
Electrochemical [3 + 2] cycloaddition of anilines and enamine ketones: construction of 3-benzoylindole derivatives†
Yangyang Hu‡
a,
Chunlei Yu‡b,
Ya'nan Rana,
Miao Yua,
Jingwen Suna,
Huiying Liua,
Jiahui Zhanga,
Haijun Wanga and
Lei Liu *a
*a
aCollege of Pharmacy, Qiqihar Medical University, Qiqihar, Heilongjiang 161006, P. R. China. E-mail: liuleiyaoxue1987@163.com
bThe Institute of Medicine, Qiqihar Medical University, Qiqihar, Heilongjiang 161006, P. R. China
First published on 18th July 2025
Abstract
This article describes an eco-friendly and atom-economical electrochemical protocol for the intermolecular [3 + 2] cycloaddition to synthesize 3-benzoylindole derivatives in synthetically viable yields. The oxidative cycloaddition proceeds under transition-metal-free and strong-oxidant-free conditions, generating H2 and (Me)2NH as the byproducts, thereby aligning with green chemistry principles. Furthermore, the methodology exhibits exceptional synthetic versatility through successful late-stage diversification and compatibility with structurally diverse substrates. Preliminary cytotoxic evaluation via CCK-8 assays against two human carcinoma cell lines revealed potent bioactivity profiles, establishing a foundation for systematic exploration of indole-based pharmacophores. This protocol offers a sustainable alternative to conventional synthetic strategies while expanding the functionalization landscape of heteroaromatic systems.
Introduction
Nitrogen-containing heterocycles represent a ubiquitous class of structural motifs that are extensively distributed throughout natural products, pharmacologically active compounds, and biologically relevant molecules, forming essential core architectures in their molecular frameworks. These structurally diverse systems hold considerable importance in driving innovation in synthetic methodology development and structure-activity relationship investigations within modern organic synthesis and medicinal chemistry research. Notably, the indole scaffold emerges as a privileged nitrogen-containing heterocyclic system that occupies a pivotal position among both naturally occurring substances and synthetic pharmaceutical agents.1–3 These derivatives play essential roles in fundamental metabolic pathways of animal systems while being ubiquitously present as bioactive constituents in botanical sources.4 Representative examples include the indole alkaloid reserpine, isolated from Rauwolfia serpentina, which has been clinically utilized as an antihypertensive therapeutic, and yohimbine, extracted from tree bark, which demonstrates significant immunosuppressive efficacy.5,6 Notably, 3-acyl-substituted indole7–9 derivatives have gained prominence as privileged structural motifs with enhanced pharmacological profiles. These compounds not only constitute