
Microwave-assisted direct amidation of thioesters: a green approach
Han Cao *,
Yijun Shi,
Peng Yan,
Fusheng Bie* and
Jie Ma*
*,
Yijun Shi,
Peng Yan,
Fusheng Bie* and
Jie Ma*
College of Chemistry, Chemical Engineering and Materials Science, Zaozhuang University, 1 Bei'an Road, Zaozhuang, Shandong 277160, China. E-mail: hcao@uzz.edu.cn; fsbie@uzz.edu.cn; jiemacn@163.com
First published on 5th August 2025
Abstract
The direct amidation of thioesters represents an essentially nonclassical route in the synthesis of amides, frequently encountering limitations due to poor compatibility between thioester and amine structures as well as poor atom economy. Herein, we describe a green, efficient and microwave-assisted protocol that not only facilitates high-yielding amide bond formation but also converts the sulfur-containing byproducts into valuable disulfides, significantly enhancing the atom economy. Mechanistic studies establish two concurrent pathways: acyl nucleophilic addition–elimination and oxygen-mediated radical oxidation of the sulfur group. Notably, the protocol has been successfully applied to the synthesis of 64 amides (including difluoromethyl amides and β-mercaptoamides) and ten disulfides with moderate to excellent yields and good functional group tolerance. This work provides a novel and sustainable alternative to traditional amide synthesis methods, offering significant potential for applications in pharmaceutical and materials chemistry.
Introduction
The amide bond is a fundamental structural motif in both biology and chemistry, serving as the key linkage in peptides, proteins, and numerous polymeric materials essential for life processes.1 Amide bonds are prevalent in small molecules, particularly in a wide range of pharmaceuticals.2 For example, difluoro- or trifluoroacetamides3 and β-mercaptoamides4 have attracted the interest of synthetic chemists over recent decades. So far, the synthesis and conversion of amides remains an alluring yet highly challenging undertaking.5The classical route to prepare amide bonds involves the coupling of carboxylic acids and amines, which typically requires activation by coupling reagents.6 These are notably prevalent, but several drawbacks, including poor atom economy and the generation of harmful or toxic waste, have led the ACS GCIPR to identify amide formation as a key area in green chemistry research within organic synthesis.7
To combat the limitations of the classical route, an alternative approach utilizing carboxylic acid surrogates with amines for the preparation of amides has gradually been developed.8 For example, the use of esters8,9 is one of the alternatives. The primary advantage of carboxylic acid esterification is that it renders the carboxyl group more electrophilic and will facilitate the amidation reaction. Recently, there has been interesting progress in utilizing oxoesters for amide construction through the application of versatile reagents or catalysts.10 Compared with oxoesters, thioesters are more susceptible to nucleophilic attack because the poor orbital overlap between the 3p orbital of sulfur and the 2p orbital of oxygen limits the extent of electron delocalization from sulfur to the carbonyl group. This makes thioesters excellent acyl transfer reagents and sulfur sources, which has led to the development of several amidation methods for thioesters, including the amidation of thioesters via thioacids,11 native chemical ligation (NCL)12 and amidation of non-typical thioesters and amines13 (Fig. 1A). These methods highlight the significant potential of thioesters in constructing amide bonds. However, several limitations are apparent: thioacids are challenging to prepare, there is a limited scope of thioesters and amines, and the atom economy is suboptimal—particularly regarding sulphur group utilization.
 | ||
| Fig. 1 Strategies for amide bond formation using thioesters, the strategy of disulfide construction, and our work for both amides and disulfides. | ||
Organic disulfides14 are among the most widely utilized organosulfur reagents, exhibiting significant value in organic synthesis. The general approach for the synthesis of disulfides involves the oxidation of thiols.14c Additionally, disulfides can be reduced to mercaptans using appropriate reducing agents (Fig. 1B).14d Interestingly, the synthesis of disulfides by the conversion of thioesters has been previously reported.15 We hypothesize that incorporating disulfide byproduct generation into thioester amidation processes may improve the atom utilization. As part of our continual interest in the development of thioesters as building blocks and green chemistry,16 we aimed to develop a more efficient strategy for the amidation of thioesters characterized by a broader substrate scope, facile access to starting materials, minimal waste production, and enhanced atom economy (Fig. 1C).
