
A carrier-free nano-photosensitizer for red light-activated NO release and combination therapy
Chaonan Lia,
Huixuan Qi b,
Jinjun Jiac,
Qingxia Lin*c and
Wenhai Lin*b
b,
Jinjun Jiac,
Qingxia Lin*c and
Wenhai Lin*b
aSchool of Environment and Biological Engineering, Wuhan Technology and Business University, Hongshan District, Wuhan 430065, China
bBiomedical Polymers Laboratory, College of Chemistry, Chemical Engineering and Materials Science, and State Key Laboratory of Radiation Medicine and Protection, Soochow University, Suzhou 215123, China. E-mail: whlin@suda.edu.cn
cDepartment of Psychiatry, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou Medical University, Wenzhou 325003, China. E-mail: 380364613@qq.com
First published on 12th August 2025
Abstract
In this work, multifunctional nanoparticles (A-PIB NPs) constructed by a nitric oxide (NO) donor, L-arginine, and a boron dipyrromethene derivative were used for red light-responsive and spatiotemporally-controllable NO release. A-PIB NPs could produce singlet oxygen and NO under 630 nm light irradiation, achieving controlled release of NO and being used for combination therapy of tumors.
Nitric oxide (NO), a ubiquitous gaseous signaling molecule, has emerged as a promising antitumor agent because of its multifaceted biological activities.1 NO can induce vasodilation, inhibit cell proliferation, and trigger apoptosis in cancer cells.2 However, the extremely short half-life of NO in biological systems poses a critical challenge for its safe, effective, and precise delivery and release.3
An ideal NO delivery system must demonstrate exceptional performance in multiple critical aspects. First, the selection of donor molecules is paramount.4 Traditional donor molecules such as organic nitrate (RONO2) and diazeniumdiolates (NONOates) lack the spatial and temporal control of NO delivery, leading to off-target effects and reduced therapeutic efficacy.5 L-Arginine is an endogenous NO donor that can undergo non-enzymatic reactions with reactive oxygen species (ROS) such as hydrogen peroxide (H2O2) and singlet oxygen (1O2) to generate NO, enabling the controlled release of NO.6 The choice of control method for NO release is crucial.7 Among the numerous triggering methods, using red light has become an attractive triggering approach because of its good tissue penetration, low toxicity, and non-invasive manipulation in biological tissues.8 The final issue is the delivery of L-arginine.9 Combining L-arginine with nano-drug delivery systems can significantly increase circulation time and improve the accumulation of L-arginine in tumors.10 However, the complexity, non-repeatability, and potential toxicity of the carrier in the preparation process of nanomaterials hinder the further application of NO therapy.11 Therefore, conjugating L-arginine with other functional molecules enables the novel design of controlled NO release.12
In this work, under red light irradiation, a carrier-free nano-photosensitizer that could control NO release and produce ROS was constructed for combination therapy (Scheme 1). The boron dipyrromethene (BODIPY) derivative (PIB) was used as a red light-responsive module for ROS generation.13 The hydrophilicity of PIB was improved by bonding with triethylene glycol (TEG) and the NO donor, L-arginine, which promoted self-assembly to form stable nanoparticles (A-PIB NPs).14 Under 630 nm light irradiation, A-PIB NPs generated 1O2 to trigger NO production for the combination of gas and photodynamic therapy (PDT).
 | ||
| Scheme 1 Schematic diagram of the formation of A-PIB NPs and the antitumor effects of 1O2 and NO. | ||
The target molecule A-PIB was successfully synthesized according to the synthesis route shown in Fig. 1. The iodinated BODIPY derivative (IB) was prepared according to our previous work.15 PIB was synthesized via a condensation reaction of IB and MEB. Subsequently, PIB was further bound to L-arginine through an esterification reaction to obtain A-PIB. The successful synthesis of PIB and A-PIB was demonstrated through 1H nuclear magnetic resonance (NMR, Fig. S1 and S2), 13C NMR (Fig. S3 and S4) and high-resolution mass spectrometry (HRMS, Fig. S5 and S6). PIB NPs and A-PIB NPs were prepared from PIB and A-PIB, respectively, through the nanoprecipitation method. After centrifugation, PIB NPs produced a large amount of precipitate, while A-PIB NPs showed good homogeneity. This result indicated that the stable nanoparticles could be obtained by introducing L-arginine.
 | ||
| Fig. 1 The synthetic route of A-PIB and the preparation of A-PIB NPs. | ||
The maximum absorption (Fig. 2a) and emission wavelengths (Fig. 2b) of A-PIB in DMF were 660 and 700 nm, respectively. However, A-PIB NPs presented an enlarged absorption spectrum, a redshift in the maximum absorption wavelength to 700 nm, and fluorescence quenching due to the aggregation effect. The average hydrated particle sizes of A-PIB NPs in phosphate-buffered saline (PBS) and Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) were measured via dynamic light scattering (DLS) as shown in Fig. 2c. The sizes were 207 and 163 nm, respectively. The difference in hydration particle size is due mainly to the influence of small particles of approximately 10 nm in size in FBS. The size and morphology of A-PIB NPs measured by transmission electron microscopy (TEM) were uniform and spherical, respectively. The sizes of A-PIB NPs in PBS and DMEM supplemented with 10% FBS remained almost unchanged for 7 days (Fig. 2d), indicating that A-PIB NPs had excellent colloidal stability in the simulated physiological environment in vitro.
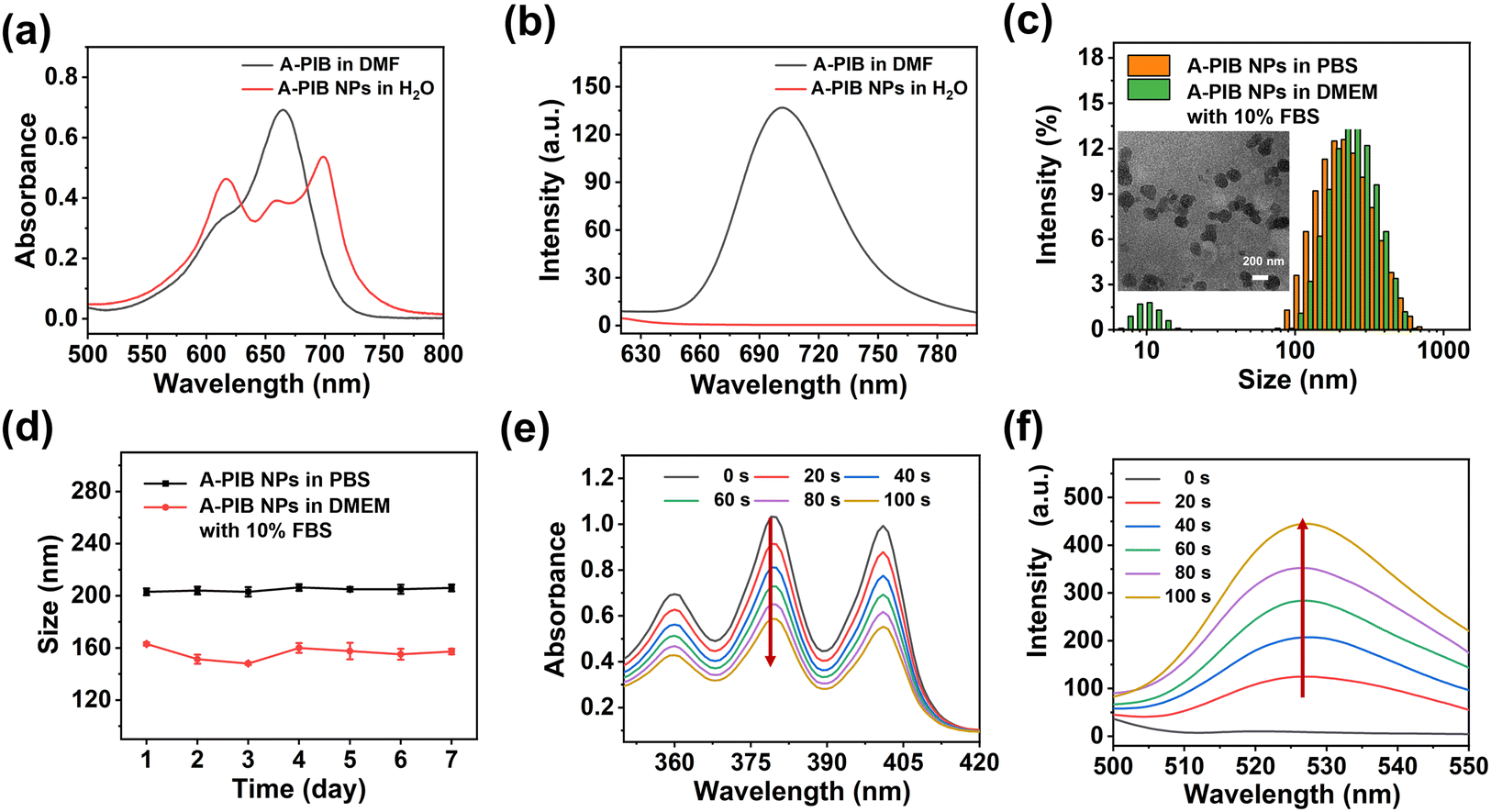 | ||
| Fig. 2 (a) Absorption and (b) emission spectra of A-PIB in DMF and A-PIB NPs in H2O. (c) Sizes measured by DLS of A-PIB NPs in PBS and DMEM supplemented with 10% FBS. Inserted image: TEM images of A-PIB NPs. (d) Average sizes of A-PIB NPs in PBS and DMEM supplemented with 10% FBS for 7 days. (e) Changes in the absorbance of ABDA and (f) fluorescence of DAF-FM DA with A-PIB NPs in PBS under red light irradiation (630 nm, 25 mW cm−2). | ||
We evaluated the photodynamic performance of A-PIB and its ability to generate NO upon red light activation. First, 1,3-diphenylisobenzofuran (DPBF) as a ROS detection reagent was used to evaluate the ROS generation ability of methylene blue (MB) and A-PIB. As shown in Fig. S7, under 630 nm light irradiation, A-PIB continuously generated ROS to degrade DPBF, while MB failed to effectively produce ROS after 20 s of irradiation due to its poor photostability. These data indicated that A-PIB exhibited superior photodynamic performance compared with the classical photosensitizer MB. To verify the types of ROS, such as 1O2, the superoxide anion (O2˙−) and the hydroxyl radical (OH−) produced by light-induced A-PIB, electron spin resonance (ESR) was employed with 2,2,6,6-tetramethylpiperidine (TEMP) and 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as the spin trapping agents. There was a strong ESR signal in the detection of 1O2 (Fig. S8a) when A-PIB was irradiated with 630 nm light for 10 min, but there was no EPR signal in the detection of O2˙− (Fig. S8b) or OH− (Fig. S8c). These results showed that A-PIB could generate 1O2 under red light irradiation. Subsequently, 9,10-anthracenediyl-bis(methylene)dimalonic acid (ABDA) was selected to detect the 1O2 generation ability of A-PIB NPs in PBS. As shown in Fig. 2e and S9a, the characteristic absorption peak at 379 nm of ABDA gradually decreased with prolonged illumination time, which indicated that A-PIB NPs could generate 1O2 under 630 nm light irradiation. Next, we evaluated the generation of NO using the Griess assay. As shown in Fig. S9b, the production of NO enhanced with increasing light irradiation time. Finally, 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate (DAF-FM DA) was used to detect the NO generation ability of A-PIB NPs. As shown in Fig. 2f and S9c, the fluorescence intensity of DAF-FM DA gradually increased under 630 nm light irradiation, which further indicated that A-PIB NPs could generate NO.
Human cervical carcinoma (HeLa) cells were incubated with A-PIB NPs for 0.5 and 2 h. As shown in Fig. 3a, a small amount of red fluorescence appeared in the cytoplasm, indicating that A-PIB NPs could be taken up by cells. As the incubation time increased, the intensity of the red fluorescence gradually increased. These results showed that the endocytosis process of A-PIB NPs by cells was time-dependent. 2,7-Dichlorodihydrofluorescein diacetate (DCFH-DA) and DAF-FM DA were commonly-used reagents for the detection of ROS and NO levels in cells, respectively. In the presence of 1O2 and NO, non-fluorescent DCFH-DA and DAF-FM DA were oxidized to green fluorescent DCF and DAF, respectively. Accordingly, there was little fluorescence in the cells in the absence of light (Fig. 3b). After red light irradiation for 5 min, the cells showed distinct green fluorescence because A-PIB NPs with light irradiation could produce 1O2 and NO in the cells. Finally, we further conducted experiments to verify the cytotoxicity of A-PIB NPs. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was used to detect the toxicity of A-PIB NPs to HeLa cells. When the concentration of A-PIB NPs was 15 μM, almost all the cells survived, indicating that A-PIB NPs had good biocompatibility (Fig. S10a). Under 630 nm light irradiation (25 mW cm−2, 10 min), the viability of the cells gradually decreased with increasing concentrations of A-PIB NPs (Fig. 3c). In contrast, the viability of the cells treated with A-PIB NPs (1 μM) was 34%, while the viability of the cells treated with PIB NPs (1 μM) was 80%, which demonstrated that A-PIB NPs with 630 nm light irradiation presented stronger phototoxicity than PIB NPs (Fig. S10b). The half maximal inhibitory concentration (IC50) of A-PIB NPs was calculated to be 0.73 μM under 630 nm light irradiation for 10 min. The results of the live/dead cell staining experiments were essentially consistent with those of the MTT assay (Fig. 3d). At an A-PIB NP concentration of 0.2 μM, all the cells showed green fluorescence, indicating that the cells were alive. Under red light irradiation, as the concentration of A-PIB NPs gradually increased, the red fluorescence representing dead cells also gradually increased, whereas the green fluorescence representing live cells gradually decreased. At an A-PIB NP concentration of 2 μM, negligible green fluorescence was observed, indicating that the cells died under red light irradiation. Finally, we evaluated the photophysical properties of A-PIB NPs before and after light illumination (630 nm, 25 mW cm−2, 10 min). The A-PIB NPs under light irradiation for 10 min were designated as A-PIB NPs(L). A-PIB NPs(L) exhibited the same absorbance as A-PIB NPs without light irradiation (Fig. S11a). The emission spectra of both NPs in DMF were basically the same (Fig. S11b). The A-PIB NPs(L) still maintained similar photodynamic performance (Fig. S11c) and photocytotoxicity (Fig. S11d) to those of A-PIB NPs. The above experimental results indicated that A-PIB NPs could be endocytosed by cells and produce 1O2 and NO under red light irradiation, ultimately resulting in good antitumor performance.
 | ||
| Fig. 3 (a) CLSM images of HeLa cells treated with A-PIB NPs at 37 °C for 0.5 and 2 h. Scale bar, 20 μm. (b) 1O2 and NO generation in HeLa cells. The concentration of A-PIB NPs was 5 μM. Scale bar, 100 μm. (c) Cell viability of HeLa cells treated with A-PIB NPs at different concentrations under red light irradiation. (d) Live/dead staining images of HeLa cells treated with A-PIB NPs at different concentrations under light irradiation (630 nm light, 25 mW cm−2, 10 min). Scale bar, 100 μm. | ||
We successfully developed carrier-free A-PIB NPs via the conjugation of BODIPY and L-arginine, achieving a combined antitumor strategy involving red light-activated release of NO and PDT. A-PIB NPs produced 1O2 under 630 nm light irradiation and further oxidized L-arginine, achieving spatiotemporal release of NO. This design overcomes the limitations of traditional NO delivery systems that lack controllable regulation while avoiding the use of complex carrier materials, significantly improving stability. A-PIB NPs exhibited excellent antitumor properties by generating 1O2 and NO under red light irradiation. This work provides a powerful tool for red light-responsive NO release and tumor therapy.
Conflicts of interest
There are no conflicts to declare.Data availability
All data are available from the corresponding authors upon request.Supplementary information available: materials, characterization methods and experiments. See DOI: https://doi.org/10.1039/d5ob00959f.
Acknowledgements
This work was supported by the Scientific Research Project of the Education Department of Hubei Province (B2023319) and the Doctoral Fund of Wuhan Business and Technology University (D2023004).References
-
(a) X. Chen, Q. Fan, K. Li, W. Li, L. Wang, W. Li and W. Hong, Biomater. Sci., 2024, 12, 964–977 RSC
; (b) Y. Tang, Q. Li, Z. Zhou, H. Bai, N. Xiao, J. Xie and C. Li, J. Nanobiotechnol., 2024, 22, 674 CrossRef PubMed
-
(a) R. Liu, B. Xu, Z. Ma, H. Ye, X. Guan, Y. Ke, Z. Xiang and Q. Shi, RSC Adv., 2022, 12, 32355–32364 RSC
-
(a) J. Hou, R. Fu, T. Yu, P. Ge, Y. Wang, M. Zhao, A. Zou and Y. Xianyu, Nano Today, 2024, 54, 102118 CrossRef CAS
-
(a) J. Cheng, G. Gan, S. Zheng, G. Zhang, C. Zhu, S. Liu and J. Hu, Nat. Commun., 2023, 14, 7510 CrossRef CAS PubMed
-
(a) Y. Duan, Y. Wang, X. Li, G. Zhang, G. Zhang and J. Hu, Chem. Sci., 2020, 11, 186–194 RSC
-
(a) Y. Zhao, Y. Liu, R. Liao, P. Ran, Y. Liu, Z. Li, J. Shao and L. Zhao, ACS Appl. Mater. Interfaces, 2024, 16, 3147–3161 CrossRef CAS PubMed
-
(a) M. Huang, J. Zhang, X. Ke, S. Gao, D. Wu, J. Chen and Y. Weng, RSC Adv., 2022, 12, 2383–2390 RSC
-
(a) L. Gao, J. Cheng, Z. Shen, G. Zhang, S. Liu and J. Hu, Angew. Chem., Int. Ed., 2022, 61, e202112782 CrossRef CAS PubMed
- H. Tan, S. Wang, X. He, G. Yang, Y. Zhu, S. Yang, S. Yan, C. Gong, W. Bai, Y. Hu, J. Song and L. Zheng, ACS Nano, 2025, 19, 9390–9411 CrossRef CAS PubMed
-
(a) Y. Huang, Z. Wu, H. Wang, H. An, J. Zhang and Z. Bao, Mater. Chem. Front., 2025, 9, 204–222 RSC
- Z. Xu, Q. Luo, Y. He, Y. He, X. Zhang, J. Wang, S. Ni, D. Gao and D. Wang, Adv. Funct. Mater., 2024, 34, 2314536 CrossRef CAS
- L. Xie, L. Wang, L. Li, C. Liu, L. Guo, Y. Liao, S. Zhou, W. Wu, Y. Duo, L. Shi and M. Yuan, ACS Appl. Mater. Interfaces, 2024, 16, 5683–5695 CrossRef CAS PubMed
-
(a) J. Wang, Q. Gong, L. Jiao and E. Hao, Coord. Chem.
Rev., 2023, 496, 215367 CrossRef CAS
- J. Xie, Y. Wang, W. Choi, P. Jangili, Y. Ge, Y. Xu, J. Kang, L. Liu, B. Zhang, Z. Xie, J. He, N. Xie, G. Nie, H. Zhang and J. S. Kim, Chem. Soc. Rev., 2021, 50, 9152–9201 RSC
- C. Li, W. Lin, S. Liu, W. Zhang and Z. Xie, J. Mater. Chem. B, 2019, 7, 4655–4660 RSC
| This journal is © The Royal Society of Chemistry 2025 |