critical pharmacophoric elements in bioactive molecules such as indomethacin and bruceolline derivatives but also serve as versatile building blocks in organic synthesis for therapeutic development, particularly demonstrating antidiabetic potential and suppressing neoplastic proliferation through the construction of diverse indole-based architectures. Exemplifying this paradigm, the 3-(3,4,5-trimethoxybenzoyl)indole derivative OXi800610 (Scheme 1a) demonstrates potent tubulin polymerization inhibition, exerting its anticancer effects through competitive displacement of radiolabeled colchicine from the β-tubulin colchicine-binding domain. This mechanistic elucidation substantiates the structural relevance of this scaffold in anticancer therapeutic development. 3-Acylindoles have established themselves as privileged medicinal chemistry scaffolds owing to their polypharmacological profiles and strategic value as versatile synthons for assembling diverse bioactive architectures, thereby serving as modular chemical platforms for modern synthetic methodology innovation and rational drug design campaigns.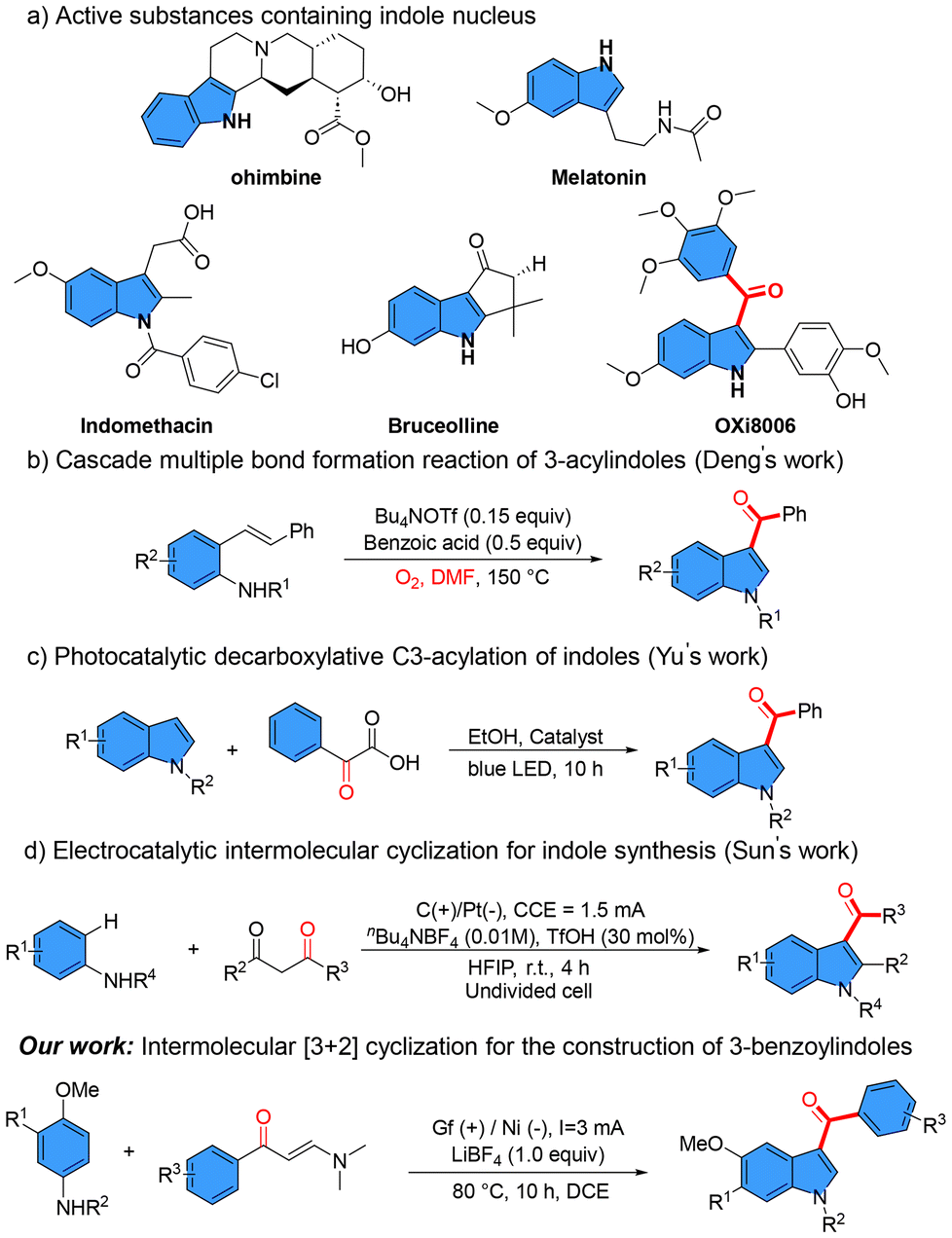 | ||
| Scheme 1 Active substances and synthetic methods of 3-acetylindoles. | ||
The imperative for methodological innovation in 3-acylindole synthesis stems from its chemically diversified architectures and polypharmacological profiles, it has attracted widespread attention in modern synthetic chemistry prompting the development of various methods to address synthesis challenges.11 It is worth noting that the Deng's12 group reported a metal-free cascade reaction strategy for constructing 3-acylindoles from 2-vinylanilines, wherein molecular oxygen serves as a terminal oxidant and dimethylformamide (DMF) acts as both solvent and carbon source, enabling sequential C–N, C–C, and C–O bond formation through oxidative cycloaddition (Scheme 1b). In parallel, the Yu's13 group demonstrated a heteroatom-directed regioselective C3-acylation protocol employing a metal-organic framework (MOF)-derived S-scheme heterojunction photocatalyst, which facilitates visible-light-driven acylation of indoles with enhanced charge separation efficiency and suppressed side reactions (Scheme 1c). Alternatively, the Sun's14 group pioneered an electrocatalytic cycloaddition strategy for synthesizing polysubstituted 3-acylindoles via redox-neutral coupling of anilines with readily available 1,3-diketones, achieving high atom economy and functional group tolerance without reliance on transition-metal catalysts or stoichiometric oxidants (Scheme 1d). Recently, Liu and co-workers achieved selective synthesis of indole and benzofuran derivatives via electrochemical cyclization reactions between para-alkoxyl or para-hydroxyphenylamine substrates and enaminone counterparts.15 In contrast to their methodology, our work encompassed not only the synthesis of the target compounds but also comprehensive biological activity evaluation studies. These methodologies collectively exemplify advancements in regiochemical control, sustainability, and operational simplicity, underscoring the evolving landscape of 3-acylindole synthesis for drug discovery and functional material development.