Results and discussion
This design concept emerged unexpectedly during our exploration of the functionalization reactions of thioesters. We serendipitously discovered that morpholine significantly catalyzes the transformation of thioesters into disulfide compounds, which piqued our interest. This unexpected finding motivated us to expand the scope of amines to encompass a wide range of primary and secondary amines, including both aromatic and aliphatic derivatives. As illustrated in Scheme 1, the data demonstrate that six-membered heterocyclic aliphatic secondary amines exhibit remarkable efficacy in inducing the conversion of thioesters to disulfides. Considering the cost of the reagents, morpholine was chosen as the focus for our subsequent investigations.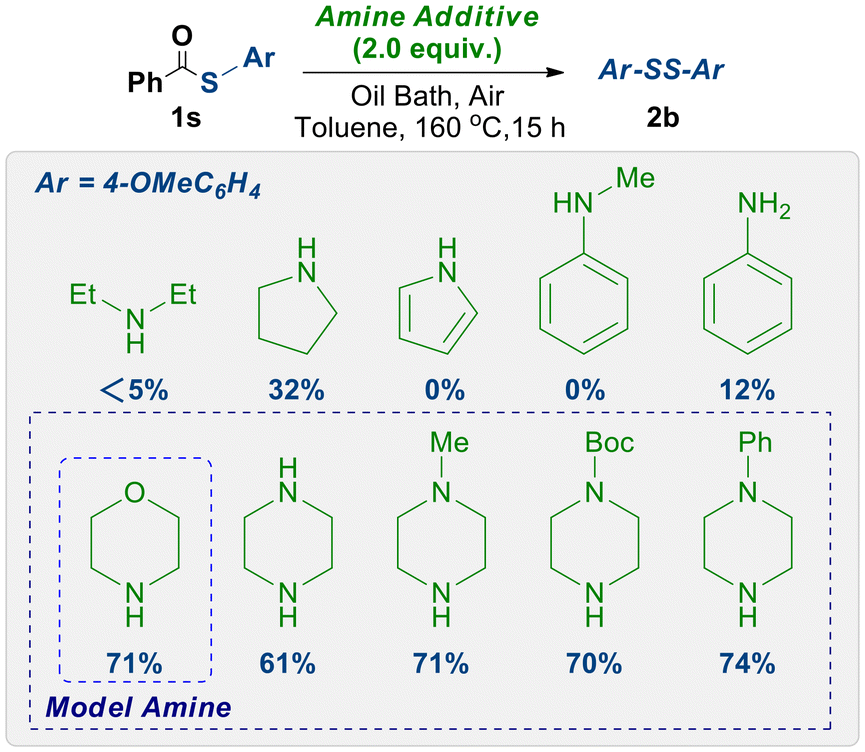 | ||
| Scheme 1 Comparison of amine additives in the synthesis of 2b. | ||
The amidation of thioesters was investigated using S-(4-methoxyphenyl) benzothioate (1s) and morpholine as model substrates. To our delight, we observed that the amidation of thioesters in the presence of morpholine (2.0 equiv.) with 4-dimethylaminopyridine (DMAP, 1.5 equiv.) as a base, via microwave radiation under solvent-free conditions in an air atmosphere at 120 °C for 5 min, delivered the desired products, both in 98% yield. The optimization results are summarized in Table 1.
|
|||||
|---|---|---|---|---|---|
| Entry | Morpholine | Solvent | Base | 2b yieldb (%) | 3a yieldb (%) |
| a Determined by 1H NMR and/or GC-MS analysis, 0.2 mmol of thioester, 0.4 mmol of morpholine, 0.5 mL of solvent at 160 °C in an oil bath in an air atmosphere for 15 h.b Microwave assisted, 0.2 mmol of thioester, 0.4 mmol of morpholine, 0.4 mmol of base, solvent-free in an air atmosphere at 120 °C for 5 min.c 0.2 mmol of morpholine.d 0.3 mmol of base.e 0.2 mmol of base.f At 100 °C. | |||||
| 1a | — | Toluene | — | 0 | 0 |
| 2a | Morpholine | Toluene | — | 72 | 79 |
| 3a | Morpholine | Dioxane | — | 59 | 76 |
| 4 | Morpholine | — | — | 77 | 87 |
| 5 | Morpholine | — | Et3N | 77 | 88 |
| 6 | Morpholine | — | Pyridine | 69 | 83 |
| 7 | Morpholine | — | DBU | 50 | 54 |
| 8 | Morpholine | — | DABCO | 81 | 98 |
| 9 | Morpholine | — | DMAP | 98 | 98 |
| 10c | Morpholine | — | DMAP | 88 | 90 |
| 11d | Morpholine | — | DMAP | 98 | 98 |
| 12e | Morpholine | — | DMAP | 82 | 93 |
| 13d,f | Morpholine | — | DMAP | 86 | 98 |

With the optimal conditions established, the scope of the reaction with thioesters was explored. As shown in Scheme 2, various substituted S-phenyl thioesters and morpholine were suitable substrates, providing both disulfide and amide in moderate to excellent yields. A wide range of electronically diverse aromatic acyl moieties underwent amidation with morpholine (3a–k), including electron-neutral (3a, 3c), electron-rich (3b), electron-deficient (3d, 3g), halide-functionalized (3e, 3f, 3k), ketone-containing (3h) and sulfonamide-containing substrates (3i). Sterically hindered substrates were also well tolerated (3j).
