
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural disorder as a key to photoprotection in eumelanin multimers†
Kavya Vinod ,
Diana Thomas and
Mahesh Hariharan*
,
Diana Thomas and
Mahesh Hariharan*
School of Chemistry, Indian Institute of Science Education and Research Thiruvananthapuram (IISER TVM), Maruthamala P.O., Vithura, Thiruvananthapuram 695551, Kerala, India. E-mail: mahesh@iisertvm.ac.in
First published on 1st July 2025
Abstract
Eumelanin, a ubiquitous pigment in animals, is known for its ability to protect against UV-induced damage by efficiently dissipating energy as heat. Despite its importance, the mechanisms underlying eumelanin's broadband absorption and ultrafast energy relaxation remain unclear, primarily due to the inherent structural complexity of the pigment. To address this, we employed model eumelanin multimers, including a monomer (DMICE), dimer (DMICE-D), and trimer (DMICE-T), and investigated their optical properties in both solution and aggregated thin films. Our results reveal that increasing multimer size and aggregation significantly broaden the absorption spectrum, a phenomenon attributed to amplified excitonic interactions. The non-radiative decay processes, governed by internal conversion and intersystem crossing, become increasingly efficient as the multimer lengthens. In thin films, the dimer and trimer exhibit ultrafast excited state relaxation (<30 picoseconds), driven predominantly by internal conversion, which closely parallels eumelanin's characteristic ultrafast energy dissipation. By quantifying the exciton interactions within the multimers, we uncover the interplay of coulombic and charge-transfer couplings in modulating the observed optical behaviour. This work provides insights into how structural organization and excitonic interactions contribute to eumelanin's unique photophysical properties and its photoprotective role, thereby advancing development of eumelanin-inspired biomimetic materials.
Introduction
Eumelanin, the most abundant type of melanin in nature, serves as a vital photoprotective pigment in animals.1 It absorbs throughout the UV-visible range of light and dissipates the absorbed energy non-radiatively to protect tissues from harmful UV-induced damage.2 Despite its critical biological function, the underlying structural factors that govern eumelanin's broadband UV-visible absorption and ultrafast energy dissipation remain poorly understood.2–5 The inherent instability and rapid polymerization of eumelanin monomers to form the insoluble black pigment hinder detailed characterization of its molecular and aggregated structures.2,3 An intricate understanding of the broadened absorption and energy dissipation mechanism in eumelanin is essential for designing synthetic materials that replicate eumelanin's unique photoprotective properties.2 Two prominent models have been proposed to explain eumelanin's broadband absorption and unique optical properties. The chemical disorder model attributes the absorption broadening to a superposition of absorption bands from a diverse pool of oligomers with varying lengths, structures, and oxidation states.4 In contrast, the geometric disorder model highlights the role of excitonic coupling between stacked molecular units in aggregates, suggesting that these interactions contribute significantly to the pigment's spectral properties.6 Recent advances in mass spectrometry,7 X-ray diffraction,8 atomic force microscopy,9,10 and computational calculations11–13 have increasingly supported that shorter multimers dominate eumelanin's composition.14–22 A careful bottom-up approach through the synthesis of stable eumelanin multimers and ordered aggregates of multimers is crucial to disentangle the contributions of chemical and geometric disorder to eumelanin's optical behaviour (Table S1†).23–27 In this context, chemical disorder encompasses variations in multimer length, functionalization, and connectivity among indole-based oligomers, while geometric disorder refers to differences in their spatial arrangement and intermolecular interactions within the aggregated state.6,28Herein, we designed and synthesized chemically stable eumelanin model systems using a blocked derivative of 5,6-dihydroxyindole carboxylic acid (DHICA), namely 5,6-dimethoxyindole carboxylic acid ethyl ester (DMICE). DMICE serves as a simplified and chemically well-defined analogue of the precursor of eumelanin. Blocking the hydroxyl and carboxylic acid groups prevent the fast polymerization of the monomer resulting in a benchtop stable molecule aiding further syntheses and experiments.29 Alkyl protection suppresses excited-state proton transfer (ESPT) and complex photochemistry in these molecules, enabling access to slower alternative relaxation pathways and offering versatile models for disentangling the excited-state dynamics of eumelanin.2,30–32 As the most probable polymerization sites on the indole ring are the second, third and seventh carbons,33 we consider a heterodimer (DMICE-D) linked at C3 and C4 and a heterotrimer (DMICE-T) with two DMICE subunits connected at the C2 and C7 positions (Fig. 1 and S1†). These stable model multimers represent the key intermediates in eumelanin biosynthesis and allow us to systematically explore their photophysical properties in monomeric and aggregated states (Fig. S2†). The steady-state and time-resolved spectroscopic features of DMICE, DMICE-D, and DMICE-T are explored to understand how multimer length and aggregation modulates the absorption spectrum as well as the efficiency of non-radiative decay, thereby enhancing photoprotection.
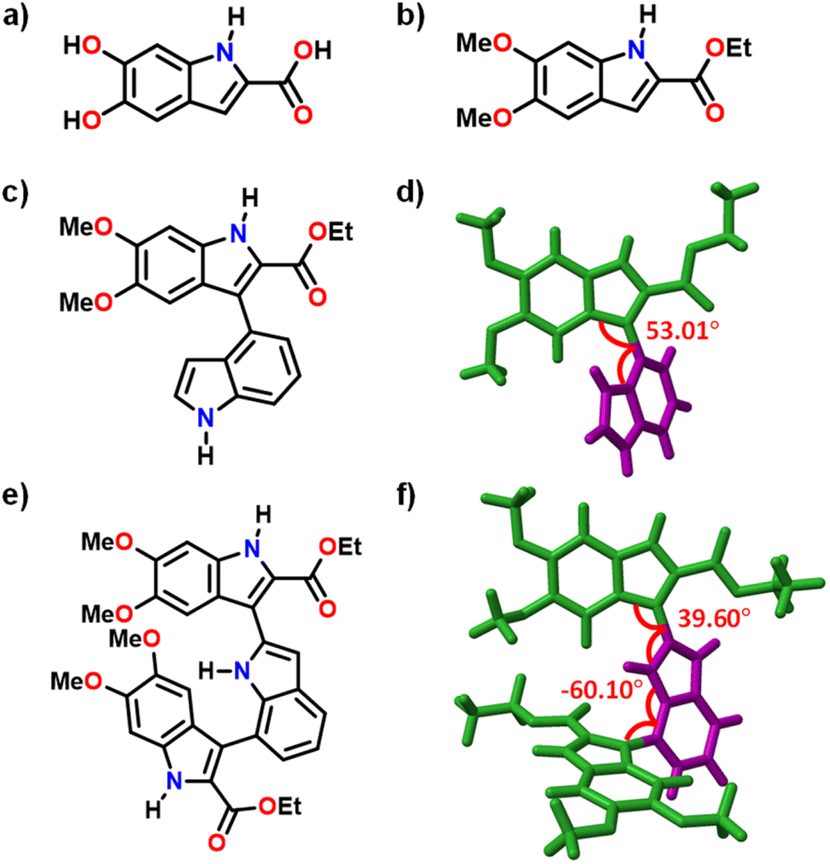 | ||
| Fig. 1 Molecular structures of (a) DHICA, (b) DMICE and (c) DMICE-D; (d) crystal structure of DMICE-D; (e) molecular structure of DMICE-T and (f) optimized structure of DMICE-T. | ||
Results and discussion
Synthesis and structural elucidation
