
Assembly of 1,3-diaryl-1H-pyrazoles via base-mediated [3 + 2] cycloaddition†
Tenglong Xu,
Zimei Zhong,
Jianwei Yan,
Yi Chen,
Aifang Wang * and
Min Wang*
* and
Min Wang*
College of Material, Chemistry and Chemical Engineering, Key Laboratory of Organosilicon Chemistry and Material Technology, Ministry of Education, Hangzhou Normal University, Hangzhou, 311121, P. R. China. E-mail: wangaf@hznu.edu.cn; mwang@hznu.edu.cn
First published on 18th July 2025
Abstract
1,3-Diaryl-1H-pyrazoles, serving as privileged heterocyclic scaffolds, demonstrate significant application potential in pharmaceutical development, functional materials, and agrochemical industries. This study presents a synthetic strategy based on [3 + 2] cycloaddition, achieving efficient construction of 1,3-diaryl-1H-pyrazoles. A novel transition metal-free protocol was established through base-catalyzed intermolecular annulation of hydrazones with vinyl sulfoxides, followed by oxidation. The reaction system exhibited excellent substrate generality and demonstrated successful gram-scale synthesis. Subsequent investigations into synthetic applications further confirmed the methodology's practicality and scalability.
The urgent demand for novel therapeutic agents has established substituted pyrazoles as highly promising candidates owing to their multifaceted pharmacological properties, including anticancer, antimalarial, anti-HIV, and antiviral activities, with notable therapeutic relevance in managing diabetes mellitus and Parkinson's disease.1 Of particular significance, 1,3-diaryl-1H-pyrazole derivatives have evolved as pivotal structural motifs in numerous pharmacologically active compounds (Fig. 1).2,3 Representative applications encompass CDPPB (1),4 a clinical-stage candidate for schizophrenia therapy, and difenamizole (2),5 utilized as an analgesic and anti-inflammatory agent. The clinical utility of this scaffold is further exemplified by commercial pharmaceuticals such as lonazolac (3).6 Furthermore, alkyl-substituted 1,3-diaryl-1H-pyrazoles have been characterized as selective ligands for estrogen receptors (4).7 Recent advancements highlight that synthetic derivatives incorporating the 1,3-diaryl-1H-pyrazole core, notably compound (5),8 demonstrate potent in vitro inhibition of AChE, underscoring their potential as neuroprotective agents. The wide-ranging applications of these compounds persistently motivate synthetic organic chemists to devise effective strategies for constructing 1,3-diaryl-1H-pyrazole architectures.
 | ||
| Fig. 1 Selected bioactive molecules containing 1,3-diaryl-1H-pyrazole. | ||
Traditional synthetic approaches to 1,3-diaryl-1H-pyrazoles typically involve condensation reactions between arylhydrazines and 1,3-biselectrophiles (e.g., 1,3-diketones and enones), though this methodology often affords regioisomeric mixtures.9 Concurrently, cycloaddition reactions have emerged as a robust and versatile strategy for constructing functionalized pyrazole scaffolds;10 however, the synthesis of diaryl-1H-pyrazoles remains challenging due to the characteristically low reactivity at the C3 position.11 Recently, Li and co-workers developed an innovative ruthenium-catalyzed acceptorless dehydrogenative coupling (ADC) protocol employing hydrazines with either 1,3-diols or allylic alcohols to access 1,3-diaryl-1H-pyrazole derivatives.12 Building upon this mechanistic paradigm, the Ghosh group subsequently established a nickel-catalyzed three-component coupling strategy,13 thereby significantly expanding the synthetic repertoire for these heterocyclic architectures. Despite their synthetic potential, the widespread implementation of these catalysts remains challenging due to economic and environmental concerns associated with their high cost and toxicity. Therefore, the development of efficient, inexpensive, and environmentally friendly metal-free catalytic systems is highly desirable for the regioselective synthesis of 1,3-diaryl-1H-pyrazoles. Building upon our group's established expertise in nitrogen heterocycle construction14 and recognizing the demonstrated versatility of sulfoxide compounds in synthesizing diverse five-membered heterocycles,15,16 we hereby introduce an innovative base-catalyzed strategy. This methodology employs a cycloaddition reaction between vinyl sulfoxides and hydrazones to strategically assemble 1,3-diaryl-4,5-dihydro-1H-pyrazole architectures (Scheme 1).