Electrochemically induced cyclization reactions have emerged as a sustainable synthetic methodology in recent years. Electrochemical organic synthesis relies on electron transfer to drive redox processes, mediated through the application of a continuous electrical current. In this paradigm, oxidation reactions occur at the anode where electrons are relinquished, while concurrent reduction processes transpire at the cathode through electron acquisition.16 Our research group has focused on electrochemical nitrogen-containing heterocycle synthesis.17 Herein, we report an electrochemical approach for synthesizing 3-benzoylindoles via a redox-neutral reaction, employing inexpensive and readily available anilines and enaminone to achieve intermolecular [3 + 2] cycloaddition. This reaction proceeds under mild conditions without requiring transition-metal catalysts or chemical oxidants, aligning with the principles of sustainable chemistry. The protocol demonstrates operational simplicity, avoids stoichiometric reagent waste, and provides an atom-economical route to access structurally diverse 3-benzoylindole derivatives through controlled electron transfer processes.
Results and discussion
The reaction employing N-(4-methoxyphenyl)-4-methylbenzene-sulfonamide (1a) and (E)-3-(dimethylamino)-1-phenylprop-2-en-1-one (2a) as substrates was conducted in an undivided single-compartment electrochemical cell. Systematic optimization studies established the following conditions as optimal: graphite felt anode, nickel sheet cathode, LiBF4 (1.0 equiv.) as electrolyte, and DCE (6 mL) as solvent under a constant current of 3 mA at 80 °C for 10 h, affording product 3aa in 80% isolated yield (Table 1, entry 1). Control experiments verified the electrochemical nature of the transformation, with no product formation observed in the absence of applied current (Table 1, entry 2). Electrode material evaluation revealed significant performance disparities: platinum cathode yielded only 30% (Table 1, entry 3), while a dual Pt electrode configuration marginally improved the yield to 40%, though still inferior to the C/Ni system (Table 1, entry 4). Alternative electrolytes (nBu4NPF6, LiClO4, nBu4NClO4) exhibited diminished efficacy compared to LiBF4 (Table 1, entries 5–7). Solvent screening demonstrated the superiority of DCE over other single-component solvents (DCM, DMSO, DMF, MeOH, MeCN; Table 1, entries 8–9).