 | ||
| Scheme 2 Amidation of different thioesters with morpholine. | ||
Subsequently, a range of aliphatic acyl moieties of thioesters underwent amidation with morpholine, including linear, branched, and cyclic substrates (3l–3o), resulting in moderate to excellent yields. Furthermore, fluorinated aliphatic structures demonstrated moderate to good tolerance (3p–3q). Notably, the amidation of a styryl-thioester produced products that retained both thiol and acyl functionality, highlighting the method's atom economy (3r). The results in Scheme 2 indicate that S-phenyl butanethioate is the optimal model for the highly efficient synthesis of diphenyl disulfide. To further demonstrate the synthetic utility of this amidation reaction to prepare disulfides, we successfully synthesized various disulfides, including aromatic (2a–2f) and aliphatic (2g–2j) disulfides, with yields ranging from moderate to excellent, by altering the thiol groups based on S-phenyl butanethioate.
Then, two sets of competitive experiments were carried out (Scheme 3). Experiment A examined the relative reactivity in the preparation of a disulfide from thioester and mercaptan substrates in an identical reaction system. Experiment B explored the selectivity of thioester conversion to a symmetric disulfide as well as the potential for forming an asymmetric disulfide. According to the results from Schemes 2 and 3, the thioester exhibits superior performance compared to the thiophenol in the synthesis of a disulfide within the same reaction system. The preparation of symmetrical aryl disulfides with electron-donating groups demonstrates greater advantages over those with electron-withdrawing groups. However, this method is not suitable for the synthesis of asymmetric disulfide compounds.
 | ||
| Scheme 3 Competition experiments for the convenient transformation of thioesters into disulfides. | ||
Next, we sought to broaden the scope of amines, encompassing both aromatic and aliphatic varieties, to further demonstrate the versatility of this amidation strategy. Subsequent experiments were conducted using 1s and aniline as a model, with reoptimized reaction conditions. Selected results are summarized in Table 2. The findings revealed that with the utilization of aniline (2.0 equiv.) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, 2.0 equiv.) as a base, the reaction proceeds efficiently at 120 °C for 15 min in an air atmosphere under microwave irradiation without solvent, yielding the desired products in an excellent 98% yield.
|
||||
|---|---|---|---|---|
| Entry | Aniline | Base | 2b yielda (%) | 3aa yielda (%) |
| a Determined by 1H NMR and/or GC-MS analysis, microwave-assisted, 0.2 mmol of thioester, 0.3 mmol of base, 0.4 mmol of aniline, solvent-free in an air atmosphere at 120 °C for 5 min.b 15 min.c 0.4 mmol of base.d At 100 °C. | ||||
| 1 | Aniline | DABCO | 4 | 6 |
| 2 | Aniline | Et3N | 12 | 15 |
| 3 | Aniline | DBU | 34 | 35 |
| 4 | Aniline | DMAP | 14 | 16 |
| 5b | Aniline | DBU | 68 | 70 |
| 6b,c | Aniline | DBU | 98 | 98 |
| 7b,d | Aniline | DBU | 68 | 71 |

Having identified the optimal conditions, we systematically explored the substrate scope of this method. Initially, 1a was chosen as the preferred substrate to investigate the reactivity of various amines (as shown in Schemes 4A and B). Anilines bearing electron-rich substituents reacted efficiently with 1a, yielding products (3ab–3ag) in moderate to excellent yields. Specifically, p-substituted anilines provided high yields of up to 92% (3ab, 3ae), while m- and o-substituted anilines resulted in moderate yields ranging from 62% to 87% (3ac, 3ad, 3af, 3ag). This discrepancy can be attributed to the fact that the p-alkoxy group enhances the nucleophilicity of the nitrogen atom. Conversely, the steric hindrance caused by the o-methoxy group and the electron-withdrawing effect of the m-methoxy groups reduce the nucleophilicity of the aniline.
 | ||
| Scheme 4 Amidation of thioesters with various amines. | ||
The secondary aromatic amine (N-methylaniline) afforded 3ah in 79% yield. Additionally, primary and secondary aliphatic amines were compatible substrates, including benzylamine, β-phenylethylamine, linear or branched butylamines, amino acid esters and pentacyclic secondary amines, delivering products (3ai–3ao) in yields ranging from 60% to 91%. Notably, N,O-dimethylhydroxylamine was also tolerated, enabling synthesis and validating the practicality of the microwave-assisted green protocol.
To comprehensively evaluate the substrate scope, we targeted the synthesis of structurally diverse α,α-difluoromethyl amides and biologically important β-mercaptoamides.
Organofluorine compounds containing a difluoromethyl group (CHF2) have attracted significant interest due to their unique biological and physical properties, including enhanced hydrogen-bonding ability, metabolic stability, and lipophilicity compared to non-fluorinated analogues. In Scheme 5A, difluoromethyl thioesters (1p) are used as acyl donors under reaction conditions B. A range of amines, including substituted anilines (3pa–3pi), secondary aromatic amines (3pj), and primary aliphatic amines (3pk, 3pl), were successfully employed for direct difluoromethylation. The yields obtained were directly correlated with the nucleophilicity of the respective amines.
 | ||
| Scheme 5 Typical synthetic applications of thioester amidation. | ||
The β-mercaptoamide structure is prevalent in bioactive compounds and holds significant importance in pharmaceutical chemistry. Traditionally, thio-Michael addition has been used to produce such compounds by adding thiols to α,β-unsaturated carbonyl compounds. Under reaction conditions A, α,β-unsaturated thioesters can undergo intramolecular thio-Michael addition to form β-mercaptoamine structures.
As illustrated in Scheme 5B, a variety of S-phenyl-3-substituted phenylprop-2-enethioates (1r) were examined with aniline. Substrates with electron-neutral (3ra, 3rf), electron-rich (3rb), electron-deficient (3re), and halide-functionalized groups (3rc, 3rd) at the para-position were well tolerated. Next, several S-substituted phenyl-3-phenylprop-2-enethioates (1r) were tested with primary anilines, yielding products (3rg–3rj) in moderate to excellent yields. Finally, S-phenyl-3-phenylprop-2-enethioates (1r) were reacted with various amines, including secondary anilines (3rk), heterocyclic aromatic amines (3rl), linear or branched butylamines (3rm–3ro), benzylamine (3rp), β-phenylethylamine (3rq) and N,O-dimethylhydroxylamine (3rr). However, the reaction yields of 4-nitroaniline, N-methylaniline, and 2-aminopyridine were notably lower, possibly due to solubility issues, steric effects, or the nucleophilicity of the amino group.
After optimizing the reaction conditions and affirming the general applicability of this thioester amidation protocol, we sought to elucidate the mechanistic pathway of the microwave-assisted one-pot synthesis of disulfides and amides via thioester amidation.
The mechanism studies (Scheme 6A) revealed that when the reaction was performed under an argon atmosphere, the yield of disulfide decreased significantly while the amide formation remained unaffected. This observation demonstrates that atmospheric oxygen is essential for disulfide generation, with the residual 16% disulfide product likely attributable to trace air remaining in the system after argon purging. Furthermore, the addition of TEMPO completely inhibited disulfide formation without affecting amide production. The radical trapping experiment indicates that a single electron transfer (SET) may be involved in this reaction.
 | ||
| Scheme 6 Mechanism study and a plausible mechanism for the amidation of thioesters. | ||
Based on the above evidence, we propose a plausible mechanism for the microwave-promoted thioester amidation (Scheme 6B). The thioester undergoes a classical nucleophilic addition–elimination process with the amine nucleophile under standard conditions (DMAP or DBU), yielding the amide products 3. The eliminated sulfur group exists as RS− in the alkaline environment, which then undergoes oxidation by oxygen via a radical process.17 The RS˙ radicals subsequently dimerize to form the disulfide compound 2. Microwave assistance accelerated the oxidation process of the RS−.