 | ||
| Scheme 1 Synthetic strategies for the construction of 1,3-diaryl-1H-pyrazoles. | ||
Our investigation began by examining the feasibility of this reaction model using 1-benzylidene-2-phenylhydrazine 1a and (vinylsulfinyl)benzene 2 as prototype substrates to explore the reaction conditions. After conducting thorough preliminary evaluations of the reaction parameters, we established sodium propoxide (C3H7ONa) as the base catalyst and 80 °C as the reaction temperature to examine other parameters of the reaction (Table 1). First, we evaluated the influence of solvents on the reaction. To our satisfaction, when DMSO was employed as the solvent, the reaction indeed proceeded smoothly, affording the desired pyrazole 3a in 43% isolated yield (entry 1). In contrast, using toluene as the solvent failed to produce the target product 3a (entry 2), while the reaction conducted in DMF yielded product 3a in 45% yield (entry 3). Notably, when DMF was used as the solvent and the reaction time was reduced to 8 hours while simultaneously increasing the temperature to 130 °C, we observed a significant improvement in yield (entries 4 and 5). Further reducing the reaction time to 6 hours continued to enhance the yield to 79% (entry 6). At this temperature, compound 3a was obtained in 65% yield using DMSO as the solvent (entry 7). Subsequently, we assessed the effect of different bases on the yield of the cycloaddition reaction (entries 8–13), while further shortening the reaction time. Ultimately, we determined that using t-BuOK as the base catalyst in DMF solvent for 3 hours could provide the pyrazole compound 3a in 87% isolated yield (entry 14). Notably, when the reaction was performed in air, a reduction in the yield of 3a was observed (entry 15).
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Solvent | Temp. (°C) | Time (h) | Yieldb (%) |
| a Reaction conditions: 1a (0.2 mmol), 2 (0.24 mmol), base (3.0 equiv.), in solvent (2.0 mL), and N2.b Isolated yields.c No Reaction.d Under air. | |||||
| 1 | n-C3H7ONa | DMSO | 80 | 12 | 43 |
| 2 | n-C3H7ONa | Toluene | 80 | 12 | Trace |
| 3 | n-C3H7ONa | DMF | 80 | 12 | 45 |
| 4 | n-C3H7ONa | DMF | 80 | 8 | 47 |
| 5 | n-C3H7ONa | DMF | 130 | 8 | 77 |
| 6 | n-C3H7ONa | DMF | 130 | 6 | 79 |
| 7 | n-C3H7ONa | DMSO | 130 | 6 | 65 |
| 8 | n-C5H11OK | DMF | 130 | 6 | 59 |
| 9 | CH3ONa | DMF | 130 | 6 | 69 |
| 10 | KOH | DMF | 130 | 6 | 62 |
| 11 | KOAc | DMF | 130 | 6 | NRc |
| 12 | K2CO3 | DMF | 130 | 6 | 19 |
| 13 | t-BuOK | DMF | 130 | 6 | 67 |
| 14 | t-BuOK | DMF | 130 | 3 | 87 |
| 15d | t-BuOK | DMF | 130 | 3 | 55 |

Subsequent exploration of the substrate scope focused on elucidating the impact of N-aryl substituents in diphenyl hydrazones 1a–1s on their cyclization with (vinylsulfinyl)benzene 2 to form 1,3-diaryl-4,5-dihydro-1H-pyrazoles 3a–3s (Scheme 2). Reactions proceeded efficiently when Ar1 bore para-electron-donating groups, delivering the corresponding 1,3-diaryl-4,5-dihydro-1H-pyrazole derivatives (3b–3e) in good to excellent yields. Notably, a clear positive correlation was observed between the electron-donating strength of para-substituents and reaction efficiency (–OMe > –Et > –Me), with the methoxy-substituted substrate delivering the desired product 3d in 89% yield. However, steric hindrance became detrimental when employing bulkier electron-donating groups (–tBu), resulting in diminished yields. para-Electron-withdrawing groups also showed reduced reaction efficiency (3f–3h), indicating that both electronic and steric factors significantly influence the reaction outcome. Next, systematic evaluation of meta-substituted (3i–3l) and ortho-substituted (3m–3p) aryl groups revealed consistently higher yields for the meta-substituted derivatives regardless of their electronic nature (3i vs. 3m, 3j vs. 3o, and 3l vs. 3p), further corroborating the critical role of steric effects in governing reaction efficiency. Finally, the methodology demonstrated good compatibility with more complex substrates, including 3,5-disubstituted aryl rings (3q and 3r) and 2-naphthyl groups (3s), all of which underwent successful cyclization to afford the desired products in satisfactory yields.