|
||
|---|---|---|
| Entry | Variations from standard conditions | Yieldb |
| a Reaction coditions: 1a (0.3 mmol), 2a (0.6 mmol), LiBF4 (0.3 mmol), DCE (6 mL), constant current = 3 mA, undivided cell, GF anode (1.5 cm × 1.0 cm) and Ni plate cathode (1.5 cm × 1.0 cm), 80 °C, 10 h.b Isolated yield. | ||
| 1 | None | 80 |
| 2 | Without electricity | ND |
| 3 | C/Pt instead of GF/Ni | 30 |
| 4 | Pt/Pt instead of GF/Ni | 40 |
| 5 | nBu4NPF6 instead of LiBF4 | 40 |
| 6 | LiClO4 instead of LiBF4 | 65 |
| 7 | nBu4NClO4 instead of LiBF4 | 22 |
| 8 | DCM instead of DCE | Trace |
| 9 | DMSO, DMF, MeOH, MeCN instead of DCE | Trace |
| 10 | 70 °C, 50 °C instead of 80 °C | 67, 43 |
| 11 | 5 mA instead of 3 mA | 55 |
| 12 | 2 mA instead of 3 mA | 27 |

The reaction showed significant thermal sensitivity, and the yield decreased with decreasing temperature (Table 1, entry 10). When exploring the effect of current intensity on the reaction, we found that increasing or decreasing the current intensity would lead to a significant decrease in yield (Table 1, entries 11 and 12).
Following optimization studies, systematic evaluation of the reaction generality demonstrated robust compatibility across electronically diverse enaminone derivatives. Electronic effect analysis revealed that both electron-withdrawing (CF3, COOMe, CN) and electron-donating (Me, Et, iPr, tBu, OMe) substituents on the aryl ring enabled efficient conversion, affording target products in 60–80% yields. It is noteworthy that substrates bearing electron-withdrawing groups exhibited enhanced reactivity, with the ester-substituted derivative 3an achieving the highest yield of 78%. Moreover, halogen substituted products (3ab–3ad) could be synthesized in good yields (70–76%). Positional studies indicated marginally superior efficiency for para-substituted substrates compared to ortho- and meta-substituted analogues, while mono- and di-substituted systems showed comparable yields (3ae–3ag). A distinct reactivity drop occurred upon replacing aryl substituents with heterocycles-the 1,3-dioxolane-containing product 3ap displayed significantly diminished yield, likely attributable to steric hindrance and destabilization of key intermediates (Table 2).

In the scope investigation of anilines derivatives, it was discovered that only potent electron-donating groups such as methoxy (3aa) and ethoxy (3ca) effectively promoted thetransformation, whereas weak electron-donating substituents (3da, 3ga) or electron-withdrawing groups (3fa, 3ha) completely inhibited reactivity. Systematic investigations on toluenesulfonyl-derived substrates revealed pronounced electronic effects governing product yields: substrates bearing electron-donating (3ia, 3oa, 3pa) consistently outperformed those with electron-withdrawing substituents (3ka). In addition, this phenomenon persists even in unsubstituted benzene ring systems (3ja), demonstrating the general applicability of the observed behavior. Halogen substitution (3la, 3ma, 3na) enabled the preparation of target products with synthetically viable yields (50–58%) under optimized reaction conditions. Notably, positional isomerism (3aa, 3oa, 3pa) exhibited negligible influence on the yield outcomes, suggesting the electronic nature rather than spatial orientation of substituents dictates the reaction efficiency.
Following the synthesis of 3aa, selective cleavage of the N-tosyl group on its indole ring was achieved, revealing a highly reactive nitrogen center. This nucleophilic nitrogen enabled versatile structural diversification via molecular hybridization, allowing direct installation of bioactive motifs through alkylation and acylation (Table 3).