Novel DMICE-T was synthesized through Suzuki coupling of iodinated DMICE with indole-2,7-boronic acid pinacol ester, using XPhos Pd G2 as the catalyst and 1,4-dioxane as the solvent at 75 °C, resulting in moderate yields of the product. Similarly, DMICE-D was synthesized by coupling with indole-2-boronic acid, producing good yields.34 Upon slow evaporation of a chloroform solution of DMICE-D layered with n-hexane, high-quality colourless single crystals suitable for X-ray crystallography were obtained. The crystal structure of DMICE-D, determined via single-crystal X-ray diffraction, confirmed its molecular structure (Fig. 1). DMICE-D crystallized in a monoclinic crystal system with a space group of P21/c and exhibited a dihedral angle of ∼53° (Fig. 1 and Table S2†). Geometry optimizations at multiple DFT levels showed comparable results with the crystal structure, with ωB97X-D/6-311G+(d,p) giving the closest agreement (Table S3†). The optimized geometry of DMICE-D revealed a dihedral angle of ∼54°, consistent with the angle determined by single-crystal X-ray diffraction (Fig. S3†). For DMICE-T, the optimized structure indicated a dihedral angle of 39.60° between the first DMICE subunit and the indole ring, and −60.10° between the second DMICE subunit and the indole ring (Fig. 1 and Table S4†).Optical properties
The electronic properties of DMICE, DMICE-D, and DMICE-T in toluene were explored through UV-visible absorption spectroscopy, fluorescence emission spectroscopy, and time-correlated single photon counting measurements (TCSPC), as illustrated in Fig. 2. The electronic absorption spectrum of DMICE in toluene revealed an absorption maximum at 325 nm, which exhibited good similarity to the absorption characteristics of the unsubstituted DHICA, as previously reported.29 This ascertains that DMICE serves as an excellent model for eumelanin monomer, offering the added advantage of benchtop stability, which is a crucial factor considering the fast oxidative polymerisation into eumelanin. Interestingly, the dimer DMICE-D displayed only slight differences in its absorption spectrum when compared to that of the monomer DMICE, suggesting that the dimer retains many of the electronic characteristics of the monomer. In contrast, DMICE-T demonstrated a more complex absorption profile, displaying a prominent absorption band with a maximum at 331 nm alongside a tail band observed till 420 nm. The observed broadening in the absorption spectra of the trimer, when compared to the dimer and monomer, demonstrates that an increase in the number of connected units leads to a decrease in the excitation energy of the molecule. Computed vertical excitation energies of DMICE, DMICE-D as well as DMICE-T also support the incremental lowering of the S1–S0 energy gap with increase in multimer length (Fig. S4 and Table S5†). This result underscores the significant influence of varying lengths of eumelanin oligomers and the consequent degree of chemical disorder on the overall electronic absorption spectrum of eumelanin.35 Furthermore, the orientations of transition dipoles within individual units, along with the extent of conjugation, could play a crucial role in the observed bathochromic shift of the trimer relative to both the dimer and the monomer (explained vide infra). | ||
| Fig. 2 (a) Normalized UV-vis absorption spectra, (b) normalized emission spectra, (c) time-resolved fluorescence decay profile of DMICE29 (green line), DMICE-D (red line) and DMICE-T (blue line) in toluene at room temperature, (d) knr plotted against the number of indole units from n = 1, 2 and 3. | ||
Upon photoexcitation, DMICE-D exhibited a fluorescence emission maximum centred at 400 nm, while DMICE-T showed a fluorescence emission maximum at 425 nm (Fig. 2b and S5†). This results in a significant bathochromic shift of ∼3868 cm−1 for DMICE-T and ∼2397 cm−1 for DMICE-D, compared to the monomeric DMICE (Table 1). The fluorescence quantum yields, determined using a relative method, revealed a marked decrease as the molecular complexity increases from DMICE to DMICE-D and further to DMICE-T. Specifically, DMICE displayed a fluorescence quantum yield (ϕf) of 49.3%, which declined to 27.5% for DMICE-D and further to 13.6% for DMICE-T, demonstrating the impact of multimer length on fluorescence efficiency. The fluorescence quantum yield (ϕf) is defined as the ratio of the radiative rate kr to the sum of both radiative and non-radiative rates, kr + knr.36 On examining the time-resolved fluorescence decay profiles of DMICE in toluene, a mono-exponential decay pattern with a lifetime of 2.72 ns was observed. In comparison, DMICE-D showed a reduced fluorescence lifetime of 1.86 ns, while DMICE-T exhibited a shorter fluorescence lifetime of 1.02 ns, indicating a substantial decrease in fluorescence lifetimes as we move from the monomer to the dimer and trimer.
| Compound | λabsa (nm) | λemib (nm) | ϕfc (%) | τfld (ns) | kre × 108 (s−1) | knrf × 108 (s−1) | λg (eV) |
|---|---|---|---|---|---|---|---|
| a UV-visible absorption band maximum.b Fluorescence maximum.c Fluorescence quantum yield.d Fluorescence lifetime measured using TCSPC.e Radiative rate constant.f Non-radiative rate constant.g Reorganization energy.h Spectroscopic parameters of DMICE are consistent with previously reported literature.29 | |||||||
| DMICEh | 325 | 365 | 49.7 ± 0.5 | 2.72 ± 0.08 | 1.82 ± 0.01 | 1.84 ± 0.01 | 0.227 |
| DMICE-D | 326 | 400 | 27.5 ± 0.9 | 1.86 ± 0.11 | 1.48 ± 0.01 | 3.90 ± 0.03 | 0.350 |
| DMICE-T | 331 | 425 | 13.6 ± 2.0 | 1.02 ± 0.16 | 1.33 ± 0.03 | 8.76 ± 0.03 | 0.404 |
Given that the radiative rates remain almost constant across the series, the observed variations in fluorescence rates are primarily linked to changes in non-radiative rates, as shown in Table 1. The non-radiative rate constant, knr, shows an increase as the number of indole units increases (Fig. 2d). As the number of indole units increases from monomer to dimer to trimer, the non-radiative decay channels for dissipating excitation energy become more efficient in the eumelanin counterparts.37 The observed increase in knr with the addition of indole units is also driven by elevated reorganization energy suggesting that structural relaxation plays a dominant role in driving the observed knr trend (Table 1).38 This trend is reflected in both the fluorescence quantum yields and fluorescence lifetimes across these molecular species (Table 1). Eumelanin precursors undergo multiple excited state decay pathways, including ESPT,2,30–32,39 electron injection,30,40 intersystem crossing,41 and charge transfer.42–44 Previous literature on DHICA and its oligomers reveal ultrafast sub-picosecond decay in acidic buffers driven by ESPT, with fluorescence decay rates independent of subunit coupling.45 On the other hand, in protected derivatives like DMICE, where ESPT is blocked,29 there is a dependence of the fluorescence lifetimes on oligomer length, allowing the deconvolution of non-radiative decay pathways beyond ESPT.
