprotonation process on the coupled intermediate after the Michael addition. The different pH preferences for the dimerization to form 21b would imply a different rate-determining step, which might be a deprotonation process that requires a basic condition. Regarding the region-selectivity on the phenolic compounds, the coupling products of phenolic compounds illustrated that HOI forms an o-QM as a Michael addition receptor for phenolic compounds, leading to the attachment of the 3-MI (HOI moiety) unit to the ortho- or para-positions of the phenolic hydroxyl. For example, products 2b and 2c are respectively the ortho- or para-substituted products of 1-hydroxynaphthalene (2a) (Scheme 2). Compounds 3b–11b also represent coupling products where 3-MI substitutes at the ortho- or para-positions of hydroxyl or amino groups (Scheme 2), indicating a preference for substitution in regions of higher electron density. Specifically, in cases where an amino group substitution is present on the hydroxynaphthalene, 3-MI can also substitute at the amino group, leading to the formation of product 7c carrying two 3-MI units. In the case of indole, preferential substitution occurs at the C-3′ of indole by 3-MI unit to form 12b (Scheme 2). Subsequently, 3-MI was substituted furtherly at the C-2′ position to form the product 12c carrying two 3-MI units (Scheme 2). For indole derivatives 13a–17a, the C2′-adducts (13b–17b) were primary products due to the presence of a substituent at the C-3′ position of the indole ring in the substrates (Scheme 2). Moreover, the reaction outcomes of indoles confirm that the C3′ side chain obviously influenced product yields. Alkyl substitution at the C-3′ position favours substitution at the C-2′ position, whereas, when electron-withdrawing groups on the side chain are in close proximity to the 3′-position carbon or when the side chain is branched, the product yield decreases to the point of being undetectable. Speculatively, the former influences the electron cloud density at C-2′, while the spatial hindrance generated by the latter obstructs the attack of the reactants on o-QM. The reaction of aromatic compounds containing heteroatoms with HOI also adheres to the principles of Michael addition reactions. In the resulting products 1b, 18b–20b, the 3-MI unit substitutes at the ortho-position to a hydroxy or amino group, while the heteroatom is typically not substituted by 3-MI (Scheme 2).
 | ||
| Scheme 2 Reactions of 3-methylene isochromanone HOI with different reactants and the structures of products. Yields were calculated under optimal conditions for each product. | ||
To explore the diversity of bioactivity, we performed a primary screening of all coupling products for α-glucosidase inhibitory activity at a concentration of 500 μM. We found that more than half exhibited higher inhibition rates (66–99%) than the monomer HOI (53%) (Table S26†). Coupling products 2b, 4b–8b, 12b–13b, 2c, 12c, and 21b, with inhibition rates over 90%, were selected for IC50 determination. Detailed evaluation revealed that they displayed inhibition with IC50 values ranging from 10.97 to 176.00 μM, which are 6 to 113-fold lower than that of the positive control voglibose (IC50 1.25 mM) (Table 1).
| Types | Compounds | IC50 (μM) | Types | Compounds | IC50 (μM) |
|---|---|---|---|---|---|
| a Each experimental value was conducted in triplicate, with the standard deviation between replicate values being less than 10%. | |||||
| Phenolic products | 2b | 54.24 | Indole products | 12b | 51.94 |
| 2c | 22.23 | 12c | 11.47 | ||
| 4b | 10.97 | 13b | 20.62 | ||
| 5b | 176.00 | Dimer of HOI | 21b | 32.36 | |
| 6b | 123.90 | ||||
| 7b | 70.42 | Positive control | Voglibose | 1253.00 | |
| 8b | 145.60 | ||||
To elucidate the lower IC50 values of 3-methylene isochromanones, molecular docking was employed to assess the binding interactions between the methylene-bridged 3-methylene isochromanones and α-glucosidase. The structure of α-D-glucosidase from Saccharomyces cerevisiae (Uniprot: P07265) was predicted using AlphaFold2 and utilized in docking simulations with MOE tools.17 The predicted catalytic triad within the active site of P07265 includes a nucleophile Asp214, a proton donor Glu276 and a transition state stabilizer Asp349. Ligand binding affinity was evaluated via docking scores (s). Results revealed that HOI had the worst score at −5.6, while coupling products scored between −6.7 and −9.7, aligning with their superior inhibitory activity over HOI. Docking results showed that over half of the simulations demonstrated key interactions between Gln350 or Arg312 and the coupling products (Fig. S3†).