Hybrid molecules bearing dual pharmacophores are strategically advantageous for multi-target engagement and toxicity mitigation, aligning with current trends in polypharmacology. Capitalizing on this reactivity, we coupled 3aa with marketed drugs through N-centered transformations. Deprotection of 3aa under optimized conditions furnished intermediate 3c in 90% yield. Subsequent, we reacted the active intermediate 3c with 3-bromopropene to obtain alkylated product 4a (86%), which was then esterified with naproxen and aspirin to obtain acylated products 4b and 4c, with yields of 74% and 69%, respectively. Based on the molecular hybridization strategy, the covalent integration of target products with privileged pharmacophores enables the exploitation of synergistic effects, thereby highlighting the substantial potential for diverse applications of this compound series in subsequent development (Scheme 2).
 | ||
| Scheme 2 Further functionalization of product. | ||
To elucidate the redox processes governing the intermolecular cycloaddition, cyclic voltammetry (CV) analyses were systematically performed, guided by prior mechanistic studies of analogous systems. Cyclic voltammetric studies unveiled divergent electrochemical signatures between the substrates. Substrate 1a demonstrated a well-defined single-electron oxidation event at Epa = 1.96 V vs. Ag/AgCl (curve b), consistent with a stepwise oxidative coupling mechanism initiated by single-electron transfer (SET). In stark contrast, substrate 2a required significantly higher activation energy, exhibiting an oxidation wave at Epa = 3.90 V, which underscores its inherent electrochemical inertness in isolated systems. Remarkably, simultaneous electrolysis of 1a and 2a (curve d) induced a substantial shift of the oxidation potential, revealing a synergistic activation mechanism. The experimental data demonstrates that the heterogeneous catalytic system exerts a pronounced catalytic effect on the oxidative transformation of compound 2a, significantly accelerating its reaction kinetics through an optimized electron transfer pathway (Fig. 1).
 | ||
| Fig. 1 Cyclic voltammograms of substrates in 0.1 M LiBF4/DCE, using a Ni wire working electrode and glassy carbon and Ag/AgCl (0.1 M in DCE) as counter and reference electrodes at 100 mV s−1 scan rate: (a) background (0.1 M LiBF4 in DCE), (b) 1a (30 mM), (c) 2a (30 mM), (d) 1a (30 mM) + 2a (30 mM) + LiBF4 (6 mM). | ||
To validate the postulated reaction mechanism, the involvement of radical intermediates was systematically investigated through complementary high-resolution mass spectrometry (HRMS) analysis and radical trapping studies (Scheme 3). HRMS characterization of the crude reaction mixture conclusively identified radical adduct 5a, corroborating the transient generation of a nitrogen-centered radical species. Additionally, the detection of adduct 6a indicates a stepwise radical propagation mechanism involving intramolecular hydrogen atom transfer (HAT) from the nitrogen-centered radical to form a stabilized carbon-centered radical intermediate. To further substantiate the radical chain hypothesis, 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO) was introduced as a stoichiometric radical inhibitor. Under optimized electrochemical conditions, TEMPO addition to the reaction system containing substrates 1a and 2a resulted in complete suppression of product formation (>99% inhibition). In the TEMPO-mediated radical trapping experiment, we detected a newly formed species, yet the mass spectrometric analysis revealed decomposition products, precluding the acquisition of high-resolution mass spectrometric data. Nevertheless, radical inhibition was unequivocally demonstrated. This definitive observation strongly supports the proposed radical-mediated pathway, as TEMPO efficiently terminates radical chain propagation through spin-trapping interactions, effectively disrupting the intermolecular cycloaddition.