Further, the influence of the local dielectric environment on the excited state optical properties of eumelanin multimers were investigated through solvent-dependent UV-visible absorption and fluorescence measurements (Table S6†). The UV-visible absorption spectra of DMICE-D and DMICE-T remained largely similar in toluene (TOL, ε = 2.38) and acetonitrile (ACN, ε = 37.5), exhibiting minimal shifts in λabs. In contrast, the fluorescence spectra showed significant solvent-dependent differences with respect to fluorescence maxima as well as fluorescence quantum yields (Fig. 3f and S6†). A notable red shift in the emission wavelength (λemi) was observed when changing the solvent from TOL to ACN, with shifts of ∼1852 cm−1 for DMICE-D and ∼1503 cm−1 for DMICE-T. Additionally, the fluorescence quantum yield (ϕf) decreased substantially in ACN compared to TOL, with values of 17.5% for DMICE-D and 4.6% for DMICE-T, both drastically lower than the ϕf of DMICE.29 The solvent-dependent variations in fluorescence quantum yields and red shifts in emission wavelengths strongly suggest the role of charge-transfer states in the excited state dynamics of DMICE-D and DMICE-T.46,47
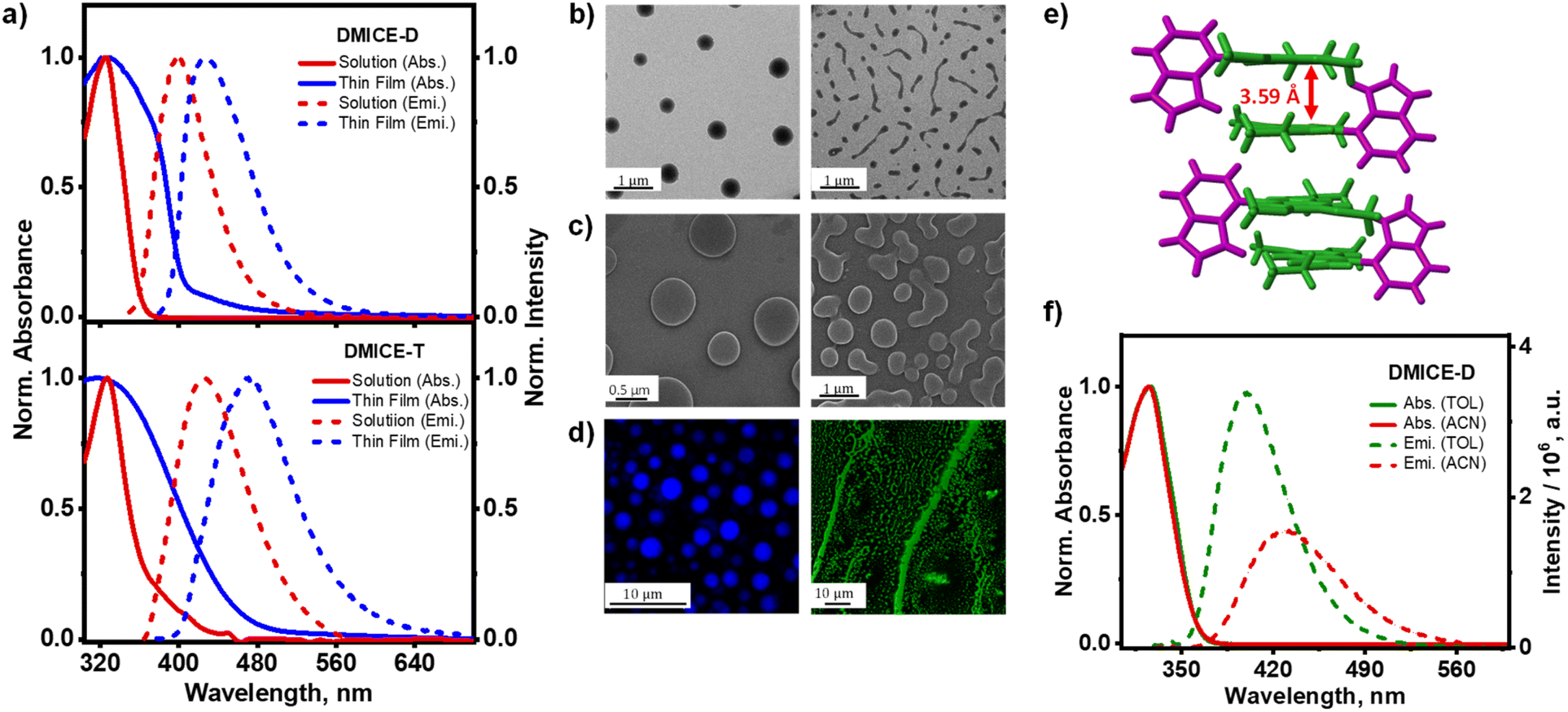 | ||
| Fig. 3 (a) Normalized absorption and emission spectra of solution and thin film aggregates of DMICE-D (top) and DMICE-T (bottom) at room temperature; (b) TEM, (c) SEM, (d) confocal fluorescence images of DMICE-D (left) and DMICE-T (right); (e) crystal packing of DMICE-D exhibiting alternate distichous assembly; (f) normalized absorption and emission spectra of DMICE-D in TOL (green line) and ACN (red line). Note: the solution-phase data in TOL from Fig. 2 are included here for ease of comparison. | ||
In the oligomeric scaffolds investigated, individual indole units are connected through single C–C bonds, positioning them in close proximity to enable electronic interactions and excited state delocalization across multiple subunits.44,48 The structural proximity within indole units provides a foundation for investigating how excitonic interactions within each unit contribute to the electronic properties observed in the dimer and trimer. To examine this, the long-range electronic coupling between the indole units was quantified (Fig. S7 and Tables S7, S8†), providing insight into the spectral characteristics of these oligomers. In the case of DMICE-D, the long-range electronic coupling was calculated to be −3.2 cm−1, which is small enough to indicate a minimal impact on the electronic spectrum of the dimer. This minimal coupling supports the similarity of the dimer's absorption spectrum with respect to that of the monomer, without significant shifts in wavelength or spectral broadening. However, for the trimer DMICE-T, the calculated long-range electronic coupling was −138.7 cm−1, a considerably higher value indicative of the relatively larger red-shifted absorption in the UV-visible absorption spectrum. This increased coupling in the trimer correlates with a modest red shift in the absorption spectrum, leading to slight extension into the visible region—unlike the dimer, whose absorption remains largely confined to the UV. While DMICE-D as well as DMICE-T exhibits limited absorption in the visible range, higher-order and more conjugated oligomers are expected to display red-shifted features that better approximate eumelanin's broadband spectrum. The transition dipole moments for the S0 → S1 transitions for DMICE, DMICE-D and DMICE-T are also shown in the ESI (Fig. S8–S10†). The dihedral angle between the two units, and consequently their transition dipoles, significantly influences the minimal red-shifts observed for DMICE-D. The simulated electronic absorption spectrum shows an incremental red-shift with changes in the orientation between the units in DMICE-D (Fig. S11 and S12†). Additional examples of geometrically optimized homo-dimer and linear trimer of DMICE exhibit minimal red-shifts, underscoring the combined effect of multimer length, connectivity, functional groups and transition dipole orientations in determining the absorption spectra for the eumelanin-based systems (Fig. S13 and S14†). These observations reinforce that both transition dipole alignment and conjugative interactions contribute significantly to the broadening and shifting of the electronic absorption spectrum, particularly as molecular complexity increases.35 Additionally, excited-state calculations on a geometry-optimized homodimer of DMICE reveal similar charge-transfer character, indicating that such behaviour is not exclusive to the heterodimer architecture (Fig. S15†).