In the results of α-glucosidase inhibition assays for phenolic products, we found that the products with a 3-MI moiety located at the para-position of the hydroxyl group (e.g.2c) demonstrate superior activity compared to those with a 3-MI group substituted at the ortho-position of the hydroxyl group (e.g.2b). Additionally, the activity is closely related to the substitution pattern of the hydroxyl groups. When a single hydroxyl or amino group is located on the ring bearing the 3-MI moiety, notable activity is observed (e.g.2b, 2c, 4b–8b), however, when two or more hydroxyl groups are present on the ring possessing 3-MI (e.g.3b, 9b–11b), the activity is reduced. For naphthol products with a single hydroxyl group on the ring where 3-MI is located, substitution of the hydroxyl group on the other ring also affects their activity. For instance, compounds 4b and 2c demonstrate higher activity than 5b. Regarding the α-glucosidase inhibition activity of indole coupling products, a methyl group or an unmodified state at the adjacent position of 3-MI substituted on the pyrrole ring confers optimal activity, while extended chains or glycosyl groups attenuate these effects. Furthermore, the product 12c bearing two 3-MI groups demonstrates better activity compared to the indole derivatives featuring only one 3-MI group.
Subsequently, the antibacterial activities of all compounds were determined against Bacillus subtilis (B. subtilis). Among the products, the phenolic products 2b, 4b–8b, 2c, 7c, and 9c, along with the indole coupling products 12b, 12c, 13b, and 16b, as well as the pyridone derivative 20b and dimer 21b, demonstrated antibacterial efficacy against B. subtilis, with MIC values ranging from 2 to 64 μg mL−1 (Table 2). HOI and other products did not show significant activity, with MIC values exceeding 64 μg mL−1 (Table 2). Similar to the results of the α-glucosidase inhibition assays, the antibacterial activity of phenolic products with a single hydroxyl or amino group located on the ring bearing the 3-MI moiety (e.g.2b, 2c, 4b–8b) is better than those with two or more hydroxyl groups on the ring possessing 3-MI (e.g.3b, 9b–11b). In addition, the antibacterial activity of 12c, carrying two 3-MI units in the pyrrole ring, was superior to that of indole products with only one 3-MI substitution (e.g.12b–17b).
| Types | Compounds | MIC (μg mL−1) | Types | Compounds | MIC (μg mL−1) |
|---|---|---|---|---|---|
| Phenolic products | 9c | 64 | Pyridinone product | 20b | 64 |
| 8b, 6b | 32 | ||||
| 2c, 4b, 7b, 7c | 16 | Dimer of HOI | 21b | 32 | |
| 2b | 8 | HOI and other products | HOI, 1b, 3b, 9b, 10b, 11b, 14b, 15b, 17b, 18b, 19b | >64 | |
| 5b | 4 | ||||
| Indole products | 12b | 16 | |||
| 13b | 8 | ||||
| 16b | 64 | Positive control | Vancomycin | <0.5 | |
| 12c | 2 |
Furthermore, to ensure structural diversity and assess the potential toxicity of the core scaffold, 6 representative compounds—including phenolic product 9b, indole products 12b and 13b, and heteroaromatic derivative 18b alongside substrate HOI and its dimer 21b—were selected for cytotoxicity evaluation. The results of the cytotoxicity assays revealed that the dimer 21b and the coupling product 13b displayed activity against the human colon cancer cell line HCT116, with IC50 values of 13.73 μM and 19.67 μM, respectively. In contrast, precursor HOI did not exhibit obvious cytotoxic activity (IC50 value >50 μM).
Conclusions
In summary, we elucidate the abiotic origin of fungal lycopodiellactone through an o-QM-mediated Michael addition. Building on this insight, we introduce the first chemical platform that harnesses isochromanone-derived o-QM intermediates for synthesis under mild conditions. By employing HOI with click-like reactivity, we constructed 24 architecturally novel bioactive PNPs through water-compatible diversification. These derivatives exhibited a multifunctional bioactivity profile, particularly with some acting as good α-glucosidase inhibitors that surpass voglibose by 6 to 113-fold.Experimental section
Experimental details for the culture of strains, analysis of the asc cluster, feeding experiments, reaction conditions, HPLC analysis of samples, isolation of compounds, NMR analysis, biological assays, and molecular docking can be found in the ESI.†Author contributions