 | ||
| Scheme 2 Scope of Ar1: 1a–1s (0.5 mmol), 2 (0.6 mmol), t-BuOK (3.0 equiv.), in DMF (5.0 mL), N2, 130 °C, and 3 h. | ||
Next, we expanded the substrate scope by investigating diverse Ar2 substituents in the synthesis of 1,3-diaryl-4,5-dihydro-1H-pyrazoles (Scheme 3). The reaction exhibited broad functional group tolerance, demonstrating excellent compatibility with various aromatic substituents including OMe, tBu, F, Cl, and Br. This versatility afforded the target products 3t–3ad in moderate to good yields (43–80%). Furthermore, hydrazones bearing 3,5-dimethyl- or 3,4-difluoro-substituted Ar2 groups proved to be viable substrates for this transformation, delivering the corresponding products 3ae and 3af in 69% and 48% yields, respectively. Extension to polycyclic systems revealed the methodology's adaptability, as evidenced by the successful transformation of a biphenyl-substituted hydrazone into the pyrazole derivative 3ag with 63% yield. Additionally, the protocol demonstrated remarkable heteroaromatic compatibility, with heteroaryl-substituted hydrazones efficiently undergoing cyclization to deliver product 3ah in 54% yield. Notably, when employing hydrazones bearing C-alkyl substituents (propyl group, 1ai), pyrazole 5ai was obtained directly without requiring oxidation.
 | ||
| Scheme 3 Scope of Ar2: 1t–1ai (0.5 mmol), 2 (0.6 mmol), t-BuOK (3.0 equiv.), in DMF (5.0 mL), N2, 130 °C, and 3 h. | ||
To elucidate the reaction mechanism, the process was quenched after 1 hour under standard conditions, enabling isolation of the uncyclized intermediate 4 in 20% yield. Crucially, when independently isolated intermediate 4 was subjected to the standard conditions, it afforded product 3a in 76% yield (Scheme 4B). Based on these experimental observations and precedent literature reports, a plausible mechanistic pathway is proposed in Scheme 4. The sequence initiates with deprotonation of hydrazone 1a to generate intermediate A, which subsequently combines with (vinylsulfinyl)benzene 2 to form intermediate B. Protonation of B yields isolable intermediate 4, followed by base-promoted intramolecular cyclization which afforded the cyclic intermediate C, which underwent protonation followed by sulfoxide elimination to yield pyrazole E. Final isomerization of E then delivered the target product 3a.
 | ||
| Scheme 4 Proposed reaction pathways. | ||
Subsequent treatment of 1,3-diaryl-4,5-dihydro-1H-pyrazole 3 with DDQ in dichloromethane consistently afforded the oxidized derivatives 1,3-diaryl-1H-pyrazoles 5 in good to excellent yields (Scheme 5).
 | ||
| Scheme 5 Oxidation of 1,3-diaryl-4,5-dihydro-1H-pyrazoles: 3 (0.2 mmol), DDQ (0.2 mmol), in DCM (5.0 mL), RT, and 3 h. | ||
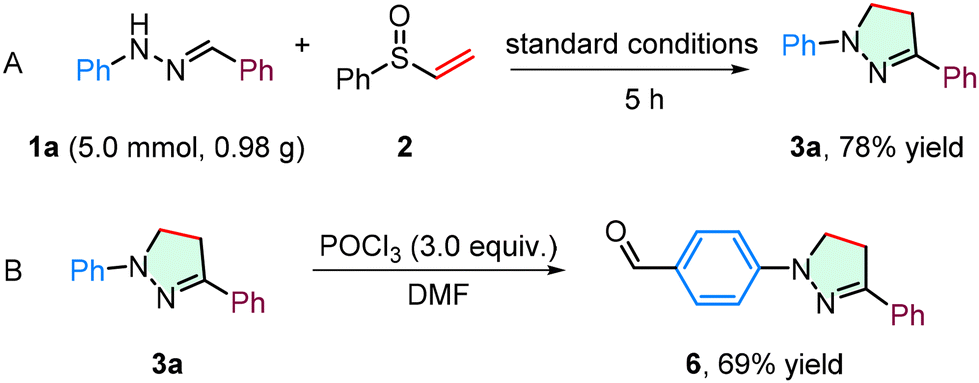
As delineated in Scheme 6A, to rigorously evaluate the scalability and synthetic utility of this methodology, we performed a gram-scale reaction using 5 mmol of starting material 1a under standard conditions. By extending the reaction time to 5 hours, the target product 3a was obtained in 78% isolated yield. Furthermore, the formylation reaction proceeded smoothly under POCl3 catalysis in DMF, successfully introducing an aldehyde functionality at the Ar1 of 3a to deliver aldehyde 6 with 69% efficiency (Scheme 6B).