Eumelanin multimer aggregates
After confirming the distinct spectroscopic differences between DMICE, DMICE-D and DMICE-T at monomeric concentrations (∼5 μM), we extended our investigation to the molecular systems in aggregated conditions. To gain insights into the optical properties in aggregated conditions, 1 mM solutions of DMICE, DMICE-D and DMICE-T were drop-casted on cleaned quartz substrates, following which the UV-visible absorption spectra and fluorescence were monitored.47 Fig. 3 displays the absorption and emission spectra of DMICE-D and DMICE-T in non-aggregated state (5 μM solution in TOL) as well as in aggregated state (thin film). A significant change in the absorption and fluorescence spectra with respect to solution and film indicates aggregate formation. In solution, DMICE-D exhibited an absorption spectrum spanning till 375 nm with absorption peak maximum at 325 nm. In contrast, the thin film of DMICE-D displayed a broadened band with significant absorption till 406 nm and tailing towards 480 nm, reflecting the intermolecular interactions resulting in aggregate formation. Similarly, for DMICE-T, the absorption spectra in TOL showed a maximum at 331 nm, while the absorption spectrum for the thin film demonstrated a broadened, red-shifted absorption band featuring till 500 nm. The distinct differences in both DMICE-D and DMICE-T at low concentrated solutions versus thin films suggest the excitonic coupling within the aggregates, leading to new, close-lying electronic energy states. To determine if aggregation was unique to DMICE-D and DMICE-T, we also examined the thin film of DMICE. In solution (5 μM in TOL), DMICE showed an absorption maximum at 325 nm, while in thin film, a broadened band emerged in the absorption spectrum featuring till 390 nm (Fig. S16†).29 This broadening and red-shift in spectral characteristics in film indicates that exciton interactions may be occurring in DMICE, DMICE-D, and DMICE-T in aggregated forms. Significant decrease in the fluorescence yields and fluorescence lifetimes are also observed in the thin films. The absolute fluorescence quantum yields for the thin-film samples are estimated to be 8.9%, 3.8%, and 2.3% for DMICE, DMICE-D, and DMICE-T, respectively. Such spectral changes, characterized by new excitonic states and aggregation-caused quenching, are commonly observed in molecular aggregates and photosynthetic pigments, where aggregation introduces new electronic interactions depending on molecular arrangement and interaction strength, impacting their optical properties.49–53To investigate the morphology of aggregates formed by DMICE, DMICE-D, and DMICE-T, transmission electron microscopy (TEM), scanning electron microscopy (SEM), and confocal microscopy imaging were performed (Fig. 3).54–56 The TEM and SEM images revealed that DMICE-D forms aggregates with a distinct disk-like morphology with diameters ranging from 0.4 to 0.6 μm (Fig. S17†). In contrast, DMICE-T aggregates displayed an elongated, fibre-like morphology, arising from the assembly of smaller, disk-like substructures (Fig. S18†). Confocal microscopy further confirmed the fluorescent nature of these aggregates, corroborating the disk-like structure of DMICE-D and the extended fibre formation observed in DMICE-T. For comparison, DMICE aggregates were also examined. Unlike DMICE-D and DMICE-T, DMICE showed a lamellar morphology, characterized by structures with an average length of 15–20 μm, as observed in both SEM and confocal fluorescence microscopy (Fig. S19 and S20†). Recent studies on allomelanin (fungal melanin) dimers underscore the value of morphology in understanding molecular assemblies and show that hydrogen bonding and π–π stacking drive aggregate formation and guide microstructure formation.57,58 This structural diversity aligns with previously reported observations of morphological heterogeneity in eumelanin pigments, which exhibit a range of aggregate forms, including spherical, rod-like, hollow-rod, and hollow-platelet structures.9,59–62
Excitonic coupling in aggregates
Examining the structural organization of eumelanin multimer aggregates sheds light on the origins of excitonic interactions within the assemblies. Detailed analysis of the crystal structures of DMICE-D and DMICE29 reveals the key intermolecular interactions that facilitate the formation of these organized assemblies. DMICE revealed a herringbone packing motif with H-bonding as the dominant interaction.29 In DMICE-D, crystal packing reveals an alternate distichous arrangement where DMICE subunits are stacked along the c-axis, stabilized by edge-to-π interactions, with indole subunits alternately protruding outward (Fig. 3e). The interlayer distance between the stacks in DMICE-D was measured to be 3.59 Å, highlighting a close-packing arrangement favourable for electronic interactions. Exciton splitting in molecular dimers occurs through long-range coulombic coupling (JCoul) via molecular transition dipole–dipole interactions and short-range charge-transfer coupling via HOMO–HOMO and LUMO–LUMO overlaps (JCT).63,64 In eumelanin multimer assemblies, excitonic coupling was assessed through both long-range dipole–dipole interactions (eqn (S1)†) and short-range charge-transfer interactions based on wave function overlap (eqn (S2)†), using crystal structures of DMICE29 and DMICE-D as references.64DMICE-D displays two distinct dimer orientations within the crystal structure, termed DMICE-D1 and DMICE-D2. Interestingly, analysis of the transition dipole moment vectors indicates an H-like alignment in DMICE-D1 and an X-like alignment in DMICE-D2 (Fig. S21†). In terms of transition dipole orientations, H-aggregates form when the transition dipoles are aligned in a near parallel fashion based on the co-facial packing of the molecules.64 On the other hand, X-aggregates (or cross-dipole stacking) result from attaining a rotational angle of 50° < α ≤ 90° between the molecular transition dipoles.65 The effective excitonic coupling values were calculated as 34.7 cm−1 for DMICE-D1 and 63.7 cm−1 for DMICE-D2, both indicating significant exciton interactions. These strong couplings are attributed to considerable coulombic coupling (JCoul) combined with non-negligible charge transfer coupling (JCT) in both types of crystal dimers (Table S9†). DMICE similarly demonstrated appreciable JCoul and JCT, as previously reported.29 The distinctive interaction network in DMICE-D and DMICE crystals indicates that the exciton splitting observed in eumelanin multimer aggregates may arise from diverse dipolar orientations and orbital overlaps within the assemblies. These results emphasize how specific molecular packing and intermolecular forces contribute to the unique optical and electronic properties that emerge in eumelanin-based assemblies.
Excited state energy dissipation of monomer versus aggregates
To investigate the excited state relaxation mechanisms responsible for non-radiative decay in the eumelanin multimers, femtosecond transient absorption (fsTA) measurements were performed (experimental details in Section 1.9 of ESI†). Fig. 4 presents the fsTA spectra and corresponding deconvoluted time constants for DMICE-D in TOL (Fig. 4a) and ACN (Fig. S22†). Upon photoexcitation of DMICE-D in TOL at λexc = 320 nm using a 100 fs pump pulse, a distinct positive absorption feature emerges between 450 and 700 nm, resembling the singlet absorption previously reported for DHICA and DMICE (Fig. 4a).2,29,66 Evolution-associated spectra (EAS) for DMICE-D in TOL were derived through global fitting of the fsTA data in a time-wavelength framework, modelled as a sequential A → B → C → GS (ground state) process. The spectral deconvolution reveals three components: the first component (A), attributed to internal conversion from the excited singlet state, decays with a time constant of τ1 = 6.7 ps. The second component (B), red-shifted by 5 nm relative to component A, decays with a time constant of τ2 = 2.43 ns. The decay time constant of component B matches closely to the fluorescence lifetime, ascertaining it to be the S1 excited state decay. With the decay of the S1 state, a non-decaying species at 440–490 nm persists, with a decay time constant of τ3 > 4 ns. The overlay of fitted traces versus experimental traces along with the deconvoluted species for DMICE-D in TOL are presented in Fig. S23.† The fsTA spectra of DMICE-D in polar solvent ACN also demonstrates similar spectral traces but different decay constants. In ACN, DMICE-D initially exhibits a well-defined absorption feature across 450–700 nm (Fig. S22†). This evolves within few picoseconds to populate a second species in the same wavelength range, followed by a persistent band at ∼440 nm. Spectral deconvolution reveals that the first component (internal conversion) decays within τ1 = 3.7 ps, subsequently populating the second component (S1 decay), which decays with a time constant of τ2 = 2.18 ns. As observed similar for DMICE-D in TOL, DMICE-D in ACN also revealed a consequent, long-lived species (>4 ns). | ||
| Fig. 4 (a) (Top) Femtosecond transient absorption (fsTA) spectra of DMICE-D in TOL; (bottom) relative population of the two components extracted from sequential global fitting; (b) (top) fsTA spectra of neat film of DMICE-D; (bottom) relative population of the two components extracted from sequential global fitting; (c) hole and electron iso-surface plots of DMICE-D at the S1 state. | ||
The solvent-dependent kinetics of the excited state species as observed from fsTA, along with steady-state fluorescence, reveals a partial charge-transfer (CT) character of the S1 state. The hole–electron iso-surfaces of the S1 state for the dimer also corroborate the experimentally observed partial charge-transfer nature, with the hole delocalized across both units and the electron localized within the DMICE unit of DMICE-D (Fig. 4c). In contrast, the S2 state exhibits Frenkel exciton character, with both the hole and electron confined to the DMICE unit of the dimer (Fig. S24†). This aligns with recent findings emphasizing the role of CT states in the excited state deactivation pathways of eumelanin and related systems.42,44,45 The CT character of S1 likely facilitates the formation of triplet states via intersystem crossing (ISC) as observed in several examples of organic chromophores like aromatic aldehydes.67–69 Nanosecond transient absorption (nsTA) measurements corroborate the presence of a long-lived species at 420–470 nm with a lifetime of 1.2 μs in TOL and 0.8 μs in ACN, further confirming ISC via the S1 state (Fig. S25 and S26†). Analysis of the natural transition orbitals (NTOs) for the S1 state and the near-degenerate T5 state shows a change of orbital character from nπ* character to ππ* character involving the non-bonding orbitals from the carbonyl group (Fig. S27 and Table S10†), following El-Sayed's rule. Prior investigations on unprotected DHI and DHICA oligomers have predominantly utilized fluorescence-based approaches, wherein excited-state proton transfer (ESPT) emerged as a principal relaxation pathway.45,70,71 However, the presence of free hydroxyl groups in the molecules facilitated rapid polymerization and the in situ generation of photoactivated species, complicating a comprehensive interpretation of the excited-state landscape. Consequently, phenomena such as charge transfer and intersystem crossing remained largely unexplored, likely obscured by the predominance of ESPT-mediated processes.
The fsTA spectra of DMICE-T in TOL closely resemble those of DMICE-D, with notable differences in decay constants. Upon photoexcitation at 320 nm, a distinct positive absorption feature between 450 and 700 nm, characteristic of singlet absorption, is observed (Fig. S28†). Spectral deconvolution identifies two components in the initial timescales followed by a consequent non-decaying component: the first component is attributed to internal conversion to the S1 state and decays with a time constant of τ1 = 7.4 ps. The second component, that is, S1 decay occurs with a longer time constant of τ2 = 0.98 ns. The similarity in excited state spectra and multi-component decay dynamics between DMICE-D and DMICE-T highlights their comparable deactivation pathways. In ACN, the fsTA spectra of DMICE-T exhibit similar features to those in TOL, with spectral deconvolution revealing an initial relaxation time constant of τ1 = 3.3 ps. The relaxed singlet state decays faster in ACN, with a time constant of τ2 = 0.34 ns (Fig. S29†). These differences in decay kinetics across solvents may be influenced by the chromophore size and the extent of electronic coupling between individual indole units in the multimers.44 Like observed in the dimer, the iso-surface analysis of the S1 state of the trimer also reveals prominent CT exciton characteristics, with the hole distributed across the indole and DMICE fragment and the electron confined to the same DMICE unit. The S2 state also exhibits CT exciton behaviour (Fig. S30†), while the higher excited state S3 demonstrates Frenkel exciton character (Fig. S31†). Consistent with the observations for DMICE-D, nsTA measurements confirm the presence of triplet excited states at 420–470 nm for DMICE-T, with a lifetime of 1.3 μs and 0.7 μs, in TOL and ACN respectively (Fig. S32 and S33†). The NTO analysis further supports the orbital character change from ππ* to nπ* in the S1 state and near-degenerate T6 state, facilitating ISC (Fig. S34 and Table S10†). A summary of the evaluated decay rate constants for the eumelanin multimers in TOL and ACN are presented in Tables S11 and S12.†
Having established the excited state decay channels of the multimers in monomeric state, we explore the excited state dynamics of the multimers in the aggregate state. Femtosecond transient absorption (fsTA) measurements were conducted on thin films of DMICE, DMICE-D, and DMICE-T following photoexcitation at 320 nm to investigate the excited state dynamics of excitonically coupled multimers in the solid state. Thin films were prepared by coating CHCl3 solutions of the samples onto cleaned quartz substrates (see Section 1.10 in ESI† for details). The fsTA spectra of the DMICE-D thin film exhibited an excited state absorption (ESA) feature spanning 440–700 nm, closely resembling the singlet absorption signature of the molecule in solution (Fig. 4b). Spectral deconvolution was modelled using a sequential kinetic scheme (A → B → GS, where GS represents the ground state; Fig. S35†). The initial decay component, characterized by a time constant of τ1 = 0.9 ps, is attributed to internal conversion between excitonically coupled singlet states. This rapid decay is followed by a slower relaxation process, with a time constant of τ2 = 21.1 ps, representing internal conversion to the ground state. In contrast to the monomeric state of DMICE-D, which exhibits singlet decay within ∼2 ns, the aggregate state demonstrates significantly faster singlet decay, occurring within <30 ps. Although excimer formation or defect-state trapping could potentially account for the observed picosecond decay in the thin films, these pathways are deemed unlikely given the absence of broad, featureless excited state absorption and red-shifted emission characteristic of excimer states.72–74 The involvement of higher-order processes such as singlet–singlet annihilation or non-radiative Förster energy transfer, were ruled out based on fluence-dependent measurements, which showed no significant changes in spectral features or kinetic decay profiles in the thin films with increasing pump fluence (Fig. S36†).2,75,76 This behaviour parallels observations in eumelanin material, where ultrafast internal conversion occurs via localized excitonic coupling without evidence of exciton migration or annihilation.2 The absence of long-lived triplet excited states in the thin films is notable, hinting at similarities with eumelanin's ultrafast relaxation dynamics (Fig. S37†). Triplet states in biomolecules like eumelanin are undesirable because they can generate reactive oxygen species (ROS), causing oxidative damage and phototoxicity.77 These findings highlight the pronounced influence of excitonic coupling on the excited state dynamics of eumelanin-like systems in the solid state, offering critical insights into their photophysical behaviour under aggregated conditions.
In the thin film, fsTA spectra of DMICE-T also initially exhibited a well-defined absorption feature across 450–700 nm. This excited state species evolved within hundreds of femtoseconds to populate a broader species in the same wavelength range (Fig. S38†). Spectral deconvolution reveals that the first component (singlet species) decays within τ1 = 0.5 ps, subsequently populating the second component, which decays with a time constant of τ2 = 19.3 ps (Fig. S39†). The ultrafast internal conversion from the S1 state (within ps) in the thin films of both DMICE-D and DMICE-T dominate over the slower intersystem crossing observed for the eumelanin analogues in solution, thereby enhancing the potential photoprotective role of the aggregates. Excited state energy calculations also indicate a denser manifold of closely spaced singlet excited states in DMICE-D and DMICE-T, which may facilitate faster internal conversion in the solid state, consistent with the observed rapid deactivation to the ground state (Table S5†). For comparison, the fsTA of DMICE thin films was also measured. In the thin film, the fsTA spectra of DMICE display a distinct absorption feature spanning 450–700 nm, which evolves within a few picoseconds, transitioning into a broader spectral species within the same wavelength range (Fig. S40†). Spectral deconvolution reveals that the first component (singlet species) decays within τ1 = 4.4 ps, subsequently populating the second component, which decays with a time constant of τ2 = 122.5 ps. The biexponential excited state absorption (ESA) signals observed in the eumelanin multimer aggregates indicate ultrafast internal conversion (IC) between electronically coupled singlet states, followed by slower IC dynamics to the ground state (S0). Such ultrafast non-radiative decay, similar to the mechanisms observed in photoprotective compounds like DNA and RNA nucleobases, serves to minimize the potential for energy transfer or photochemical reactions. Time-resolved studies on nucleosides and single-stranded DNA have shown that their lowest singlet excited state decays within picoseconds via internal conversion to the ground state, driven by efficient non-radiative pathways.78,79 This behaviour, effectively replicated by the monomer, dimer as well as trimer aggregates, aligns with eumelanin's intrinsic photoprotective properties. Future studies employing advanced spectroscopic techniques may further elucidate excitonic coupling pathways in the aggregated films.
The observed behaviour of DMICE-D and DMICE-T in monomeric and aggregate states highlight the interplay of intramolecular exciton interactions within the eumelanin multimers, along with intermolecular electronic coupling between states of varying energies in the solid-state aggregates. This duality aligns with eumelanin's complex excited state dynamics, where both intra- and inter-molecular effects contribute to its optical properties and photoprotective functions. By capturing these interactions in simplified model systems, our findings provide valuable insights into the fundamental processes at play in eumelanin. Fig. 5 illustrates a schematic representation of the energy dissipation mechanisms within the chemically and geometrically disordered eumelanin multimers. The distinctive optical properties of eumelanin arise from its structural features, which combine elements of both chemical and geometric order and disorder. Chemical order–disorder refers to the defined molecular structure and resulting interactions within the moiety.28 In contrast, geometric order–disorder enables strong excitonic coupling (JCoul and JCT) among eumelanin multimers through stacking, which disperses the electronic absorption across the exciton energy levels.6 Heterogeneous stacking and diverse chromophore orientations are manifestations of structural disorder, which enhance excitonic coupling and facilitate non-radiative decay via internal conversion.6,80 A comparison of the structural and photophysical features of DMICE-based multimers with respect to natural eumelanin is provided in Table S13.† Table S14† highlights how chemical protection in DMICE-based systems enables identification of excited-state processes compared to unprotected DHI/DHICA oligomers. A comprehensive understanding of the involved interactions reveals the complexity and richness of eumelanin's optical properties, opening up new avenues for research into its potential applications in areas such as photonics and biomimetic materials.
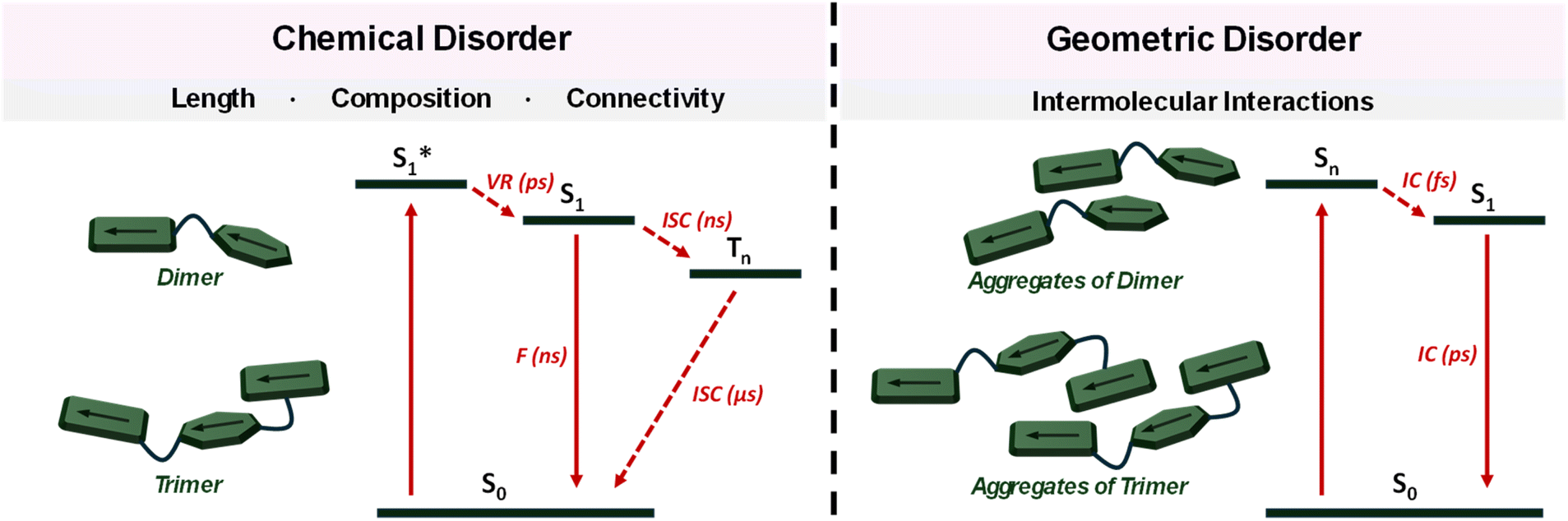 | ||
| Fig. 5 Energy deactivation profile of (left) monomeric forms of DMICE-D and DMICE-T; (right) aggregates of DMICE-D and DMICE-T (F = fluorescence, ISC = intersystem crossing, VR = vibronic relaxation, IC = internal conversion). | ||
Conclusions
In summary, we have synthesized and examined model eumelanin multimers in their monomeric, crystalline and aggregated films to decipher the origin of the unique photophysical properties of eumelanin. A significant decrease in the fluorescence quantum yield and fluorescence lifetime is observed in DMICE-T compared to DMICE-D and DMICE indicating that the non-radiative decay pathways are much faster and more efficient in the trimer than the dimer and monomer. Our spectroscopic investigations of eumelanin multimer aggregates reveal that chemical as well as geometric disorders are both crucial factors contributing to the broadened absorption and ultrafast energy dissipation of eumelanin. In solution, the multimers exhibit nanosecond-scale excited state lifetimes, driven by internal conversion and intersystem crossing. In contrast, excitonically coupled molecules in thin films demonstrate ultrafast excited state deactivation (<30 ps), dominated by internal conversion, mimicking eumelanin's ultrafast energy dissipation. These findings highlight the importance of structural order–disorder and excitonic interactions in modulating eumelanin's optical behaviour and photoprotection mechanisms, paving way for future innovations in sustainable energy, bioelectronics, and photoprotective coatings.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
All authors contributed to the preparation of the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
M. H. acknowledges the Science and Engineering Research Board (CRG/2023/005859), Department of Science and Technology, Govt. of India, for financial support. K. V. acknowledges CSIR for financial assistance. D. T. acknowledges INSPIRE for financial support. We thank Mr Alex P. Andrews for the X-ray diffraction analyses. We thank Mr Gowtham Raj and Dr Reji Varghese for CLSM measurements. We thank Mr Mohammed Raees for PLQY in solid state. We thank the Padmanabha HPC cluster at IISER TVM for the computing time.References
- M. d'Ischia, A. Napolitano, A. Pezzella, P. Meredith and T. Sarna, Angew. Chem., Int. Ed., 2009, 48, 3914–3921 CrossRef PubMed
.