Yi Wang: writing – original draft, methodology, investigation, data curation, formal analysis, visualization. Dan Li: writing – original draft, methodology, investigation, formal analysis, data curation. Hongping Long: formal analysis, investigation, data curation. Furong Liu: resources, data curation, validation. Guitao Bai: resources, data curation, validation. Kangping Xu: data curation, validation. Xia Yu: writing – review & editing, supervision, funding acquisition, conceptualization, project administration.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
We gratefully acknowledge financial support by the Natural Science Foundation of Hunan Province (No. 2024JJ5415), Central South University Innovation-Driven Research Program (No. 2023CXQD057), and the Postgraduates Innovation Program of Central South University (No. 1053320241592). We also thank the Institute for Advanced Study of Central South University for NMR measurement.References
- A. G. Atanasov, S. B. Zotchev, V. M. Dirsch, I. E. Orhan, M. Banach, J. M. Rollinger, D. Barreca, W. Weckwerth, R. Bauer, E. A. Bayer, M. Majeed, A. Bishayee, V. Bochkov, G. K. Bonn, N. Braidy, F. Bucar, A. Cifuentes, G. D'Onofrio, M. Bodkin, M. Diederich, A. T. Dinkova-Kostova, T. Efferth, K. El Bairi, N. Arkells, T.-P. Fan, B. L. Fiebich, M. Freissmuth, M. I. Georgiev, S. Gibbons, K. M. Godfrey, C. W. Gruber, J. Heer, L. A. Huber, E. Ibanez, A. Kijjoa, A. K. Kiss, A. Lu, F. A. Macias, M. J. S. Miller, A. Mocan, R. Müller, F. Nicoletti, G. Perry, V. Pittalà, L. Rastrelli, M. Ristow, G. L. Russo, A. S. Silva, D. Schuster, H. Sheridan, K. Skalicka-Woźniak, L. Skaltsounis, E. Sobarzo-Sánchez, D. S. Bredt, H. Stuppner, A. Sureda, N. T. Tzvetkov, R. A. Vacca, B. B. Aggarwal, M. Battino, F. Giampieri, M. Wink, J.-L. Wolfender, J. Xiao, A. W. K. Yeung, G. Lizard, M. A. Popp, M. Heinrich, I. Berindan-Neagoe, M. Stadler, M. Daglia, R. Verpoorte and C. T. Supuran, Nat. Rev. Drug Discovery, 2021, 20, 200–216 CrossRef CAS PubMed
.
- L. Wu, Q. Zhang, Z. Deng and Y. Yu, Nat. Prod. Rep., 2022, 39, 919–925 RSC
- G. Karageorgis, D. J. Foley, L. Laraia and H. Waldmann, Nat. Chem., 2020, 12, 227–235 CrossRef CAS PubMed
- G. Karageorgis, D. J. Foley, L. Laraia, S. Brakmann and H. Waldmann, Angew. Chem., Int. Ed., 2021, 60, 15705–15723 CrossRef CAS PubMed
- E. A. Crane and K. Gademann, Angew. Chem., Int. Ed., 2016, 55, 3882–3902 CrossRef CAS PubMed
- T. El-Elimat, H. A. Raja, M. Figueroa, J. O. Falkinham 3rd and N. H. Oberlies, Phytochemistry, 2014, 104, 114–120 CrossRef CAS PubMed
- G. H. Silva, H. L. Teles, H. Trevisan, V. d. S. Bolzani, M. C. M. Young, L. H. Pfenning, M. N. Eberlin, R. Haddad, C. M. Costa-Neto and Â. R. Araújo, J. Braz. Chem. Soc., 2005, 16, 1463–1466 CrossRef CAS
- S. Niu, D. Liu, Z. Shao, J. Huang, A. Fan and W. Lin, RSC Adv., 2021, 11, 29661–29667 RSC
- K. Krohn, I. Kock, B. Elsässer, U. Flörke, B. Schulz, S. Draeger, G. Pescitelli, S. Antus and T. Kurtán, Eur. J. Org. Chem., 2007, 2007, 1123–1129 CrossRef
- J. Li, F. Fang, R. Wang, Y. Li, B. Xu, H. Liu and Y. Zhou, Org. Chem. Front., 2021, 8, 3876–3882 RSC
- H. Guo, J. Liu, G. Wu, W. Yao and F. Zhong, Green Chem., 2022, 24, 5598–5603 RSC
- L. Cai, Q. Lai, L. Zhang, G. Xue, Y. Zhang, N. He, M. Huang, S. Hu and S. Cai, Org. Lett., 2023, 25, 7126–7131 CrossRef CAS PubMed
- G. Bai, D. Li, Y. Wang, J. Yi, K. Xu, W. Wang, J. Li, G. Tan and X. Yu, Org. Lett., 2024, 26, 6303–6308 CrossRef CAS PubMed
- C. Li, B. Yang, R. Fenstemacher, J. Turkson and S. Cao, Tetrahedron Lett., 2015, 56, 1724–1727 CrossRef CAS
- J. Fan, G. Liao, F. Kindinger, L. Ludwig-Radtke, W.-B. Yin and S.-M. Li, J. Am. Chem. Soc., 2019, 141, 4225–4229 CrossRef CAS PubMed
- G. Liao, J. Fan, L. Ludwig-Radtke, K. Backhaus and S.-M. Li, J. Org. Chem., 2020, 85, 1298–1307 CrossRef CAS PubMed
- Chemical Computing Group ULC Molecular Operating Environment (MOE) 2019.01, Chemical Computing Group ULC, Montreal, QC, Canada, H3A 2R7, 2021.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ob00815h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |