
Tuning valence-variable single atomic metal for efficient antibiotic degradation and in situ chlorinated byproduct elimination under current pulsation†
Yang Yuac,
Yibo Linac,
Binyao Wangac,
Yangqi Eac,
Qian Liac,
Huachang Jinb,
Raúl Muñoz Torred,
Zhao Huange,
Jianmeng Chenf and
Dongzhi Chen *ac
*ac
aZhejiang Key Laboratory of Pollution Control for Port-Petrochemical Industry, Zhejiang Ocean University, Zhoushan 316022, China. E-mail: chendz@zjou.edu.cn
bNational and Local Joint Engineering Research Center, College of Life and Environmental Science, Wenzhou University, Wenzhou 325035, China
cNational & Local Joint Engineering Research Center of Harbor Oil & Gas Storage and Transportation Technology, Zhejiang Ocean University, Zhoushan 316022, China
dInstitute of Sustainable Processes, University of Valladolid, Valladolid, 47011, Spain
eChina National Offshore Oil Corporation Limited Shanghai Branch, Shanghai, 200051, China
fCollege of Environmental and Resources Science, Zhejiang University of Science & Technology, Hangzhou, 310032, China
First published on 8th July 2025
Abstract
Electrocatalytic oxidation is a promising technique for antibiotic-contaminated wastewater treatment; however, the concomitant production of toxic chlorinated byproducts remains an obstacle. Herein, a dual-functional single-atom heterojunction, Rh/Mn-SAH, was tailored to create an alternating oxidation–reduction environment within an identical electrode for current pulsation. Importantly, an interlayered Rhn+–O–Mnn+ pseudocapacitive electronic bridge was constructed, which reversibly electro-triggered the formation of metastable high-valent and low-valent metal species during anodic and cathodic cycles, respectively, with an increase in their steady-state concentration by over 2–3 orders of magnitude, thereby inducing alternate antibiotic degradation and byproduct elimination. Consequently, in anodic cycles, a normalized tetracycline degradation kinetic constant of 8.1 × 10−7 m s−1 was achieved, superior to those of the reported state-of-the-art electrodes, and tetracycline degradation shifted from the reactive chlorine species (RCS)-dominated pathway toward the RCS-free pathway along with alleviated byproduct generation, while in cathodic cycles, byproducts could be in situ eliminated by up to 40 times.
1. Introduction
Antibiotics, characterized by easy enrichment, poor biodegradability, and selective pressure on bacteria, have gradually become popular as a typical class of emerging contaminants.1 With the increasing discharge of antibiotic-containing wastewater from the medical, livestock, and aquatic industries, various antibiotics, such as tetracyclines, sulfonamides, and quinolones, have been frequently detected in natural water.2 Recently, antibiotics have been listed in China as one of the emerging contaminants that require urgent control.3 Despite extensive efforts and elegant progress, most existing technologies, such as biodegradation, adsorption, membrane separation, chemical oxidation, and wet oxidation, suffer from low efficiency, harsh conditions, and non-environmental friendliness.4–6Electrocatalytic oxidation offers significant advantages, including eco-friendliness, rapid reaction rates, and mild conditions, displaying broad prospects in antibiotic-contaminated wastewater treatment.7 Furthermore, the inherent salts (mainly NaCl) present in such wastewater not only favorably reduce ohmic resistance, thereby saving energy consumption, but also easily induce chlorine evolution, thereby producing reactive chlorine species (RCS) to enhance antibiotic degradation.8 However, one daunting challenge is that RCS can also react with contaminants via electrophilic substitution to generate organic chlorinated byproducts (OCBs), particularly chloroacetic acids and chloromethanes, or can be directly oxidized into inorganic chlorinated byproducts (ICBs), particularly ClO3− and ClO4−.9 Owing to their considerable toxicity, these chlorinated byproducts need to be minimized before discharging.
During the electrocatalytic oxidation of antibiotics, chlorinated byproducts generated at the anode will diffuse toward the cathode. There is a possibility that the chlorinated byproducts will be reduced at the cathode if some reductive species are produced at the cathode.10 Notwithstanding, owing to the electronegativity of Cl, chlorinated byproducts tend to be electrostatically attracted to the positively charged anode.11 Such an electrostatic interaction limits the negative diffusion of chlorinated byproducts. However, a certain gap always exists between the anode and the cathode. All these factors lead to the serious mass transfer limitation of chlorinated byproducts from the anode to the cathode, accordingly lowering chlorinated byproduct reduction efficiency.12
Current pulsation may provide a viable solution to overcome such a mass transfer limitation. If a pair of identical electrodes is used as both the anode and cathode, whose polarity is periodically reversed by imposing pulsating current, an alternate oxidation–reduction atmosphere can be periodically created on the same electrode. Specifically, under anodic cycles, oxidative species are generated to degrade antibiotics, while switching to cathodic cycles, reductive species are generated to in situ eliminate chlorinated byproducts. However, current pulsation has only been applied to detach deposits from the scaled cathode during electrochemical water softening.13 Utilizing current pulsation for the reduction of chlorinated byproducts has not been reported in the literature. Although the electrodes undergo current pulsation similarly in these two processes, the specific principles and electrode functionalities are essentially different. The former focuses on cathode–anode switching for scale detachment and requires the electrode to exhibit good water electrolysis, whereas the latter focuses on anode–cathode switching for chlorinated byproduct reduction and requires the electrode to possess both significant oxidation and reduction properties. This motivates us to seek a specially tailored electrode for use in current pulsation in this work.