 | ||
| Scheme 3 Investigation of reaction mechanism. | ||
Building upon established mechanistic precedents,18 we propose a plausible cascade cycloaddition pathway involving sequential bond reorganization events (Scheme 4). The reaction initiates with the anodic oxidation of benzenesulfonamide derivative 1a, which undergoes deprotonation to generate a nitrogen-centered radical intermediate I exhibiting single-electron oxidation characteristics. This radical intermediate achieves resonance stabilization through intramolecular electron density redistribution, thereby evolving into a carbon-centered radical II. Subsequent regioselective radical addition to the olefinic substrate 2a forms adduct III, which undergoes a second anodic oxidation to trigger intramolecular cycloaddition. This step proceeds via a concerted electron transfer and anodic oxidation, yielding the cationic intermediate IV. Finally, the target product 3aa was obtained through further cycloaddition and dehydrogenation aromatization. Interestingly, concurrent proton reduction at the cathode generates hydrogen gas as a stoichiometric byproduct while synchronously mediating the cleavage of the dimethylamino protecting group. This dual redox synergy ensures efficient proton management and drives the overall transformation by suppressing undesired proton accumulation.
 | ||
| Scheme 4 Proposed mechanism. | ||
The synthesized 3-benzoylindole derivatives19 were evaluated for in vitro cytotoxic activity against MCF-7 (breast adenocarcinoma) and 5637 (bladder carcinoma) cell lines via the CCK-8 assay. These compounds exhibited dose-dependent antiproliferative effects, displaying marked selectivity against bladder carcinoma cells. The complete series demonstrated enhanced cytotoxic potency toward 5637 cells relative to the clinical chemotherapeutic agent 5-fluorouracil (5-FU), with IC50 values comparable to or lower than those of 5-FU (23.25 ± 1.11 μM). Notably, derivatives 3ba and 3ap exhibited pronounced cytotoxic selectivity, demonstrating IC50 values of 7.63 ± 1.85 μM and 8.92 ± 0.91 μM, respectively, corresponding to a 3.1-fold improvement in potency relative to 5-FU in bladder carcinoma models. Conversely, the compound series manifested pan-cellular cytotoxic profiles against MCF-7 cells, indicative of limited tissue-specific targeting efficacy for mammary adenocarcinoma systems (Table 4).
| Compound | 5637 | Compound | 5637 |
|---|---|---|---|
| 3ak | 16.19 ± 0.73 | 3ga | 16.59 ± 0.73 |
| 3ap | 8.922 ± 0.91 | 3la | 14.67 ± 1.62 |
| 3ba | 7.627 ± 1.85 | 5-Fu | 23.25 ± 1.11 |
| Compound | MCF-7 | Compound | MCF-7 |
| 3ea | 29.43 ± 0.92 | 3na | 47.84 ± 0.53 |
| 3ma | 45.51 ± 0.87 | 5-Fu | 28.95 ± 1.40 |
Conclusions
In summary, we have established an atom-economical and eco-conscious electrochemical protocol for the efficient construction of 3-benzoylindole derivatives. This methodology utilizes an intermolecular [3 + 2] cycloaddition strategy between benzenesulfonamides and enaminones under transition metal-free and stoichiometric oxidant-free conditions, delivering target products in moderate to excellent yields with extensive substrate scope. The oxidative dehydrogenative cycloaddition operates efficiently under mild electrochemical parameters, eliminating reliance on harsh reagents or exogenous oxidants. The in vitro antitumor potential of these derivatives was validated through CCK-8 assays against 5637 (bladder carcinoma) and MCF-7 (breast adenocarcinoma) human cancer cell lines. Significantly, select derivatives demonstrated enhanced cytotoxic potency against 5637 cells compared to the clinical benchmark 5-fluorouracil (5-FU). This work not only establishes a versatile platform for indole scaffold diversification but also reveals promising lead candidates for advanced pharmacological evaluation in oncology-focused drug discovery pipelines.Conflicts of interest
There are no conflicts to declare.Data availability
The datasets generated during this study are fully available within the article and supplementary materials.Acknowledgements
We thank the Natural Science Foundation of Heilongjiang Province of China (LH2024B026), the Program for Young Talents of Basic Research in Universities of Heilongjiang Province (YQJH2023115), the Program for Heilongjiang Provincial Department of Education Science and Technology Research Project (2022-KYYWF-0799) and the Construction Project of Dominant Characteristic Disciplines of Qiqihar Medical University (QYZDXK-001) for financial support.References
- For selected reviews, see: B. P. Kaur, J. Kaur and S. S. Chimni, RSC Adv., 2021, 11, 2126–2140 RSC
.