 | ||
| Scheme 6 Gram-scale synthesis and synthetic transformations. | ||
Conclusions
In conclusion, this study establishes a base-catalyzed [3 + 2] cycloaddition strategy employing hydrazones and vinyl sulfoxides. The methodology enables synthesis of 1,3-diaryl-1H-pyrazoles through a concerted C–C/C–N bond-forming pathway. Experimental evaluation demonstrated broad substrate scope compatibility and gram-scale feasibility, while subsequent synthetic applications confirmed operational practicality and scalability. This protocol provides a versatile platform for rapid assembly of 1,3-diaryl-1H-pyrazole architectures, demonstrating its significant potential for accelerating discovery processes in pharmaceutical development, materials science, and agrochemical research.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
We thank the National Natural Science Foundation of China (No. 22178079) for financial support.References
- S. Fustero, M. S. Rosello, P. Barrio and A. S. Fuentes, Chem. Rev., 2011, 111, 6984–7034 CrossRef CAS PubMed
; G. R. Bebernitz, G. Argentieri, B. Battle, C. Brennan, B. Balkan, B. F. Burkey, M. Eckhardt, J. Gao, P. Kapa, R. J. Strohschein, H. F. Schuster, M. Wilson and D. D. Xu, J. Med. Chem., 2001, 44, 2601–2611 CrossRef PubMed
- T. de Paulis, K. Hemstapat, Y. Chen, Y. Zhan, S. Saleh, D. Alagille, R. M. Baldwin, G. D. Tamagnan and P. J. Conn, J. Med. Chem., 2006, 49, 3332–3344 CrossRef CAS PubMed
- L. Sun, P. Wang, L. Xu, L. Gao, J. Li and H. Piao, Bioorg. Med. Chem., 2019, 29, 1187–1193 CrossRef CAS PubMed
- K. Karrouchi, S. Radi, Y. Ramli, J. Taoufik, Y. N. Mabkhot, F. A. Aizari and M. Ansar, Molecules, 2018, 23, 134–220 CrossRef PubMed
- R. Kumar, R. Sharma and D. K. Sharma, Curr. Top. Med. Chem., 2023, 23, 2097–2115 CrossRef CAS PubMed
- M. Elkady, R. Nie, A. M. Schaible, J. Bauer, S. Luderer, G. Ambrosi, O. Werz and S. A. Laufer, J. Med. Chem., 2012, 55, 8958–8962 CrossRef CAS PubMed
- S. R. Stauffer and J. A. Katzenellenbogen, J. Comb. Chem., 2000, 2, 318–329 CrossRef CAS PubMed
- S. Shaikh, P. Dhavan, G. Pavale, M. M. V. Ramana and B. L. Jadhav, Bioorg. Chem., 2020, 96, 103589 CrossRef CAS PubMed
- B. E. Fink, D. S. Mortensen, S. R. Stauffer, Z. D. Aron and J. A. Katzenellenbogen, Chem. Biol., 1999, 6, 205–219 CrossRef CAS PubMed
- S. T. Heller and S. R. Natarajan, Org. Lett., 2006, 8, 2675–2678 CrossRef CAS PubMed
- L. O'Sullivan, K. V. Patel, B. C. Rowley, D. K. Brownsey, E. Gorobets, B. S. Gelfand, J. F. Van Humbeck and D. J. Derksen, J. Org. Chem., 2022, 87, 846–854 CrossRef PubMed
- Y. Zheng, Y. Long, H. Gong, J. Xu, C. Zhang, H. Fu, X. Zheng, H. Chen and R. Li, Org. Lett., 2022, 24, 3878–3883 CrossRef CAS PubMed
- P. Kukreti, R. Chauhan, A. Panwar and K. Ghosh, Org. Biomol. Chem., 2024, 22, 8273–8278 RSC
- W. Xu, H. Dang, H. Sheng, J. Shen and M. Wang, Org. Biomol. Chem., 2025, 23, 3836–3840 RSC
- P. Zhou, W. T. Yang, W. J. Hao and B. Jiang, Org. Lett., 2023, 25, 8495–8500 CrossRef CAS PubMed
- On the reactivity of phenyl vinylsulfoxide, see: O. De Lucchi, G. Licini, L. B. Krasnova and A. K. Yudin, in eEROS Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Chichester, UK, 2008, DOI:10.1002/047084289X.rp103.pub2
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ob00907c |
| This journal is © The Royal Society of Chemistry 2025 |