Metal species with variable valence states, mainly including metal oxides, metal nitrides, and metal prussiates, can be ideal candidates.14 The emerging single-atom (SA) catalyst, which exhibits nearly 100% metal-atom utilization, a flexible electronic structure, and reduced cost, has recently attracted growing attention. Capitalizing on their pseudocapacitive nature, the high-valent state (Mhigh) and low-valent state (Mlow) metals can be possibly produced in anodic and cathodic cycles of current pulsation, respectively, as demonstrated below:15
| M − ne− → Mhigh | (1) |
| M + ne− → Mlow | (2) |
The produced Mhigh and Mlow may serve as oxidative species for antibiotic degradation and reductive species for the elimination of chlorinated byproducts, respectively. Nevertheless, currently available metal species are unable to generate sufficient Mhigh/Mlow. Noble metal species exhibit fast electron transfer; however, their valence state variation windows are narrow, limiting the redox potential. Transition metal species are characterized by broad valence variation windows; however, they always face sluggish electron transfer, lowering the oxidation/reduction rates.16
The incorporation of noble metal and transition metal species can combine the advantages of both and may resolve the bottleneck above. However, the direct intermixture only results in a simple average of the two components, and there is barely a synergistic effect. We prepared Ti/RhOX–TaOX by simply mixing RhOX and TaOX previously, and found that the electrochemical characteristic of Ti/RhOX–TaOX was between RhOX and TaOX.17 Consequently, the effective synergism of the advantages of noble metal and transition metal species is the key to designing an electrode favorable to producing Mhigh/Mlow. We were inspired by the concept of the multilayer heterojunction catalyst.18 Xie et al. developed a CeO2–Pt@mSiO2–Co core–shell structured catalyst, which sequentially connected water-gas shift and Fischer–Tropsch synthesis in the core and shell, respectively, significantly improving the selectivity of CO2 conversion to C2–C4 carbon hydrides.19 Thus, it is rational to conjecture that if we use noble SA (Rh-SA herein) and transition SA (Mn-SA herein) as the inner layer and the outer layer, respectively, and form a Rh/Mn-SA heterojunction (Rh/Mn-SAH), then a synergism is expected to be created between Rh-SA and Mn-SA; the presence of the inner-layered Rh-SA can accelerate the electron transfer from the outer-layer Mn-SA, and the coverage of the outer-layered Mn-SA can enhance the redox potential of the inner-layered Rh-SA, enhancing the Mhigh/Mlow production.
Actually, the treatment of antibiotic-containing wastewater under this special current pulsation scenario involves a series of sub-reactions, including the production of oxidative/reductive species, antibiotic degradation, organic polymerization, generation of chlorinated byproducts, and the in situ elimination of byproducts. The interaction between inner-layered Rh-SA and outer-layered Mn-SA will induce mutually synergistic/inhibitory effects between sub-reactions. To make things more complicated, the operational conditions of current pulsation will significantly affect both antibiotic degradation and the balance between chlorinated byproduct generation and elimination. Accordingly, a systematic study should be undertaken to achieve both satisfactory antibiotic degradation and elimination of chlorinated byproducts.
The main objectives of this study are: (1) to authenticate the interlayered Rhn+–O–Mnn+ pseudocapacitive electronic bridge between inner-layered Rh-SA and outer-layered Mn-SA; (2) to investigate tetracycline degradation and concomitant chlorinated byproduct generation pattern; (3) to disclose the Mhigh-dominated degradation mechanism; (4) to decipher the current pulsation-generated Mlow-mediated byproduct elimination.
2. Experimental section
2.1 Chemicals
Chemicals are listed in Text S1.†2.2 Preparation of the electrode
The Rh/Mn-SAH was prepared using the hydrothermal-ion exchange-pyrolysis three-step procedures, as shown in Fig. S1.†Then, single-atomic Mn or Rh-anchored MIL-125(Ti) was prepared using the ion exchange method. MIL-125(Ti) was suspended in Rh(NO3)3 (10 mg) or Mn(NO3)2 (10 mg) solution, vigorously stirred for 24 h, and then the resultant single-atomic Mn or Rh-anchored MIL-125(Ti) was centrifuged, washed with DI water, and vacuum-dried.
The aforementioned single-atom electrodes (Rh-SA, Mn-SA, and Rh/Mn-SAH), whose preparation details are provided in Section 2.2.2, were used directly as the working electrode.
2.3 Characterization
The morphology of the electrode was observed using field emission scanning electron microscopy (FE-SEM, GEMINI 300, Zeiss, Germany) and three-dimensional interference microscopy (TDIM, NT9100, Veeco, Germany). Single atomic Mn or Rh was characterized using high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM, JEM-ARM300F, JEOL, Japan). The crystal structure and composition of the electrode were identified using X-ray diffraction (XRD, D8 Advance, Bruker, Germany) and Raman spectroscopy (DXR, Thermo Fisher Scientific, USA). Detection of metal content in single-atom catalysts was performed using ICP-MS (7800, Agilent, USA). The elemental compositions and chemical states of the electrode were analyzed using X-ray photoelectron spectroscopy (XPS, K-Alpha, Thermo Fisher Scientific, USA). The coordination environments of Mn and Rh were measured by extended X-ray absorption fine structure (EXAFS) and X-ray absorption near-edge structure (XANES) (BL11B beamlines, China). The surface functional group of the electrode was analyzed by Fourier transform infrared spectroscopy (FTIR, Nexus 670, Thermo Fisher Scientific, USA). The reactive species produced by the electrode were identified using electron paramagnetic resonance (EPR, A300, Bruker, Germany).Electrochemical characterizations, including cyclic voltammetry (CV), Tafel polarization, electrochemical impedance spectroscopy (EIS), charging–discharging, and accelerated life test, were performed in an electrochemical workstation (VSP300, Biologic, France) in a standard membrane-divided three-electrode cell (Fig. S2†), in which a Pt foil and an Ag/AgCl electrode served as the counter and reference electrodes, respectively. More details can be found in Text S2.†
2.4 Antibiotic degradation and in situ chlorinated byproduct elimination
The details of the automatic control strategy for anode–cathode pulsation are presented in Text S3.† Antibiotic degradation and chlorinated byproduct in situ elimination were carried out in an undivided cell (Fig. S3†). Typical antibiotics, such as tetracycline, cephalexin, cloxacillin, levofloxacin, sulfadiazine, and cephalosporin, were selected as model pollutants. Scavenging test, wherein tert-butanol (TBA), p-benzoquinone (p-BQ), L-histidine, urea, dimethyl sulfoxide (DMSO), I2, and K2Cr2O7 served as scavengers for ˙OH, ˙O2−, 1O2, RCS, Mhigh, ˙H, and Mlow, respectively,20,21 was conducted to semi-quantify the roles of the reactive species in antibiotic degradation/chlorinated byproduct in situ elimination. Furthermore, the chemical probing test was also undertaken to quantify the relative contribution of the reactive species. Response surface methodology (RSM) was employed to optimize the antibiotic degradation and in situ elimination of chlorinated byproducts under various operational variables, including current density, pulsation frequency, and NaCl concentration. The details of the RSM are presented in Text S4.†2.5 Analysis
Chemical oxygen demand (COD) was analyzed using a COD analyzer (DRB200/DR6000, Hach, USA). Total organic carbon (TOC) and total nitrogen (TN) were analyzed by a TOC analyzer (1000e, Shimadzu, Japan). RCS and Cl− were determined by the titration method. NH4+–N, NO2−–N, and NO3−–N were determined by spectrophotometry. Antibiotics were quantified using high-performance liquid chromatography (HPLC, 1260, Agilent, USA). Aromatic compounds were identified by liquid chromatography-mass spectrometry (LC-MS, 1290-6550qtof, Agilent, USA). Carboxylic acids and ICBs (ClO3− and ClO4−) were quantified by ion chromatography (IC, Aquion RFIC, Dionex, USA). OCBs (chloroacetic acids and chloromethanes) were quantified by gas chromatography-mass spectrometry (GC-MS, GC7890B-MS5977B, Agilent, USA). Toxicity assessment of degradation intermediates was conducted using a toxicity estimation software tool. More details can be found in Text S5.†2.6 Density functional theory (DFT) calculation
The Vienna Ab initio Simulation Package was used for DFT calculations. Three models, Mn-SA, Rh-SA, and Rh/Mn-SAH, were developed using ab initio molecular dynamics. The Gibbs free energy diagrams for the oxygen evolution reaction and RCS production were determined using the nudged elastic band. The details are shown in Text S6.†3. Results and discussion
3.1 Construction of interlayered Rhn+–O–Mnn+ pseudocapacitive electronic bridge within Rh/Mn-SAH
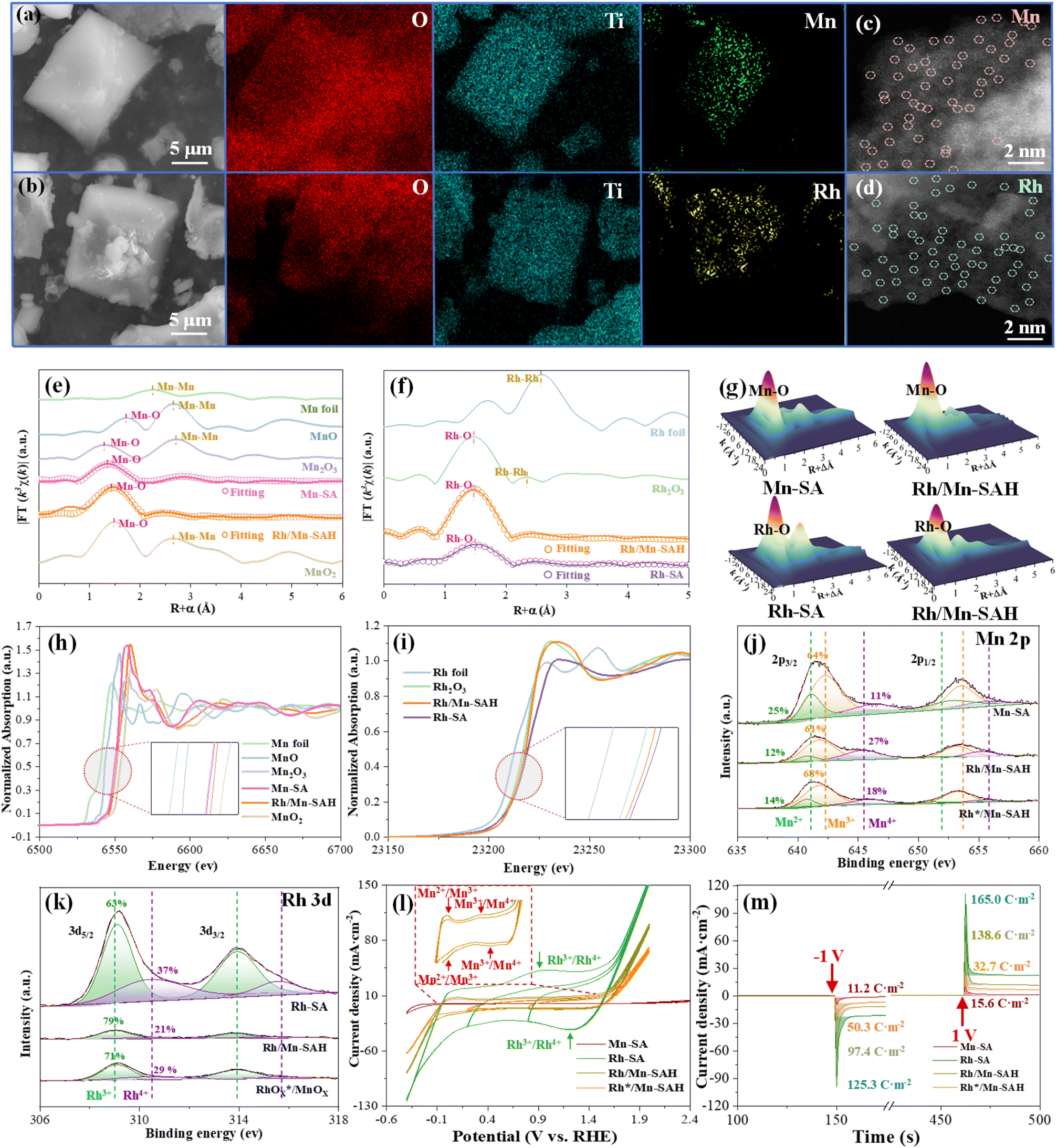 | ||
| Fig. 1 Interlayered Rhn+–O–Mnn+ pseudocapacitive electronic bridge within Rh/Mn-SAH. SEM images and corresponding elemental mapping of (a) Mn-SA and (b) Rh-SA. High-resolution HAADF-STEM images of (c) Mn-SA and (d) Rh-SA. (e) Mn K-edge FT-EXAFS spectra, (f) Rh K-edge FT-EXAFS spectra, and (g) wavelet transform analysis of Mn-SA, Rh-SA, and Rh/Mn-SAH. (h) Normalized Mn K-edge XANES spectra of Mn-SA, Rh/Mn-SAH, and standards. (i) Normalized Rh K-edge XANES spectra of Rh-SA, Rh/Mn-SAH, and standards. (j) Mn 2p XPS spectra of Mn-SA, Rh/Mn-SAH, and Rh*/Mn-SAH. (k) Rh 3d XPS spectra of Rh-SA, Rh/Mn-SAH, and Rh*/Mn-SAH. (l) Pseudocapacitive behavior of Mn-SA, Rh-SA, Rh/Mn-SAH, and Rh*/Mn-SAH from redox CV with stepwise decreased potential limits. (m) Pulsed voltage-induced charge–discharge plots of Mn-SA, Rh-SA, Rh/Mn-SAH, and Rh*/Mn-SAH. | ||
Aside from the internal pseudocapacitive improvement, the inner-layered Rh-SA might also affect the interfacial electron transfer between the electrode and electrolyte. The EIS plots indicated that the interfacial charge transfer resistance could be reduced by up to 115 times, with Rh-SA serving as the inner layer (Fig. S11b and Table S4†). CV in 10 mM [Fe(CN)6]3−/4− was performed to examine the reversibility of the interfacial electron transfer. No obvious oxidation or reduction peak was found in Mn-SA, indicating the inferior interfacial electron transfer between Mn-SA and [Fe(CN)6]3−/4−. This was comprehensive because the Mn-SA showed extremely large Rct semicircles (high-frequency region) with high interfacial charge-transfer resistance in the EIS plots (Fig. S11b†), leading to the sluggish electron transfer between Mn-SA and the interfacial [Fe(CN)6]3−/4−. However, a pair of oxidation and reduction peaks corresponding to the [Fe(CN)6]4− − e− → [Fe(CN)6]3− and reversible [Fe(CN)6]3− + e− → [Fe(CN)6]4− conversion appeared in Rh-SA and Rh/Mn-SAH, indicating their surface-sensitive electron transfer (Fig. S11c†). The separation between the oxidation and reduction peaks represented the reversibility of the [Fe(CN)6]4− ↔ [Fe(CN)6]3− conversion. An apparent reversibility decline was observed only after the loading of the inner-layered Rh-SA was halved (Rh*/Mn-SAH), which corroborated that the Rh-SA was also capable of accelerating the interfacial electron transfer.
3.2 Tetracycline degradation and concomitant chlorinated byproduct generation
Upon confirming the construction of the interlayered Rhn+–O–Mnn+ pseudocapacitive electronic bridge, we further investigated its effect on tetracycline degradation. Tetracycline can be completely removed within just 2 h by Rh/Mn-SAH, which outperformed Rh-SA and Mn-SA, even being comparable to commercial boron-doped diamond (BDD), which is generally recognized as highly efficient in pollutant degradation (Fig. 2a). The corresponding COD and TOC removal showed a similar variation pattern to that of tetracycline removal (Fig. 2b and S12†). Fig. 2c compares the performance of our Rh/Mn-SAH with other state-of-the-art electrodes available in the literature.25–36 Impressively, Rh/Mn-SAH was considerably superior to most of the existing electrodes, e.g., Ti4O7, SnOx–CeOx, Ti/α-PbO2/β-PbO2, since it possessed higher current efficiency and simultaneously lower energy consumption (Text S7†). Notably, the current efficiency and energy consumption of Rh/Mn-SAH were found to be comparable to those of BDD, which is typically considered highly efficient in pollutant degradation and thus selected as a fair comparative sample, herein, indicating its excellent catalytic efficiency. For a fair comparison, the pseudo-first-order reaction rate constants (kobs) were normalized by the electrochemical active surface areas (Text S8 and Fig. S13†), and it was found that the normalized kobs of Rh/Mn-SAH was close to that of BDD, and still exceeded those of other electrodes. | ||
| Fig. 2 Tetracycline degradation and concomitant chlorinated byproduct generation. (a) Tetracycline and (b) TOC variations during the electrocatalytic oxidation of tetracycline. (c) Pollutant removal performance comparison between Rh/Mn-SAH and other state-of-the-art electrodes reported in terms of normalized kobs, current efficiency, and energy consumption. (d) Cl−; (e) RCS; (f) chlorinated byproducts; and (g) C balance, (h) Cl balance, and (i) N balance variations during the electrocatalytic oxidation of tetracycline. Reaction conditions: initial tetracycline concentration = 320 mg L−1, initial NaCl concentration = 65 mM, current density = 20 mA cm−2. | ||
Cl−, an inherent component in pharmaceutical wastewater, was also monitored, and the Cl− content decreased with time for all electrodes, with Rh-SA showing the most rapid consumption (Fig. 2d). The consumed Cl− may be converted to various species, e.g., RCS or even chlorinated byproducts. Evidently, a prominent RCS was produced by Rh-SA, which exceeded other electrodes (Fig. 2e). Though Rh-SA produced the highest RCS, which was generally recognized as one of the oxidative species contributing to the degradation of tetracycline, the tetracycline and TOC removal efficiency was much inferior to those of Rh/Mn-SAH, as shown in Fig. 2a and b, again confirming the excellent catalytic activity of dual-atom catalysts. However, such an effective production of RCS may intensify the generation of OCB since it can easily attack contaminants via electrophilic substitution. Seven typical OCBs, including monochloroacetic acid (MCAA), dichloroacetic acid (DCAA), trichloroacetic acid (TCAA), monochloromethane (MCM), dichloromethane (DCM), trichloromethane (TCM), and perchloromethane (PCM), were quantified (Fig. S14†). As expected, due to the intensified production of RCS, the highest concentration of OCBs was observed in Rh-SA. We also quantified the generation of ICBs, including ClO3− and ClO4−, because the produced RCS was possibly deeply oxidized at the anode; however, it was found that the Rh-SA was relatively inactive in generating ICBs, while BDD exhibited the highest activity (Fig. S14†). Collectively, Rh-SA exacerbated the OCB generation while BDD exacerbated the ICB generation (Fig. 2f). This is a detrimental phenomenon because OCBs and ICBs are considered to be toxic. Stoichiometric C balance was then analyzed, and it was observed that most of the C remained as tetracycline for Mn-SA, which was in accordance with its inferior degradation performance (Fig. 2g). In sharp contrast, the proportion of tetracycline was extremely low for other electrodes. Subsequently, the proportion of aromatic compounds was calculated by subtracting tetracycline, defined as a carboxylic acid (Fig. S15†), and CO2 from the total carbon, and Rh/Mn-SAH and BDD showed a promoted ring cleavage with minimized aromatic compounds. The Cl balance validated that the OCBs (11.9%) and ICBs (18.5%) were the major byproducts for Rh-SA and BDD, respectively (Fig. 2h). Furthermore, a large proportion of undefined Cl species were found in Rh-SA, which could be attributed to the unquantified organic chlorinated intermediates, implying that the actual OCB concentrations were underestimated. Notably, Rh/Mn-SAH displayed the lowest proportions of OCBs (3.9%) and ICBs (3.8%), along with the decreased undefined Cl species, which indicated that the generation of chlorinated byproducts was alleviated to some extent. Apart from the C and Cl species, the N species contained in tetracycline could also be oxidized. As shown in Fig. S16,† the N-containing products were mainly composed of total organic N (TON), NH3+, N2, NO2−, and NO3−. Similar to the C balance, Rh/Mn-SAH and BDD showed preferable TON oxidation performance. Nonetheless, their major ultimate products were distinguished. The N2 species dominated in Rh/Mn-SAH while NO3− dominated in BDD (Fig. 2i). This implied that the N-related byproducts could be considered negligible for Rh/Mn-SAH and hence will not be considered in the subsequent byproduct in situ elimination experiment.
3.3 Mhigh-dominated degradation mechanism
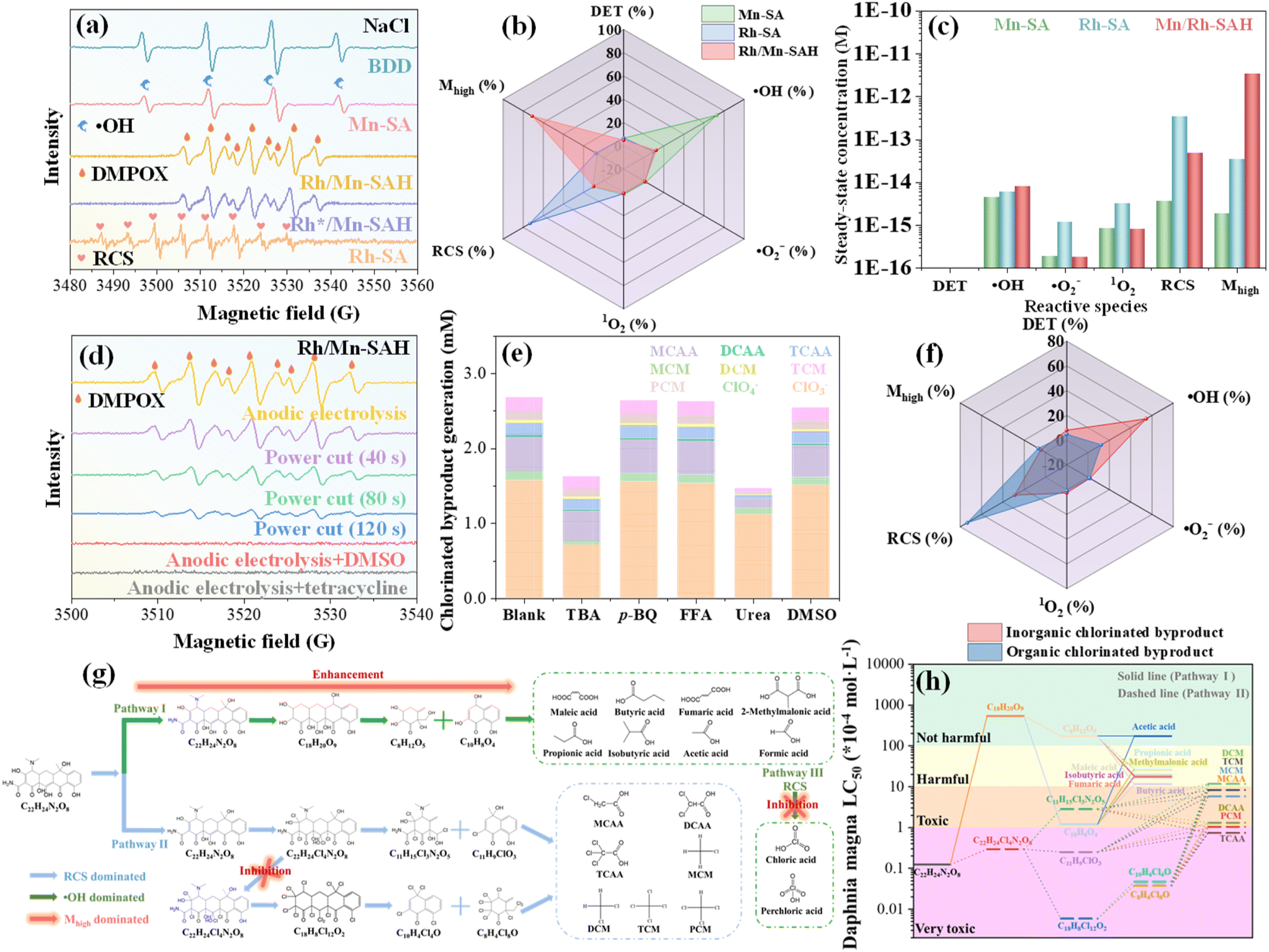 | ||
| Fig. 3 Mhigh-dominated degradation. (a) EPR spectra for different electrodes. (b) The corresponding contribution of each oxidative species to tetracycline degradation in the scavenging test. (c) Steady-state concentrations of different oxidative species during tetracycline degradation in the chemical probing test. (d) EPR spectra of DMPOX in the presence of scavengers or after stopping anodic electrolysis. (e) Chlorinated byproduct generation in the presence of various scavengers and (f) the corresponding contribution of each oxidative species to chlorinated byproduct generation. Reaction conditions: initial tetracycline concentration = 320 mg L−1, initial NaCl concentration = 60 mM, current density = 20 mA cm−2. (g) Schematic illustration of the Mhigh-dominated tetracycline degradation pathway. (h) LC50 of degradation intermediates. | ||
The aforementioned oxidative species also contributed to the generation of chlorinated byproducts, which were inhibited in the presence of certain scavengers for Rh/Mn-SAH; however, the most efficient scavenger varied depending on the types of byproducts (Fig. S20†). It is observed that the OCBs were significantly inhibited by urea, while ICBs were significantly inhibited by TBA (Fig. 3e), indicating that the RCS and ˙OH were respectively responsible for the generation of OCB and ICB; the detailed contributions were calculated to be 73.8% (RCS toward OCB) and 54.5% (˙OH toward ICB) (Fig. 3f), further explaining why Rh-SA and BDD favored the generation of OCBs and ICBs. Notably, Mhigh did not contribute significantly to either OCB or ICB generation, which explains the generation of fewer chlorinated byproducts by Rh/Mn-SAH.
The above results lead us to envision that Mhigh can alter the degradation pathway of tetracycline. As shown in Fig. S21,† due to the prevailing RCS in Rh-SA, various organic chlorinated intermediates were detected. This was comprehensive because RCS can easily attack the electronegative sites of tetracycline, as verified by the electron accumulation environment along with the high Fukui index (f−) of these atoms (Fig. S22†). In sharp contrast, as Mhigh functioned as a major oxidative species in Rh/Mn-SAH, extremely limited organic chlorinated intermediates were detected (Fig. S23†). Consequently, the emergence of strong-oxidative Mhigh beneficially enhanced chlorine-free degradation (pathway I) while circumventing the RCS-dominated electrophilic substitution (pathway II) and the ˙OH-dominated deep oxidation of RCS (pathway III), thus mitigating the generation of chlorinated byproducts, as depicted in the degradation pathway (Fig. 3g). Furthermore, the toxicity of these chlorinated byproducts/intermediates was assessed, with their lethal concentration 50 (LC50) values classifying them as toxic or very toxic, whereas the chlorine-free intermediates were classified as either harmful or barely harmful (Fig. 3h). The bioconcentration factor displayed a similar pattern with the chlorinated intermediates/byproducts possessing much higher indexes than chlorine-free intermediates (Fig. S24†). In addition, the survival of zebrafish in treated wastewater was investigated to evaluate the comprehensive toxicity, and it was observed that the treated wastewater posed almost no influence on the survival of zebrafish (Fig. S25†). Summarily, the dual-atom heterojunction beneficially triggered the evolution of high-valent metals, enhancing the mineralization of tetracycline; meanwhile, the production of RCS was alleviated, further inhibiting the generation of chlorinated byproducts.
 | ||
| Fig. 4 Theoretical exploration of metastable Mhigh. XPS spectra of Rh/Mn-SAH before electrolysis and anodic electrolysis in terms of (a) Mn 2p and (b) Rh 3d. (c) The relative proportion of Mnn+ and Rhn+ before electrolysis and anodic electrolysis for Rh/Mn-SAH. (d) Gibbs free energy and corresponding electron transfer for H2O adsorption. (e) Gibbs free energy for the oxygen evolution reaction. (f) Electron transfer for Cl− adsorption and (g) Gibbs free energy for RCS production. | ||
DFT calculations were performed, and Rh/Mn-SAH had a more negative H2O adsorption energy (−2.02 eV) than Rh-SA (−1.14 eV) and Mn-RA (−0.60 eV), which suggested that it was easier to adsorb H2O; meanwhile, Rh/Mn-SAH showed the fewest electron transfer toward surface-adsorbed H2O, which facilitated its further electrolysis (Fig. 4d). Oxygen evolution reaction activity is a crucial indicator to evaluate electrochemical oxidation ability, and the ˙OH, ˙O, ˙OOH, and O2 were formed on the catalyst surface through successive one-electron transfer steps, wherein the ˙O implied the generation of Mhigh. Importantly, the Gibbs free energy for ˙O formation decreased from 4.24 eV (Mn-SA) and 4.28 eV (Rh-SA) to 1.28 eV (Rh/Mn-SAH), which authenticated that Rh/Mn-SAH was more inclined to generate Mhigh thermodynamically (Fig. 4e). Furthermore, the rate-determining step, which was recognized as the highest energy barrier during the four steps, changed from ˙O formation in Mn-SA (2.15 eV) and Rh-SA (3.63 eV) toward ˙OOH formation (represented by Mhigh quenching) in Rh/Mn-SAH (2.64 eV), which indicated that the subsequent quenching of Mhigh was inhibited. Additionally, the Gibbs free energy for RCS formation was calculated, and it was found that Rh/Mn-SAH displayed the weakest electron extraction capability from surface-adsorbed Cl−, which indicated its inferior activation ability (Fig. 4f). Rh-SA had a tremendously negative ˙Cl generation value (−0.87 eV), which again explained why Rh-SA favored OCB generation; while Rh/Mn-SAH elevated the ˙Cl generation free energy to a positive value of 0.54 eV, which was close to that of Mn-SA (0.67 eV) (Fig. 4g). This indicated that RCS production was inhibited in Rh/Mn-SAH.
3.4 Current pulsation-generated Mlow-mediated byproduct in situ elimination
Though Rh/Mn-SAH generated fewer chlorinated byproducts than other electrodes, it would have been better if those toxic chlorinated byproducts had been eliminated. Considering the highly reversible nature of the Rhn+–O–Mnn+ electronic bridge, Rh/Mn-SAH may also possibly function as the cathode to reduce chlorinated byproducts. Therefore, Rh/Mn-SAH was subjected to cathodic electrolysis, and specific concentrations of chlorinated byproducts were dissolved in water. Both OCBs and ICBs could be reduced (Fig. S27†), which verified our hypothesis. Similar to the tetracycline degradation, chlorinated byproduct reduction may involve various reductive species, such as ˙H, Mlow, and DET. The scavenging test indicated that the inhibition degree of byproduct reduction was low on I2 (scavenger of ˙H) but predominantly high on K2Cr2O7 (scavenger of adsorbed reductive species, including ˙H and Mlow) (Fig. 5a), which implied that the Mlow played a crucial role. Further calculations proved that Mlow contributed more than 70% to the reduction of chlorinated byproducts (Fig. 5b). XPS was used to probe the transformation of Mn3+/4+ and Rh4+ into Mn2+ and Rh3+ in Rh/Mn-SAH during cathodic electrolysis, with their contents increasing from pristine 12% and 79% to 59% and 91%, respectively (Fig. 5c, d and S28†). In accordance with the anodic electrolysis results, Mn-SA showed inferior valence variation during cathodic electrolysis (Fig. S29†). | ||
| Fig. 5 Current pulsation-generated Mlow-mediated byproduct in situ elimination. (a) Chlorinated byproduct reduction performance and (b) the corresponding contribution of each reductive species in the scavenging test. XPS spectra of Rh/Mn-SAH before electrolysis and cathodic electrolysis in terms of (c) Mn 2p and (d) Rh 3d. (e) In situ elimination of chlorinated byproducts in different modes. (f) FTIR spectra of Rh/Mn-SAH in the as-prepared, without current pulsation, and with current pulsation conditions. (g) CV plots and (h) current recovery of electrodes in the as-prepared, without current pulsation, NaOH immersion, and with current pulsation conditions. Reaction conditions: initial tetracycline concentration = 320 mg L−1, initial NaCl concentration = 60 mM, current density = 20 mA cm−2, pulsation period = 20 min. | ||
This suggests that if a pair of Rh/Mn-SAH is used, chlorinated byproducts generated at the Rh/Mn-SAH anode can then be in situ reduced at another Rh/Mn-SAH cathode. However, after substituting an inert Ti cathode (entry I in Fig. 5e) with a Rh/Mn-SAH cathode (entry II in Fig. 5e), only a minor decrease was found for both OCBs and ICBs, which is not as expected. This insufficient reduction efficiency was mainly attributed to the following two factors: (1) mass transfer limitation. Before cathodic reduction, chlorinated byproducts need to diffuse from the anode toward the cathode, and the gap between the anode and the cathode usually results in mass transfer limitation. This was counter-evidenced by setting different electrode gaps, and it was found that as the electrode gap shortened from 10 to 2 mm, the amount of chlorinated byproducts gradually decreased by up to 2.7 times (entry III–IV in Fig. 5e), demonstrating that the mass transfer improvement enhanced the reduction of chlorinated byproducts. However, as the electrode gap further decreased from 2 to 1 mm (entry IV–V in Fig. 5e), the chlorinated byproducts no longer decreased but remained nearly stable, which indicated that the mass transfer improvement had reached a limit. This suggested that other factors influenced the reduction of chlorinated byproducts. (2) Anode poisoning. To verify the anode poisoning effect, FTIR analysis was conducted. Compared with the as-prepared Rh/Mn-SAH anode, a series of intensive peaks assigned to the ring-like compound and the carboxylic acid emerged in the Rh/Mn-SAH anode without current pulsation (Fig. 5f), which was ascribed to the production of the polymeric film during tetracycline degradation on the anode.23 With the gradual coverage of the polymeric film, anode poisoning occurred, as verified by the decline in current density of the Rh/Mn-SAH anode without current pulsation (Fig. 5g). However, such poisoning intensified the generation of chlorinated byproducts because it inhibited the dilution of RCS, thereby exacerbating the electrophilic substitution of RCS directly toward the polymeric film and RCS deep oxidation (Fig. S30†).
As a proof of concept, the current pulsation was adopted herein to enhance the in situ reduction efficiency. It turned out that only a trace amount of chlorinated byproducts was detected, with their concentration reduced by up to 40 times, confirming the superiority of the current pulsation (entry VI in Fig. 5e). On the one hand, current pulsation could be regarded as two electrodes with infinitely small gaps, thus overcoming the mass transfer limitation between the anode and cathode. On the other hand, we surprisingly found that these polymeric film peaks nearly disappeared when the current pulsation was conducted, and the current density of Rh/Mn-SAH almost recovered to the initial value (Fig. 5f and g). This de-poisoning was attributed to the OH− and H2 produced by cathodic water electrolysis, wherein the OH− could dissolve the acidic groups of the polymeric film while the H2 was capable of stripping the polymeric film while releasing from the cathode. To distinguish the contribution of OH− and H2, the poisoned Rh/Mn-SAH was immersed into a NaOH solution, whose pH was similar to that of the cathodic vicinity (pH = 12.3), and it was found that 65% of the current density recovered, with the remaining attributed to the H2 stripping (35%) (Fig. 5h). Subsequently, RSM was conducted, and the response surfaces revealed that the tetracycline removal was positively correlated with current density and NaCl concentration, while it was negatively correlated with pulsation frequency (Fig. S31a–c†). Nonetheless, chlorinated byproducts showed an opposite trend, i.e., low current density, low NaCl concentration, and high pulsation frequency favored the generation of minimized chlorinated byproducts (Fig. S31d–f†), implying that a trade-off should be made between tetracycline removal and chlorinated byproduct minimization. To this end, empirical equations relating tetracycline removal and chlorinated byproduct minimization with the current density, NaCl concentration, and pulsation frequency were established (Text S10†). This is encouraging because by using these equations, the effect of operational parameters can be well extrapolated, thereby optimizing operational parameters to achieve both efficient removal of tetracycline and minimization of chlorinated byproducts.
3.5 Stability and universality
A good electrode should not only be efficient but also be stable. For this reason, the as-prepared electrode was subjected to the accelerated life test, wherein the service life was defined as working hours at which the voltage variation exceeded 5 V. From Fig. S32a,† the presence of inner-layered Rh-SA appeared to significantly prolong the stability of the electrode, since the service life of Mn-SA was merely around 20 h while those of Rh-SA and Rh/Mn-SAH were 1400 and 1250 h, respectively. What should be emphasized here is that a significantly longer service life could be obtained in normal applications since unexpectedly harsh conditions were applied in the accelerated life test. By determining the service life of Rh/Mn-SAH in different current densities and pulsation periods (Fig. S32b and c†), a regression model was established (Text S11†). Assuming that the current density and pulsation period are 20 mA cm−2 and 20 min−1 under normal conditions, respectively, the service life of Rh/Mn-SAH is predicted to be over 16 years. Therefore, Rh/Mn-SAH is believed to be stable enough for practical applications.The operational stability was also evaluated, and it was found that almost all tetracycline was removed during the 10 cycles, accompanied by a low and stable byproduct concentration (≤0.25 mM) (Fig. S33a and b†). The leaching contents of Mn and Rh were analyzed, and appropriate leaching was found only in the first 2 cycles, which was mainly attributed to the surficial loosely bonded metal. Thereafter, the leaching seemed to be negligible (Fig. S33c†). The total leaching contents of Rh and Mn were calculated to be less than 0.05%. Furthermore, other typical antibiotics, such as cephalexin, cloxacillin, levofloxacin, sulfadiazine, and cephalosporin, were found to be effectively degraded with an average removal efficiency higher than 90%, and the byproduct concentration was controlled within 0.35 mM under periodic current pulsation (Fig. S33d†). In addition, Rh/Mn-SAH demonstrated stable tetracycline degradation efficacy across various water matrices and degradation efficacy on different antibiotics, which again confirmed its robustness in diverse application scenarios (Fig. S33e†).
4. Conclusion
A dual-functional Rh/Mn-SAH heterojunction has been designed to achieve antibiotic degradation and in situ byproduct elimination through current pulsation. The single-atom heterojunction beneficially constructed an interlayered Rhn+–O–Mnn+ pseudocapacitive electronic bridge, electro-triggering metastable Mhigh and Mlow during anodic and cathodic cycles, respectively, with their steady-state concentrations promoted by over 2–3 orders of magnitude. This facilitated the creation of an alternating oxidation–reduction atmosphere in the same electrode, inducing the periodic elimination of antibiotic degradation byproducts. In consequence, in anodic cycles, Rh/Mn-SAH achieved a normalized tetracycline degradation kinetics constant of 8.1 × 10−7 m s−1, outcompeting those of the reported state-of-the-art electrodes; meanwhile, tetracycline degradation was shifted from the RCS-dominated pathway toward the RCS-free pathway, alleviating byproduct generation; while in cathodic cycles, byproducts could be in situ eliminated by up to 40 times. This study presents a novel protocol for electrocatalytic oxidation by introducing a variable-valent metal to achieve efficient antibiotic degradation and in situ byproduct elimination.Data availability
The data generated and analyzed during this work are available from the corresponding author upon reasonable request.Author contributions
Yang Yu conceived the idea. Yibo Lin, Binyao Wang and Yangqi E performed the experiments and analyzed the data. Qian Li, Huachang Jin, Raúl Muñoz Torre, Zhao Huang, and Jianmeng Chen provided constructive suggestions and discussion. Yang Yu wrote the manuscript. Dongzhi Chen supervised the work.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was financially supported by the Natural Science Foundation of China (No. 52200133 and 52100108) and the Exploration Program of Zhejiang Provincial Natural Science Foundation (Q23E080060).References
- L. Jin, S. You, N. Ren, B. Ding and Y. Liu, Environ. Sci. Technol., 2022, 56, 11750–11759 CrossRef CAS PubMed
.