- D. Pala, A. Lodola, A. Bedini, G. Spadoni and S. Rivara, Int. J. Mol. Sci., 2013, 14, 8093–8121 CrossRef PubMed
-
(a) M. Shiri, Chem. Rev., 2012, 112, 3508–3549 CrossRef CAS PubMed
-
(a) N. E. Golantsov, A. A. Festa, A. V. Karchava and M. A. Yurovskaya, Chem. Heterocycl. Compd., 2013, 49, 203–225 CrossRef CAS
- M. Srivastava, S. Kesharwani, R. Kesharwani, D. K. Patel and S. N. Singh, Int. J. Res. Ayurveda Pharm., 2021, 12, 106–109 CrossRef CAS
- Y. Liu, H. Y. Yu, H. Z. Xu, J. J. Liu, X. G. Meng, M. Zhou and H. L. Ruan, J. Nat. Prod., 2018, 81, 1841–1849 CrossRef CAS PubMed
-
(a) S. A. Patil, R. Patil and D. D. Miller, Curr. Med. Chem., 2009, 16, 2531–2565 CrossRef CAS PubMed
- C. G. Veale, R. Zoraghi, R. M. Young, J. P. Morrison, M. Pretheeban, K. A. Lobb and M. T. Davies-Coleman, J. Nat. Prod., 2015, 78, 355–362 CrossRef CAS PubMed
- A. J. Rago and G. Dong, Green Synth. Catal., 2021, 2, 216–227 Search PubMed
-
(a) M. B. Hadimani, M. T. MacDonough, A. Ghatak, T. E. Strecker, R. Lopez, M. Sriram and K. G. Pinney, J. Nat. Prod., 2013, 76, 1668–1678 CrossRef CAS PubMed
- For selected examples, see:
(a) W. Wu and W. J. Su, J. Am. Chem. Soc., 2011, 133, 11924–11927 CrossRef CAS PubMed
- J. B. Wang, Y. L. Li and J. Deng, Adv. Synth. Catal., 2017, 359, 3460–3467 CrossRef CAS
- Z. Gao, Y. Jian, S. Yang, Q. Xie, C. J. Ross Mcfadzean, B. Wei and G. Yu, Angew. Chem., Int. Ed., 2023, 62, e202304173 CrossRef CAS PubMed
- X. Chang, X. Chen, S. Lu, Y. Zhao, Y. Ma, D. Zhang and P. Sun, Adv. Synth. Catal., 2022, 364, 2865–2871 CrossRef CAS
- Z. Zhong, Y. Liu, L. Liao and J.-P. Wan, Org. Lett., 2025, 27, 2537–2541 CrossRef CAS PubMed
-
(a) Y. Jiang, K. Xu and C. Zeng, Chem. Rev., 2018, 118, 4485–4540 CrossRef CAS PubMed
-
(a) J. Zhang, Y. Hu, Y. N. Ran, H. Liu, J. Sun, H. Wang and L. J. Liu, Org. Chem., 2025, 90, 2869–2878 CrossRef CAS PubMed
-
(a) W. Hu, X. Diao, J. Yuan, W. Liang, W. Yang, L. Yang and S. J. Zhang, Org. Chem., 2023, 89, 644–655 CrossRef PubMed
-
(a) S. Mahboobi, H. Pongratz, H. Hufsky, J. Hockemeyer, M. Frieser, A. Lyssenko and T. J. Beckers, Med. Chem., 2001, 44, 4535–4553 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data of all the compounds. See DOI: https://doi.org/10.1039/d5ob00831j |
| ‡ These authors share co-first authorship. |
| This journal is © The Royal Society of Chemistry 2025 |