- A. Ilina, K. E. Thorn, P. A. Hume, I. Wagner, R. R. Tamming, J. J. Sutton, K. C. Gordon, S. K. Andreassend, K. Chen and J. M. Hodgkiss, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2212343119 CrossRef CAS PubMed
- W. Cao, N. C. McCallum, Q. Z. Ni, W. Li, H. Boyce, H. Mao, X. Zhou, H. Sun, M. P. Thompson, C. Battistella, M. R. Wasielewski, A. Dhinojwala, M. D. Shawkey, M. D. Burkart, Z. Wang and N. C. Gianneschi, J. Am. Chem. Soc., 2020, 142, 12802–12810 CrossRef CAS PubMed
- Z. E. Siwicka, F. A. Son, C. Battistella, M. H. Moore, J. Korpanty, N. C. McCallum, Z. Wang, B. J. Johnson, O. K. Farha and N. C. Gianneschi, J. Am. Chem. Soc., 2021, 143, 3094–3103 CrossRef CAS PubMed
- M. d'Ischia, A. Napolitano, A. Pezzella, P. Meredith and M. Buehler, Angew. Chem., Int. Ed., 2020, 59, 11196–11205 CrossRef
- C.-T. Chen, C. Chuang, J. Cao, V. Ball, D. Ruch and M. J. Buehler, Nat. Commun., 2014, 5, 3859 CrossRef CAS PubMed
- A. Pezzella, A. Napolitano, M. d'Ischia, G. Prota, R. Seraglia and P. Traldi, Rapid Commun. Mass Spectrom., 1997, 11, 368–372 CrossRef CAS
- V. Capozzi, G. Perna, P. Carmone, A. Gallone, M. Lastella, E. Mezzenga, G. Quartucci, M. Ambrico, V. Augelli, P. F. Biagi, T. Ligonzo, A. Minafra, L. Schiavulli, M. Pallara and R. Cicero, Thin Solid Films, 2006, 511–512, 362–366 CrossRef CAS
- C. M. R. Clancy, J. B. Nofsinger, R. K. Hanks and J. D. Simon, J. Phys. Chem. B, 2000, 104, 7871–7873 CrossRef CAS
- C. M. R. Clancy and J. D. Simon, Biochemistry, 2001, 40, 13353–13360 CrossRef CAS PubMed
- K. B. Stark, J. M. Gallas, G. W. Zajac, J. T. Golab, S. Gidanian, T. McIntire and P. J. Farmer, J. Phys. Chem. B, 2005, 109, 1970–1977 CrossRef CAS PubMed
- S. Meng and E. Kaxiras, Biophys. J., 2008, 94, 2095–2105 CrossRef CAS PubMed
- E. Kaxiras, A. Tsolakidis, G. Zonios and S. Meng, Phys. Rev. Lett., 2006, 97, 218102 CrossRef PubMed
- D. Tuna, A. Udvarhelyi, A. L. Sobolewski, W. Domcke and T. Domratcheva, J. Phys. Chem. B, 2016, 120, 3493–3502 CrossRef CAS PubMed
- A. Pezzella, A. Iadonisi, S. Valerio, L. Panzella, A. Napolitano, M. Adinolfi and M. d'Ischia, J. Am. Chem. Soc., 2009, 131, 15270–15275 CrossRef CAS PubMed
- F. R. Kohl, C. Grieco and B. Kohler, Chem. Sci., 2020, 11, 1248–1259 RSC
- M. Gaeta, R. Randazzo, V. Villari, N. Micali, A. Pezzella, R. Purrello, M. d’Ischia and A. D’Urso, Front. Chem., 2020, 8, 616961 CrossRef CAS PubMed
- D. N. Peles, E. Lin, K. Wakamatsu, S. Ito and J. D. Simon, J. Phys. Chem. Lett., 2010, 1, 2391–2395 CrossRef CAS
- C. Battistella, N. C. McCallum, K. Gnanasekaran, X. Zhou, V. Caponetti, M. Montalti and N. C. Gianneschi, ACS Cent. Sci., 2020, 6, 1179–1188 CrossRef CAS PubMed
- Y. Liu, K. Ai and L. Lu, Chem. Rev., 2014, 114, 5057–5115 CrossRef CAS PubMed
- J. D. Simon and D. N. Peles, Acc. Chem. Res., 2010, 43, 1452–1460 CrossRef CAS PubMed
- Y. Huang, Y. Li, Z. Hu, X. Yue, M. T. Proetto, Y. Jones and N. C. Gianneschi, ACS Cent. Sci., 2017, 3, 564–569 CrossRef CAS PubMed
- P. Meredith, B. J. Powell, J. Riesz, S. P. Nighswander-Rempel, M. R. Pederson and E. G. Moore, Soft Matter, 2006, 2, 37–44 RSC
- M. d'Ischia, A. Napolitano, V. Ball, C.-T. Chen and M. J. Buehler, Acc. Chem. Res., 2014, 47, 3541–3550 CrossRef
- W. Xie, A. Dhinojwala, N. C. Gianneschi and M. D. Shawkey, Chem. Rev., 2024, 124, 7165–7213 CrossRef CAS PubMed
- W. Xie, E. Pakdel, Y. Liang, Y. J. Kim, D. Liu, L. Sun and X. Wang, Biomacromolecules, 2019, 20, 4312–4331 CrossRef CAS PubMed
- X. Wang, L. Kinziabulatova, M. Bortoli, A. Manickoth, M. A. Barilla, H. Huang, L. Blancafort, B. Kohler and J.-P. Lumb, Nat. Chem., 2023, 15, 787–793 CrossRef CAS PubMed
- M. L. Tran, B. J. Powell and P. Meredith, Biophys. J., 2006, 90, 743–752 CrossRef CAS PubMed
- K. Vinod, S. D. Jadhav and M. Hariharan, Chem.–Eur. J., 2024, 30, e202400499 CrossRef CAS PubMed
- J. J. Nogueira, A. Corani, A. El Nahhas, A. Pezzella, M. d'Ischia, L. González and V. Sundström, J. Phys. Chem. Lett., 2017, 8, 1004–1008 CrossRef CAS PubMed
- S. Olsen, J. Riesz, I. Mahadevan, A. Coutts, J. P. Bothma, B. J. Powell, R. H. McKenzie, S. C. Smith and P. Meredith, J. Am. Chem. Soc., 2007, 129, 6672–6673 CrossRef CAS PubMed
- A. Corani, A. Pezzella, T. Pascher, T. Gustavsson, D. Markovitsi, A. Huijser, M. d'Ischia and V. Sundström, J. Phys. Chem. Lett., 2013, 4, 1383–1388 CrossRef CAS PubMed
- P. Meredith and T. Sarna, Pigm. Cell Res., 2006, 19, 572–594 CrossRef CAS
- A. H. Aebly, J. N. Levy, B. J. Steger, J. C. Quirke and J. M. Belitsky, RSC Adv., 2018, 8, 28323–28328 RSC
- A. Choudhury, R. Ramakrishnan and D. Ghosh, Chem. Commun., 2024, 60, 2613–2616 RSC
- C. Albrecht, Anal. Bioanal. Chem., 2008, 390, 1223–1224 CrossRef CAS
- Y. R. Poh, S. Pannir-Sivajothi and J. Yuen-Zhou, J. Phys. Chem. C, 2023, 127, 5491–5501 CrossRef CAS
- P. Ghosh, A. M. Alvertis, R. Chowdhury, P. Murto, A. J. Gillett, S. Dong, A. J. Sneyd, H.-H. Cho, E. W. Evans, B. Monserrat, F. Li, C. Schnedermann, H. Bronstein, R. H. Friend and A. Rao, Nature, 2024, 629, 355–362 CrossRef CAS
- B. Marchetti and T. N. V. Karsili, Phys. Chem. Chem. Phys., 2016, 18, 3644–3658 RSC
- C. Grieco, F. R. Kohl and B. Kohler, Photochem. Photobiol., 2023, 99, 680–692 CrossRef CAS PubMed
- M. Gauden, A. Pezzella, L. Panzella, A. Napolitano, M. d'Ischia and V. Sundström, J. Phys. Chem. B, 2009, 113, 12575–12580 CrossRef CAS PubMed
- V. Petropoulos, A. Mavridi-Printezi, A. Menichetti, D. Mordini, P. Kabacinski, N. C. Gianneschi, M. Montalti, M. Maiuri and G. Cerullo, J. Phys. Chem. Lett., 2024, 15, 3639–3645 CrossRef CAS PubMed
- M. Matta, A. Pezzella and A. Troisi, J. Phys. Chem. Lett., 2020, 11, 1045–1051 CrossRef CAS PubMed
- C. Grieco, F. R. Kohl, A. T. Hanes and B. Kohler, Nat. Commun., 2020, 11, 4569 CrossRef CAS
- A. Corani, A. Huijser, T. Gustavsson, D. Markovitsi, P.-Å. Malmqvist, A. Pezzella, M. d'Ischia and V. Sundström, J. Am. Chem. Soc., 2014, 136, 11626–11635 CrossRef CAS PubMed
- A. Mazumder, K. Vinod, P. D. Maret, P. P. Das and M. Hariharan, J. Phys. Chem. Lett., 2024, 15, 5896–5904 CrossRef CAS PubMed
- A. R. Mallia, P. S. Salini and M. Hariharan, J. Am. Chem. Soc., 2015, 137, 15604–15607 CrossRef CAS PubMed
- J. Wang and L. Blancafort, Angew. Chem., Int. Ed., 2021, 60, 18800–18809 CrossRef CAS PubMed
- N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069–7163 CrossRef CAS PubMed
- S. Marguet, D. Markovitsi, P. Millié, H. Sigal and S. Kumar, J. Phys. Chem. B, 1998, 102, 4697–4710 CrossRef CAS
- V. Sundström, T. Pullerits and R. van Grondelle, J. Phys. Chem. B, 1999, 103, 2327–2346 CrossRef
- K. Vinod, R. Mathew, C. Jandl, B. Thomas and M. Hariharan, Chem. Sci., 2024, 15, 16015–16024 RSC
- W. Z. Yuan, P. Lu, S. Chen, J. W. Y. Lam, Z. Wang, Y. Liu, H. S. Kwok, Y. Ma and B. Z. Tang, Adv. Mater., 2010, 22, 2159–2163 CrossRef CAS PubMed
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed
- C. Naranjo, A. Doncel-Giménez, R. Gómez, J. Aragó, E. Ortí and L. Sánchez, Chem. Sci., 2023, 14, 9900–9909 RSC
- D. S. Philips, K. K. Kartha, A. T. Politi, T. Krüger, R. Q. Albuquerque and G. Fernández, Angew. Chem., Int. Ed., 2019, 58, 4732–4736 CrossRef CAS PubMed
- X. Zhou, X. Gong, W. Cao, C. J. Forman, J. Oktawiec, L. D'Alba, H. Sun, M. P. Thompson, Z. Hu, U. Kapoor, N. C. McCallum, C. D. Malliakas, O. K. Farha, A. Jayaraman, M. D. Shawkey and N. C. Gianneschi, Angew. Chem., Int. Ed., 2021, 60, 17464–17471 CrossRef CAS
- X. Zhou, N. C. McCallum, Z. Hu, W. Cao, K. Gnanasekaran, Y. Feng, J. F. Stoddart, Z. Wang and N. C. Gianneschi, ACS Nano, 2019, 13, 10980–10990 CrossRef CAS PubMed
- M. d'Ischia, K. Wakamatsu, F. Cicoira, E. Di Mauro, J. C. Garcia-Borron, S. Commo, I. Galván, G. Ghanem, K. Kenzo, P. Meredith, A. Pezzella, C. Santato, T. Sarna, J. D. Simon, L. Zecca, F. A. Zucca, A. Napolitano and S. Ito, Pigm. Cell Melanoma Res., 2015, 28, 520–544 CrossRef PubMed
- J. B. Nofsinger, S. E. Forest, L. M. Eibest, K. A. Gold and J. D. Simon, Pigm. Cell Res., 2000, 13, 179–184 CrossRef CAS
- R. Maia, D. R. Rubenstein and M. D. Shawkey, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10687–10692 CrossRef CAS PubMed
- K.-Y. Ju, M. C. Fischer and W. S. Warren, ACS Nano, 2018, 12, 12050–12061 CrossRef CAS PubMed
- M. Kasha, H. R. Rawls and M. A. El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CAS
- N. J. Hestand and F. C. Spano, Acc. Chem. Res., 2017, 50, 341–350 CrossRef CAS PubMed
- S. Ma, Y. Liu, J. Zhang, B. Xu and W. Tian, J. Phys. Chem. Lett., 2020, 11, 10504–10510 CrossRef CAS PubMed
- K. Vinod, L. Hakeem K, D. Thomas, P. P. Das and M. Hariharan, Org. Chem. Front., 2025, 12, 33–41 RSC
- M. Lv, X. Wang, D. Wang, X. Li, Y. Liu, H. Pan, S. Zhang, J. Xu and J. Chen, Phys. Chem. Chem. Phys., 2021, 23, 25455–25466 RSC
- P. A. Robertson, H. M. Bishop and A. J. Orr-Ewing, J. Phys. Chem. Lett., 2021, 12, 5473–5478 CrossRef CAS PubMed
- N. Ohmori, T. Suzuki and M. Ito, J. Phys. Chem., 1988, 92, 1086–1093 CrossRef CAS
- A. Corani, A. Huijser, A. Iadonisi, A. Pezzella, V. Sundström and M. d'Ischia, J. Phys. Chem. B, 2012, 116, 13151–13158 CrossRef CAS PubMed
- M. Gauden, A. Pezzella, L. Panzella, M. T. Neves-Petersen, E. Skovsen, S. B. Petersen, K. M. Mullen, A. Napolitano, M. d'Ischia and V. Sundström, J. Am. Chem. Soc., 2008, 130, 17038–17043 CrossRef CAS PubMed
- R. M. Young and M. R. Wasielewski, Acc. Chem. Res., 2020, 53, 1957–1968 CrossRef CAS PubMed
- S. Kang, W. Choi, J. Ahn, T. Kim, J. H. Oh and D. Kim, ACS Appl. Mater. Interfaces, 2024, 16, 18134–18143 CrossRef CAS PubMed
- S. Wang, H. Liu, S. Zhao, Q. Wu, Z. Yang, D. Yang, Y. Lv, Q. Su, S.-T. Zhang and B. Yang, Chem. Sci., 2025, 16, 3275–3284 RSC
- M. Morgenroth, S. Mirko, G. Laure, O. Kawon and T. Lenzer, Mol. Phys., 2021, 119, e1959072 CrossRef
- M. Schlosser and S. Lochbrunner, J. Phys. Chem. B, 2006, 110, 6001–6009 CrossRef CAS PubMed
- P. D. Wood and R. W. Redmond, J. Am. Chem. Soc., 1996, 118, 4256–4263 CrossRef CAS
- B. Bauer, R. Sharma, M. Chergui and M. Oppermann, Chem. Sci., 2022, 13, 5230–5242 RSC
- J.-M. L. Pecourt, J. Peon and B. Kohler, J. Am. Chem. Soc., 2000, 122, 9348–9349 CrossRef CAS
- S. Meng and E. Kaxiras, Biophys. J., 2008, 95, 4396–4402 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2389911. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc00920k |
| This journal is © The Royal Society of Chemistry 2025 |