Conclusions
In summary, we have developed a novel microwave-assisted thioester amidation strategy that enables simultaneous amide bond construction and generation of valuable disulfide byproducts under metal-free, solvent-free conditions. This unique approach addresses two critical limitations of conventional amide synthesis: (1) it eliminates the need for coupling reagents while achieving excellent yields (up to 98%) within 5–15 minutes through a single electron transfer pathway; (2) it transforms traditionally wasted sulfur groups into synthetically useful disulfides, thereby significantly improving the atom economy. The protocol demonstrates exceptional versatility, enabling efficient C–N bond formation across diverse thioesters and amine partners, with particular success in constructing α,α-difluoromethyl amides and β-mercaptoamides. Mechanistic studies reveal an oxygen-dependent radical process for disulfide formation that is uniquely accelerated by microwave irradiation. Beyond its operational simplicity and environmental compatibility, this dual-functional methodology opens new possibilities for peptide chemistry applications and further exploration of radical-mediated amidation mechanisms, establishing a promising platform for sustainable amide bond formation. We will continue to focus on the amidation of thioesters and anticipate making further discoveries in the functional applications of thioesters through this novel strategy.Author contributions
H. Cao: conceived and designed the study, writing original draft and editing; F. Bie and J. Ma: writing-review and editing; H. Cao, Y. Shi, F. Bie and J. Ma: performed the synthetic experiments and analyzed the data for all compounds; P. Yan: performed the NMR, LCMS and HRMS analyses and data analysis.Conflicts of interest
There are no conflicts to declare.Data availability
Supplementary Information (SI) available: Experimental procedures and characterization data (NMR and HRMS spectra). See DOI: https://doi.org/10.1039/d5ob01014d.Acknowledgements
This research received no external funding. The Bruker 400 MHz spectrometer used in this study was supported by College of Chemistry, Chemical Engineering and Materials Science at Zaozhuang University.References
-
(a) T. Wieland and M. Bodanszky, The World of Peptides: A Brief History of Peptide Chemistry, Springer, Berlin, 1991 CrossRef
; (b) A. Greenberg, C. M. Breneman and J. F. Liebman, The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science, Wiley, New York, 2000 Search PubMed
-
(a) H. Laronha, I. Carpinteiro, J. Portugal, A. Azul, M. Polido, K. T. Petrova, M. Salema-Oom and J. Caldeira, Biomolecules, 2020, 10, 717 CrossRef CAS PubMed
-
(a) L. E. Zimmer, C. Sparr and R. Gilmour, Angew. Chem., Int. Ed., 2011, 50, 11860–11871 CrossRef CAS PubMed
-
(a) E. Oueis, F. Nachon, C. Sabot and P. Y. Renard, Chem. Commun., 2014, 50, 2043–2045 RSC
-
(a) Y. Cai, Y. Zhao, K. Tang, H. Zhang, X. Mo, J. Chen and Y. Huang, Nat. Commun., 2024, 15, 496 CrossRef CAS PubMed
-
(a) V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS PubMed
-
(a) D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer, Jr., R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaksh and T. Y. Zhang, Green Chem., 2007, 9, 411–420 RSC
- R. M. de Figueiredo, J. S. Suppo and J. M. Campagne, Chem. Rev., 2016, 116, 12029–12122 CrossRef CAS PubMed
- A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557–6602 CrossRef CAS PubMed
-
(a) T. B. Halima, J. K. Vandavasi, M. Shkoor and S. G. Newman, ACS Catal., 2017, 7, 2176–2180 CrossRef
-
(a) S. Ning, K. Sreenivas, G. Rachel and J. W. Lawrence, J. Am. Chem. Soc., 2003, 125, 7754–7755 CrossRef PubMed
-
(a) P. E. Dawson, T. W. Muir, L. Clark-Lewis and S. B. H. Kent, Science, 1994, 266, 776–779 CrossRef CAS PubMed
-
(a) M. Ueda, K. Seki and Y. Imai, Synthesis, 1981, 991–993 CrossRef CAS
-
(a) Y. Xu, S. Zhang, D. Huang and X. Wu, Org. Biomol. Chem., 2024, 22, 6443–6484 RSC
-
(a) B. W. Yoo, H. S. Baek, S. R. Keum, C. M. Yoon, G. S. Nam, S. H. Kim and J. H. Kim, Synth. Commun., 2000, 30, 4317–4322 CrossRef CAS
-
(a) H. Cao, F. Bie, X. Liu, Y. Han, J. Ma, Y. Shi, P. Yan, C. Sun and H. Wang, Tetrahedron, 2020, 76, 131205 CrossRef CAS
- T. J. Wallace, A. Schriesheim and W. Bartok, J. Org. Chem., 1963, 28, 1311–1314 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2025 |