- L. Wang, L. Rao, M. Ran, Q. Shentu, Z. Wu, W. Song, Z. Zhang, H. Li, Y. Yao, W. Lv and M. Xing, Nat. Commun., 2023, 14, 7841 CrossRef CAS PubMed
- J. Guo, Y. Wang, Y. Shang, K. Yin, Q. Li, B. Gao, Y. Li, X. Duan and X. Xu, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e1981580175 Search PubMed
- Y. Yu, H. Liu, H. Jin, J. Chen and D. Chen, Water Res., 2023, 245, 120578 CrossRef CAS PubMed
- M. Li, P. Wang, K. Zhang, H. Zhang, Y. Bao, Y. Li, S. Zhan and J. C. Crittenden, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e1989262176 Search PubMed
- Y. Kang, Z. Gu, B. Ma, W. Zhang, J. Sun, X. Huang, C. Hu, W. Choi and J. Qu, Nat. Commun., 2023, 14, 6590 CrossRef CAS PubMed
- Y. Gao, S. Liang, B. Liu, C. Jiang, C. Xu, X. Zhang, P. Liang, M. Elimelech and X. Huang, Nat. Commun., 2023, 14, 2059 Search PubMed
- M. R. Haider, W. Jiang, J. Han, A. Mahmood, R. Djellabi, H. Liu, M. B. Asif and A. Wang, Environ. Sci. Technol., 2023, 57, 18668–18679 CrossRef CAS PubMed
- M. Lin, D. M. Bulman, C. K. Remucal and B. P. Chaplin, Environ. Sci. Technol., 2020, 54, 12673–12683 CrossRef CAS PubMed
- S. Almassi, C. Ren, J. Liu and B. P. Chaplin, Environ. Sci. Technol., 2022, 56, 3267–3276 CrossRef CAS PubMed
- S. Almassi, P. R. V. Samonte, Z. Li, W. Xu and B. P. Chaplin, Environ. Sci. Technol., 2020, 54, 1982–1991 CrossRef CAS PubMed
- S. Wang, S. Pei, J. Zhang, J. Huang and S. You, Water Res., 2022, 218, 118454 Search PubMed
- Y. Yu, H. C. Jin, X. D. Jin, R. X. Yan, L. Zhang and X. M. Chen, Ind. Eng. Chem. Res., 2018, 57, 6585–6593 CrossRef CAS
- T. C. Liu, S. Z. Xiao, N. Li, J. B. Chen, X. F. Zhou, Y. J. Qian, C. H. Huang and Y. L. Zhang, Nat. Commun., 2023, 14, 2881 CrossRef CAS PubMed
- Z. Yang, Y. Cui, B. Pan and J. J. Pignatello, Environ. Sci. Technol., 2023, 57, 18918–18928 CrossRef CAS PubMed
- H. Dong, W. Yu and M. R. Hoffmann, J. Phys. Chem. C, 2021, 125, 20745–20761 CrossRef CAS
- H. C. Jin, Y. Zhang, X. J. Zhang, Y. Yu and X. M. Chen, Ind. Eng. Chem. Res., 2021, 60, 4310–4320 CrossRef CAS
- K. Liu, M. L. Yu, H. Y. Wang, J. Wang, W. P. Liu and M. R. Hoffmann, Environ. Sci. Technol., 2019, 53, 6474–6482 CrossRef CAS PubMed
- C. L. Xie, C. Chen, Y. Yu, J. Su, Y. F. Li, G. A. Somorjai and P. D. Yang, Nano Lett., 2017, 17, 3798–3802 CrossRef CAS PubMed
- Y. Yang, N. C. Ramos, J. A. Clark and H. W. Hillhouse, Water Res., 2022, 221, 118722 CrossRef CAS PubMed
- Q. Y. Wu, Z. W. Yang, Z. W. Wang and W. L. Wang, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2075044176 Search PubMed
- Y. Luo, Y. Deng, W. Mao, X. Yang, K. Zhu, J. Xu and Y. Han, J. Phys. Chem. C, 2012, 116, 20975–20981 CrossRef CAS
- Q. Li, D. Z. Chen, H. C. Jin, Q. G. Chen, Y. Yu and X. M. Chen, Chem. Eng. J., 2022, 448, 137497 Search PubMed
- D. Guo, S. Jiang, L. Jin, K. Huang, P. Lu and Y. Liu, J. Mater. Chem. A, 2022, 10, 15981–15989 RSC
- Y. Yu, H. C. Jin, Q. Li, X. J. Zhang, Y. Zhang and X. M. Chen, Sep. Purif. Technol., 2021, 263, 118395 CrossRef CAS
- H. C. Jin, X. J. Zhang, Y. Yu and X. M. Chen, Chem. Eng. J., 2022, 435, 135167 CrossRef CAS
- Q. Li, D. Z. Chen, H. C. Jin, Q. G. Chen, Y. Yu and X. M. Chen, Chem. Eng. J., 2022, 448, 137497 CrossRef CAS
- D. Z. Chen, H. Y. Liu, J. M. Chen and Y. Yu, Appl. Catal., B, 2023, 335, 122874 Search PubMed
- H. Lin, H. Peng, X. Feng, X. Li, J. Zhao, K. Yang, J. Liao, D. Cheng, X. Liu, S. Lv, J. Xu and Q. Huang, Water Res., 2021, 190, 116790 CrossRef CAS PubMed
- H. Shi, Y. Wang, C. Li, R. Pierce, S. Gao and Q. Huang, Environ. Sci. Technol., 2019, 53, 14528–14537 CrossRef CAS PubMed
- L. Wang, J. Lu, L. Li, Y. Wang and Q. Huang, Water Res., 2020, 170, 115254 CrossRef CAS PubMed
- W. Li, R. Xiao, J. Xu, H. Lin, K. Yang, W. Li, K. He, L. Tang, J. Chen, Y. Wu and S. Lv, Water Res., 2022, 216, 118287 CrossRef CAS PubMed
- Q. Chen, L. Wang, Y. Xu, P. Li, X. Wang, M. Wang and Y. Kong, Int. J. Electrochem. Sci., 2018, 13, 7301–7309 CrossRef CAS
- Y. Sun, P. Li, H. Zheng, C. Zhao, X. Xiao, Y. Xu, W. Sun, H. Wu and M. Ren, Chem. Eng. J., 2017, 308, 1233–1242 Search PubMed
- H. Jin, X. Xu, R. Liu, X. Wu, X. Chen, D. Chen, X. Zheng, M. Zhao and Y. Yu, Water Res., 2024, 252, 121229 Search PubMed
- H. Jin, X. Xu, R. Liu, X. Wu, X. Chen, X. Zheng, M. Zhao and Y. Yu, Chem. Eng. J., 2024, 483, 149195 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Texts S1–S11, Fig. S1–S33 and Tables S1–S5. See DOI: https://doi.org/10.1039/d5ta03432a |
| This journal is © The Royal Society of Chemistry 2025 |